
A New Oxygen Containing Pyclen-Type Ligand as a Manganese(II) Binder for MRI and 52Mn PET Applications: Equilibrium, Kinetic, Relaxometric, Structural and Radiochemical Studies

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
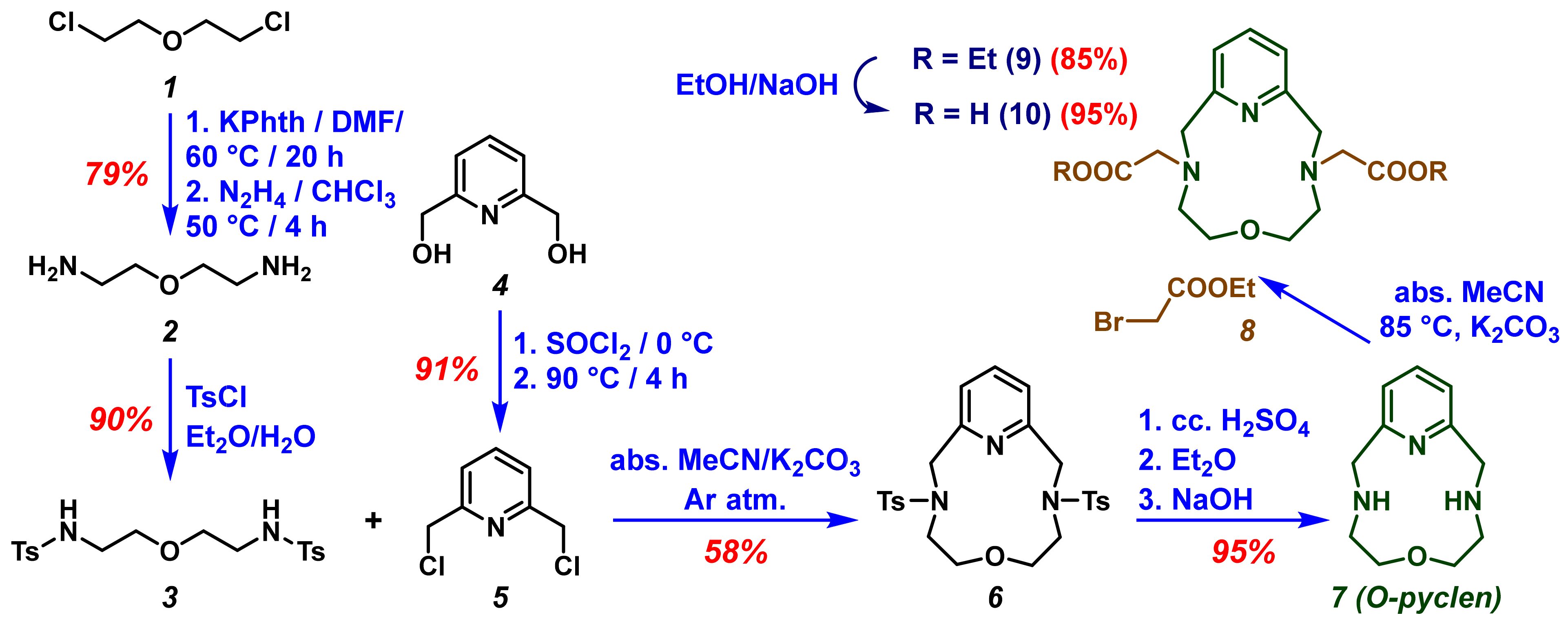
2.1. Synthesis of the Ligand
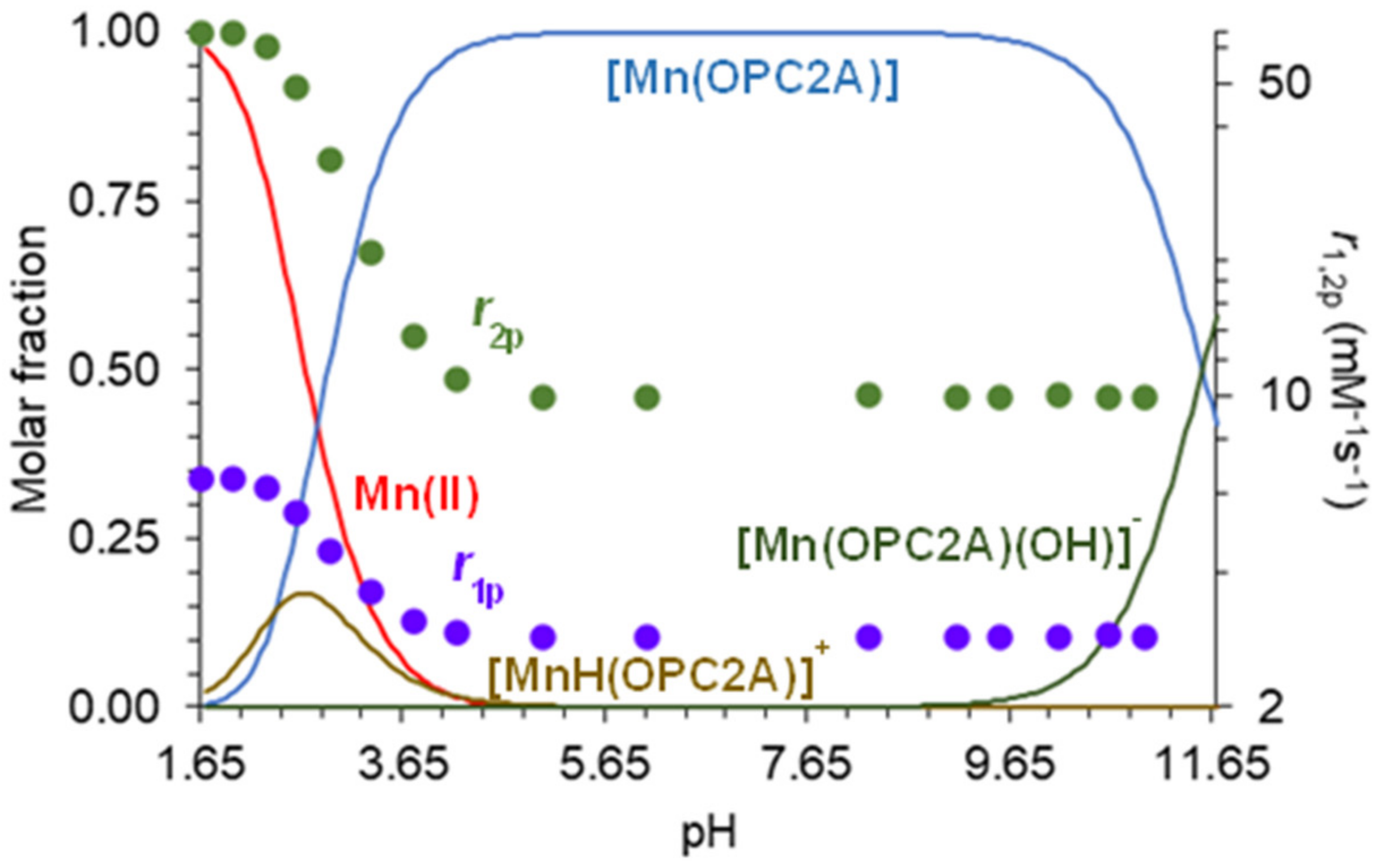
2.2. Equilibrium Studies
2.3. Relaxivity of the Mn(II) Complex
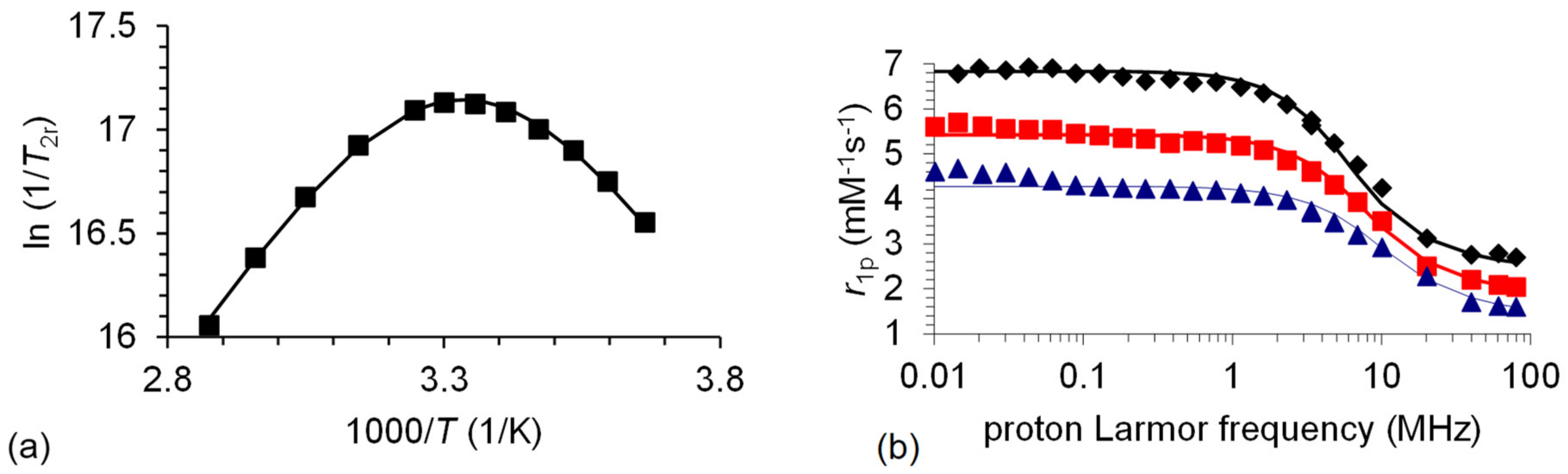
2.4. 17. O and NMRD Measurements
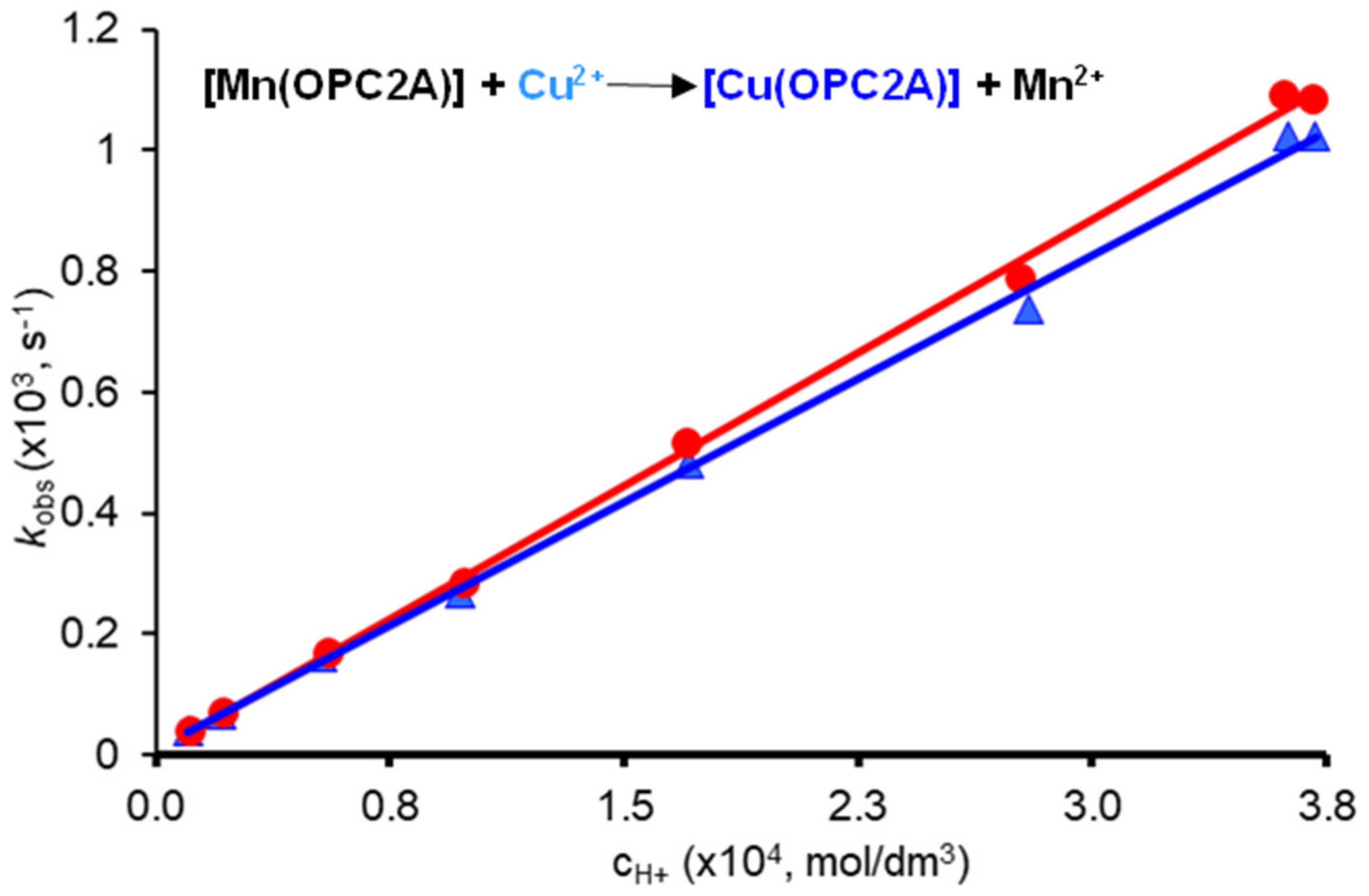
2.5. Kinetic Studies
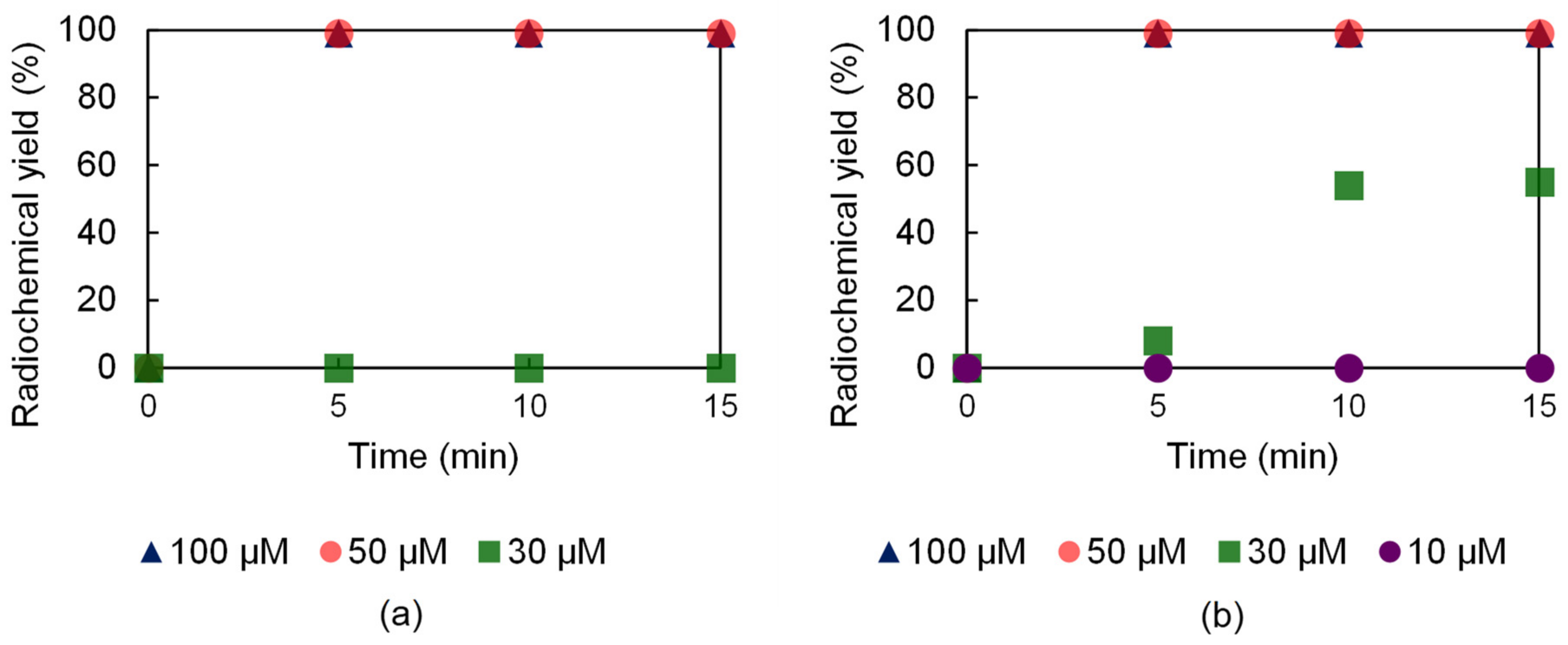
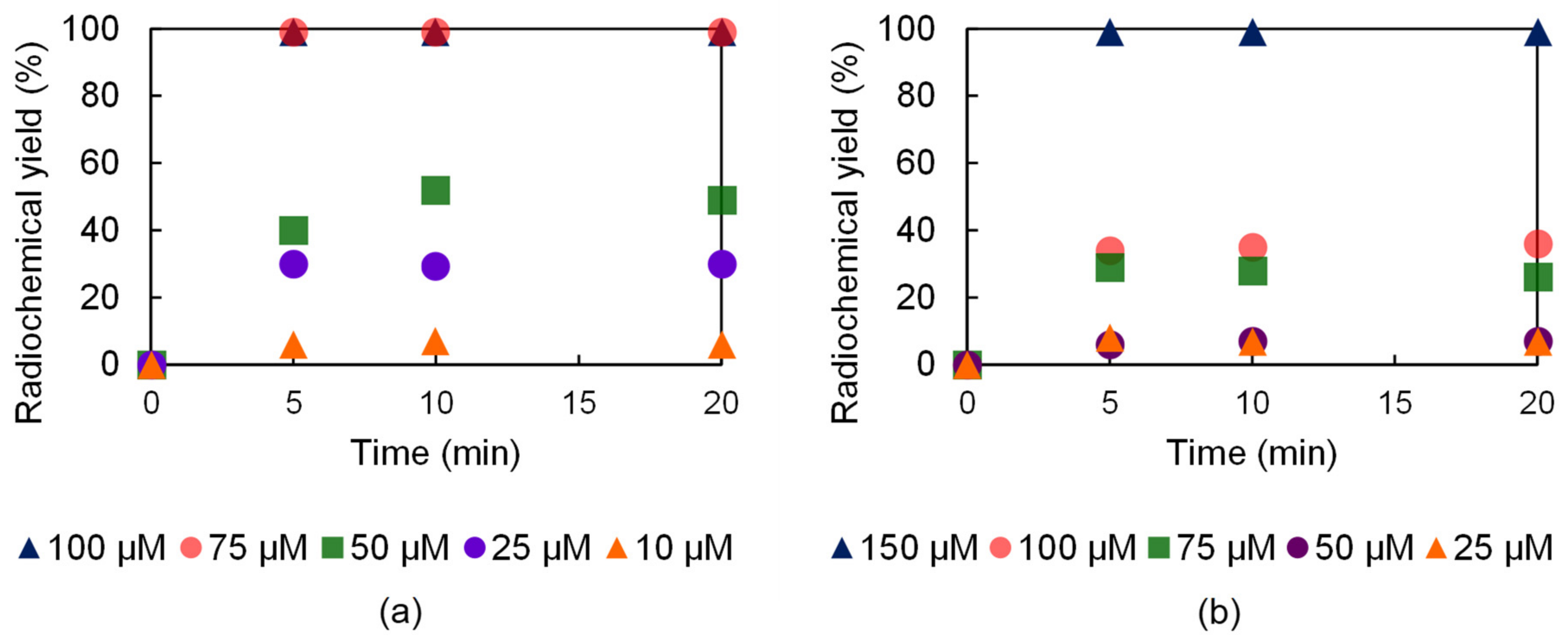
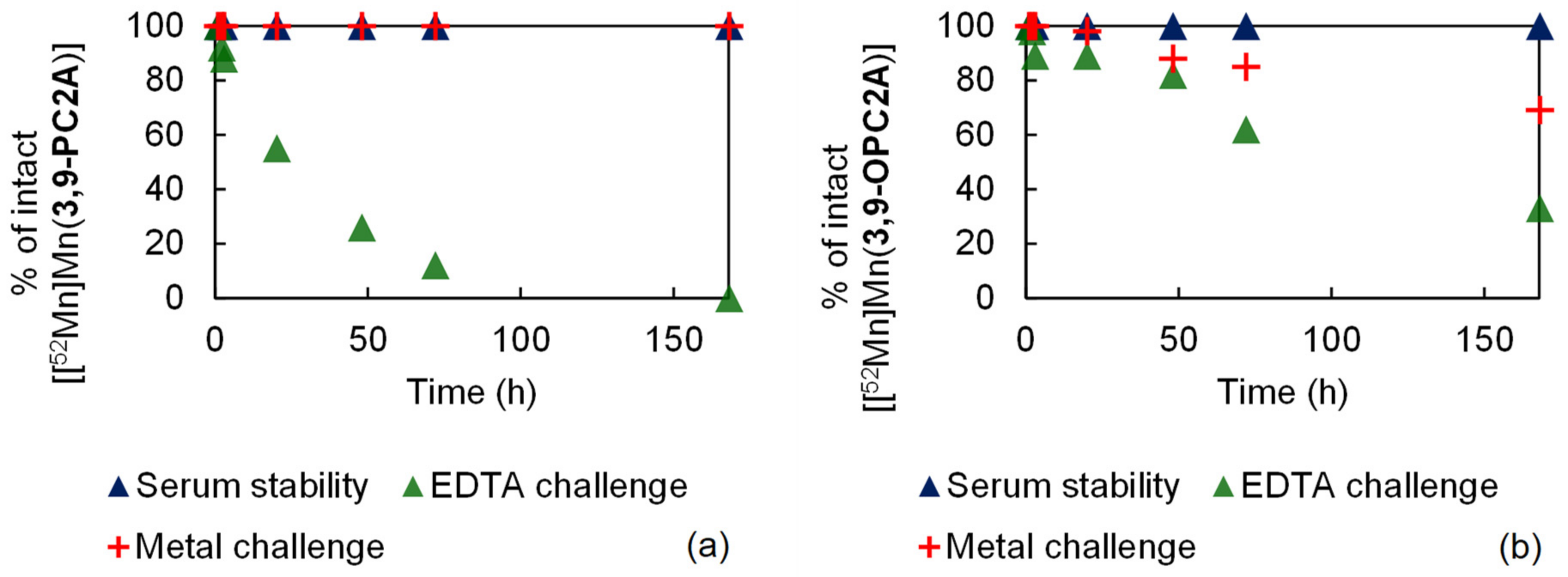
