Abstract
Understanding the interactions of organic donor and acceptor molecules in binary associates is crucial for design and control of their functions. Herein, we carried out a theoretical study on the properties of charge transfer complexes of 1,3,6-trinitro-9,10-phenanthrenequinone (PQ) with 23 aromatic π-electron donors. Density functional theory (DFT) was employed to obtain geometries, frontier orbital energy levels and amounts of charge transfer in the ground and first excited states. For the most effective donors, namely, dibenzotetrathiafulvalene, pentacene, tetrathiafulvalene, 5,10-dimethylphenazine, and tetramethyl-p-phenylenediamine, the amount of charge transfer in the ground state was shown to be 0.134−0.240 e−. Further, a novel charge transfer complex of PQ with anthracene was isolated in crystalline form and its molecular and crystal structure elucidated by single-crystal synchrotron X-ray diffraction.
1. Introduction
Organic π-π charge transfer complexes (CTCs) form a special class of binary compounds stabilized by partial electron transfer between noncovalently interacting donor (D) and acceptor (A) molecules. The degree of electron transfer in CTCs is governed by the difference between the donor ionization potential and the acceptor electron affinity which can be approximated as the difference between the donor highest occupied molecular orbital (HOMO) and the acceptor lowest unoccupied molecular orbital (LUMO) [1]. The HOMO-LUMO energy gap can be obtained from DFT calculations.
Individual CTCs may undergo self-assembly and form crystalline or supramolecular structures [2]. The properties of such assemblies depend on the stoichiometric composition of the complexes [3,4] and their polymorphism [5,6]. CTCs in crystals tend to form one of two types of molecular stacks: (a) mixed-type stacks with alternating donor and acceptor molecules {-D-A-D-A}∞ or {-D-A-D-D-A-D}∞ and (b) segregated stacks of donor and acceptor molecules {-D-D-D-}∞ ǁ {-A-A-A-}∞ [3,7].
CTCs exhibit a wide range of physical properties therefore the search for new effective electron donors, acceptors, and synthesis of new CTCs on this basis is of high relevance [8]. At the same time, quantum-chemical modeling is one of the main approaches to study structure and properties of CTCs. Computer modeling allows a large number of complexes to be examined in a short period of time to select only a few of the most promising for further experimental research [9].
There have been only a few studies on CTCs with 9,10-phenanthrenequinone nitro derivatives as acceptors [10,11,12]. In [11,12], a series of CTCs based on anthracene, phenanthrene, and 9,10-phenanthrenequinone derivatives was studied. Of all the derivatives considered, 1,3,6-trinitro-9,10-phenanthrenequinone showed the strongest acceptor properties.
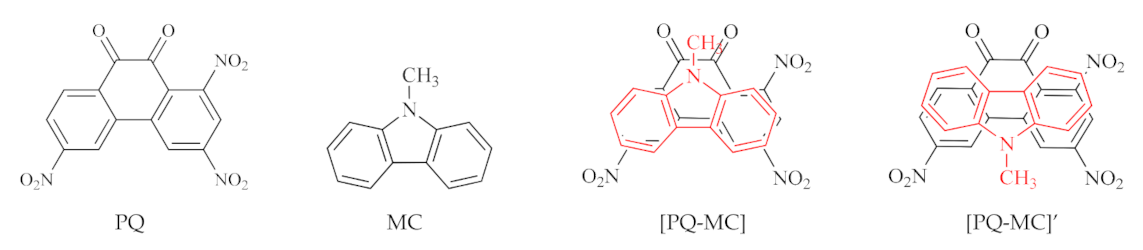
The purpose of the present work was to study CTCs based on 1,3,6-trinitro-9,10-phenanthrenequinone (PQ) as electron acceptor and different donors. We selected 23 donors with a varying number of π-electrons (from 6 to 26) and a different aromatic system structure; some of them had N and S heteroatoms and substituents. The following donors were used: benzene (BZ), pyridine (PD), N,N,N′,N′-tetramethyl-p-phenylenediamine (TMDA), naphthalene (NA), quinoline (QN), isoquinoline (IQN), acenaphthene (ACN), azulene (AZU), tetrathiafulvalene (TTF), anthracene (AN), phenanthrene (PA), acridine (ACR), 9-methylcarbazole (MC), 5,10-dimethylphenazine (DMPZ), tetracene (TET), tetraphene (TPH), chrysene (CRS), pyrene (PYR), triphenylene (TPL), dibenzotetrathiafulvalene (DBTTF), pentacene (PEN), porphyrin (POR), coronene (COR) (Figure 1 and Supplementary Material Figure S1).
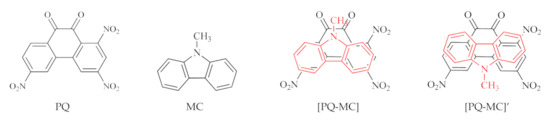
Figure 1.
Configurations of acceptor PQ, one of the donors MC, and two possible CTCs [PQ-MC] and [PQ-MC]’.
Asymmetrical molecules such as PD, QN, IQN, PA, ACR, CCN, MC, TPH, and AZU produce different stable arrangements of donor and acceptor in CTCs, therefore, the quantum-chemical calculation was performed for 32 possible models (Figure 1 and Figure S2, Table 1).

