
Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review

 , , ,
, , , 
 ,
, 

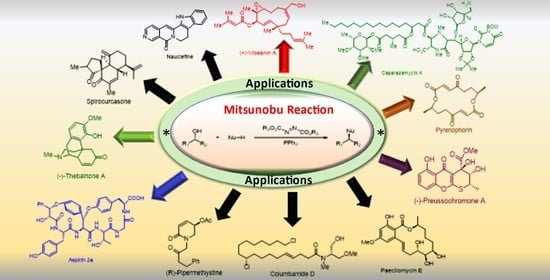
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Review of the Literature
2.1. Alkaloids-Based Natural Product Synthesis
2.1.1. Yuzurimine Alkaloids
2.1.2. Phenanthrene Alkaloids
2.1.3. Indole Alkaloids
2.1.4. Discorhabdin Alkaloids
2.1.5. Lycopodium Alkaloids
2.1.6. Pyrrole Alkaloids
2.1.7. Oroidin Alkaloids
2.1.8. Communesin Alkaloids
2.1.9. Pyrroloquinoline Alkaloids
2.1.10. Morphine Alkaloids
2.1.11. Calciphylline Alkaloids
2.1.12. Aspidosperma Alkaloids
2.1.13. Amaryllidaceae Alkaloids
2.2. Neolignans-Based Natural Product Synthesis
Sesquineolignans
2.3. Synthesis of Polyketides-Based Natural Products
2.4. Natural Products-Based Terpenes Synthesis
2.5. Synthesis of Lactone-Based Natural Products
2.5.1. Synthesis of Glutamate Receptors
2.5.2. Synthesis of Steroid-Based Natural Products
2.5.3. Synthesis of Chromone-Based Natural Products
2.5.4. Synthesis of Acetylenic-Based Natural Products
2.5.5. Synthesis of Piperidinones-Based Natural Products
2.5.6. Synthesis of Peptide-Based Natural Products
2.5.7. Synthesis of Amino Acids-Based Natural Products
2.5.8. Synthesis of Natural Products Containing Carbohydrate Derivatives
2.5.9. Synthesis of Natural Products-Based Fatty Acid Amides
2.5.10. Synthesis of Chlorosulpholipids-Based Natural Products
2.5.11. Synthesis of Dihydropyran-Based Natural Products
2.5.12. Synthesis of Enediynes-Based Natural Products
2.6. Synthesis of Epidithiodiketopiprazine-Based Natural Products
2.7. Liponucleoside-Based Natural Product Synthesis
Miscellaneous
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- But, T.Y.S.; Toy, P.H. The Mitsunobu reaction: Origin, mechanism, improvements and applications. Chem. Asian J. 2007, 2, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. The Mitsunobu reaction in 21st century. Org. Chem. Front. 2015, 2, 739–752. [Google Scholar] [CrossRef]
- Beddoe, R.H.; Sneddon, H.F.; Denton, R.M. The catalytic Mitsunobu reaction: A critical analysis of current state of art. Org. Biomol. Chem. 2018, 16, 7774–7781. [Google Scholar] [CrossRef] [PubMed]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef]
- Hughes, D.L. Progress in Mitsunobu reaction: A review. Org. Prep. Proced. Int. 1996, 28, 127–164. [Google Scholar] [CrossRef]
- Dandapani, S.; Curran, D.P. Fluorous Mitsunobu reagents and reactions. Tetrahedron 2002, 58, 3855–3864. [Google Scholar] [CrossRef]
- Sun, Z.; Shang, Z.; Forelli, N.; Po, K.H.L.; Chen, S.; Brady, S.F.; Li, X. Total synthesis of malacidin A by β-hydroxyaspartic acid ligation mediated cyclization and absolute structure establishment. Angew. Chem. Int. Ed. 2020, 59, 19868–19872. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Miura, T.; Oikawa, T.; Qin, X.Y.; Kojima, S.; Kagechika, H. Design, synthesis and antitumor activity of phthalazine-1,4-dione-based menaquinone analogs. Biorg. Med. Chem. Lett. 2021, 43, 128065. [Google Scholar] [CrossRef]
- Ziegler, T.; Cosky, E. Mitsonobu reaction of cannabidiol. Synthesis of water-soluble cannabidiol derivatives. J. Org. Chem. 2021, part iv, 198–205. [Google Scholar] [CrossRef]
- Bhangare, D.N.; Mahale, V.B.; Shinde, R.G.; Nikalje, M.D.; Duthade, G.S.; Lokhande, M.N. Synthesis of potent calcimimetics (+)-NPS R-568 by palladium-catalyzed oxidative kinetic resolution. Malays. J. Chem. 2021, 23, 1–6. [Google Scholar] [CrossRef]
