
Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective


 , ,
, ,  and
and
Abstract
:
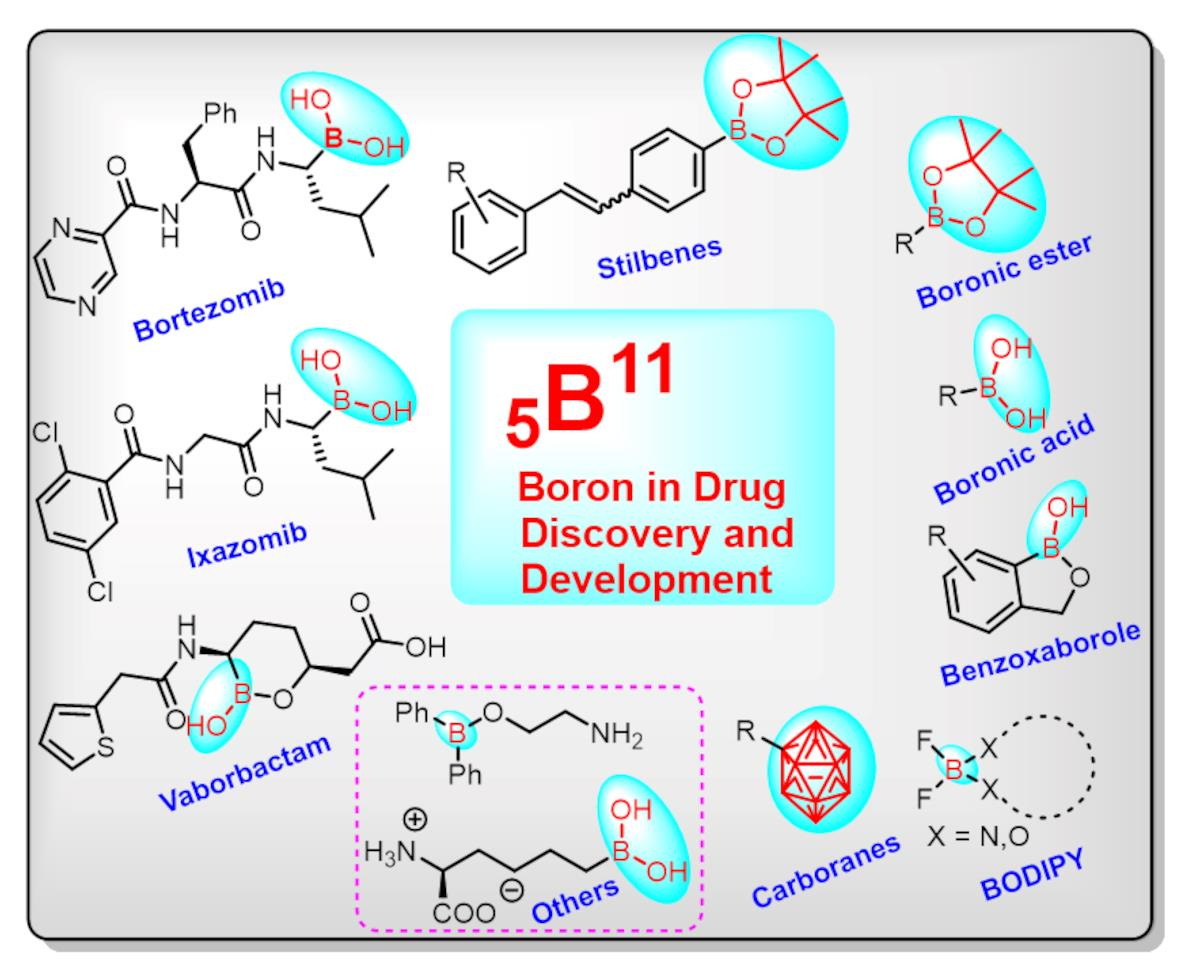
1. Introduction
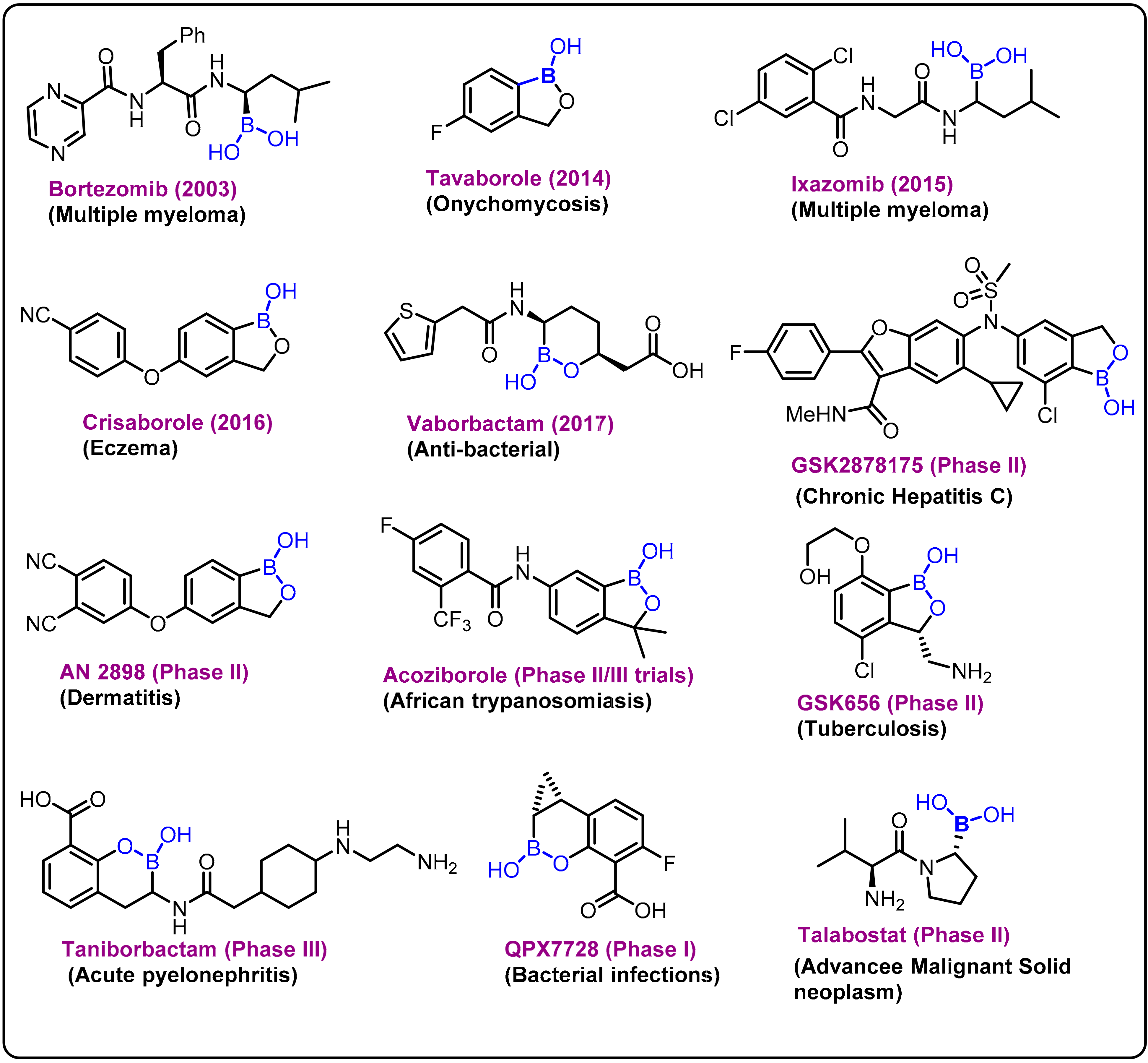
2. Boron-Based Therapeutics
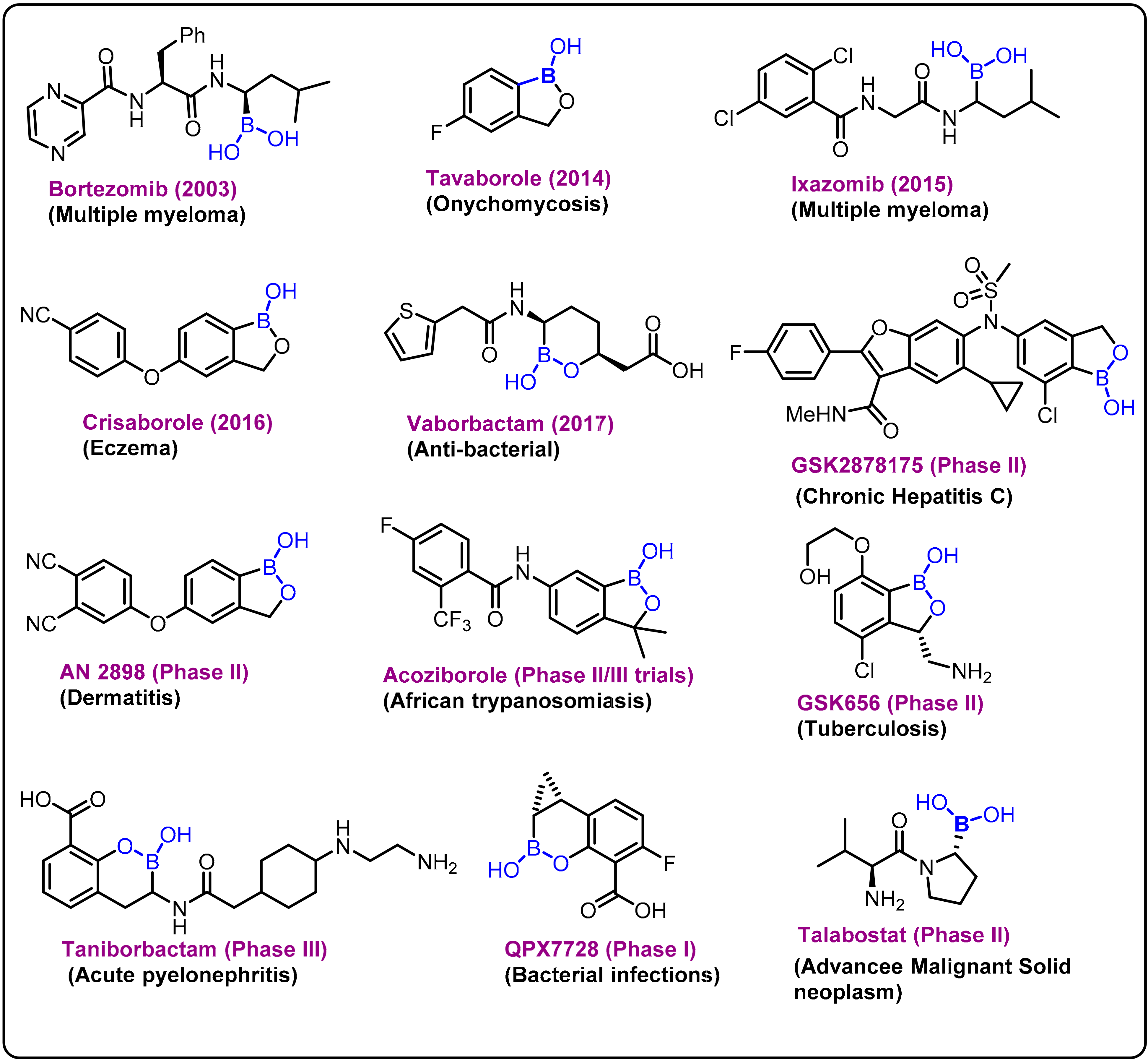