Table 1.
The calculated energies (eV) of frontier molecular orbitals of donors and CTCs (DEHOMO, CTCEHOMO, CTCELUMO), energy gaps (eV) of isolated and interacting donor and acceptor (ΔEMO, Δ CTCEMO), partial NPA charges (e−) of complex ground and first excited states (qNPA, q*NPA), and association energy (ΔEass, kJ/mol).
2. Results and Discussion
2.1. Theoretical
The formation of charge transfer complexes is controlled by the energy difference (ΔEMO) between the LUMO of the isolated acceptor (AELUMO) and the HOMO of the isolated donor (DEHOMO) [3,13]. Since AELUMO is constant for all the considered CTCs and is equal to −4.57 eV, ΔEMO depends only on DEHOMO which varies from −6.97 to −4.37 eV for selected donors. Based on the DEHOMO values we can expect an increase in donor properties in the following series: PD < BZ < QN < IQN < TPL < NA < PA < ACR < CRS < ACN < COR < PYR < MC < TPH < AN < AZU < POR < TET < DBTTF < PEN < TTF < DMPZ < TMDA (Table 1).
The most important structural features that determine electron donor properties of a molecule are the number and position of the condensed aromatic rings and also the presence of heteroatoms and functional groups. It is evident from Table 1 that donors become stronger as the number of aromatic rings grows. The ΔEMO value decreases in the series of donors with linear arrangement of rings: BZ (2.39 eV), NA (1.47 eV), AN (0.90 eV), TET (0.53 eV), PEN (0.28 eV). The same goes for ΔCTCEMO: the HOMO–LUMO energy difference in CTCs decreases from 3.10 eV to 1.37 eV for complexes of PQ with benzene and pentacene, respectively.
ΔCTCEMO values for anthracene [PQ-AN] (1.83 eV) and phenanthrene [PQ-PA] (2.43 eV)/[PQ-PA]’ (2.57 eV) complexes demonstrate that donors with a linear arrangement of aromatic rings are stronger than those with a non-linear arrangement. Similarly, ΔCTCEMO values increase when replacing tetracene [PQ-TET] (1.45 eV) to, tetraphene [PQ-TPH] (1.99 eV), pyrene [PQ-PYR] (2.00 eV), chrysene [PQ-CRS] (2.15 eV), or triphenylene [PQ-TPL] (2.37 eV). ΔCTCEMO for the pentacene complex [PQ-PEN] (1.37 eV) is lower than that for coronene [PO-COR] (1.92 eV) (Table 1).
Introduction of a nitrogen heteroatom into donor molecules leads to an increase of ΔCTCEMO values for the corresponding complexes, which points to a decrease of donor properties. The same trend for ΔCTCEMO holds true when changing BZ for PD, NA for QA/IQA, and AN for ACR (Table 1).
ΔCTCEMO values for complexes with acridine (1.88 and 1.72 eV) and azulene (1.57 and 2.10 eV) are found to be less than those of the complex with naphthalene (2.19 eV). Therefore, ACN and AZU are more active donors than NA. It is worth noting that the relative spatial arrangement of AZU and PQ molecules in CTCs (Figure S3) determines not only ΔCTCEMO values, but also the formation energies ΔEass (−63.5 and −73.3 kJ/mol) as well as the mean distance between donor and acceptor planes R (3.16 and 3.11 Å).
Substitution of benzene [PQ-BZ] for tetramethyl-p-phenylenediamine [PQ-TMDA] in the complex changes ΔCTCEMO from 3.10 to 1.29 eV. When TTF is replaced with DBTTF ΔCTCEMO of the corresponding complexes increases from 1.45 to 1.53 eV. The strongest electron donor in the series considered in this work is DMPZ. ΔCTCEMO of [PQ-DMPZ] complex has the lowest value of 1.04 eV.