- Dong, M.; Zhang, M.L.; Shi, Q.W.; Gu, Y.C.; Kiyota, H. The daphniphyllum alkaloids. Curr. Org. Chem. 2009, 13, 646–682. [Google Scholar] [CrossRef]
- Hayakawa, I.; Nagatani, R.; Ikeda, M.; Yoo, D.E.; Saito, K.; Kigoshi, H.; Sakakura, A. Toward the synthesis of yuzurimine-type alkaloids: Stereoselective construction of the heterocyclic portions of deoxyyuzurimine and macrodaphnine. Org. Lett. 2019, 21, 6337–6341. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.D.; Barancyk, S.V.; Burch, E.L. Lewis acid catalysis of photochemical reactions. 11. Conformations, spectroscopy, and photochemistry of methyl phenanthrene-9-carboxylate, and phenanthrene-9-carboxamides, and their Lewis acid complexes. J. Am. Chem. Soc. 1992, 114, 3866–3870. [Google Scholar] [CrossRef]
- Zhang, H.C.; Liu, R.; An, Z.P.; Li, H. Aristolactam-type alkaloids and aristolochic acids from Aristolochia moupinensis and Aristolochia cathcartii. Biochem. Syst. Ecol. 2016, 65, 198–201. [Google Scholar] [CrossRef]
- Luong, T.M.; Pilkington, L.I.; Barker, D. Stereoselective total synthesis of (+)-aristolactam GI. J. Org. Chem. 2019, 84, 5747–5756. [Google Scholar] [CrossRef] [PubMed]
- Boucherle, B.; Haudecoeur, R.; Queiroz, E.F.; Waard, M.D.; Wolfender, J.L.; Robins, R.J.; Boumendjel, A. Nauclea latifolia: Biological activity and alkaloid phytochemistry of a west african tree. Nat. Prod. Rep. 2016, 33, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Hotellier, F.; Delaveau, P.; Pousset, J.L. Nauclefine et naucletine deux nouveaux alcaloides de type indoloquinolizidine isoles du Nauclea latifolia. Phytochem 1975, 14, 1407–1409. [Google Scholar] [CrossRef]
- Chen, K.; Dong, H.; Wang, J.; Lei, X. Concise total synthesis of nauclefine: A regioselective Rhodium (III)-catalyzed oxidative C-H activation approach. Tetrahedron 2021, 87, 132120. [Google Scholar] [CrossRef]
- Li, F.; Peifer, C.; Janussen, D.; Tasdemir, D. New Discorhabdin Alkaloids from the Antarctic Deep-Sea Sponge Latrunculia biformis. Mar. Drugs 2019, 17, 439. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Sakata, J.; Tokuyama, H. Synthetic studies on discorhabdin V: Construction of the A–F hexacyclic framework. Tetrahedron Lett. 2021, 81, 153333. [Google Scholar] [CrossRef]
- Ma, X.; Gang, D.R. The lycopodium alkaloids. Nat. Prod. Rep. 2020, 21, 752–772. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Jia, Z.; Qiu, F.G. A concise asymmetric total synthesis of (+)-fawcettimine. Tetrahedron Lett. 2020, 61, 152329. [Google Scholar] [CrossRef]
- Katakawa, K.; Mito, H.; Kogure, N.; Kitajima, M.; Wongseripipatana, S.; Arisawa, M.; Takayama, H. Ten New fawcettimine-related alkaloids from three species of lycopodium. Tetrahedron 2021, 67, 6561–6567. [Google Scholar] [CrossRef]
- Kaneko, H.; Takahashi, S.; Kogure, N.; Kitajima, M.; Takayama, H. Asymmetric total synthesis of fawcettimine-type lycopodium alkaloid, lycopoclavamine-A. J. Org. Chem. 2019, 84, 5645–5654. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Chen, Y.; Ching, Y.C.; Lee, C.S.; He, S. An approach towards the tetracyclic skeleton of palhinine alkaloids. Org. Chem. Front. 2020, 7, 2243–2246. [Google Scholar] [CrossRef]
- Matsuo, R.; Miyashita, A.; Kuwabara, M.; Adachi, S.; Matsuzawa, A.; Sugita, K. Concise diastereoselective total synthesis of (±)-parvistemonine A. Synlett 2020, 31, 1800–1804. [Google Scholar] [CrossRef]
- Lindal, T. Chemistry and biology of the pyrrole-imidazole alkaloids. Alkaloids Chem. Biol. 2017, 77, 117–219. [Google Scholar] [CrossRef]