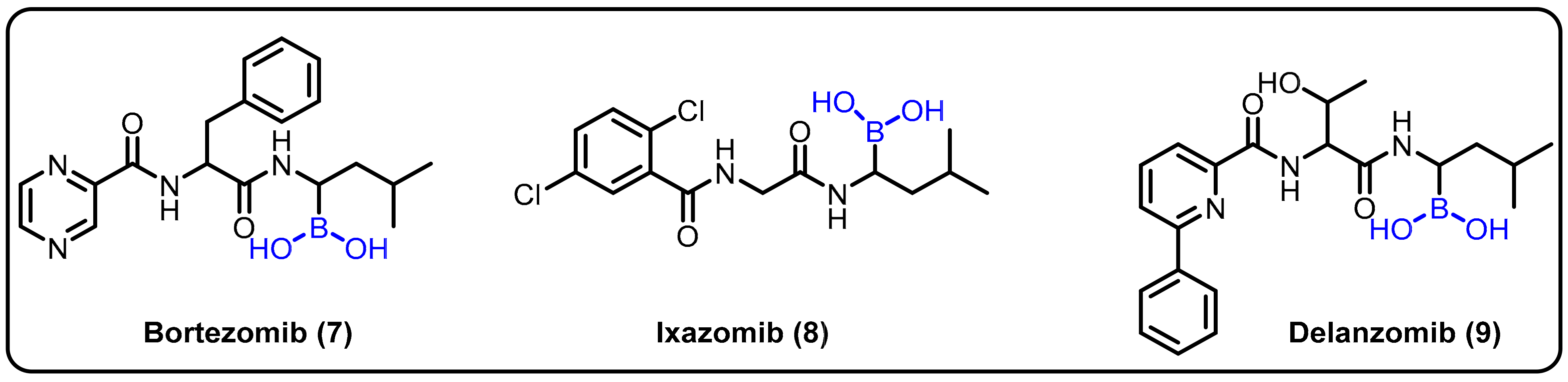
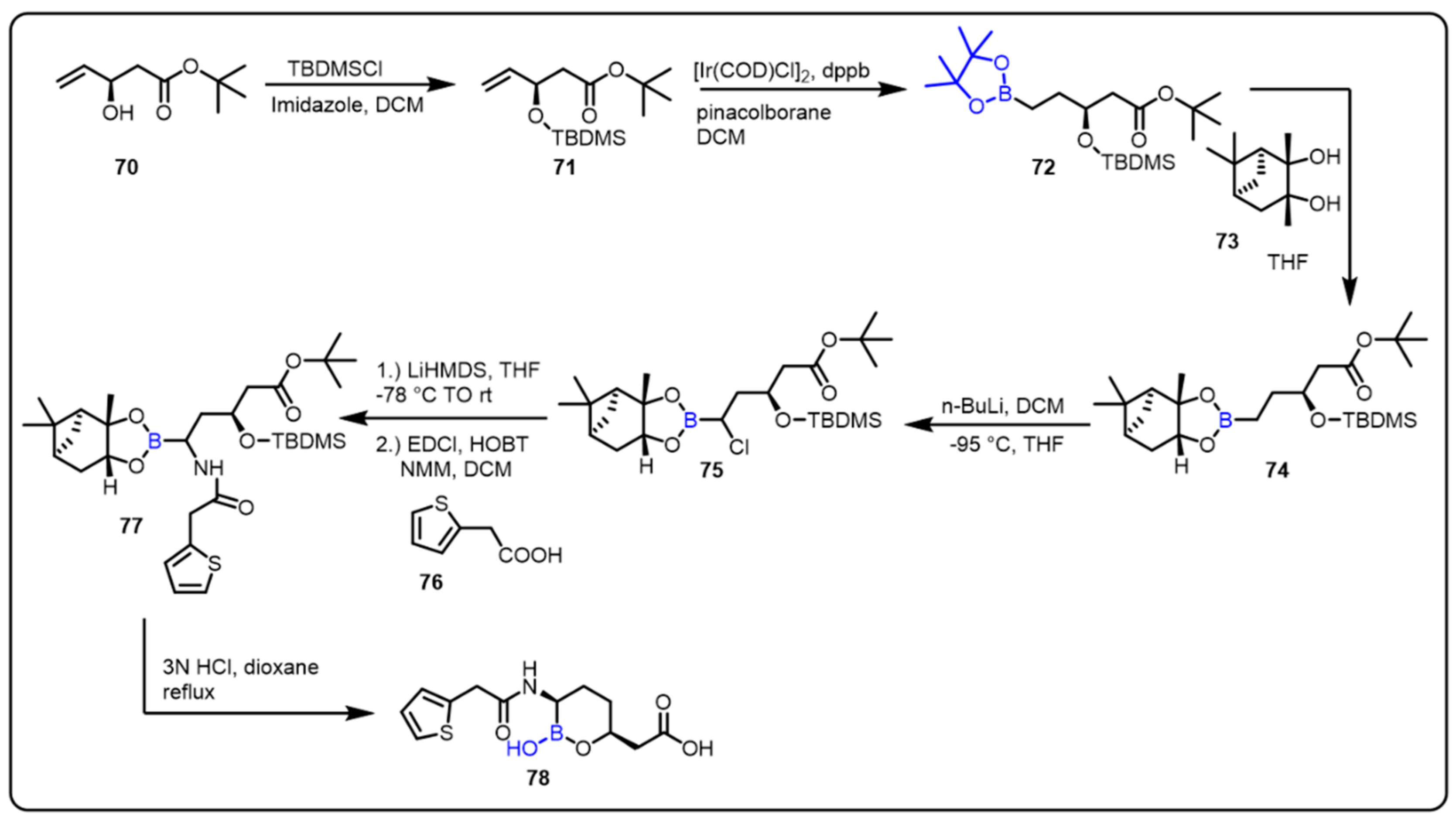
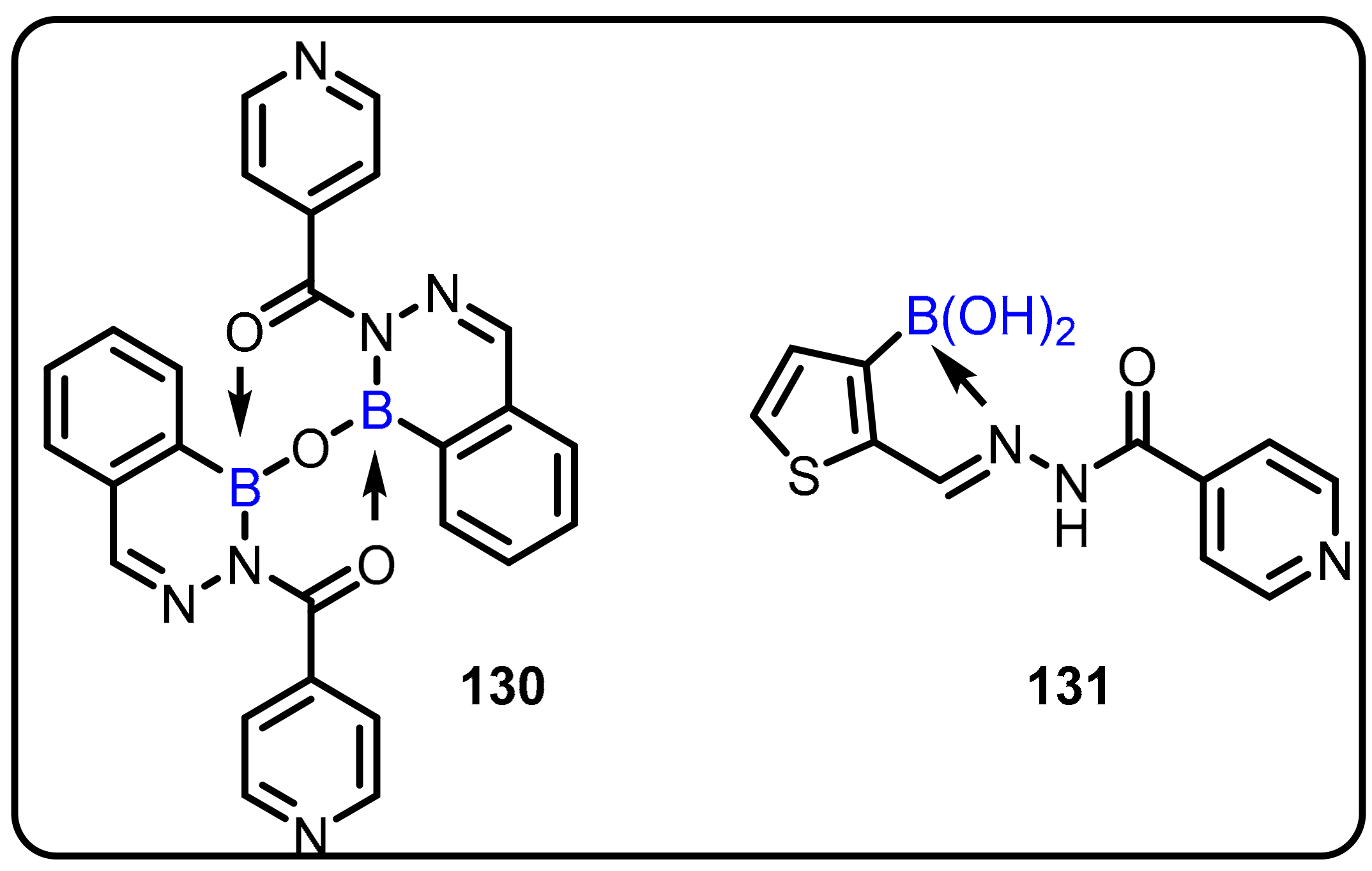
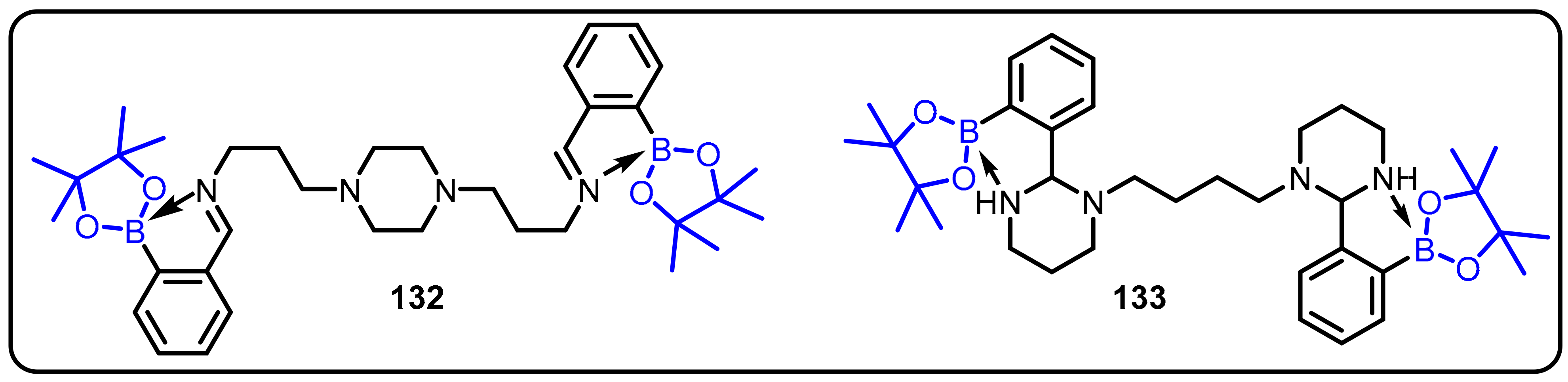
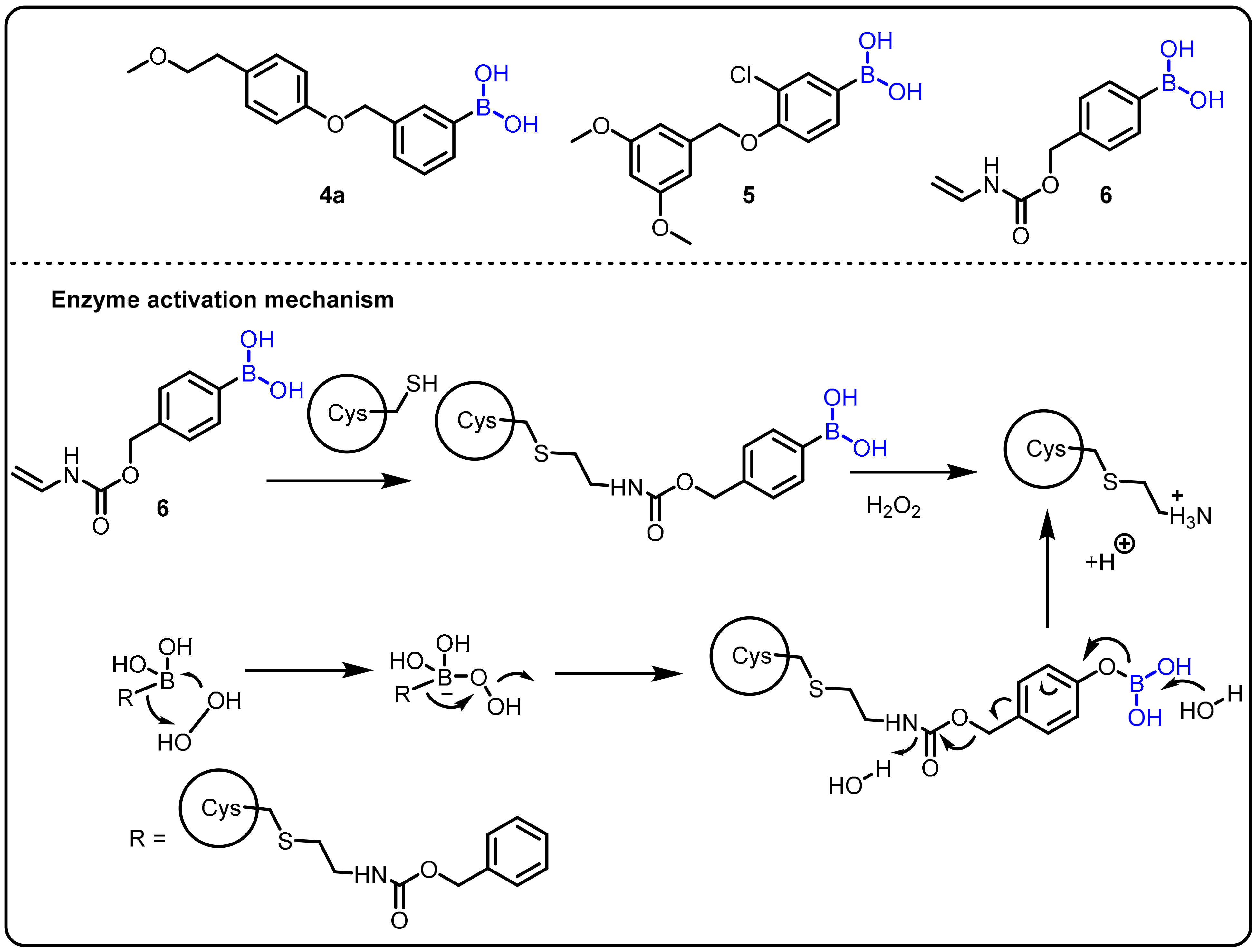
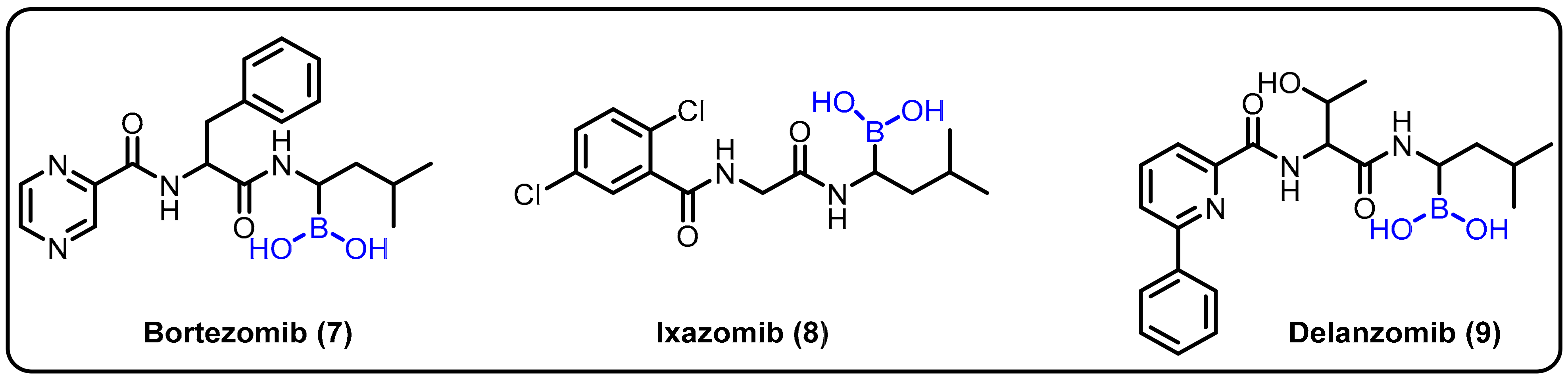