2.6. Radiochemistry
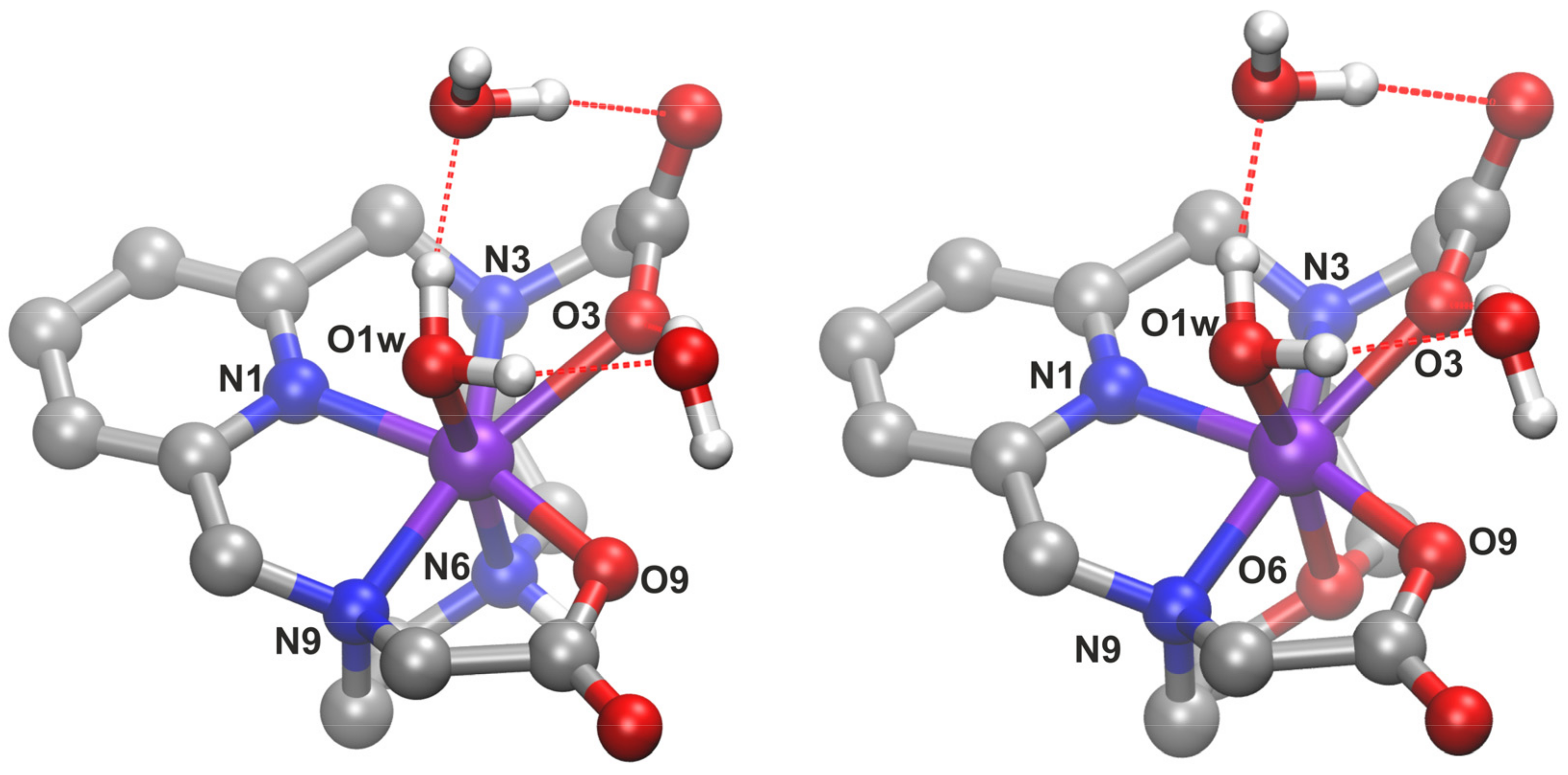
2.7. DFT Calculations
3. Summary/Conclusions
4. Materials and Methods
4.1. General Methods
4.2. Synthesis and Characterization of New Compunds
4.3. Equilibrium Studies
4.4. Relaxation Properties
4.5. Kinetic Studies
4.6. H- and 17O-NMR Relaxometry
4.7. Computational Details
4.8. Radiochemistry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
List of Abbreviations
| MRI | Magnetic Resonance Imaging |
| PET | Positron Emission Tomography |
| CAs | Contrast Agents |
| GBCAs | Gadolinium-Based Contrast Agents |
| NSF | Nephrogenic Systemic Fibrosis |