- Bhandari, M.R.; Herath, A.K.; Rasapalli, S.; Yousufuddin, M.; Lovely, C.J. Total Synthesis of the Nagelamides—Synthetic studies towards the reported structure of nagelamide D and nagelamide E framework. J. Org. Chem. 2020, 85, 12971–12987. [Google Scholar] [CrossRef]
- Numata, A.; Takahashi, C.; Ito, Y.; Takada, T.; Kawai, K.; Usami, Y.; Matsumura, E.; Imachi, M.; Ito, T.; Hasegawa, T. Communesins, cytotoxic metabolites of a fungus isolated from a marine alga. Tetrahedron Lett. 1993, 34, 2355–2358. [Google Scholar] [CrossRef]
- Pompeo, M.M.; Cheah, J.H.; Movassaghi, M. Total Synthesis and Anti-Cancer Activity of All Known Communesin alkaloids and related derivatives. J. Am. Chem. Soc. 2019, 141, 14411–14420. [Google Scholar] [CrossRef]
- Sun, H.H.; Sakemi, S.; Bureas, N.; MacCarthy, P. Isobatzellines A, B, C, and D. Cytotoxic and antifungal pyrroloquinoline alkaloids from the marine sponge Batzella sp. J. Org. Chem. 1990, 55, 4964–4966. [Google Scholar] [CrossRef]
- Yamashita, Y.; Poignant, L.; Sakata, J.; Tokuyama, H. Divergent total synthesis of Isobatzellines A/B and batzelline A. Org. Lett. 2020, 22, 6239–6243. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, P.R.; White, J.D. Morphine, the proteus of organic molecules. Chem. Commun. 2002, 7, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, F.M.; Zhang, C.S.; Liu, S.Z.; Tian, J.M.; Wang, S.H.; Zhang, X.M.; Tu, Y.Q. Enantioselective synthesis of cis-hydrobenzofurans bearing all-carbon quaternary stereocenters and application to total synthesis of (−)-morphine. Nat. Commun. 2019, 10, 2507. [Google Scholar] [CrossRef]
- Rautschek, J.; Metz, P. Toward the Synthesis of (−)-Codeine by Chiral Auxiliary-Mediated Nitrone Cycloaddition. Heterocycl 2017, 95, 1106–1120. [Google Scholar] [CrossRef]
- Wang, Y.; Hennig, A.; Küttler, T.; Hahn, C.; Jäger, A.; Metz, P. Total Synthesis of (±)-Thebainone A by Intramolecular nitrone cycloaddition. Org. Lett. 2020, 22, 3145–3148. [Google Scholar] [CrossRef]
- Hou, S.H.; Prichina, A.Y.; Dong, G. Deconstructive asymmetric total synthesis of morphine-family alkaloid (−)-thebainone A. Angew. Chem. Int. Ed. 2021, 60, 13057–13064. [Google Scholar] [CrossRef]
- Morita, H.; Kobayashi, J.I. Calyciphyllines A and B, two novel hexacyclic alkaloids from daphniphyllum calycinum. Org. Lett. 2003, 5, 2895–2898. [Google Scholar] [CrossRef]
- Kumar, B.S.; Raghavan, S. Stereoselective synthesis of the A, E-ring bicyclic core of calyciphylline B-type alkaloids. Synlett 2019, 30, 2157–2160. [Google Scholar] [CrossRef]
- Gan, C.Y.; Low, Y.Y.; Thomas, N.F.; Kam, T.S. Rhazinilam-leuconolam-leuconoxinealkaloids from leuconotis griffithii. J. Nat. Prod. 2013, 76, 957–964. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H. Unified enantioselective total syntheses of (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine and (−)-mersicarpine. Chem. Commun. 2019, 55, 3544–3547. [Google Scholar] [CrossRef]
- Saxton, J.E. Alkaloids of the aspidospermine group. In The Alkaloids: Chemistry and Biology; Academic Press: San Diego, CA, USA, 1998; Volume 51, pp. 1–197. [Google Scholar] [CrossRef]
- Zhou, Y.G.; Wong, H.N.C.; Peng, X.S. Total syntheses of (−)-deoxoapodine, (−)-kopsifoline D and (−)-beninine. J. Org. Chem. 2019, 85, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, F.S. A total synthesis of (±)-leuconodines D and E. J. Org. Chem. 2019, 84, 13890–13896. [Google Scholar] [CrossRef]