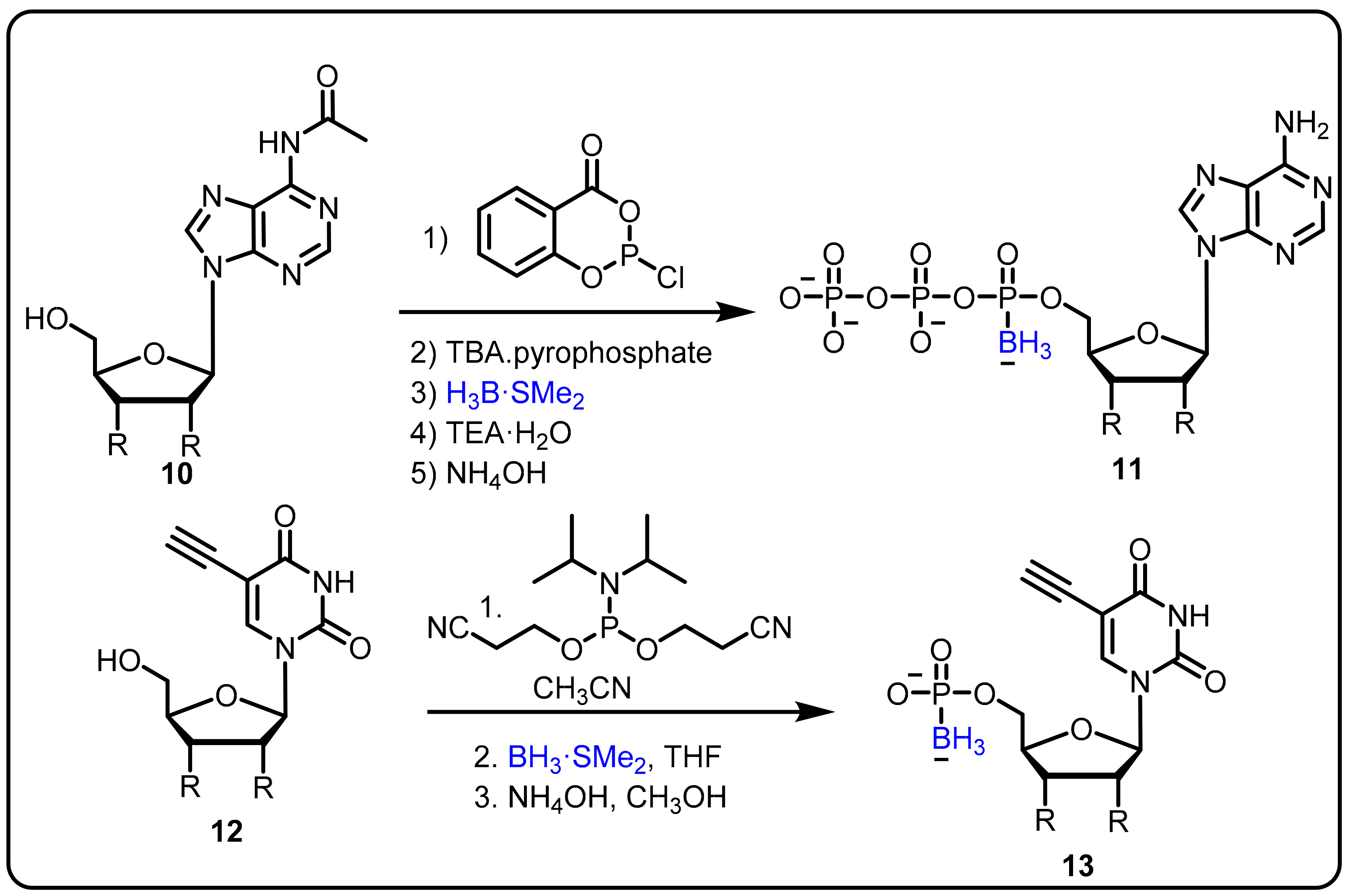
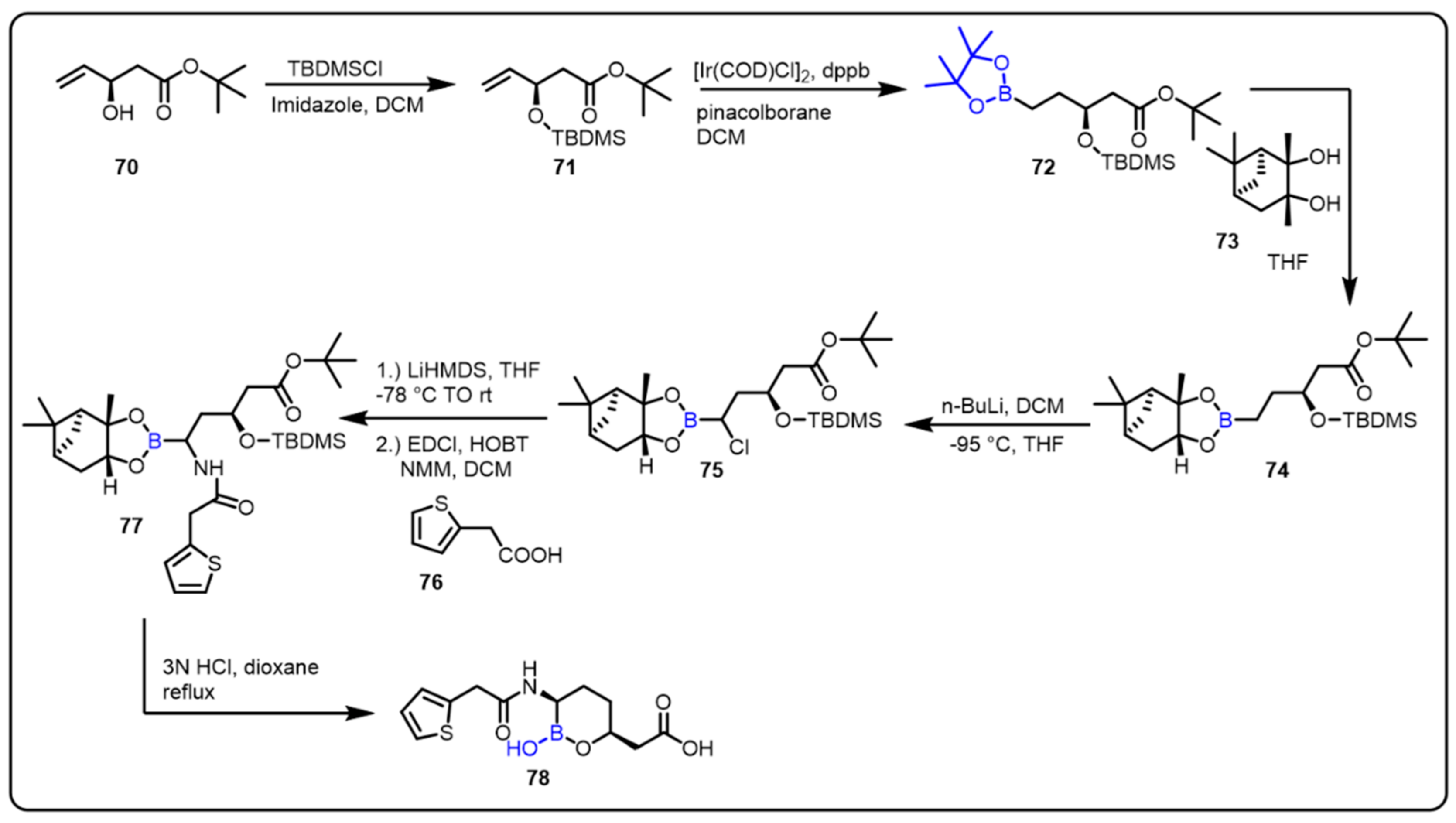
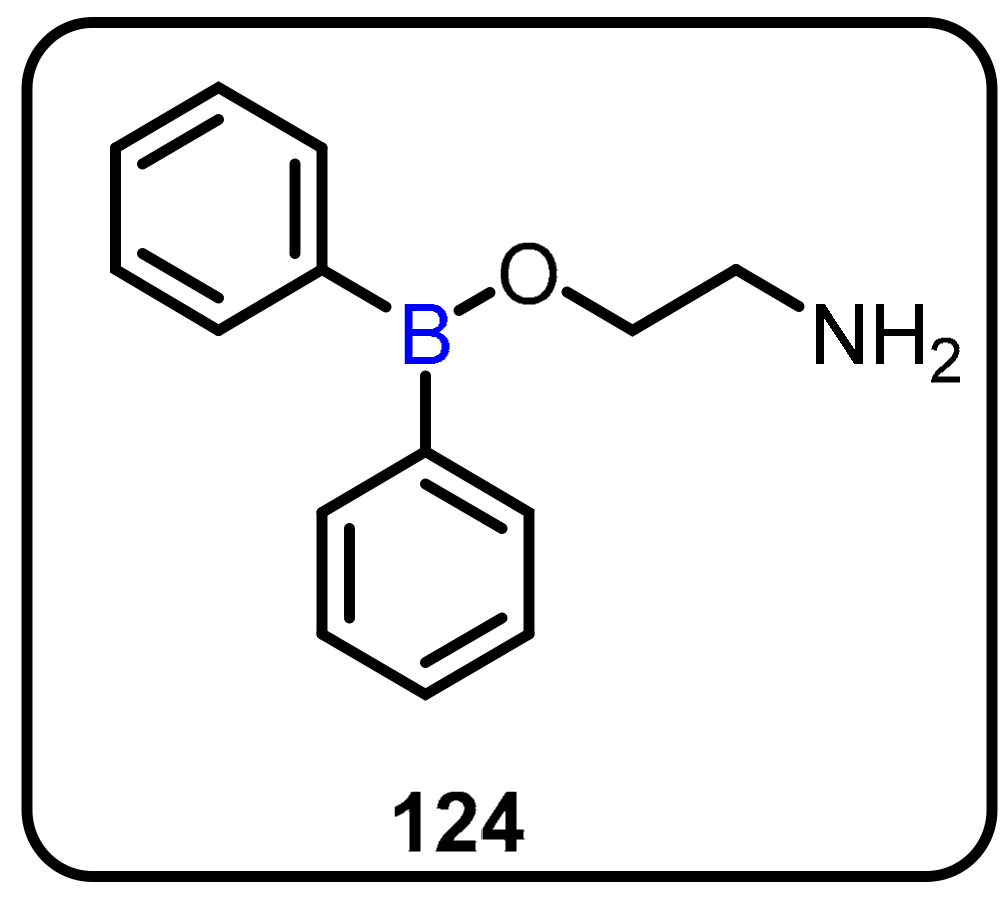
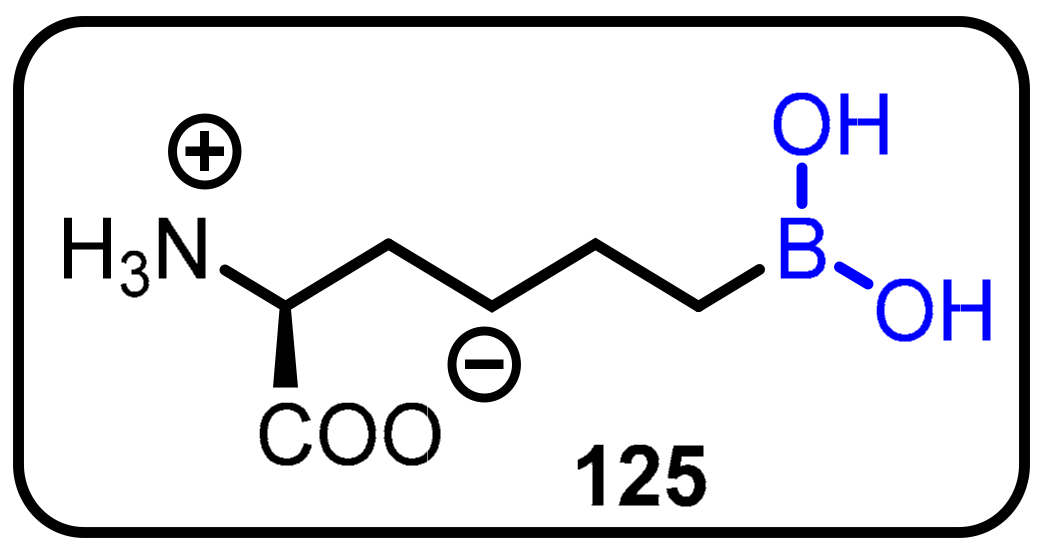
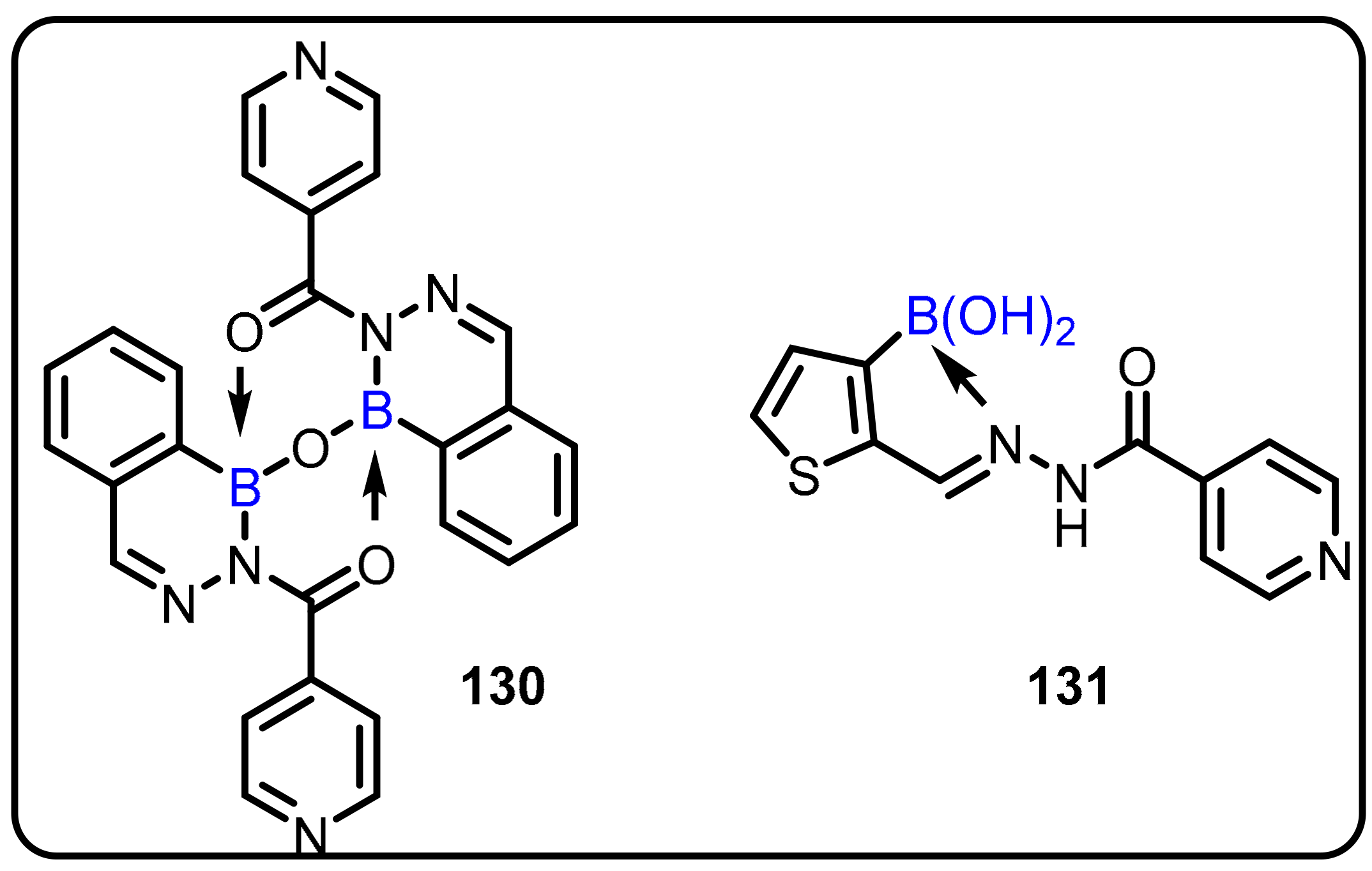
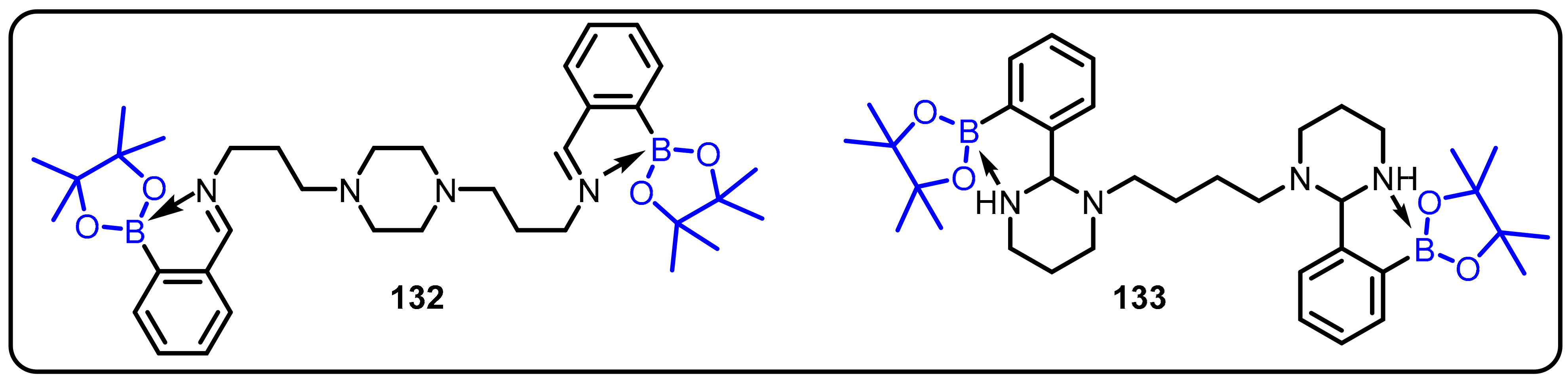
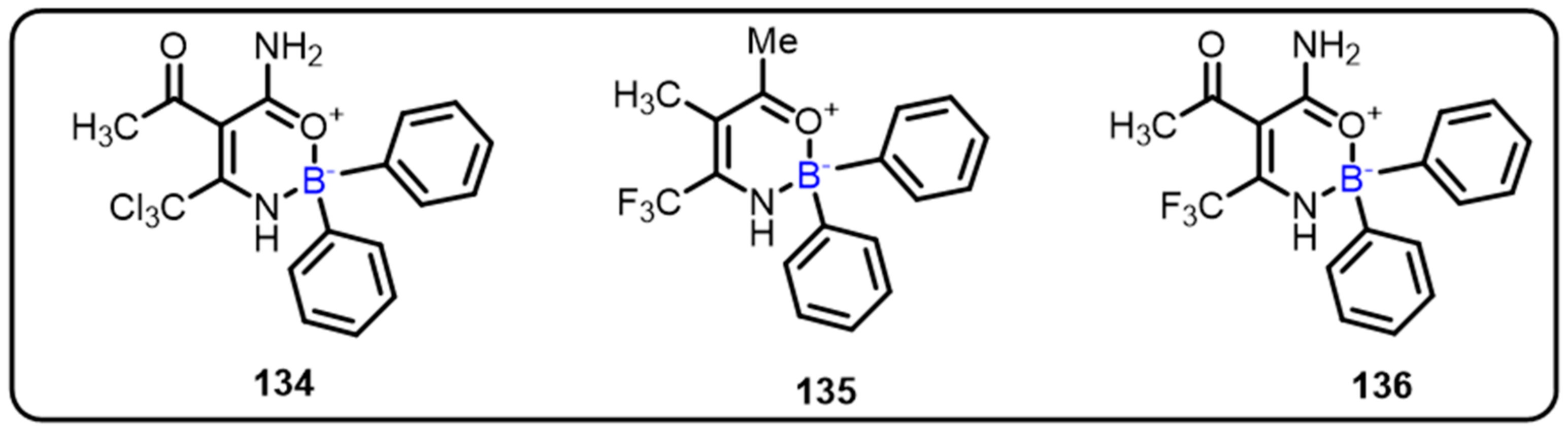