| BFCs | Bifunctional Chelators |
| EMA | European Medicines Agency |
| FDA | US Food and Drug Administration |
| HSA | Human Serum Albumin |
| DFT | Density Functional Theory |
| KPhth | Potassium Phthalimide |
| TsCl | 4-Methylbenzene-1-sulfonyl Chloride |
| DMF | N,N-Dimethylformamide |
| DIPEA | N,N-Diisopropylethylamine |
| TEA | Triethylamine |
| TFA | Trifluoroacetic Acid |
| HEPES | N-(2-Hydroxyethyl)piperazine-N′-(2-Ethanesulfonic Acid) |
| mAb | Monoclonal Antibody |
| MBO | Mayer Bond Orders |
| MBV | Mayer Bonded Valence |
| MFV | Mayer Free Valence |
References
- Grobner, T. Gadolinium—A specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol. Dial. Transplant. 2006, 21, 1104–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marckmann, P.; Skov, L.; Rossen, K.; Dupont, A.; Damholt, M.B.; Heaf, J.G.; Thomsen, H.S. Nephrogenic systemic fibrosis: Suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J. Am. Soc. Nephrol. 2006, 17, 2359–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency EMA’s final opinion confirms restrictions on use of linear gadolinium agents in body scans Recommendations conclude EMA’s Scientific Review of Gadolinium Deposition. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/gadolinium-containing-contrast-agents (accessed on 29 December 2021).