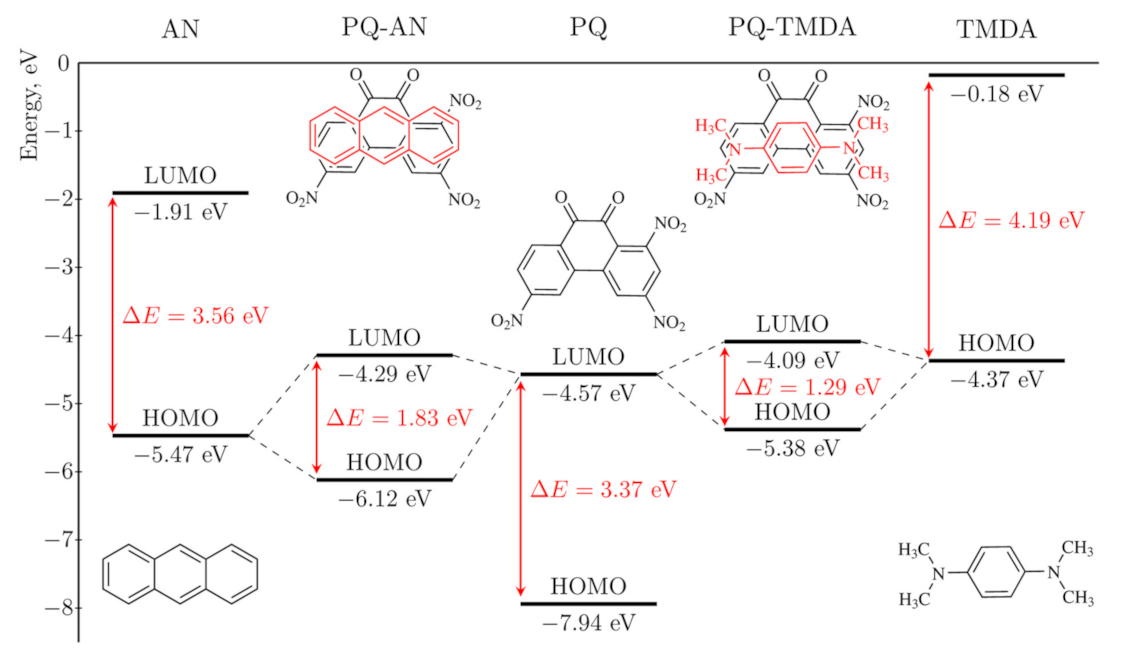
Donor and acceptor orbitals constitute the HOMO and LUMO in complexes (Figure S4). However, upon complexation their energy levels change: CTCEHOMO lies below DEHOMO, while CTCELUMO is higher than AELUMO (Figure 2). For the CTCs having the highest degree of charge transfer in the series, namely, [PQ-TTF] and [PQ-TMDA], the magnitude of these changes reaches 1.50 eV and 1.49 eV, respectively. As a result, ΔCTCEMO values are significantly larger than the corresponding ∆EMO but less than the HOMO-LUMO gaps of isolated PQ or the donors. Figure 2 illustrates this difference for CTCs of PQ with AN and TMDA: ΔCTCEMO values (1.83 and 1.29 eV) are smaller than the HOMO-LUMO gaps of PQ (3.37 eV), AN (3.56 eV) and TMDA (4.19 eV). The energy difference values ΔEMO of isolated acceptor and donors in the series decrease from 2.40 to −0.20 eV (Table 1). Notably, DEHOMO of the most pronounced donors, e.g., TTF, TMPZ, and TMDA, lies higher than AELUMO as graphically exemplified in Figure 2.
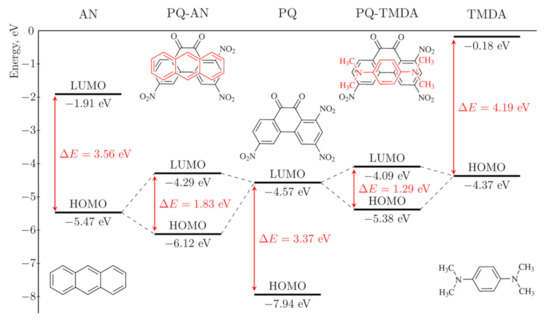
Figure 2.
HOMO-LUMO energy levels for complexes of PQ with AN and TMDA according to DFT calculations.
The calculated wavelengths of electronic transitions based on the ∆CTCEMO values vary from 468 nm for the [PQ-QN]’ complex, to 509 and 481 nm for the [PQ-PA] and [PQ-PA]’ complexes, to 676 nm for the [PQ-AN] complex and further to 959 and 1186 nm for the [PQ-TMDA] and [PQ-DMPZ] complexes, respectively. Two sets of experimentally determined CT absorption bands for complexes [PQ-PA] and [PQ-AN] measured in CH2Cl2 and toluene are available in the literature. Solutions of complex with phenanthrene absorb at 490 and 495 nm [12], while [PQ-AN] spectra show peaks at 658 and 641 nm for dichloromethane and toluene, respectively [11]. The presence of the charge-transfer bands in the absorption spectra of the complexes is a reliable confirmation of the CTC formation. For a series of CTCs based on the same acceptor, the shift of charge-transfer bands to long wavelengths indicates an increase in donor power. According to this criterion, donor properties increase in the following sequence of molecules: QN, IQN, PA, TPL, NA, ACR, CRS, PYR, TPH, MC, COR, ACN, AZU, AN, POR, DBTTF, TET, TTF, PEN, TMDA, DMPZ.
The amounts of ground state charge transfer in CTCs qNPA, calculated as the sum of NPA charges on donor atoms in CTC, is in the range from −0.004 to 0.249 e−. Assuming alternating molecular stacking in crystals, these CTCs can be classified as neutral and mixed-valence CT solids [14]. Even a small charge transfer amount of 0.2 is enough for materials to exhibit conducting properties and neutral ionic phase transition in the mixed valent state [14]. For the first excited states the amounts of charge transfer q*NPA lie between 0.967 and 1.089 e− (Table 1). This indicates that the electron transitions upon excitation in the CTCs are mainly associated with the transfer of electron density from the donor to the acceptor atoms.