2.1. Boronic Acids/Esters/Aminoboronic Acids as Drug Motifs
Breakthrough 1
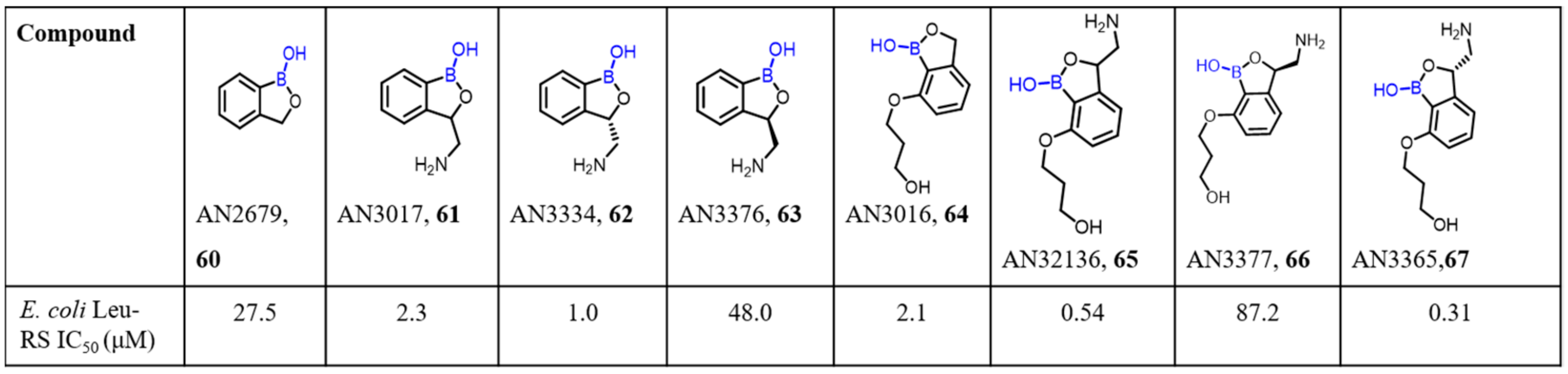
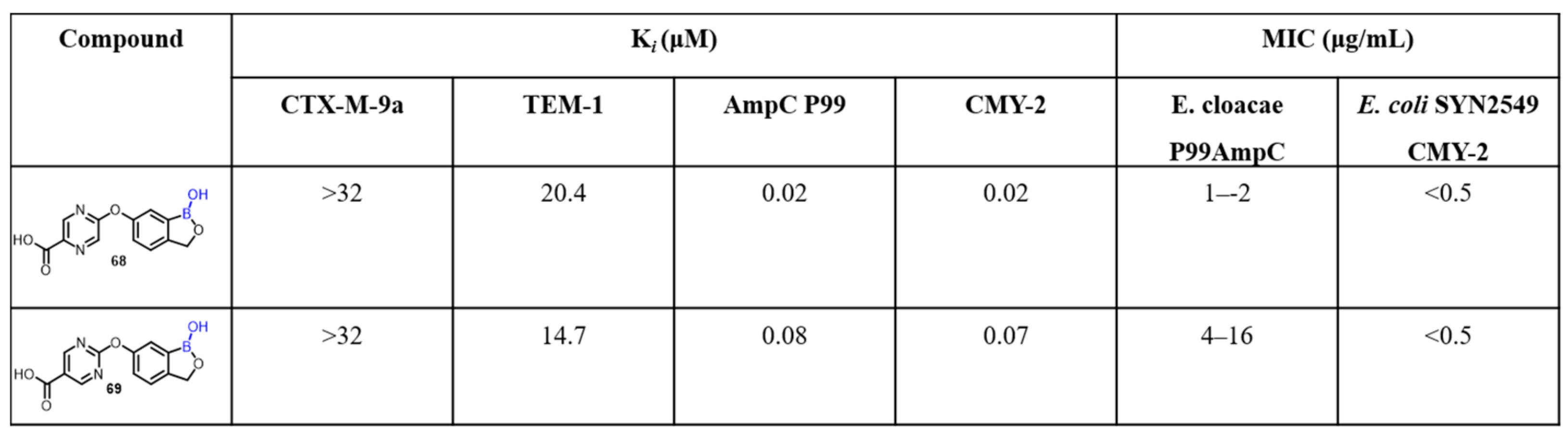
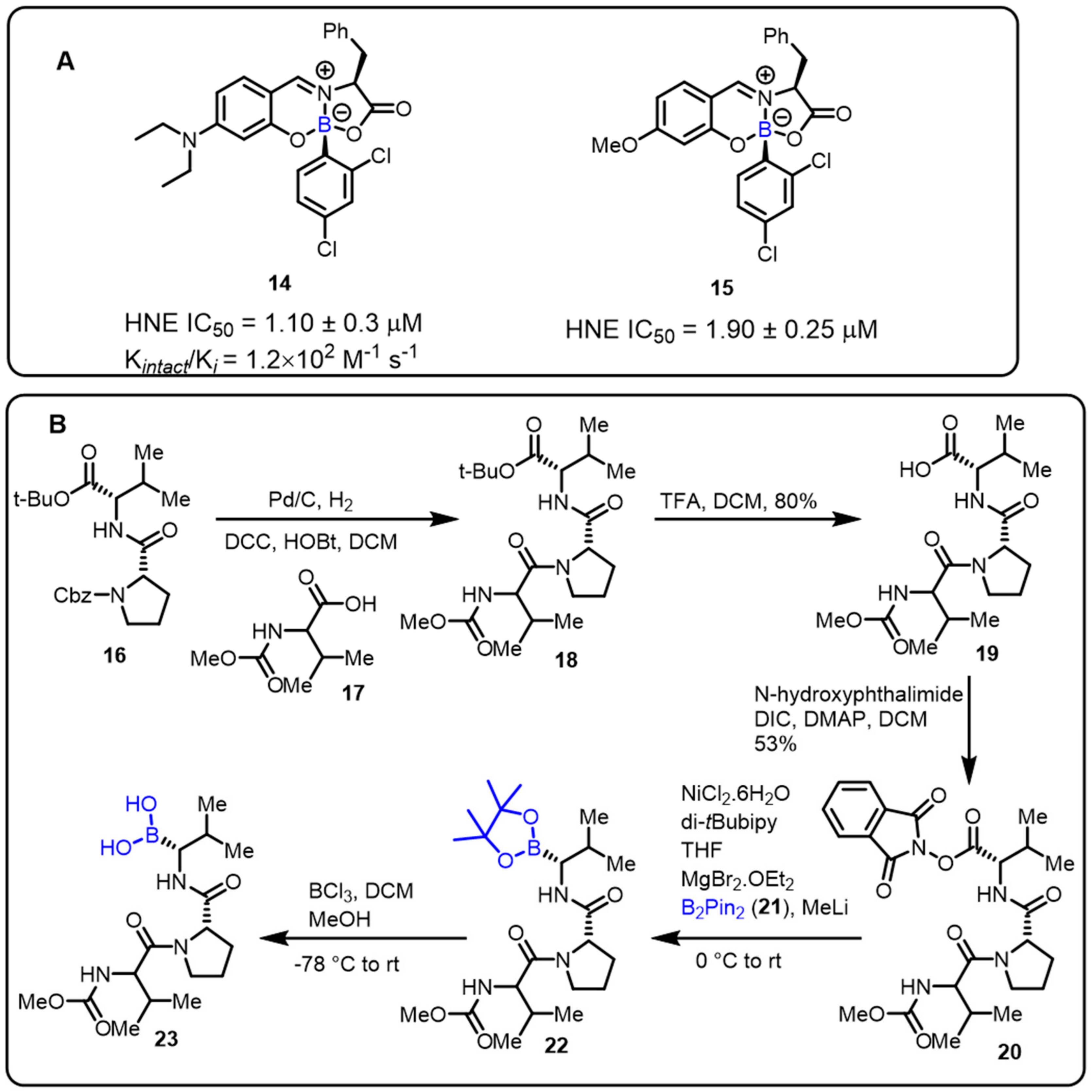
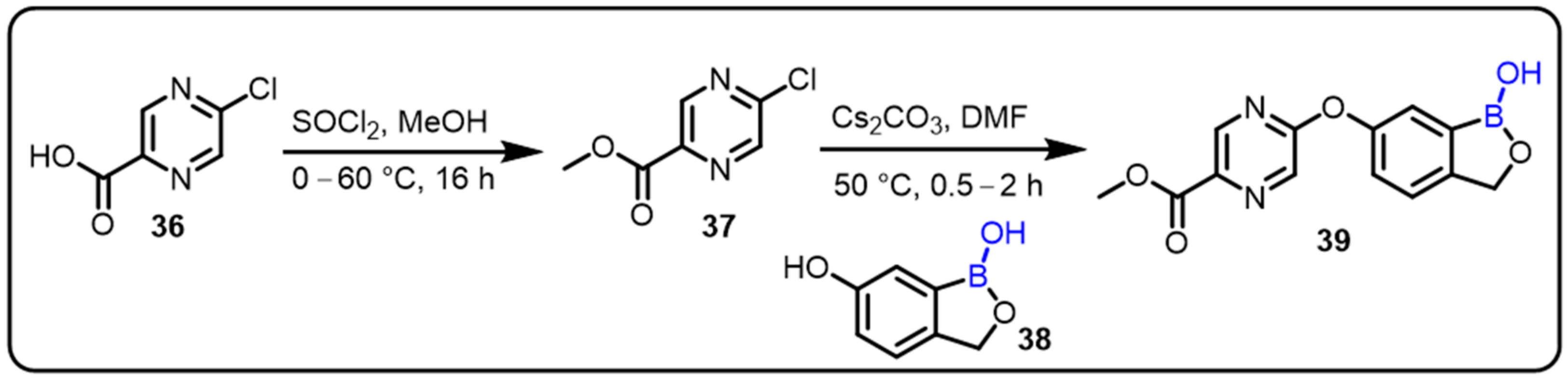