- Fraga, C.G. Relevance, essentiality and toxicity of trace elements in human health. Mol. Aspects Med. 2005, 26, 235–244. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Tircsó, G. Kinetic inertness of the Mn2+ complexes formed with AAZTA and some open-chain EDTA derivatives. Inorg. Chem. 2012, 51, 10065–10067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, E.M.; Atanasova, I.P.; Blasi, F.; Ay, I.; Caravan, P. A Manganese Alternative to Gadolinium for MRI Contrast. J. Am. Chem. Soc. 2015, 137, 15548–15557. [Google Scholar] [CrossRef] [Green Version]
- Pota, K.; Garda, Z.; Kálmán, F.K.; Barriada, J.L.; Esteban-Gómez, D.; Platas-Iglesias, C.; Tóth, I.; Brücher, E.; Tircsó, G. Taking the next step toward inert Mn2+ complexes of open-chain ligands: The case of the rigid PhDTA ligand. New J. Chem. 2018, 42, 8001–8011. [Google Scholar] [CrossRef] [Green Version]
- Vanasschen, C.; Molnár, E.; Tircsó, G.; Kálmán, F.K.; Tóth, É.; Brandt, M.; Coenen, H.H.; Neumaier, B. Novel CDTA-based, Bifunctional Chelators for Stable and Inert MnII Complexation: Synthesis and Physicochemical Characterization. Inorg. Chem. 2017, 56, 7746–7760. [Google Scholar] [CrossRef]
- Drahoš, B.; Kubíček, V.; Bonnet, C.S.; Hermann, P.; Lukeš, I.; Tóth, É. Dissociation kinetics of Mn2+ complexes of NOTA and DOTA. Dalt. Trans. 2011, 40, 1945–1951. [Google Scholar] [CrossRef]
- Garda, Z.; Molnár, E.; Kálmán, F.K.; Botár, R.; Nagy, V.; Baranyai, Z.; Brücher, E.; Kovács, Z.; Tóth, I.; Tircsó, G. Effect of the nature of donor atoms on the thermodynamic, kinetic and relaxation properties of Mn(II) complexes formed with some trisubstituted 12-membered macrocyclic ligands. Front. Chem. 2018, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Drahoš, B.; Kotek, J.; Hermann, P.; Lukeš, I.; Tóth, É. Mn2+ complexes with pyridine-containing 15-membered macrocycles: Thermodynamic, kinetic, crystallography, and1H/17O relaxation studies. Inorg. Chem. 2010, 49, 3224–3238. [Google Scholar] [CrossRef]
- Garda, Z.; Forgács, A.; Do, Q.N.; Kálmán, F.K.; Timári, S.; Baranyai, Z.; Tei, L.; Tóth, I.; Kovács, Z.; Tircsó, G. Physico-chemical properties of MnII complexes formed with cis- and trans-DO2A: Thermodynamic, electrochemical and kinetic studies. J. Inorg. Biochem. 2016, 163, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Forgács, A.; Tei, L.; Baranyai, Z.; Tóth, I.; Zékány, L.; Botta, M. A Bisamide Derivative of [Mn(1,4-DO2A)]–Solution Thermodynamic, Kinetic, and NMR Relaxometric Studies. Eur. J. Inorg. Chem. 2016, 2016, 1165–1174. [Google Scholar] [CrossRef]
- Forgács, A.; Tei, L.; Baranyai, Z.; Esteban-Gómez, D.; Platas-Iglesias, C.; Botta, M. Optimising the relaxivities of Mn2+ complexes by targeting human serum albumin (HSA). Dalt. Trans. 2017, 46, 8494–8504. [Google Scholar] [CrossRef]
- Ndiaye, D.; Sy, M.; Pallier, A.; Même, S.; de Silva, I.; Lacerda, S.; Nonat, A.M.; Charbonnière, L.J.; Tóth, É. Unprecedented Kinetic Inertness for a Mn2+-Bispidine Chelate: A Novel Structural Entry for Mn2+-Based Imaging Agents. Angew. Chem. Int. Ed. 2020, 59, 11958–11963. [Google Scholar] [CrossRef]
- Garda, Z.; Molnár, E.; Hamon, N.; Barriada, J.L.; Esteban-Gómez, D.; Váradi, B.; Nagy, V.; Pota, K.; Kálmán, F.K.; Tóth, I.; et al. Complexation of Mn(II) by Rigid Pyclen Diacetates: Equilibrium, Kinetic, Relaxometric, Density Functional Theory, and Superoxide Dismutase Activity Studies. Inorg. Chem. 2021, 60, 1133–1148. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Nagy, V.; Váradi, B.; Garda, Z.; Molnár, E.; Trencsényi, G.; Kiss, J.; Même, S.; Même, W.; Tóth, É.; et al. Mn(II)-Based MRI Contrast Agent Candidate for Vascular Imaging. J. Med. Chem. 2020, 63, 6057–6065. [Google Scholar] [CrossRef]
- Botár, R.; Molnár, E.; Trencsényi, G.; Kiss, J.; Kálmán, F.K.; Tircsó, G. Stable and Inert Mn(II)-Based and pH-Responsive Contrast Agents. J. Am. Chem. Soc. 2020, 142, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Botár, R.; Molnár, E.; Garda, Z.; Madarasi, E.; Trencsényi, G.; Kiss, J.; Kálmán, F.K.; Tircsó, G. Synthesis and characterization of a stable and inert Mn(II)-based Zn(II) responsive MRI probe for molecular imaging of glucose stimulated zinc secretion (GSZS)No Title. Inorg. Chem. Front. 2022. [Google Scholar] [CrossRef]