Absolute values of the calculated association energies ∆Eass increase as the π-conjugated system grows. In the series BZ, NA, AN, TET, and PEN ∆Eass values are −47.0, −63.5, −82.3, −106.1, and −118.0 kJ/mol, respectively. The stability of CTCs with TTF, TMDA, DMPZ, POR, and DBTTF is determined not by the size of molecule, but by the presence of heteroatoms (Table 1).
The calculated intermolecular separation distances (R) in CTCs lie in the range from 2.87 to 3.25 Å. The complexes with the lowest distance values exhibit substantial deviation from a planar structure (Figure S5). The calculated R for [PQ-AN] complex (3.24 Å) agrees with the interplanar distance of the X-ray structure (3.49 ± 0.26 Å). The bond lengths of [PQ-AN] predicted by DFT are close to those of both calculated structures of isolated PQ and AN and the X-ray structure (Table 2).
Table 2.
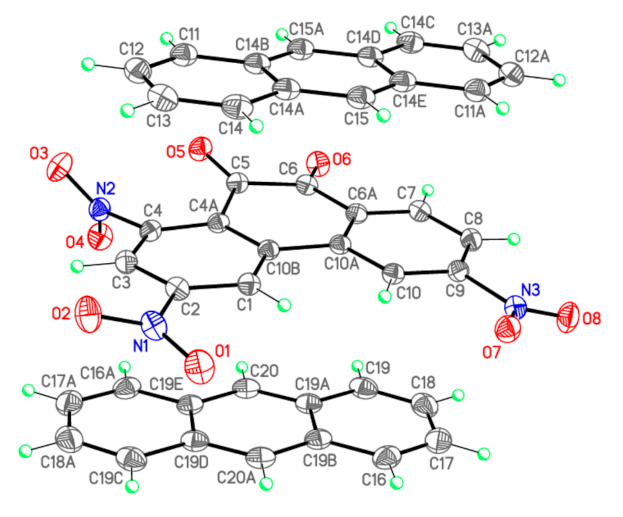
Bond lengths d (Å) and valence angles ω (deg.) of complex I (X-ray diffraction data), complex [PQ-AN] and isolated PQ and AN molecules (DFT calculations). Atom numbering scheme is given in Figure 3.
Table S1 demonstrates the NPA charges on atoms of complex [PA-AN] and isolated molecules PQ and AN in both ground and first excited states. In the ground state of [PQ-AN] complex the amount of charge transferred from AN to PQ equals 0.046 e−. Interestingly, this value hides the fact that AN carbon atoms gain −0.062 e− while hydrogen atoms lose 0.111 e− when the complex forms. At the same time oxygen and nitrogen atoms are the acceptor centers of PQ.
Upon excitation the charge on the donor increases to 0.990 e− indicating the formation of [D+-A−] complex. This is accompanied by the electron density transfer from AN carbon atoms to PQ oxygen and nitrogen atoms (0.369 e−) and C-atoms (0.624 e−).
2.2. Experimental
Dark green single prism-shaped crystals of [PQ-AN] complex (I) were grown by slow evaporation from equimolar solution of PQ and AN in CH2Cl2. The X-ray diffraction study confirmed the 1:1 ratio of PQ and AN in complex I and revealed the monoclinic structure (space group P21/c).
The molecular structure of PQ was determined for the first time, although in a complex with AN. It is interesting to discuss and compare the main geometric features of PQ and 2,4,7-trinitro-9,10-phenathrenequinone (TNPQ), especially in complexes with AN (II [11]) and PA (III [12]).
The C=O bond lengths in I (1.2103(13) and 1.2135(13) Å, Table 2, Figure 3) do not differ from those in II (1.211(2) и 1.217(2) Å) [11] and III (1.211(2) и 1.216(2) Å) [12]. The bond C5–C6 in I (1.5379(15) Å) is significantly longer than the standard single bond of C(sp2)–C(sp2) type (1.479 Å) [15]. This deviation is determined by anti-bonding interactions of oxygen atoms in o-quinones as was shown earlier in II (1.528(2) Å) [11] and III (1.526(2) Å) [12]. Valence angles of C5 and C6 in I have values close to those of II and III. In complex I atoms O5 и O6 are found to deviate notably from the plane of the central ring of PQ (torsion angle O5–C5–C6–O6 is 15.54(16)°), which is different from TNPQ where atoms O5 and O6 lie on the plane of the central ring (0.4(2)° for II [11] and 0.8(2)° for III [12]).
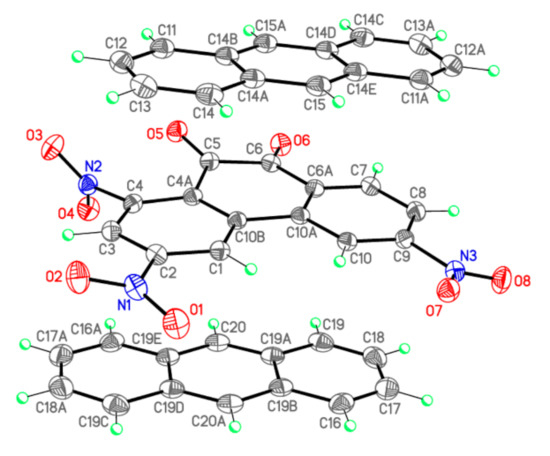
Figure 3.