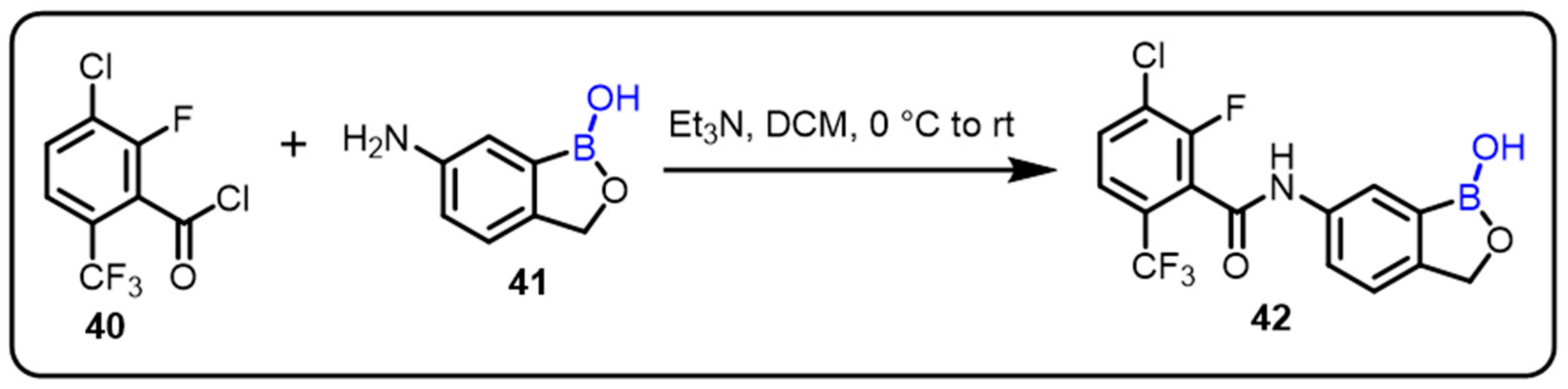
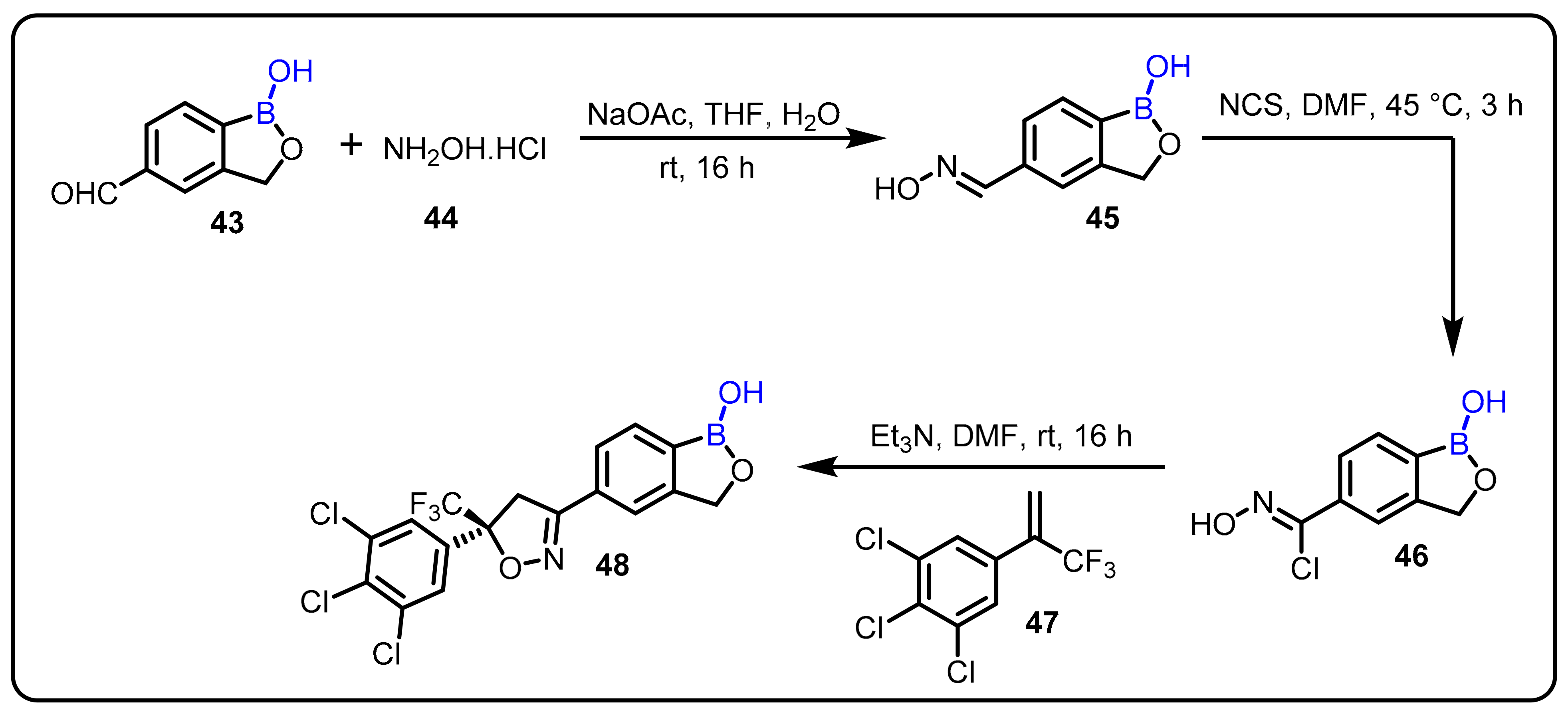
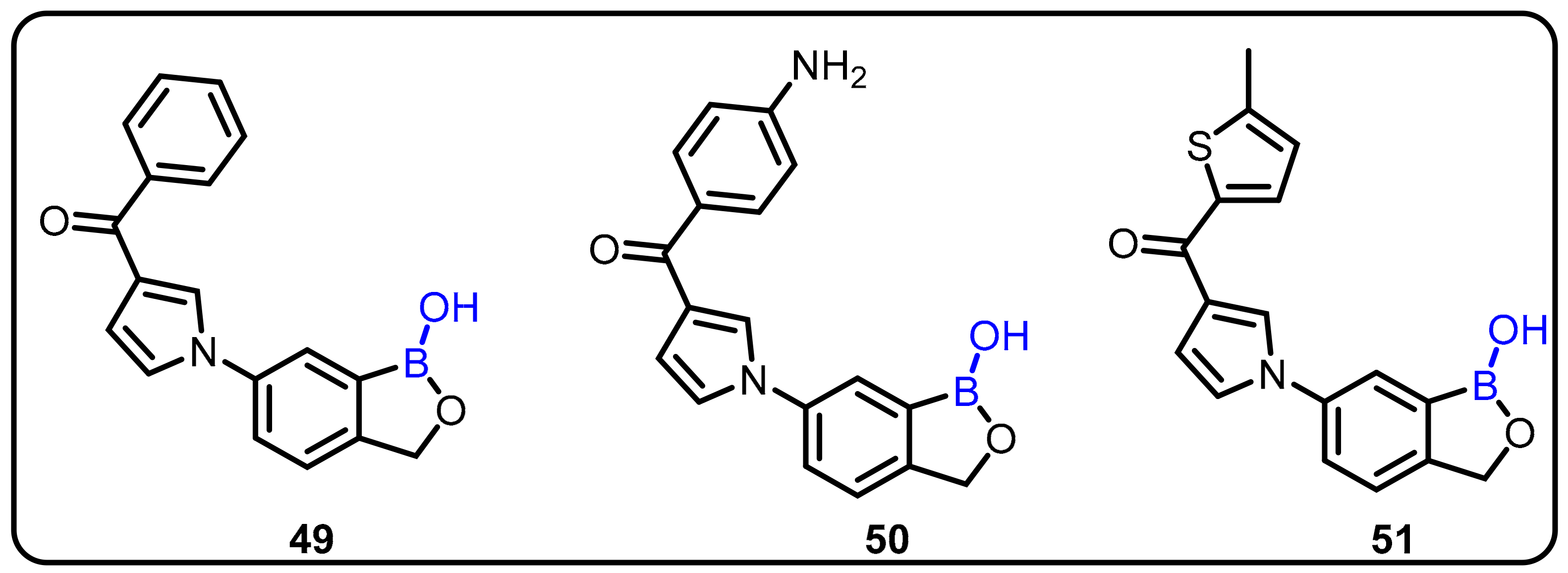
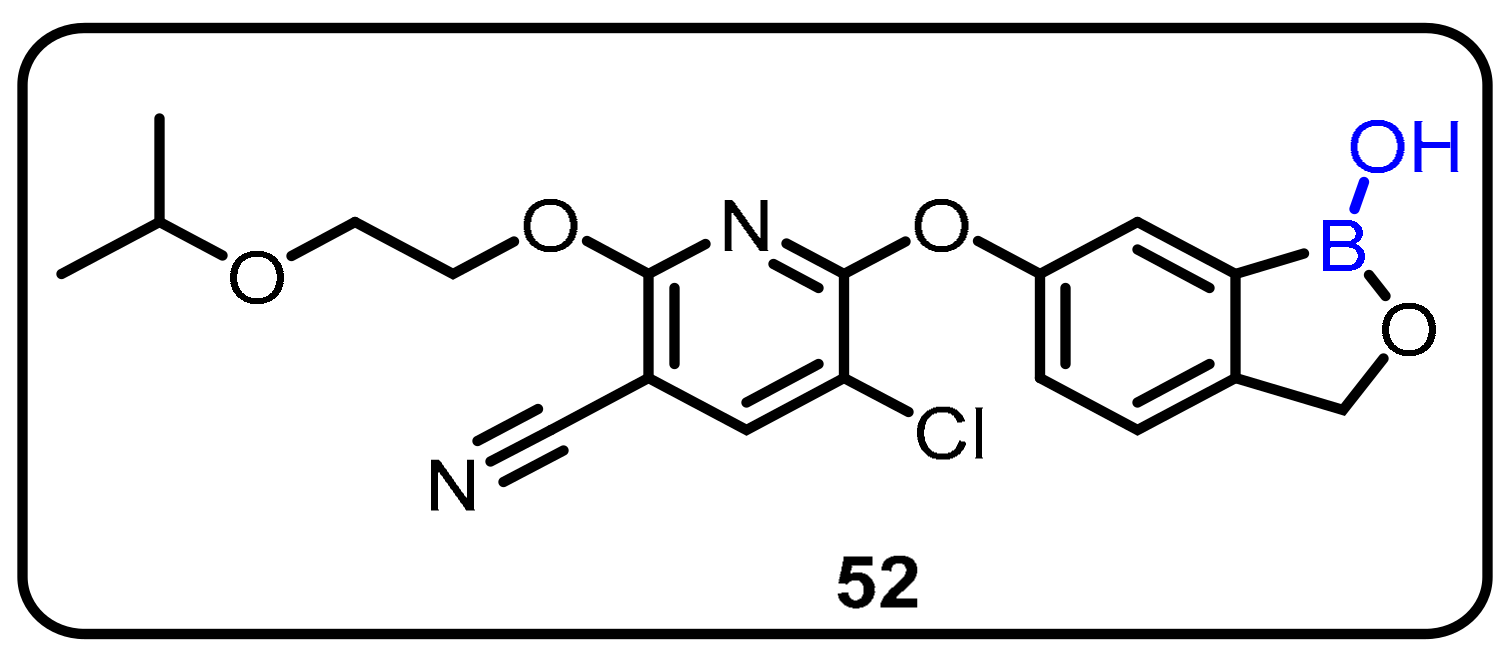
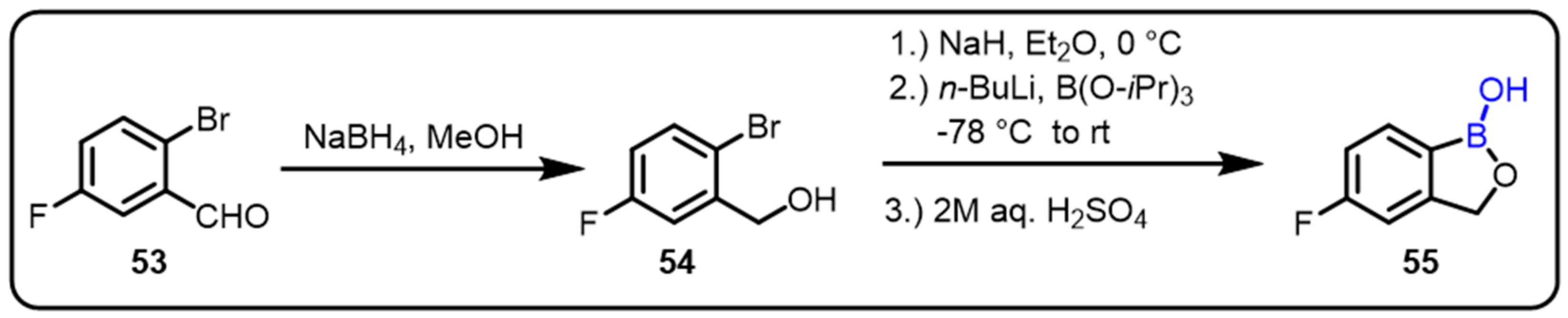
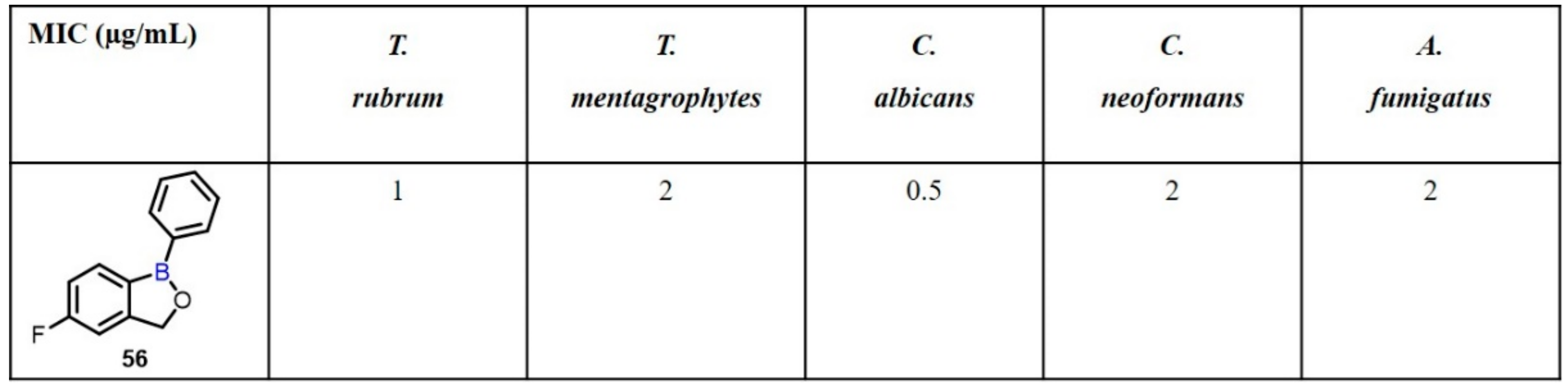
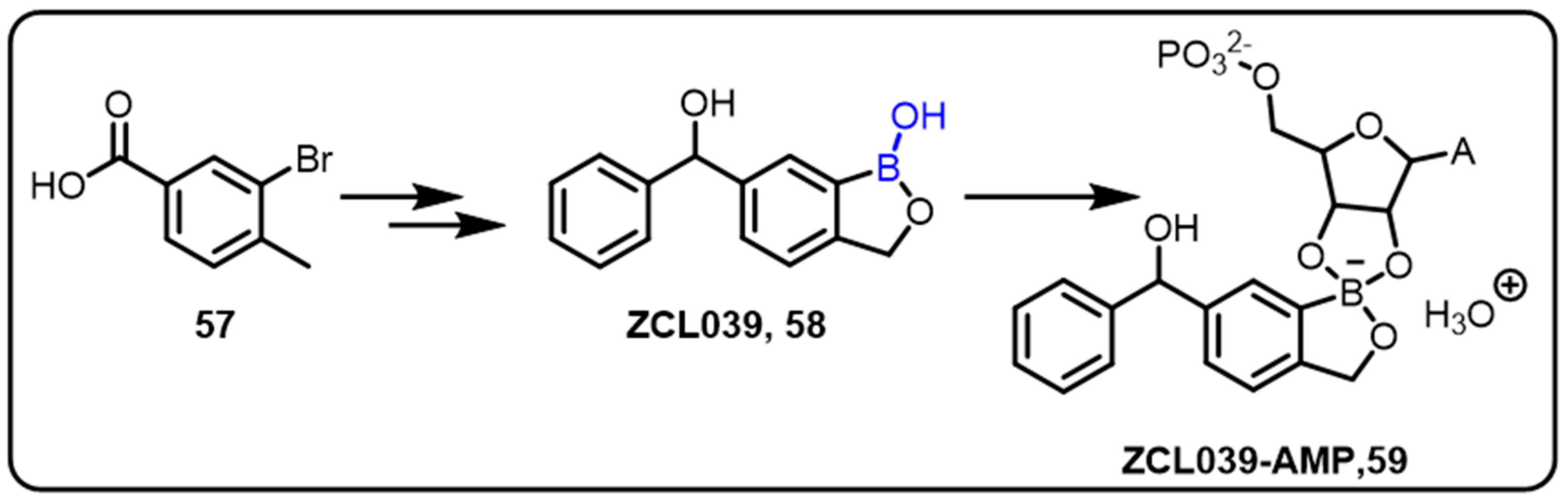