- Kálmán, F.K.; Nagy, V.; Uzal-Varela, R.; Pérez-Lourido, P.; Esteban-Gómez, D.; Garda, Z.; Pota, K.; Mezei, R.; Pallier, A.; Tóth, É.; et al. Expanding the ligand classes used for mn(Ii) complexation: Oxa-aza macrocycles make the difference. Molecules 2021, 26, 1524. [Google Scholar] [CrossRef] [PubMed]
- Rolla, G.A.; Platas-Iglesias, C.; Botta, M.; Tei, L.; Helm, L. 1H and 17O NMR relaxometric and computational study on macrocyclic Mn(II) complexes. Inorg. Chem. 2013, 52, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Cardinale, J.; Rausch, I.; Mindt, T.L. Manganese in PET imaging: Opportunities and challenges. J. Label. Compd. Radiopharm. 2019, 62, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Gawne, P.; Man, F.; Fonslet, J.; Radia, R.; Bordoloi, J.; Cleveland, M.; Jimenez-Royo, P.; Gabizon, A.; Blower, P.J.; Long, N.; et al. Manganese-52: Applications in cell radiolabelling and liposomal nanomedicine PET imaging using oxine (8-hydroxyquinoline) as an ionophore. Dalt. Trans. 2018, 47, 9283–9293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.; Vögtle, F. Ligandstruktur und Komplexierung, V. Neue Kronenäther und ihre Alkalimetallion-Komplexe. Chem. Ber. 1976, 109, 1803–1831. [Google Scholar] [CrossRef]
- Kato, A.; Nagatsuka, Y.; Hiratsuka, T.; Kiuchi, S.; Iwase, Y.; Okuno, Y.; Tsukamoto, T.; Kiran, Y.B.; Sakai, N.; Konakahara, T. Synthesis and cytotoxic activity of novel 11-methyl-6H-pyrido[4,3-b]carbazole derivatives linked to amine, N-methylurea, and N-methyl-N-nitrosourea moieties with various types of carbamoyl tethers at the C-5 atom. Tetrahedron 2016, 72, 4258–4272. [Google Scholar] [CrossRef]
- Su, H.; Wu, C.; Zhu, J.; Miao, T.; Wang, D.; Xia, C.; Zhao, X.; Gong, Q.; Song, B.; Ai, H. Rigid Mn(II) chelate as efficient MRI contrast agent for vascular imaging. Dalt. Trans. 2012, 41, 14480–14483. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V. Design and Synthesis of Macrocyclic Ligands and Their Complexes of Lanthanides and Actinides. Chem. Rev. 1995, 95, 273–342. [Google Scholar] [CrossRef]
- Costisor, O.; Linert, W. The Template Effect. In Metal Mediated Template Synthesis of Ligands; World Scientific Pub. Co.: Singapore, 2004; pp. 1–17. ISBN 9789812794819. [Google Scholar]
- Aime, S.; Botta, M.; Crich, S.G.; Giovenzana, G.B.; Jommi, G.; Pagliarin, R.; Sisti, M. Synthesis and NMR Studies of Three Pyridine-Containing Triaza Macrocyclic Triacetate Ligands and Their Complexes with Lanthanide Ions. Inorg. Chem. 1997, 36, 2992–3000. [Google Scholar] [CrossRef]
- Kim, W.D.; Hrncir, D.C.; Kiefer, G.E.; Dean Sherry, A. Synthesis, Crystal Structure, and Potentiometry of Pyridine-Containing Tetraaza Macrocyclic Ligands with Acetate Pendant Arms. Inorg. Chem. 1995, 34, 2225–2232. [Google Scholar] [CrossRef]
- Swift, T.J.; Connick, R.E. NMR-relaxation mechanisms of17O in aqueous solutions of paramagnetic cations and the lifetime of water molecules in the first coordination sphere. J. Chem. Phys. 1964, 37, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Bloembergen, N.; Morgan, L.O. Proton relaxation times in paramagnetic solutions. Effects of electron spin relaxation. J. Chem. Phys. 1961, 34, 842–850. [Google Scholar] [CrossRef] [Green Version]
- Freed, J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. II. Finite jumps and independent T 1 processes. J. Chem. Phys. 1978, 68, 4034–4037. [Google Scholar] [CrossRef]
- Gale, E.M.; Zhu, J.; Caravan, P. Direct Measurement of the Mn (II) Hydration State in Metal Complexes and Metalloproteins Through 17 O NMR Line widths. J. Am. Chem. Soc. 2013, 135, S1–S15. [Google Scholar] [CrossRef] [Green Version]
- Pujales-Paradela, R.; Carniato, F.; Esteban-Gómez, D.; Botta, M.; Platas-Iglesias, C. Controlling water exchange rates in potential Mn2+-based MRI agents derived from NO2A2−. Dalt. Trans. 2019, 48, 3962–3972. [Google Scholar] [CrossRef]
- Drahoš, B.; Pniok, M.; Havlíčková, J.; Kotek, J.; Císařová, I.; Hermann, P.; Lukeš, I.; Tóth, É. Mn2+ complexes of 1-oxa-4,7-diazacyclononane based ligands with acetic, phosphonic and phosphinic acid pendant arms: Stability and relaxation studies. Dalt. Trans. 2011, 40, 10131–10146. [Google Scholar] [CrossRef]