X-ray crystallographic structure of PQ and AN in complex I.
The main structural features of NO2 groups in I (Table 2), II [11], and III [12] differ only slightly and are close to the average values [16]: the N–O bond lengths in I are in the range from 1.2198(13) to 1.2289(13) Å and the O–N–O valence angles are from 123.79(10)° to 125.46(10)°. The nitro groups in structure I at atoms C2, C4, and C9 are rotated out of the aromatic plane by 16.84(13), 62.75(13) and 13.56(12)°, respectively, which significantly distinguishes them from similar values at atoms C1, C3, and C8 in II (69.02(19), 0.25(18), and 19.93(18)°) [11] and III (73.8(2), 1.35(15), and 0.95(15)°) [12]. Unlike structures II and III, where the greatest rotation angle was observed in the NO2 group at the C1 atom, experiencing significant steric repulsion from atoms C10 and H10, in structure I steric difficulties arise between the nitro group at the atom C4 and the carbonyl group O5–C5, which causes a ~63° rotation of the NO2 group and a significant non-planarity of the carbonyl. It should be noted that the C–N bonds near the heavily rotated nitro groups are somewhat elongated relative to other similar bonds: N2-C4 1.4791(13) Å in I (Table 2), N1-C1 1.480(2) Å in II [11], and N1–C1 1.483(2) Å in III [12].
The AN molecule in complex I has C-C bond lengths in the range from 1.359(2) to 1.4400(15) Å and valence angles between 118.32(12)–122.62(11)°, which match with the corresponding values in II (bonds from 1.360(3) to 1.443(2) Å and angles within 118.38(14)−122.50(15)° [11]), in the free AN molecule [15] and in its CTC with 2,3,5,6-tetrachloro-1,4-dicyanobenzene [17].
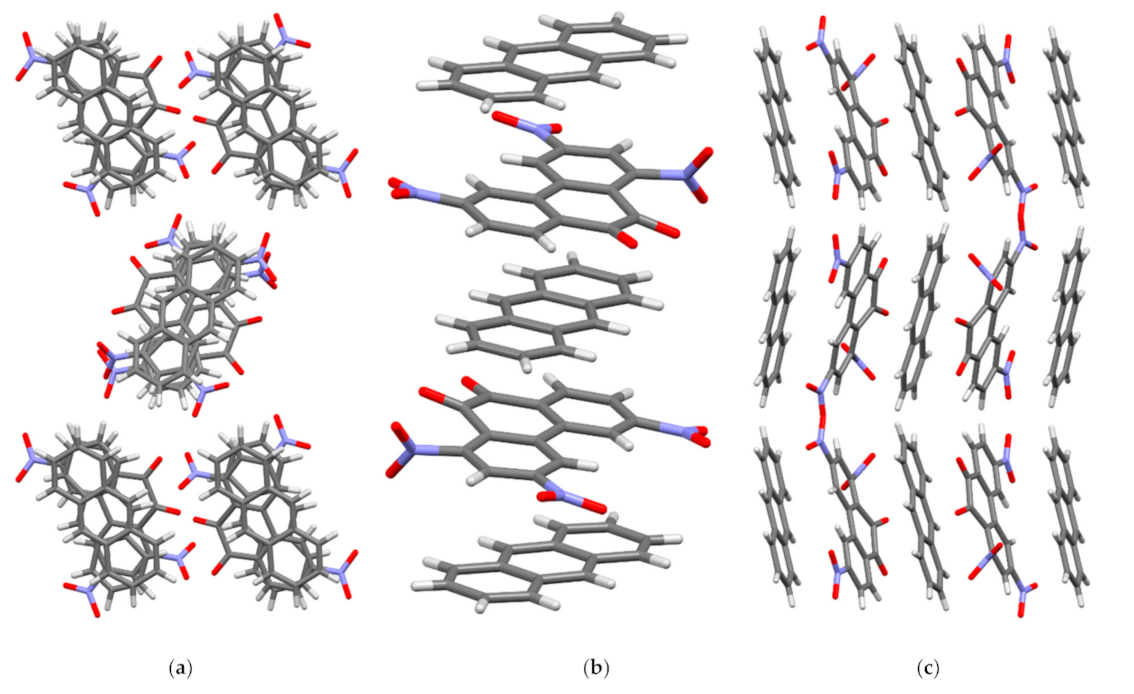
In crystal I, the molecules of the acceptor PQ and the donor AN are arranged parallel to each other and form stacks of mixed type {⋅⋅⋅[A-D]⋅⋅⋅[A-D]’⋅⋅⋅}∞ along the crystallographic axis a (Figure 4). Every second PQ molecule in the stack is rotated in a plane by 180° relative to the previous one (A and A’), which was observed for TNPQ molecules in II [11]. The AN molecules in I are displaced relative to each other (D and D’) only slightly and their central ring practically overlaps, which distinguishes them significantly from II, where the AN molecules are rotated by 60° relative to each other [11]. As a result of this mutual arrangement of the molecules PQ and AN in I, peculiar triads [D-A]⋅⋅⋅[D-]’ and [D-A]’⋅⋅⋅[D-] are formed, in which the π systems of the donor and the acceptor overlap almost to the same degree (Figure 4).