- Jin, Z. Amaryllidaceae and sceletium alkaloids. Nat. Prod. Rep. 2003, 30, 849–868. [Google Scholar] [CrossRef]
- He, M.; Qu, C.; Gao, O.; Hu, X.; Hong, X. Biological and pharmacological activities of amaryllidaceae alkaloids. Rsc Adv. 2015, 5, 16562–16574. [Google Scholar] [CrossRef]
- Lo, H.J.; Chang, Y.K.; Ananthan, B.; Lih, Y.H.; Liu, K.S.; Yan, T.H. Total synthesis of (+)-lycoricidine, conduramine B-1, ent-C-1, C-4, D-1, ent-F-1, ent-F-4, and formal synthesis of (−)-laminitol—A C2-symmetric chiral pool-based flexible strategy. J. Org. Chem. 2019, 84, 10065–10075. [Google Scholar] [CrossRef]
- Fürst, R. Narciclasine—An amaryllidaceae alkaloid with potent antitumor and anti-inflammatory properties. Planta Med. 2016, 16, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.S. Lignans, neolignans and related compounds. Nat. Prod. Rep. 1999, 16, 75–96. [Google Scholar] [CrossRef]
- Apers, S.; Vlietinck, A.; Pieters, L. Lignans and neolignans as lead compounds. Phytochem. Rev. 2003, 2, 201–217. [Google Scholar] [CrossRef]
- Kim, K.H.; Moon, E.; Kim, H.K.; Oh, J.Y.; Kim, S.Y.; Choi, S.U.; Lee, K.R. Alkaloids from Acorus gramineus rhizomes and their biological activity. Bioorg. Med. Chem. Lett. 2012, 22, 6155–6161. [Google Scholar] [CrossRef]
- Avula, S.K.; Das, B.; Csuk, R.; Al-Rawahi, A.; Al-Harrasi, A. Total synthesis of surinamensinols A and B. SynOpen 2020, 4, 84–88. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, W.; Yao, H.; Shen, Y.; Zhang, Y.; Li, Y.Z.; QI, Q.; Wongpraset, K.; Tang, Y.J. Discover 4,6-O-thenylidene-#-D-glucopyranoside-(2#-acetamido,3-acetyl-di-S-5-fluorobenzothizole/5-fluorobenzoxazole)-4’ demethylepipodophyllotoxin as potential less toxic antitumor candidate drugs by reducing DNA damage and less inhibition of PI3K. J. Med. Chem. 2020, 63, 2877–2893. [Google Scholar] [CrossRef]
- Xu, M.; Hou, M.; He, H.; Gao, S. Asymmetric total synthesis of aglacins A, B and E. Angew. Chem. Int. Ed. 2021, 60, 16655–16660. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Kim, H.J.; Ali, M.S.; Lee, Y.S. An update on bioactive plant lignans. Nat. Prod. Rep. 2005, 22, 696–716. [Google Scholar] [CrossRef]
- Colombo, E.; Paladino, G.; Ciriello, U.; Passarella, D. Convenient Preparation and Spectroscopic Characterization of 7R-Hydroxymatairesinol. Molecules 2021, 26, 5838. [Google Scholar] [CrossRef]
- Lee, J.J.; Choi, H.J.; Yun Misun Kang Yingin Jung, J.I.; Ryu Yiseul Kim, T.Y.; Cha, Y.J.; Cho, H.S.; Min, J.J.; Chung, C.W.; Kim, H.S. Enzymatic prenylation and oxime ligation for the synthesis of stable and homogeneous protein–drug conjugates for targeted Therapy. Angew. Chem. Int. Ed. 2015, 54, 12020–12024. [Google Scholar] [CrossRef] [PubMed]
- Lozinski, O.A.; Bistodeau, J.; Pelissero, C.B.; Khilya, V.P.; Shinkaruk, S. Assembling the prenylneofavone system through a Pechmann condensation/Mitsunobu reaction/Claisen rearrangement/olefn cross-metathesis sequence. Monatsh. Chem. 2020, 151, 605–610. [Google Scholar] [CrossRef]
- Teponno, R.B.; Kusari, S.; Spiteller, M. Recent advances in research on lignans and neolignans. Nat. Pro. Rep. 2016, 33, 1044–1092. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, T.; Cohick, W.; Raskin, I. Dietary phytoestrogens and health. Phytochem 2004, 65, 995–1016. [Google Scholar] [CrossRef] [PubMed]
- Mane, B.B.; Kumbhar, D.D.; Waghmode, B.B. Enantioselective total synthesis of ligraminol D and ligraminol E. Synlett 2019, 30, 2285–2289. [Google Scholar] [CrossRef]