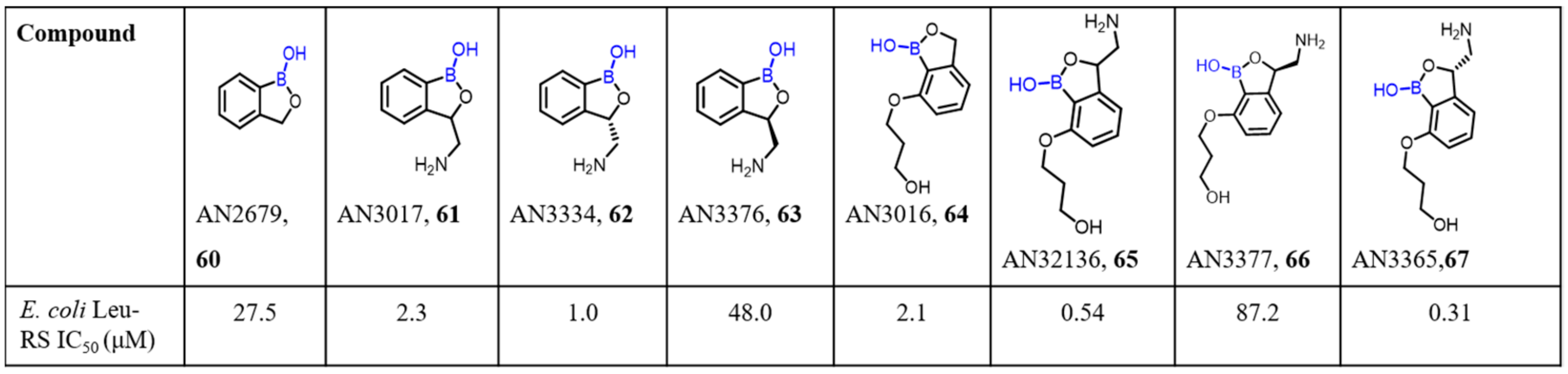
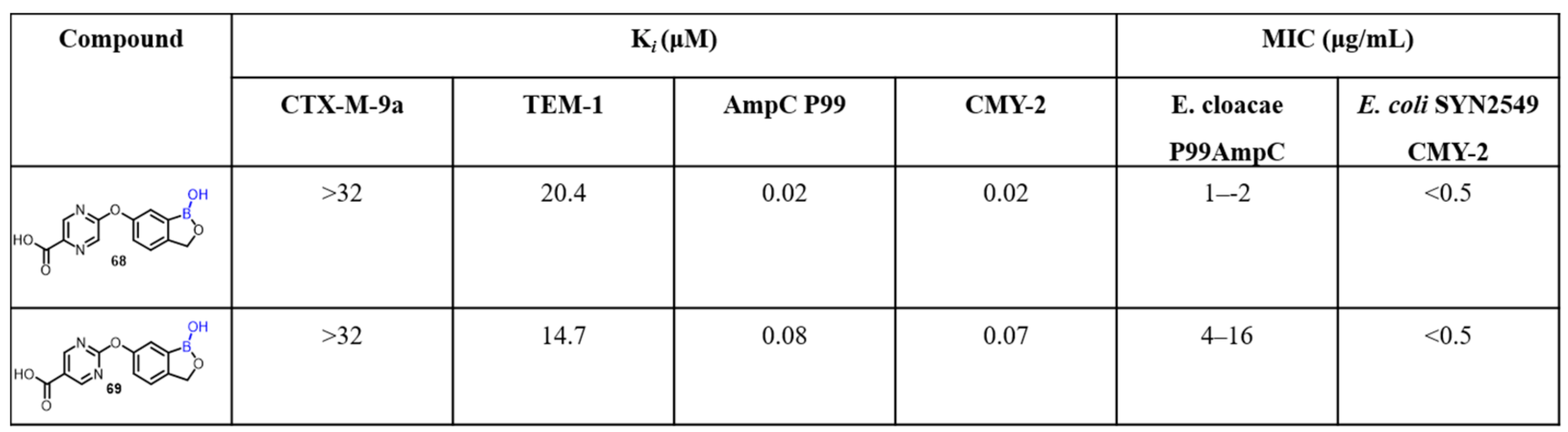
2.2. Benzoxaborole as Drug Motifs
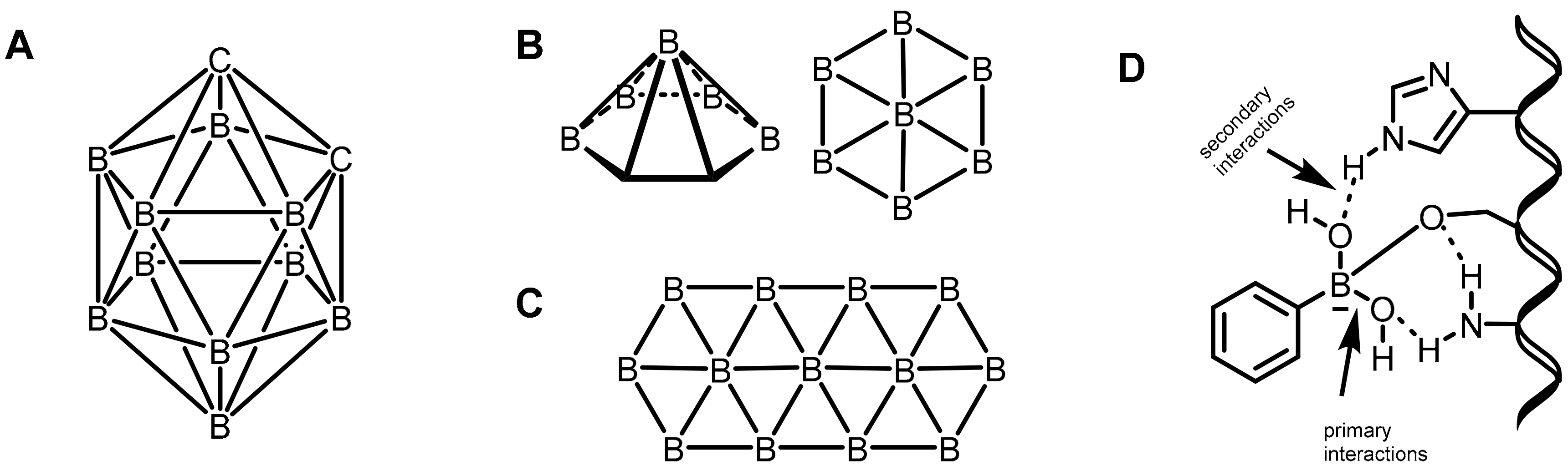
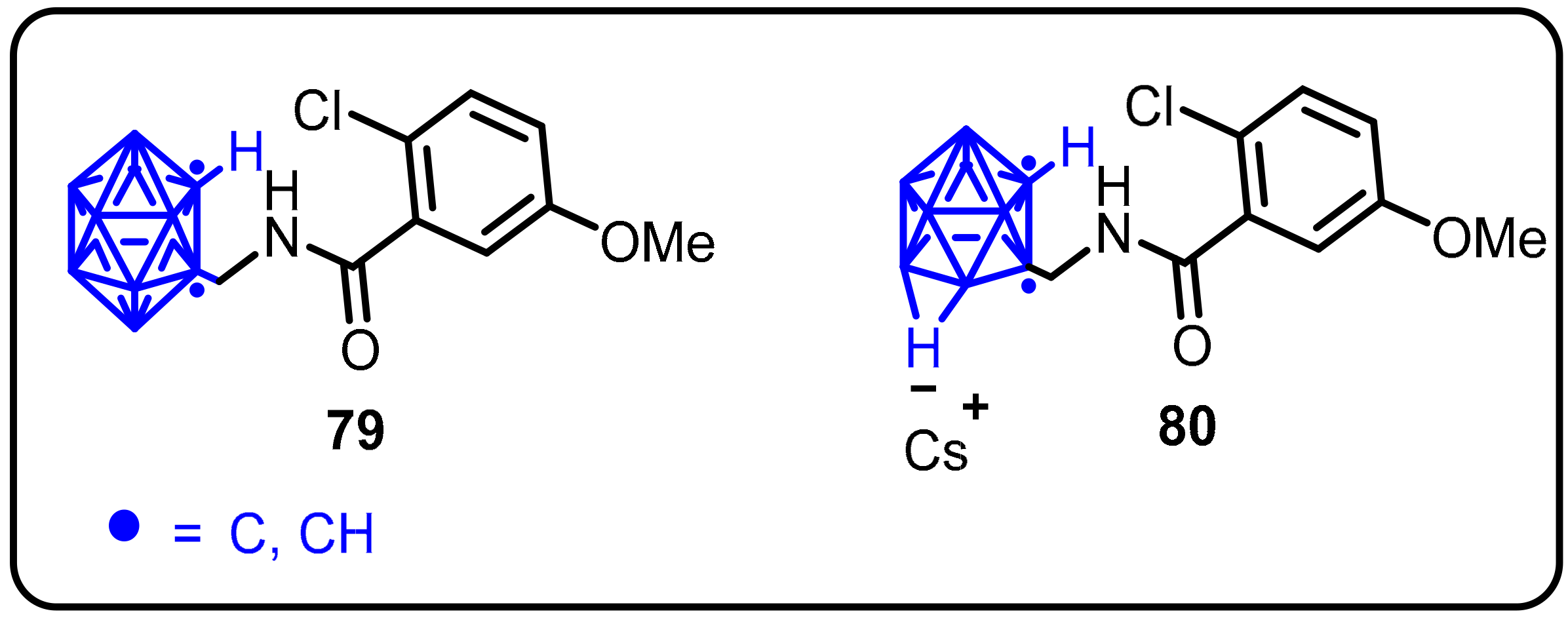
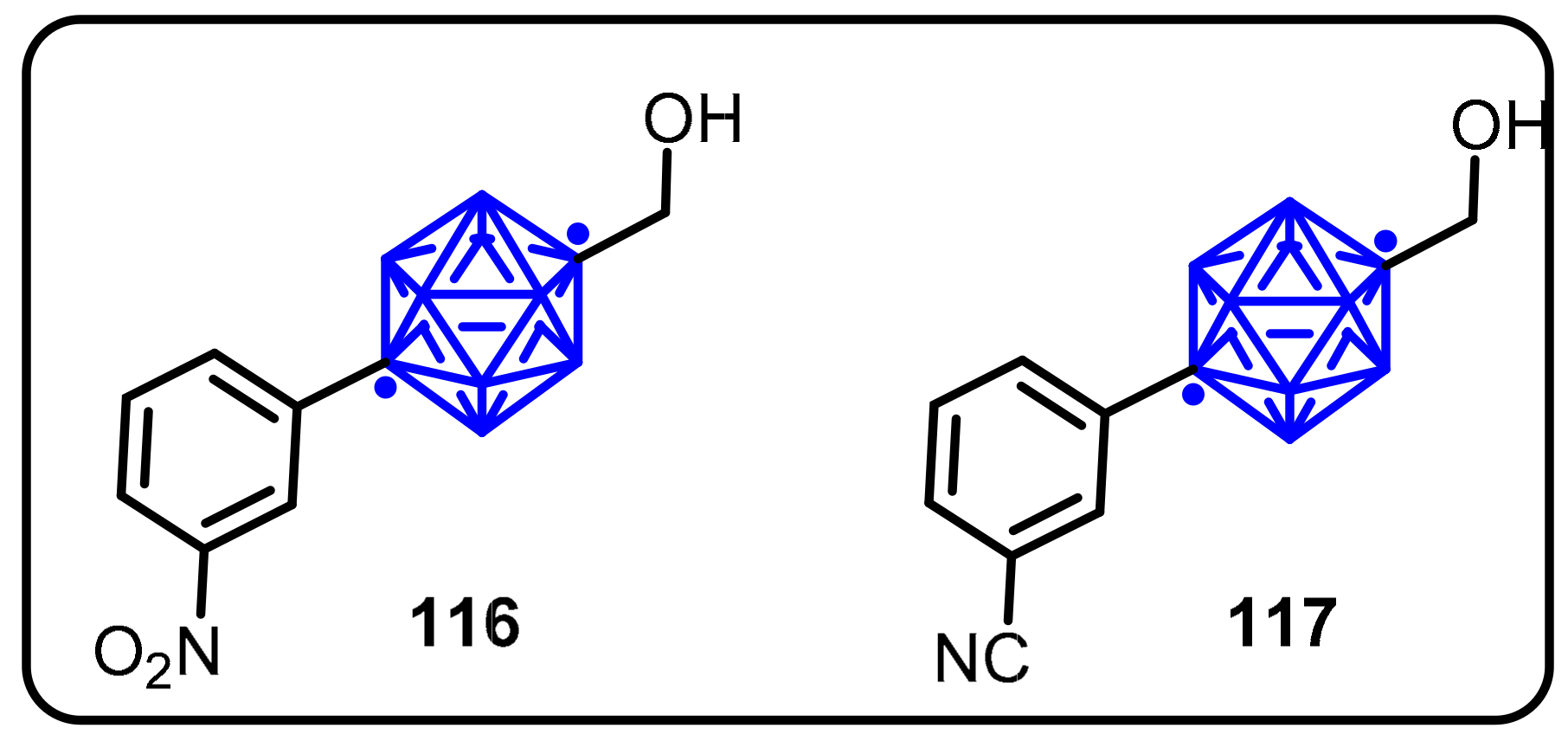