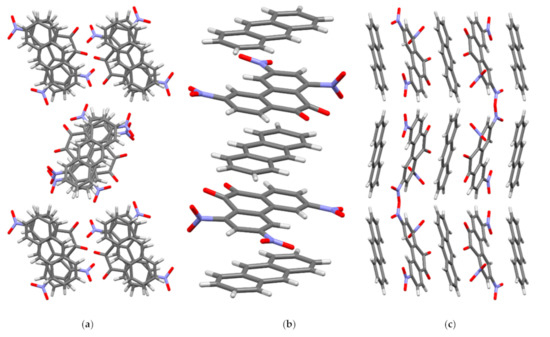
Figure 4.
Crystal structure of complex I. (a) View along crystallographic a axis; (b) view along crystallographic b axis; (c) view along crystallographic c axis.
In I, the PQ and AN molecules form two types of shortened contacts which are less than the sum of the van der Waals radii (Table S2). [A-D] and, [D-A]’ contacts are found in the same stack while A⋅⋅⋅D and A⋅⋅⋅A’ contacts are between the adjacent stacks. In one stack, each acceptor molecule establishes six C⋅⋅⋅C contacts with molecules D and D’ in the range from 3.263(2) to 3.363(2) Å, which may indicate strong π-π interactions between the molecules. The molecules D and D’ form a different number of shortened C⋅⋅⋅C contacts in the stack: each D molecule has four C⋅⋅⋅C contacts with acceptor molecules A and A’, while each D’ has only two such contacts.
The average interplanar distances D⋅⋅⋅A in I are about 3.434(5) Å, which is close to those in CTCs of AN with 2,3,5,6-tetrachloro-1,4-dicyanobenzene (3.427(3) Å) [17] or with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (3.379(2) Å) [18]. The calculated charge transfer values for the CTCs [PQ-AN] (0.046 e−) (Table 1) and [TNPQ-AN] (0.091 e−) [11] are consistent with the interplanar distances.
Each PQ molecule in I interacts with AN and PQ molecules from adjacent stacks via O⋅⋅⋅H-C shortened contacts in the range of 2.45−2.64 Å. There are also O⋅⋅⋅C contacts between the PQ molecules from adjacent stacks—from 2.921(2) to 3.097(2) Å (Table S2). The presence of such a significant number of various intermolecular interactions in I, the number of which for each PQ molecule reaches twenty-five, and the observed geometric characteristics of PQ can be due to its high acceptor capacity.
3. Materials and Methods
3.1. Synthesis
1,3,6-trinitro-9,10-phenathrenequinone (PQ, melting point 261−263 °C) was obtained by nitration and subsequent decomposition of 9,10-sulfonyldioxyphenanthrene in concentrated nitric acid (d = 1.51) [19]. Pure-grade anthracene was used without additional purification. The solvent, namely, pure-grade CH2Cl2 was purified by standard methods. To obtain CTC in the crystalline state, the solutions of acceptor (PQ, 0.2 mmol in 12 mL of CH2Cl2) and donor (AN, 0.2 mmol in 5 mL of CH2Cl2) were mixed in equimolar amounts. Single crystals of the [PQ-AN] complex suitable for the X-ray diffraction studies were grown by slow evaporation of the solvent.
3.2. X-ray Crystallography and Structure Refinement
The X-ray diffraction study of [PQ-AN] complex was carried out at the “BELOK” beamline of the Kurchatov Institute Synchrotron Radiation Source. The parameters of the unit cell and the reflection intensities were measured using a Rayonix SX165 CCD two coordinate detector (λ = 0.79272 Å, φ-scanning in 1.0° steps) (Rayonix LLC, 1880 Oak Ave UNIT 120, Evanston, IL 60201, USA). The structure was solved by direct methods and refined by the full-matrix least squares technique on F2 with anisotropic displacement parameters for all non-hydrogen atoms using the iMOSFLM (CCP4) [20], SCALA [21], and SHELXL [22] programs. All hydrogen atoms were placed in calculated positions and included in the refinement within the riding model with fixed isotropic displacement parameters Uiso(H) = 1.5Ueq(O), 1.2Ueq(N), and 1.2Ueq(C). The crystallographic data as well as the experimental and refinement parameters are summarized in Table 3. Crystallographic data is available online at the Cambridge Crystallographic Data Centre (CCDC 2099997).
Table 3.
Summary of crystallographic experiment data and structure refinement parameters for I.
3.3. Quantum Chemical Calculations
Quantum chemical simulation of the electronic structure of donor, acceptor, and CTC molecules was performed in the framework of the density functional theory using the B3LYP hybrid functional and the def2-SV(P) basis set. TDDFT methodology was used to explore the low-lying excited states. The Boys–Bernardi method was used for BSSE correction [23]. All D4 dispersion correction was used in all calculations [24]. The amount of charge transfer from a donor to an acceptor was calculated using the natural populations analysis (NPA) [25] as the difference between the sum of charges on the acceptor atoms in free state and in complex for both the ground (ΔqNPA, e−) and first excited (Δq*NPA, e−) states. The CTC association energies are defined as follows:
where CTCEtot, AEtot and DEtot are total energies (in kJ/mol) of the CTC, acceptor, and donor, respectively. All the calculations were performed using the Firefly 8.20 software package [26].