- Luecha, P.; Umehara, K.; Miyasi, T.; Noguchi, H. Antiestrogenic constituents of the Thai medicinal plants Capparis flavicans and Vitex glabrata. J. Nat. Prod. 2009, 72, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Ueno, H.; Yoshida, N.; Ouchi, H.; Asakawa, T.; Yoshimura, F.; Inai, M.; Kan, T. Diastereo- and regiodivergent total synthesis of princepin and isoprincepin in both (7″R,8″R) and (7″S,8″S) Isomers. J. Org. Chem. 2019, 84, 14227–14240. [Google Scholar] [CrossRef]
- Winssinger, N.; Barluenga, S. Chemistry and biology of resorcylic acid lactones. Chem. Commun. 2007, 1, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, V.; Mayer-Bartschmid, A.; Muller, H.; Greif, G.; Kleymann, G.; Zitzmann, W.; Tichy, H.T.; Stadler, M. Pochonins A-F, new antiviral and anti-parasitic resorcylic acid lactones from Pochonia chlamydosporia var. catenulate. J. Nat. Prod. 2003, 66, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Bhavani, G.; Jonnaya, S.; Bantu, R.; Reddy, B.V.S. A concise and stereoselective total synthesis of paecilomycin E. Nat. Prod. Commun. 2019, 14, 131–133. [Google Scholar] [CrossRef]
- Goh, S.H.; Ee, G.C.L.; Chuah, C.H.; Mak, T.C.W. 5β-hydroxygoniothalamin, a styrylpyrone derivative from Goniothalamus dolichocarpus (Annonaceae). Nat. Prod. Lett. 1995, 5, 255–259. [Google Scholar] [CrossRef]
- Mereyala, H.J.; Joe, M. Cytotoxic activity of styryl lactones and their derivatives. Curr. Med. Chem. Anti-Cancer Agents 2001, 1, 293–300. [Google Scholar] [CrossRef]
- Kotammagri, T.K.; Paul, S.; Bhattacharya, A.K. Unusual Epimerization in Styryllactones: Synthesis of (−)-5-hydroxygoniothalamin, (−)-5-acetylgoniothalamin, and O-TBS-goniopypyrone. ACS Omega 2019, 4, 22549–22556. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.J.R. The biology of dinoflagellates. In Botanical Monographs, 1st ed.; Blackwell: Oxford, UK, 1987; Volume 21. [Google Scholar]
- Hess, S.N.; Mo, X.; Wirtz, C.; Furstner, A. Total synthesis of limaol. J. Am. Chem. Soc. 2021, 143, 2464–2469. [Google Scholar] [CrossRef]
- Liaw, C.C.; Wu, T.Y.; Chang, F.R.; Wu, Y.C. Historic perspectives on annonaceous acetogenins from the chemical bench to preclinical trials. Planta Med. 2010, 76, 1390–1404. [Google Scholar] [CrossRef]
- Mullapudi, V.; Ahmad, I.; Senapati, S.; Ramana, C.V. Total synthesis of (+)-petromyroxol, (−)-iso-petromyroxol, and possible diastereomers. ACS Omega 2020, 5, 25334–25348. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Dwyer, B.G.; Liu, C.; Abegg, D.; Cai, Z.J.; Hoch, D.G.; Yin, X.; Qui, N.; Liu, J.Q.; Adebekian, A.; et al. Total synthesis and target Identification of the curcusone diterpenes. J. Am. Chem. Soc. 2021, 143, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.I.; Ogura, A.; Yoshida, K.; Simizu, S. Total synthesis of natural products using intramolecular Nozaki–Hiyama–Takai–Kishi reactions. Synlett 2020, 31, 421–433. [Google Scholar] [CrossRef]
- Zhang, P.; Li, Y.H.; Yan, Z.; Gong, J.; Yang, Z. Asymmetric Total synthesis of (−)-pavidolide B via a thiyl-radical-mediated [3+2] annulation reaction. J. Org. Chem. 2019, 84, 15958–15971. [Google Scholar] [CrossRef]
- Khatua, A.; Niyogi, S.; Bisai, V. Total synthesis of (+)-ar-macrocarpene. Org. Biomol. Chem. 2019, 17, 7140–7143. [Google Scholar] [CrossRef]
- Connelly, J.D.; Hill, R.A. Dictionary of Terpenoids, 1st ed.; Chapman & Hall: London, UK, 1991; Available online: https://www.routledge.com/Dictionary-of-Terpenoids/onnollyHill/p/book/9780412257704# (accessed on 12 December 1991).