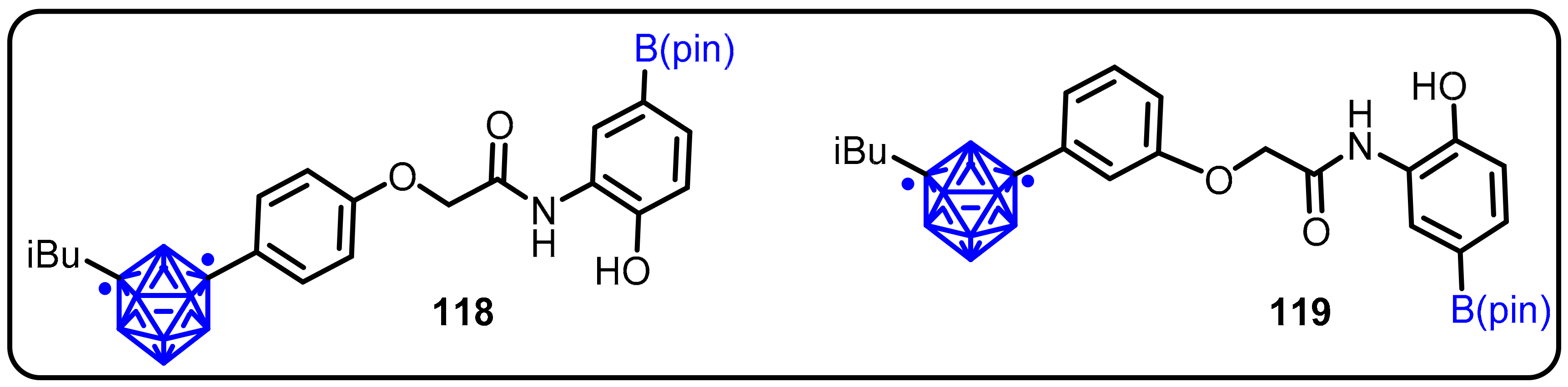
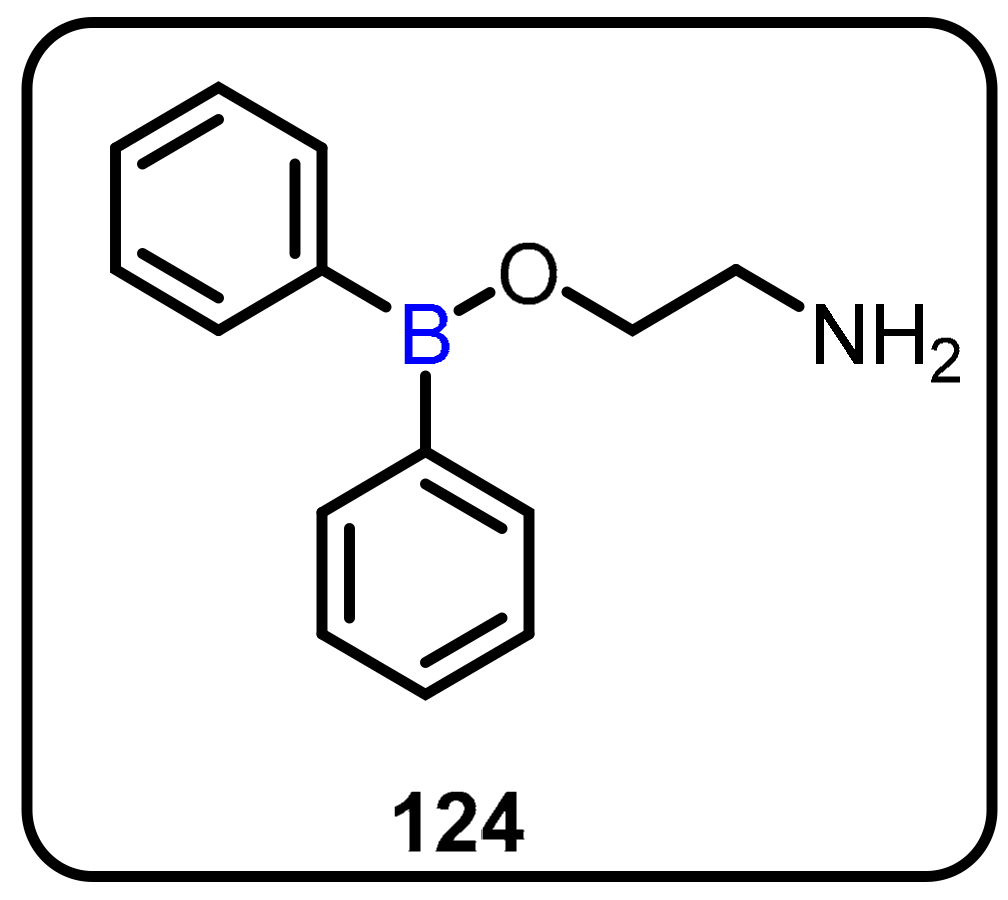
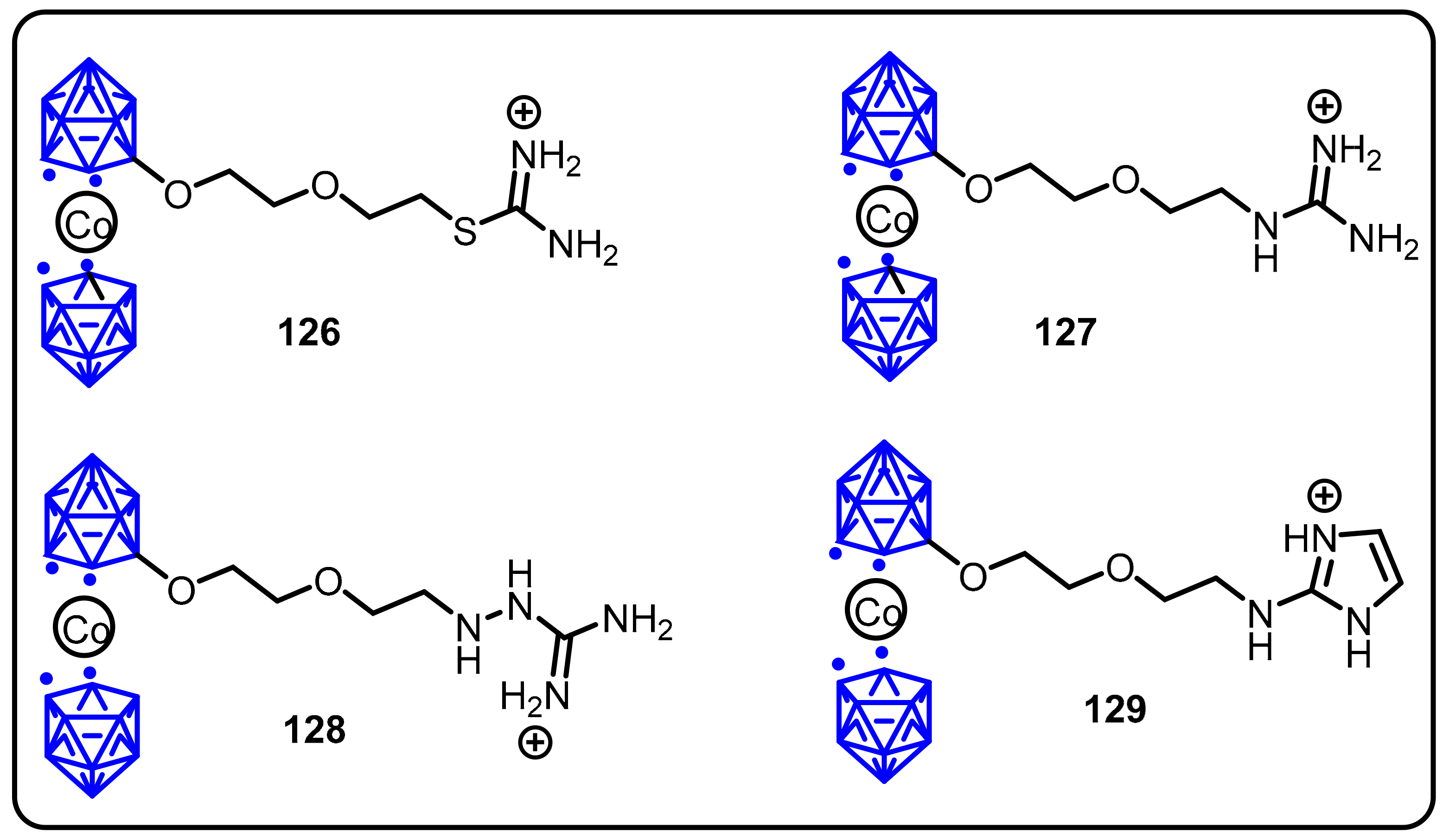
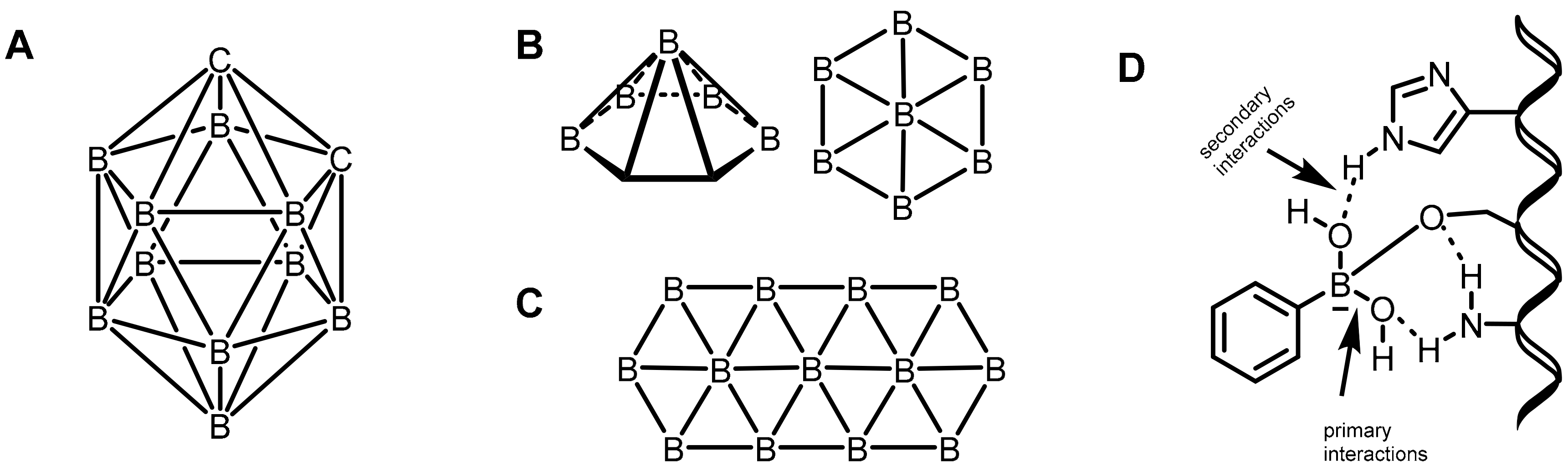
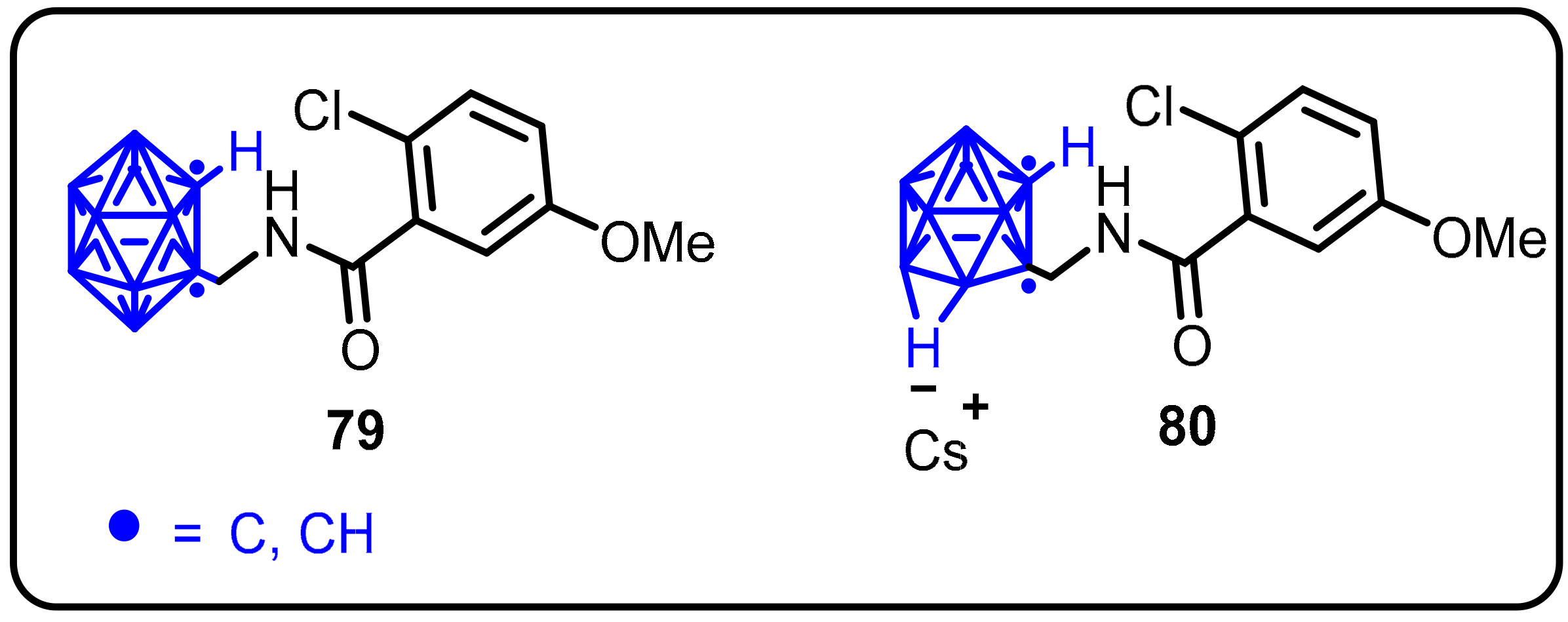
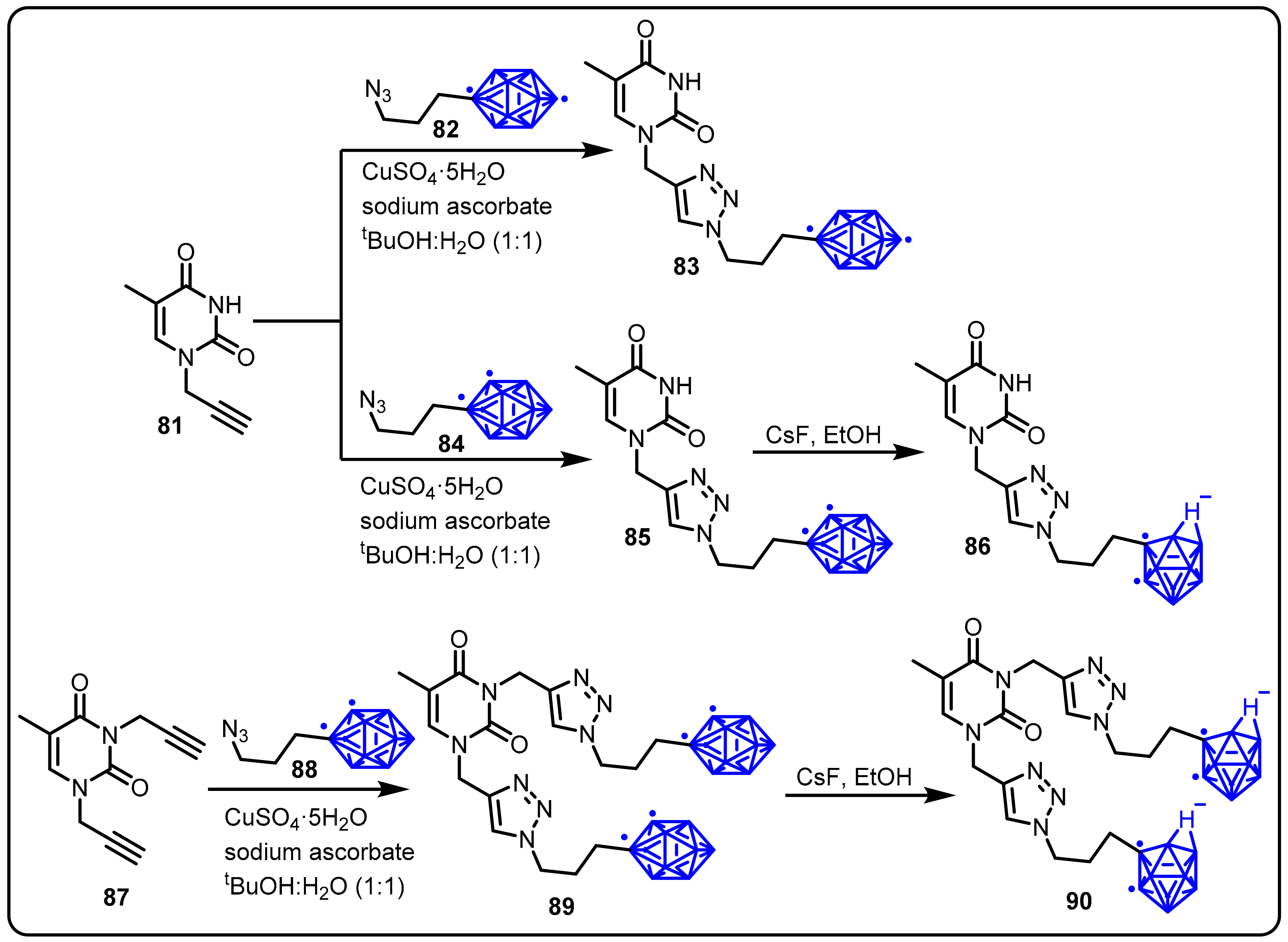
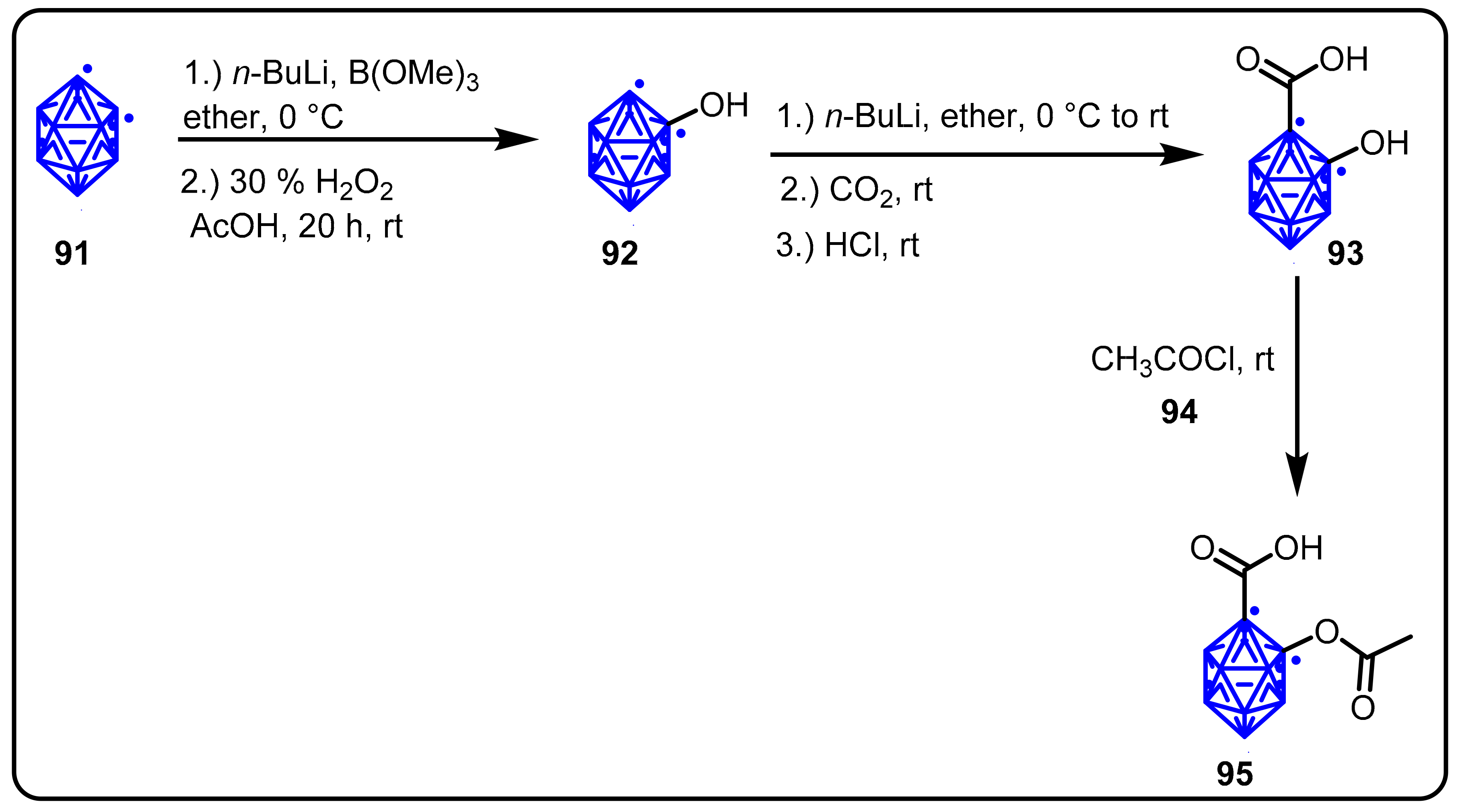