ΔEass = CTCEtot − AEtot − DEtot
4. Conclusions
In this study we explored a series of 23 charge transfer complexes based on 1,3,6-trinitro-9,10-phenanthrenequinone and different electron donors by means of density functional theory. Complexes with dibenzotetrathiafulvalene, pentacene, tetrathiafulvalene, 5,10-dimethylphenazine, and tetramethyl-p-phenylenediamine were shown to be in a mixed-valence state with a ground state charge transfer degree of 0.134–0.240 e−. A charge transfer complex with anthracene was synthesized, isolated as a single crystal, and the structure determined by X-ray diffraction experiment. Geometric and electronic structure features and their influence on the charge transfer properties of the complexes are discussed.
Supplementary Materials
The following are available online. Figure S1: Chemical structure depiction of PQ and 23 donors used in this work, Figure S2: Configurations of charge transfer complexes considered in this work, Figure S3: DFT optimized molecular structure of complexes: (a) [PQ-AZU] and (b) [PQ-AZu]’, Figure S4: Electron density corresponding to (a) HOMO and (b) LUMO of [PQ-AN] complex, Figure S5: DFT optimized molecular structure of complexes: (a) [PQ-DBTTF] (R = 2.87 Å) and (b) [PQ-DMPZ] (R = 3.02 Å), Table S1: Calculated NPA partial charges on atoms of complex [PQ-AN] in ground and first ex-cited ([PQ-AN]*) states and of isolated PQ and AN molecules, Table S2: Selected shortened contacts d (Å) between PQ and AN in complex I.
Author Contributions
Conceptualization, R.L.; methodology, R.L., M.R., V.D., and P.S.; validation, R.L., M.R., and P.S.; formal analysis, M.R.; investigation, R.L., M.R., P.S., P.D., and V.K.; writing—original draft preparation, R.L., M.R. and P.S. writing—review and editing, R.L., M.R., and P.S.; visualization, R.L., M.R., and P.S. supervision, R.L.; project administration, R.L.; funding acquisition, R.L. All authors have read and agreed to the published version of the manuscript.
Funding
This paper was supported by the RUDN University Strategic Academic Leadership Program.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Abbreviations
| ΔEMO | the energy difference between AELUMO and DEHOMO |
| DEHOMO | energy level of the HOMO of isolated donor molecule |
| AELUMO | energy level of the LUMO of isolated acceptor molecule |
| ΔCTCEMO | the energy difference between CTCELUMO and CTCEHOMO |
| CTCEHOMO | energy level of the HOMO of complex |
| CTCELUMO | energy level of the LUMO of complex |
| qNPA | partial NPA charges in the ground state |
| q*NPA | partial NPA charges in the first excited state |
| ΔEass | energy of association of donor and acceptor |
| A | acceptor |
| ACN | acenaphthene |
| ACR | acridine |
| AN | anthracene |
| AZU | azulene |
| BZ | benzene |
| COR | coronene |
| CRS | chrysene |
| CTCs | charge transfer complexes |
| D | donor |
| DBTTF | dibenzotetrathiafulvalene |
| DFT | density functional theory |
| DMPZ | 5,10-dimethylphenazine |
| HOMO | highest occupied molecular orbital |
| IQN | isoquinoline |
| LUMO | lowest unoccupied molecular orbital |
| MC | 9-methylcarbazole |
| NA | naphthalene |
| PA | phenanthrene |
| PD | pyridine |
| PEN | pentacene |
| POR | porphyrin |
| PQ | 1,3,6-trinitro-9,10-phenanthrenequinone |
| PYR | pyrene |
| QN | quinoline |
| TET | tetracene |
| TMDA | N,N,N’,N’-tetramethyl-p-phenylenediamine |
| TPH | tetraphene |
| TPL | triphenylene |
| TTF | tetrathiafulvalene |
References
- Pavelyev, V.G.; Parashchuk, O.D.; Krompiec, M.; Orekhova, T.V.; Perepichka, I.F.; Van Loosdrecht, P.H.M.; Paraschuk, D.Y.; Pshenichnikov, M.S. Charge transfer dynamics in donor-acceptor complexes between a conjugated polymer and fluorene acceptors. J. Phys. Chem. C 2014, 118, 30291–30301. [Google Scholar] [CrossRef] [Green Version]
- Al Kobaisi, M.; Bhosale, R.S.; El-Khouly, M.E.; La, D.D.; Padghan, S.D.; Bhosale, S.V.; Jones, L.A.; Antolasic, F.; Fukuzumi, S.; Bhosale, S.V. The sensitivity of donor – acceptor charge transfer to molecular geometry in DAN–NDI based supramolecular flower-like self-assemblies. Sci. Rep. 2017, 7, 16501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Du, K.; Wei, F.; Jiang, H.; Kloc, C. Crystal Growth, HOMO–LUMO Engineering, and Charge Transfer Degree in Perylene-Fx TCNQ (x = 1, 2, 4) Organic Charge Transfer Binary Compounds. Cryst. Growth Des. 2016, 16, 3019–3027. [Google Scholar] [CrossRef]