- Yang, L.; Wurm, T.; Poudel, B.S.; Kriche, M.J. Enantioselective total synthesis of andrographolide and 14-hydroxy-colladonin: Carbonyl reductive Coupling and trans-decalin formation via hydrogen transfer. Angew. Chem. Int. Ed. 2020, 59, 23169–23173. [Google Scholar] [CrossRef]
- Alkofahi, A.; Ma, W.W.; Mckenzie, A.T.; Byrn, S.R.; McLaughlin, J.L. Goniotriol from Goniathalamus giganteus. J. Nat. Prod. 1989, 52, 1371–1373. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Sugimoto, M.; Oda, A.T.; Makabe, H. Synthesis of (+)-goniopypyrone and (+)-goniotriol using Pd-catalyzed carbonylation. Tetrahedron Lett. 2019, 60, 151039. [Google Scholar] [CrossRef]
- Li, C.; Lee, D.; Graf, T.N.; Phifer, S.S.; Nakanishi, Y.; Riswan, S.; Setyowati, F.M.; Saribi, A.M.; Soejarto, D.D.; Farnsworth, N.R.; et al. Bioactive constituents of the stem bark of mitrephora glabra. J. Nat. Prod. 2009, 72, 1949–1953. [Google Scholar] [CrossRef]
- Doda, S.R.; Raghvendor, A.; Haridasyam, S.B.; Putta, C.S.; Rao, B.K.; Kadari, S. Asymmetric total synthesis of filamentous fungi related resorcylic acid lactones 7-epi-zeaenol and zeaenol. Heterocycl. Commun. 2019, 25, 78–84. [Google Scholar] [CrossRef]
- Thomas, R. A biosynthetic classification of fungal and streptomycete fused-ring aromatic polyketides. ChemBioChem 2001, 2, 612–627. [Google Scholar] [CrossRef]
- Das, P.; Reddy, D.S. Total synthesis of twelve membered resorcyclic acid lactones, (R)-penicimenolide A, (R)-resorcyclide and (R)-dihydroresorcyclide. Tetrahedron 2021, 85, 132059. [Google Scholar] [CrossRef]
- Edukondalu, P.; Sreenivasulu, R.; Raju, R.R. Stereoselective total synthesis of (−)-pyrenophorin. Chem. Pap. 2020, 74, 2945–2950. [Google Scholar] [CrossRef]
- Yamada, Y.; Gilliland, K.; Xiang, Z.; Haymer, D.; Crocker, K.E.; Loch, M.T.; Schulte, M.L.; Rodriguez, A.L.; Niswender, C.M.; Conn, P.J.; et al. Positive allosteric modulators (PAMs) of the group II metabotropic glutamate receptors: Design, synthesis, and evaluation as ex-vivo tool compounds. Bioorg. Med. Chem. Lett. 2021, 50, 128342. [Google Scholar] [CrossRef]
- Gao, W.; Chai, C.; He, Y.; Li, F.; Hao, X.; Cao, F.; Gu, L.; Liu, J.; Hu, Z.; Zhang, Y. Periconiastone A, an antibacterial ergosterol with a pentacyclo [8.7.0.01,5.02,14.010,15]heptadecane system from Periconia sp. TJ403-rc01. Org. Lett. 2019, 21, 8469–8472. [Google Scholar] [CrossRef]
- Chen, P.; Wang, C.; Yang, R.; Xu, H.; Wu, J.; Jiang, H.; Chen, K.; Ma, Z. Asymmetric total synthesis of dankasterones A and B and periconiastone a through radical cyclization. Angew. Chem. Int. Ed. 2021, 60, 5512–5518. [Google Scholar] [CrossRef]
- Zhang, F.; Li, L.; Niu, S.; Si, Y.; Guo, L.; Jiang, X.; Che, Y.J. A thiopyranchromenone and other chromone derivatives from an endolichenic fungus, Preussia Africana. Nat. Prod. 2012, 75, 230–237. [Google Scholar] [CrossRef]
- Beller, M.P.; Harms, C.; Koert, U. Total synthesis of (−)-preussochromone A. Org. Lett. 2020, 22, 6127–6131. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Anticancer activity of natural and synthetic acetylenic lipids. Lipids 2006, 41, 883–924. [Google Scholar] [CrossRef]
- Casotti, G.; Fusini, G.; Ferriri, M.; Pardini, F.P.; Evangelisti, C.; Angelici, G.; Carpita, A. Total synthesis of asparenydiol by two Sonogashira cross-coupling reactions promoted by supported Pd and Cu catalysts. Synthesis 2020, 52, 1795–1803. [Google Scholar] [CrossRef]
- Arrayas, R.G.; Alcudia, A.; Liebeskind, L.S. Facile enantiodivergent approach to 5-hydroxy-5,6-dihydro-2(1H)-pyridones. First total synthesis of both enantiomers of pipermethystine. Org. Lett. 2001, 3, 3381–3383. [Google Scholar] [CrossRef] [PubMed]