- Atanasijevic, T.; Zhang, X.A.; Lippard, S.J.; Jasanoff, A. MRI sensing based on the displacement of paramagnetic ions from chelated complexes. Inorg. Chem. 2010, 49, 2589–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanali, G.; Cao, Y.; Ascenzi, P.; Fasano, M. Mn(II) binding to human serum albumin: A 1H-NMR relaxometric study. J. Inorg. Biochem. 2012, 117, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Canton, S.; Crich, S.G.; Terreno, E. 1H and 17O relaxometric investigations of the binding of Mn(II) ion to human serum albumin. Magn. Reson. Chem. 2002, 40, 41–48. [Google Scholar] [CrossRef]
- Wen, J.; Geng, Z.; Yin, Y.; Wang, Z. A mononuclear Mn 2+ complex based on a novel tris-(ethyl acetate) pendant-armed tetraazamacrocycle: Effect of pyridine on self-assembly and weak interactions. Inorg. Chem. Commun. 2012, 21, 16–20. [Google Scholar] [CrossRef]
- Mayer, I. Bond Order and Valence Indices: A Personal Account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef]
- Bridgeman, A.J.; Cavigliasso, G.; Ireland, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalt. Trans. 2001, 2095–2108. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. PSEQUAD. In Computational Method for Determination of Formation Constants; Springer: Boston, MA, USA; New York, NY, USA, 1985; pp. 291–353. ISBN 978-1-4684-4936-5. [Google Scholar]
- Molnár, E.; Camus, N.; Patinec, V.; Rolla, G.A.; Botta, M.; Tircsó, G.; Kálmán, F.K.; Fodor, T.; Tripier, R.; Platas-Iglesias, C. Picolinate-containing macrocyclic Mn2+ complexes as potential MRI contrast agents. Inorg. Chem. 2014, 53, 5136–5149. [Google Scholar] [CrossRef]
- Merbach, A.; Helm, L.; Tóth, É. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Raiford, D.S.; Fisk, C.L. Calibration of methanol and ethylene glycol nuclear magnetic resonance thermometers—Analytical Chemistry (ACS Publications). Anal. Chem. 1979, 51, 2050–2051. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Yerly, F. VISUALISEUR 3.3.7 and OPTIMISEUR 3.3.7. Lausanne, Switzerland, 2006. [Google Scholar]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracyElectronic supplementary information (ESI) available:[DETAILS]. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Stefan, G.; Stephan, E.; Lars, G. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Pantazis, D.A. Assessment of double-hybrid density functional theory for magnetic exchange coupling in manganese complexes. Inorganics 2019, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Wooten, A.L.; Lewis, B.C.; Lapi, S.E. Cross-sections for (p,x) reactions on natural chromium for the production of 52,52m,54Mn radioisotopes. Appl. Radiat. Isot. 2015, 96, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, M.; Spahn, I.; Coenen, H.H. Optimized separation procedure for production of no-carrier-added radiomanganese for positron emission tomography. Radiochim. Acta 2015, 103, 893–899. [Google Scholar] [CrossRef]
- Fonslet, J.; Tietze, S.; Jensen, A.I.; Graves, S.A.; Severin, G.W. Optimized procedures for manganese-52: Production, separation and radiolabeling. Appl. Radiat. Isot. 2017, 121, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Equivalent Amount of Compound 8 | O-pyclen (7) tR = 2.77 min | 3-OPCAOEt (11) tR = 4.74 min | 3,9-OPC2AOEt (9) tR = 7.86 min |
|---|---|---|---|
| 0.4 | 52.1% | 47.9% | - |
| 0.8 | 17.5% | 66.7% | 15.8% |
| 1.2 | - | 54.1% | 45.9% |
| 1.6 | - | 16.2% | 83.8% |
| 2.0 | - | 1.9% | 98.1% |
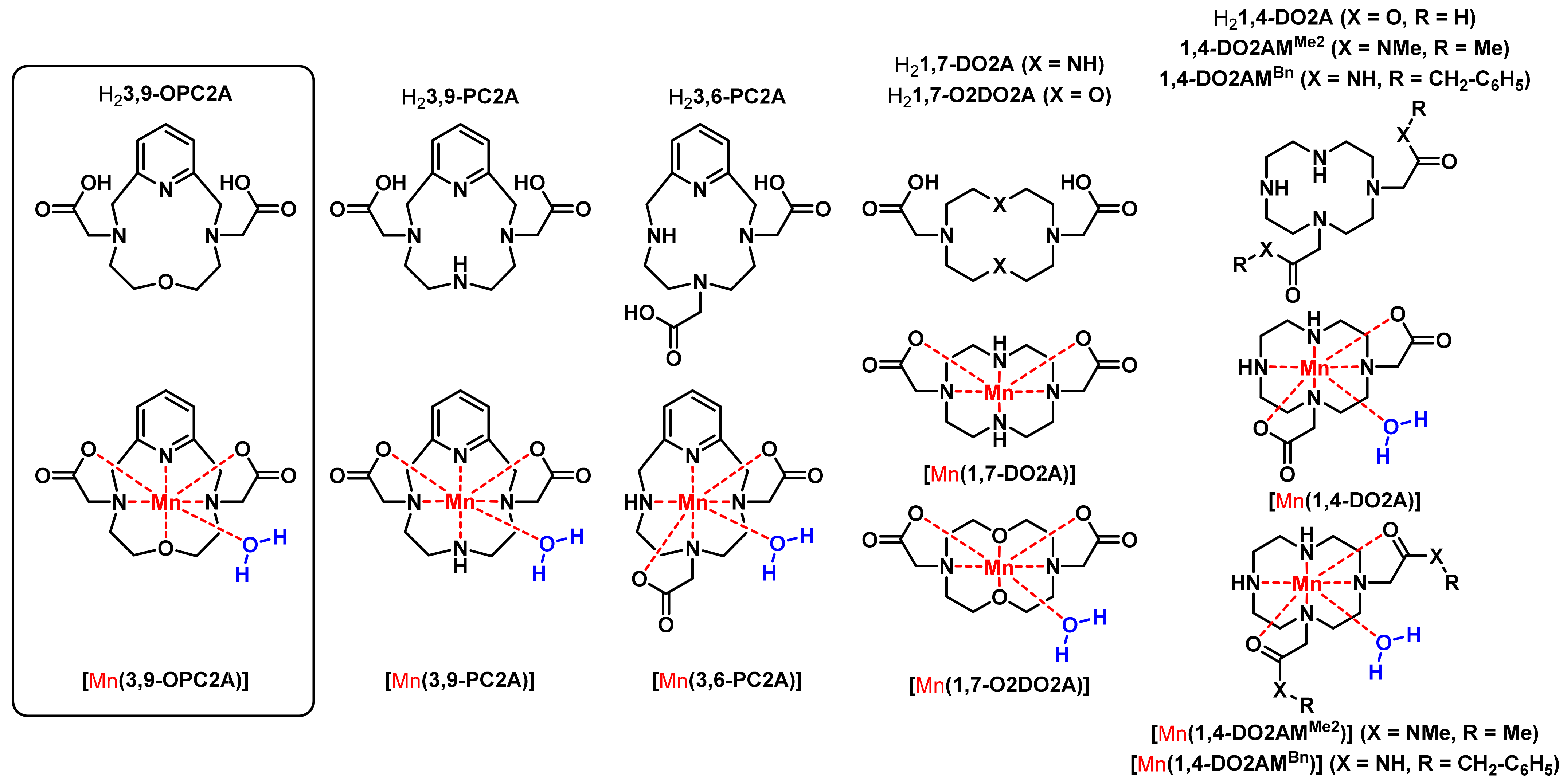
| H23,9-OPC2A | H23,9-PC2A a | H23,6-PC2A a | H21,4-DO2A b | H21,7-DO2A b | H21,7-O2DO2A c | |
| log K1H | 7.73(2) | 12.25 | 10.72 | 11.44 | 11.69 | 8.05 |
| log K2H | 7.66(1) | 5.97 | 8.37 | 9.51 | 9.75 | 7.43 |
| log K3H | 2.13(1) | 3.47 | 3.81 | 4.14 | 3.97 | 2.06 |
| log K4H | - | 1.99 | 1.26 | 1.55 | 2.68 | - |
| Σlog Κ2H | 15.39 | 18.22 | 19.09 | 20.95 | 21.44 | 15.48 |
| H23,9-OPC2A | H23,9-PC2A a | H23,6-PC2A a | H21,4-DO2A b | H21,7-DO2A b | H21,7-O2DO2A c | ||
| Σlog Κ2H | 15.39 | 18.22 | 19.09 | 20.95 | 21.44 | 15.48 | |
| Mg2+ | log KMnL | 7.02(1) | 9.84;(8.4) d | 8.11 | - | - | 3.91 |
| log KMnHL | - | 5.91 | 5.87 | - | - | - | |
| Ca2+ | log KMnL | 8.27(1) | 9.92;(10.0) d | 9.57 | 8.62 | 8.86 | 6.96 |
| log KMnHL | - | 5.08 | 5.27 | - | - | - | |