- Singh, M.; Chopra, D. Diversity in Mechanical Response in Donor–Acceptor Coupled Cocrystal Stoichiomorphs Based on Pyrene and 1,8-Dinitroanthraquinone Systems. Cryst. Growth Des. 2018, 18, 6670–6680. [Google Scholar] [CrossRef]
- Averkiev, B.; Isaac, R.; Jucov, E.V.; Khrustalev, V.N.; Kloc, C.; McNeil, L.E.; Timofeeva, T.V. Evidence of Low-Temperature Phase Transition in Tetracene–Tetracyanoquinodimethane Complex. Cryst. Growth Des. 2018, 18, 4095–4102. [Google Scholar] [CrossRef]
- Henderson, J.; Masino, M.; Hatcher, L.E.; Kociok-Köhn, G.; Salzillo, T.; Brillante, A.; Raithby, P.R.; Girlando, A.; Da Como, E. New Polymorphs of Perylene:Tetracyanoquinodimethane Charge Transfer Cocrystals. Cryst. Growth Des. 2018, 18, 2003–2009. [Google Scholar] [CrossRef]
- Starodub, V.A.; Starodub, T.N. Radical anion salts and charge transfer complexes based on tetracyanoquinodimethane and other strong π-electron acceptors. Russ. Chem. Rev. 2014, 83, 391. [Google Scholar] [CrossRef]
- Wang, W.; Luo, L.; Sheng, P.; Zhang, J.; Zhang, Q. Multifunctional Features of Organic Charge-Transfer Complexes: Advances and Perspectives. Chem.-A Eur. J. 2021, 27, 464–490. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Aquino, A.J.A.; Sue, A.C.-H.; Lischka, H. Analysis of charge transfer transitions in stacked π-electron donor–acceptor complexes. Phys. Chem. Chem. Phys. 2018, 20, 26957–26967. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.K. Electron affinities of polynuclear acceptors. Dinitro- and trinitrophenanthrenequinones. J. Phys. Chem. 1967, 71, 2277–2282. [Google Scholar] [CrossRef]
- Linko, R.V.; Ryabov, M.A.; Strashnov, P.V.; Polyanskaya, N.A.; Davydov, V.V.; Dorovatovskii, P.V.; Lin’ko, I.V.; Khrustalev, V.N. Charge transfer complexes of nitro derivatives of 9,10-phenanthrenequinone with anthracene. crystal and molecular structures of the (1:1) complex of 2,4,7-trinitro- 9,10-phenanthrenequinone with anthracene. J. Struct. Chem. 2021, 62, 137–146. [Google Scholar] [CrossRef]
- Linko, R.V.; Ryabov, M.A.; Strashnov, P.V.; Polyanskaya, N.A.; Davydov, V.V.; Dorovatovskii, P.V.; Khrustalev, V.N. Quantum-Chemical Simulation of the Structure of Charge-Transfer Complexes of 9,10-Phenanthrenequinone Nitro-Derivatives with Phenanthrene. Crystal and Molecular Structure of 1:1 Complex of 2,4,7-Trinitro−9,10-phenanthrenequinone with Phenanthrene. Russ. J. Gen. Chem. 2020, 90, 1869–1877. [Google Scholar] [CrossRef]
- Winget, P.; Brédas, J.-L. Ground-State Electronic Structure in Charge-Transfer Complexes Based on Carbazole and Diarylamine Donors. J. Phys. Chem. C 2011, 115, 10823–10835. [Google Scholar] [CrossRef]
- Saito, G.; Murata, T. Mixed valency in organic charge transfer complexes. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2008, 366, 139–150. [Google Scholar] [CrossRef]
- Sinclair, V.C.; Robertson, J.M.; McL Mathieson, A. The crystal and molecular structure of anthracene. II. Structure investigation by the triple Fourier series method. Acta Crystallogr. 1950, 3, 251. [Google Scholar] [CrossRef]
- Sadova, N.I.; Vilkov, L.V. The Molecular Geometry of Nitro-compounds. Russ. Chem. Rev. 1982, 51, 87–105. [Google Scholar] [CrossRef]
- Britton, D. 2,3,5,6-Tetrachloro−1,4-dicyanobenzene–anthracene (1/1). Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, o1707–o1708. [Google Scholar] [CrossRef]
- Hu, P.; Li, H.; Li, Y.; Jiang, H.; Kloc, C. Single-crystal growth, structures, charge transfer and transport properties of anthracene-F4 TCNQ and tetracene-F4 TCNQ charge-transfer compounds. CrystEngComm 2017, 19, 618–624. [Google Scholar] [CrossRef]
- Andrievskii, A.M.; Linko, R.V.; Grachev, M.K. Synthesis and reactions of trinitro−9,10-phenanthrenequinone derivatives. Russ. J. Org. Chem. 2013, 49, 1025–1030. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 dispersion coefficient model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2004. [Google Scholar]
- Granovsky, A.A. FireFly Version 8.20. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 20 May 1997).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).