- Amaya, L.Y.V.; Quintero, L.; Molina, B.R.; Piscil, F.S. Transition-metal-free total synthesis and revision of the absolute configuration of pipermethystine. J. Org. Chem. 2020, 85, 3949–3953. [Google Scholar] [CrossRef] [PubMed]
- Shabani, S.; White, J.M.; Hutton, C.A. Total synthesis of the putative structure of asperipin-2a and stereochemical reassignment. Org. Lett. 2020, 22, 7730–7734. [Google Scholar] [CrossRef] [PubMed]
- Garnier-Amblard, E.C.; Mays, S.G.; Arrendale, R.F.; Baillie, M.T.; Bushnev, A.S.; Culver, D.G.; Evers, T.J.; Holt, J.J.; Howard, R.B.; Liebeskind, L.S. Novel synthesis and biological evaluation of enigmols as therapeutic agents for treating prostate cancer. ACS Med. Chem. Lett. 2011, 2, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.A.; Haque Ashanul Yadav, J.S. Stereoselective total synthesis of ()-galantinic acid and 1-deoxy-5-hydroxysphingolipids via prins cyclization. Tetrahedron Lett. 2020, 61, 152149. [Google Scholar] [CrossRef]
- Igarashi, Y.; Yu, L.; Ikeda, M.; Oikawa, T.; Kitani, S.; Nihira, T.; Bayanmunkh, B.; Panbangred, W. Jomthonic acid A, a modified amino acid from a soil-derived Streptomyces. J. Nat. Prod. 2012, 75, 986–990. [Google Scholar] [CrossRef]
- Dumpala, M.; Srinivas, B.; Krishna, P.R. First total synthesis of jomthonic Acid A. Synlett 2019, 31, 69–72. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, J.; Wu, P.; Xu, L.; Xie, H.; Wei, X. Polyoxygenated methyl cyclohexanoids from a terrestrial Ampelomyces fungus. J. Nat. Prod. 2009, 72, 265–269. [Google Scholar] [CrossRef]
- Brindisi, C.; Vazquez, S.; Suescun, L.; Seoane, G.A.; Martin, V.S.; Brovetto, M. Chemoenzymatic total synthesis and structural revision of ampelomins B, D, E, and epi-ampelomin B. J. Org. Chem. 2019, 84, 15997–16007. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Pereira, A.R.; Debonsi, H.M.; Ligresti, A.; Marzo, V.D.; Gerwick, W.H. Cannabinomimetic lipid from a marine cyanobacterium. J. Nat. Prod. 2011, 74, 2313–2317. [Google Scholar] [CrossRef]
- Lopez, J.A.V.; Petitbois, J.G.; Vairappan, C.S.; Umezawa, T.; Matsuda, F.; Okino, T. Columbamides D and E: Chlorinated fatty acid amides from the marine cyanobacterium Moorea bouillonii collected in Malaysia. Org. Lett. 2017, 19, 4231–4234. [Google Scholar] [CrossRef] [PubMed]
- Ghotekar, G.S.; Mujahid, M.; Muthukrishnan, M. Total synthesis of marine natural products serinolamide A and columbamide D. ACS Omega 2019, 4, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Elovson, J.; Vagelos, P.R. A new class of lipids: Chlorosulfolipids. Proc. Natl. Acad. Sci. USA 1969, 62, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Carriera, E.M. Stereochemical revision, total synthesis and solution state conformation of the complex chlorosulfolipid mytilipin B. J. Am. Chem. Soc. 2019, 141, 10510–10519. [Google Scholar] [CrossRef]
- Nakayama, A.; Sato, H.; Nagano, S.; Karanjit, S.; Imagawa, H.; Namba, K. Asymmetric total syntheses and structure elucidations of (+)-eurotiumide F and (+)-eurotiumide G. Chem. Pharm. Bull. 2019, 67, 953–958. [Google Scholar] [CrossRef]
- Lam, K.S.; Hesler, G.A.; Gustavson, D.R.; Crosswell, A.R.; Veitch, J.M.; Forenza, S.; Tomita, K. Kedarcidin, a new chromoprotein antitumor antibiotic. I. Taxonomy of producing organism, fermentation and biological activity. J. Antibiot. 1991, 5, 472–478. [Google Scholar] [CrossRef][Green Version]
- Lear, M.J.; Hirai, K.; Ogawa, K.; Yamashita, S.; Hirama, M. A convergent total synthesis of the kedarcidin chromophore: 20-years in the making. J. Antibiot. 2019, 72, 350–363. [Google Scholar] [CrossRef]