| Mn2+ | log KMnL | 13.03(1) | 17.09 | 15.53 | 15.68 | 14.64 | 9.38 |
| log KMnHL | 2.40(2) | 2.14 | 3.06 | 4.15 | 4.40 | - | |
| log KMnLOH | 11.49(1) | - | - | - | - | 12.38 | |
| pMn e | 8.69 | 8.64 | 8.09 | 7.27 | 6.52 | 6.67 | |
| Zn2+ | log KMnL | 14.81(1) | 19.49 | 20.37 | 18.03 | 18.86 | 10.55 |
| log KMnHL | 2.08(3) | 2.74 | 2.36 | 3.58 | 4.23 | - | |
| log KMnLOH | 10.95(2) | - | - | 1.65 f | 1.78 f | 11.40 | |
| Cu2+ | log KMnL | 18.41(4) g | 23.58 g | 24.09 g | 24.43 g | 24.24 | 14.56 |
| log KMnHL | 1.92(4) g | 2.12 g | 2.37 g | 2.95 | 3.06 | 2.46 | |
| log KMnLOH | 10.50(5) | - | - | - | - | 11.95 |
| H23,9-OPC2A (25 °C/37 °C) | H23,9-PC2A a | H23,6-PC2A a | H21,4-DO2A b | H21,7-DO2A b | H21,7-O2DO2A c | ||
| 0.49 T | r1p | 3.13(2)/2.54(1) | 2.91 | 2.72 | 2.10 | 1.50 | 2.86 |
| r2p | 5.15(3)/4.17(4) | 3.96 | 3.49 | - | - | - | |
| 1.41 T | r1p | 2.72(2)/2.06(1) | 2.24 | 2.40 | - | - | - |
| r2p | 9.90(5)/7.37(3) | 4.82 | 4.20 | - | - | - |
| Parameter | [(Mn(3,9-OPC2A)] | [Mn(3,9-PC2A)] a | [Mn(1,4-DO2A)] b | [Mn(1,4-DO2AMMe2)]2+ c | [Mn(1,7-O2DO2A)] d |
| r129820 MHz/mM−1s−1 | 3.09 | 2.91 | 2.1 | 2.5 | 2.86 |
| kex298/107s−1 | 5.3 ± 0.4 | 12.6 | 113 | 11.5 | 5.3 |
| ΔH‡/kJmol−1 | 28.5 ± 1.7 | 37.5 | 29.4 | 36.9 | 29.7 |
| ΔS‡/JK−1mol−1 | −1.9 ± 0.8 | – | – | - | 2.8 |
| τR298/ps | 40.0 ± 1.1 | – | 46 | 53 | 46.7 |
| ER/kJmol−1 | 14.8 ± 0.6 | – | 19.1 | 19.1 | 20 e |
| Δ2/1018s−2 | 17.8 ± 3.6 | – | 481 | 510 | 111 |
| τv298/ps | 19.3 ± 3.0 | – | 4.4 | 5.5 | – |
| q e | 1 | 1 | 0.87 | 0.87 | 1 |
| [Mn(3,9-OPC2A)] | [Mn(3,9-PC2A)] a | [Mn(3,6-PC2A)] a | [Mn(1,4-DO2A)] b | [Mn(1,4-DO2AMMe2)]2+ c | [Mn(1,7-O2DO2A)] d | |
| k0 (s−1) | (8.6 ± 1.1) × 10−6 | – e | – e | – e | – e | – e |
| k1 (M−1s−1) | 2.81 ± 0.07 | 221 | 70 | 99 | 8.7 | 85 |
| k2 (M−2s−1) | - | - | 1.5 × 105 | 1.4 × 106 | - | 3.0 × 106 |
| k3 (M−1s−1) | - | 3.6 × 10−2 | 2.6 × 10−2 | - | - | - |
| KMnL x H | - | 3.6 × 103 | 1.15 × 103 | - | - | - |
| KML x Cu | 7 ± 4 | 26 | 16 | - | - | - |
| t1/2 (h) at pH = 7.4 | 21.9 f 1625 | 21.0 | 63.2 | 48 | 556 | 56.8 |
| [Mn(3,9-PC2A)] | [Mn(3,9-OPC2A)] | |||
| Bond | R | MBO | R | MBO |
| Mn–N1 | 2.041 | 0.56 | 2.041 | 0.56 |
| Mn–N3 | 2.610 | 0.24 | 2.692 | 0.22 |
| Mn–N6/O6 | 2.077 | 0.48 | 2.190 | 0.26 |
| Mn–N9 | 2.101 | 0.48 | 2.067 | 0.50 |
| Mn–O3 | 2.252 | 0.19 | 2.099 | 0.26 |
| Mn–O9 | 2.051 | 0.45 | 2.016 | 0.44 |
| Mn–O1w | 2.114 | 0.35 | 2.151 | 0.34 |
| [Mn(3,9-PC2A)] | [Mn(3,9-OPC2A)] | |||||
| atom | q | MBV | MFV | q | MBV | MFV |
| Mn | 0.297 | 3.46 | 1.50 | 0.434 | 3.19 | 1.77 |
| N1 | −0.118 | 3.15 | 0.00 | −0.108 | 3.15 | 0.00 |
| N3 | −0.238 | 2.97 | 0.00 | −0.254 | 2.97 | 0.00 |
| N6/O6 | −0.019 | 3.22 | 0.00 | −0.277 | 2.07 | 0.00 |
| N9 | −0.086 | 3.24 | 0.00 | −0.081 | 3.31 | 0.00 |
| O3 | −0.520 | 1.85 | 0.00 | −0.516 | 1.87 | 0.00 |
| O9 | −0.464 | 1.89 | 0.00 | −0.456 | 1.92 | 0.00 |
| O1w | −0.447 | 2.08 | 0.00 | −0.455 | 2.05 | 0.00 |
| Solvent A (MeCN) | Solvent B (Aqueous 5 mM TFA Solution) | |
|---|---|---|
| 0.0 min | 0% | 100% |
| 15.0 min | 90% | 10% |
| 16.0 min | 0% | 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csupász, T.; Szücs, D.; Kálmán, F.K.; Hollóczki, O.; Fekete, A.; Szikra, D.; Tóth, É.; Tóth, I.; Tircsó, G. A New Oxygen Containing Pyclen-Type Ligand as a Manganese(II) Binder for MRI and 52Mn PET Applications: Equilibrium, Kinetic, Relaxometric, Structural and Radiochemical Studies. Molecules 2022, 27, 371. https://doi.org/10.3390/molecules27020371
Csupász T, Szücs D, Kálmán FK, Hollóczki O, Fekete A, Szikra D, Tóth É, Tóth I, Tircsó G. A New Oxygen Containing Pyclen-Type Ligand as a Manganese(II) Binder for MRI and 52Mn PET Applications: Equilibrium, Kinetic, Relaxometric, Structural and Radiochemical Studies. Molecules. 2022; 27(2):371. https://doi.org/10.3390/molecules27020371
Chicago/Turabian StyleCsupász, Tibor, Dániel Szücs, Ferenc Krisztián Kálmán, Oldamur Hollóczki, Anikó Fekete, Dezső Szikra, Éva Tóth, Imre Tóth, and Gyula Tircsó. 2022. "A New Oxygen Containing Pyclen-Type Ligand as a Manganese(II) Binder for MRI and 52Mn PET Applications: Equilibrium, Kinetic, Relaxometric, Structural and Radiochemical Studies" Molecules 27, no. 2: 371. https://doi.org/10.3390/molecules27020371