- Williams, D.E.; Bombuwala, K.; Lobkovsky, E.; de Silva, E.D.; Karunatne, V.; Allen, T.M.; Clardy, J.; Andersen, R.J. Ambewelamides A and B, antineoplastic epidithiapiperazinediones isolated from the lichen Usnea sp. Tetrahedron Lett. 1998, 39, 9579–9582. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Banne, S.; Guo, J.; He, Y. Towards the Total synthesis of scabrosins: Synthesis of desulfur-scabrosins skeleton and its stereoisomers. J. Org. Chem. 2019, 84, 5838–5845. [Google Scholar] [CrossRef]
- Igarashi, M.; Nakagawa, N.; Doi, N.; Hattori, S.; Naganawa, H.; Hamada, M. Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J. Antibiotic. 2003, 56, 580–583. [Google Scholar] [CrossRef]
- Nakamura, H.; Tsukano, C.; Yoshida, T.; Yasui, M.; Yokouchi, S.; Kobayashi, Y.; Igarashi, M.; Takemoto, Y. Total synthesis of caprazamycin a: Practical and scalable synthesis of syn-β-hydroxyamino acids and introduction of a fatty acid side chain to 1,4-diazepanone. J. Org. Chem. Soc. 2019, 141, 8527–8540. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Y.; Yan, Y.M.; Meng, X.H.; Nafie, L.A.; Xu Te Dukor, R.K.; Qin, H.B.; Cheng, Y.X. Isolation, total synthesis, and absolute configuration determination of renoprotective dimeric N-acetyldopamine- adenine hybrids from the insect Aspongopus chinensis. Org. Lett. 2020, 22, 5726–5730. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Miyasaka, T.; Kudo, Y.; Sugimoto, K.; Yamashita, M.Y.; Nishikawa, T. Total syntheses and determination of absolute configurations of cep-212 and cep-210, predicted biosynthetic intermediates of tetrodotoxin isolated from toxic newt. Org. Lett. 2019, 21, 780–784. [Google Scholar] [CrossRef]
- Springer, J.P.; Cutler, H.G.; Crumley, F.G.; Cox, R.H.; Davis, E.E.; Thean, J.E. Plant growth regulatory effects and stereochemistry of cladosporin. J. Agric. Food Chem. 1981, 29, 853–855. [Google Scholar] [CrossRef]
- Das, P.; Mankad, Y.; Reddy, D.S. Scalable synthesis of cladosporin. Tetrahedron Lett. 2019, 60, 831–833. [Google Scholar] [CrossRef]
- Wright, A.D.; König, G.M.; Nys, R.D.; Sticher, O. Seven new metabolites from the marine red alga Laurencia majuscule. J. Nat. Prod. 1993, 56, 394–401. [Google Scholar] [CrossRef]
- Shephard, E.D.; Dyson, B.S.; Hak, W.E.; Nguyen, Q.N.N.; Lee, M.; Kim, M.J.; Sohn, T.I.; Kim, D.; Burton, J.W.; Paton, R.S. Structure determination of a chloroenyne from Laurencia majuscule using computational methods and total synthesis. J. Org. Chem. 2019, 84, 4971–4991. [Google Scholar] [CrossRef]










































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munawar, S.; Zahoor, A.F.; Ali, S.; Javed, S.; Irfan, M.; Irfan, A.; Kotwica-Mojzych, K.; Mojzych, M. Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review. Molecules 2022, 27, 6953. https://doi.org/10.3390/molecules27206953
Munawar S, Zahoor AF, Ali S, Javed S, Irfan M, Irfan A, Kotwica-Mojzych K, Mojzych M. Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review. Molecules. 2022; 27(20):6953. https://doi.org/10.3390/molecules27206953
Chicago/Turabian StyleMunawar, Saba, Ameer Fawad Zahoor, Shafaqat Ali, Sadia Javed, Muhammad Irfan, Ali Irfan, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2022. "Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review" Molecules 27, no. 20: 6953. https://doi.org/10.3390/molecules27206953
APA StyleMunawar, S., Zahoor, A. F., Ali, S., Javed, S., Irfan, M., Irfan, A., Kotwica-Mojzych, K., & Mojzych, M. (2022). Mitsunobu Reaction: A Powerful Tool for the Synthesis of Natural Products: A Review. Molecules, 27(20), 6953. https://doi.org/10.3390/molecules27206953