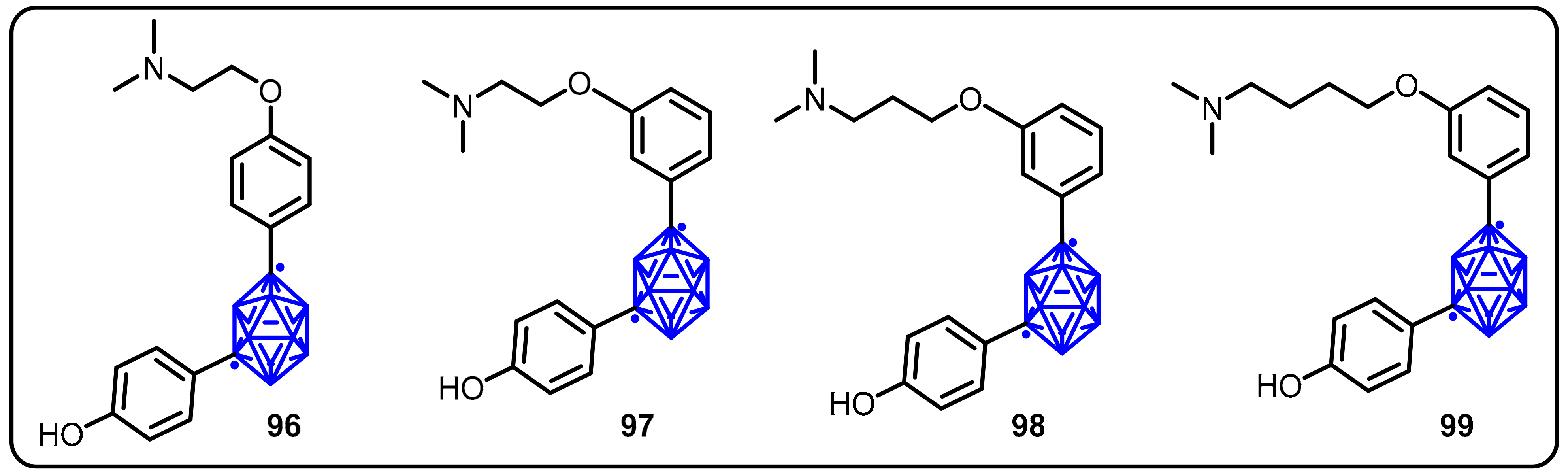
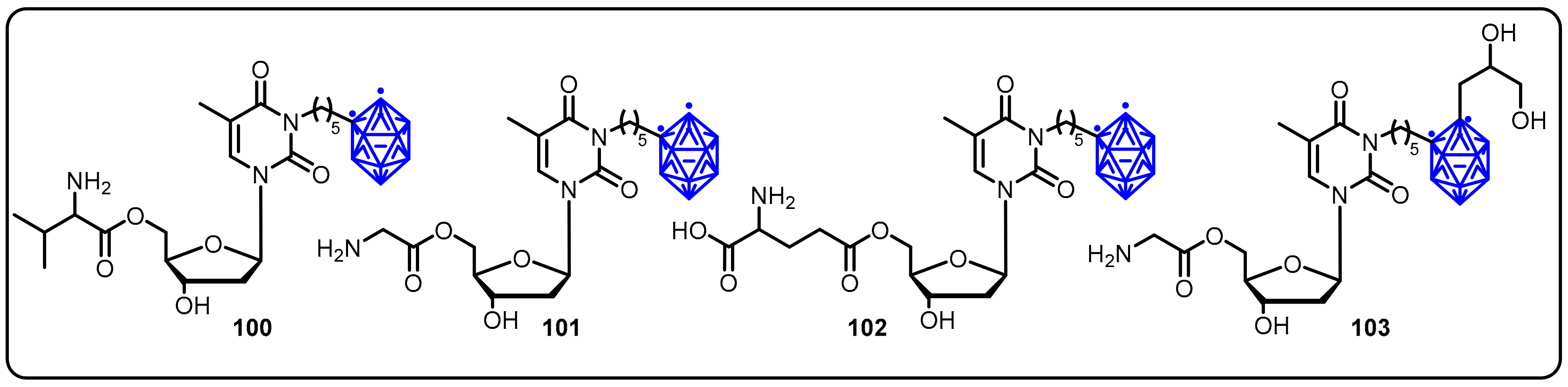
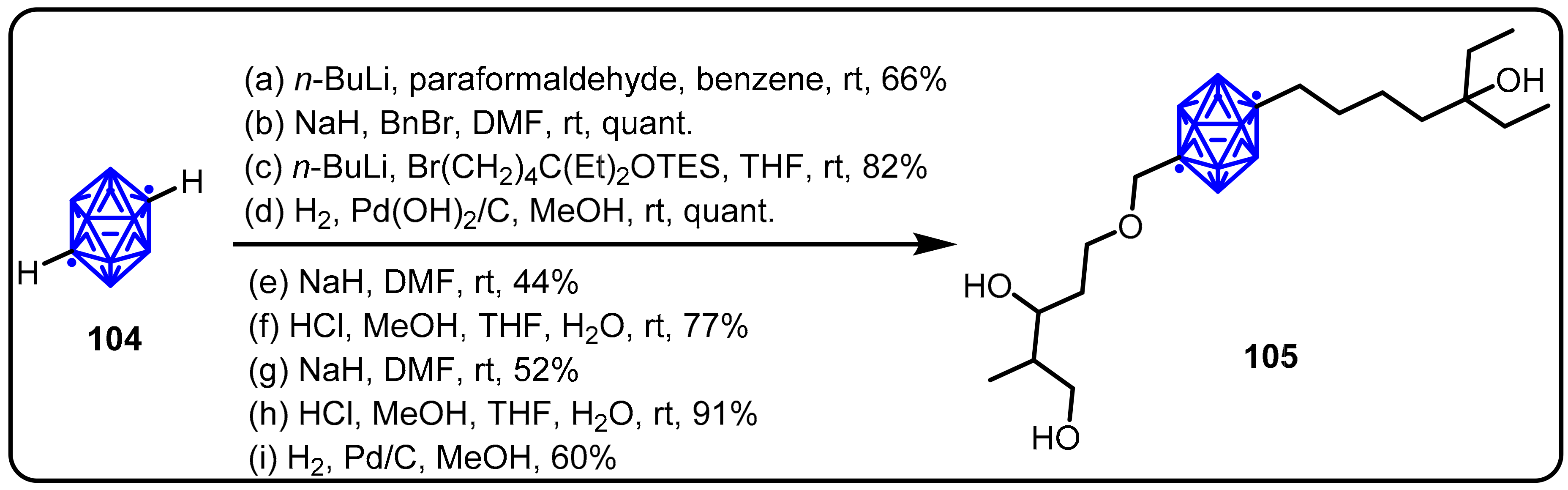
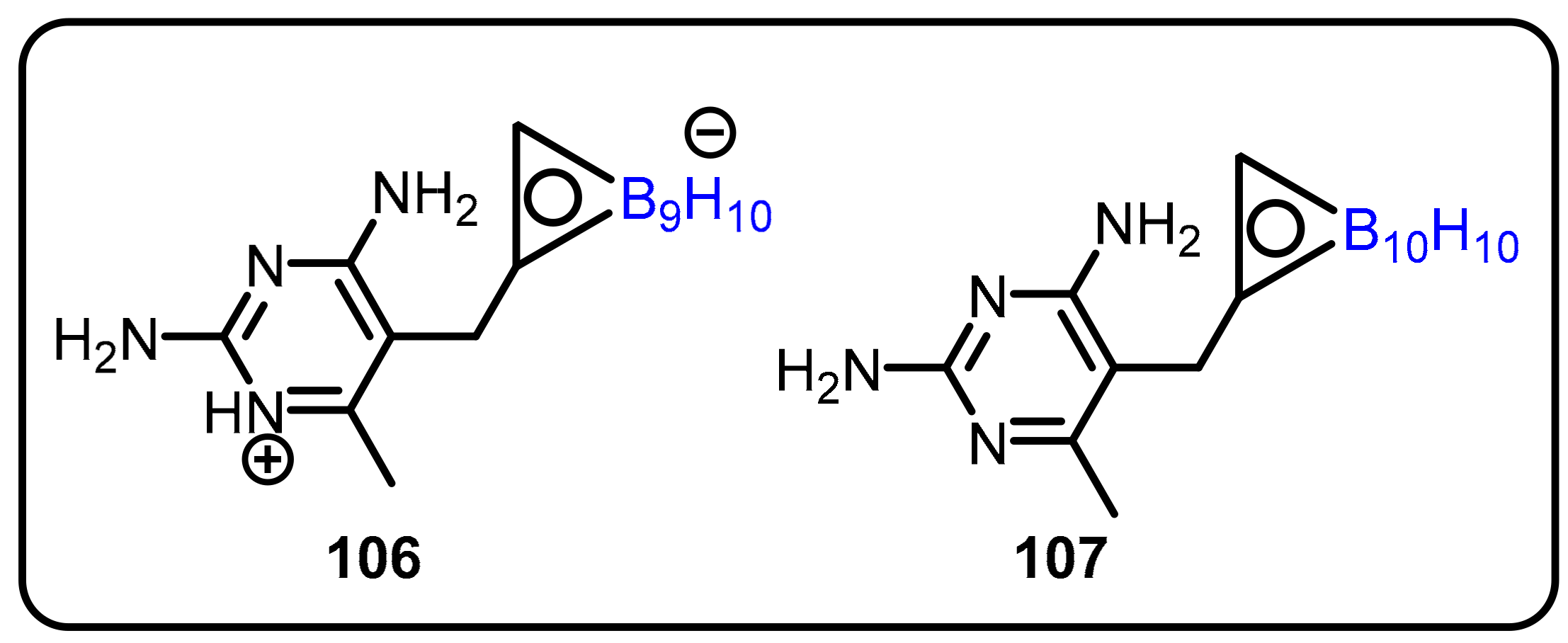
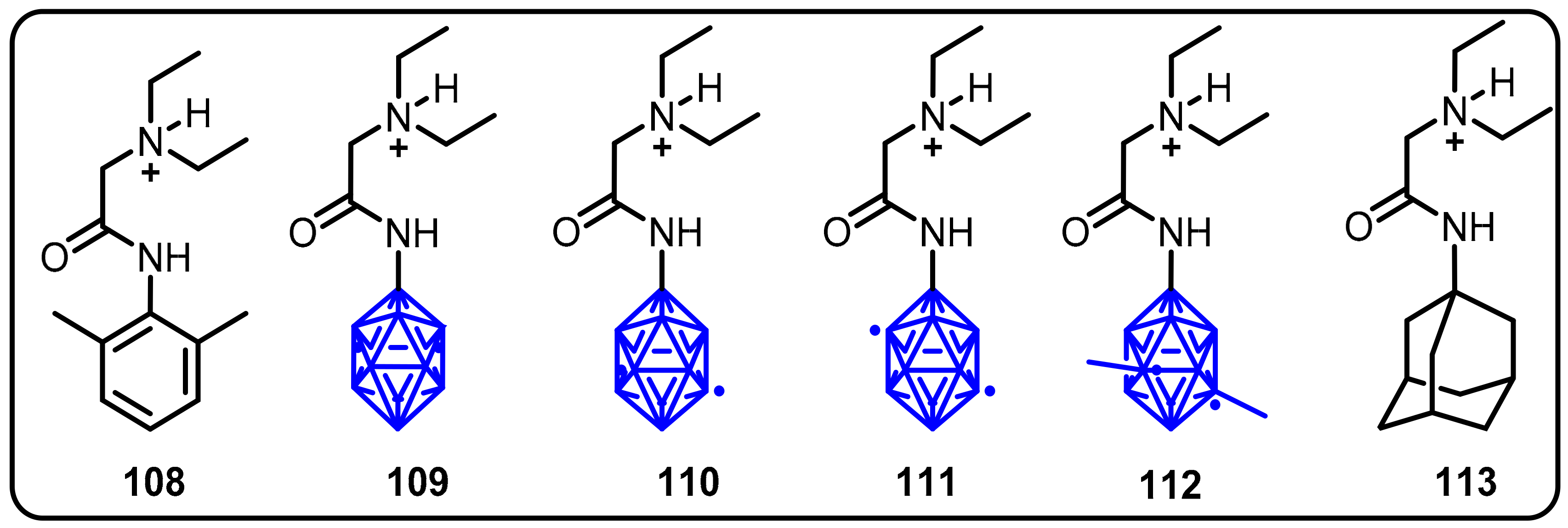
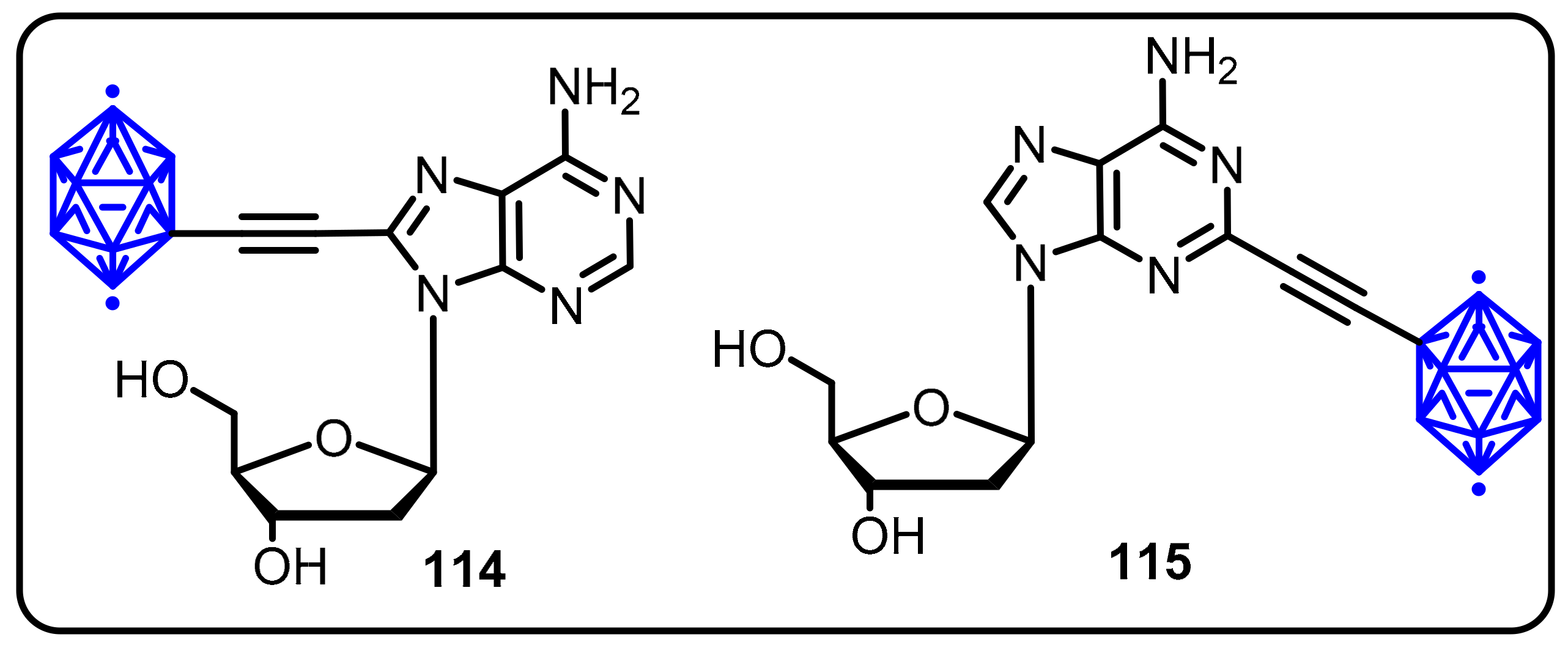
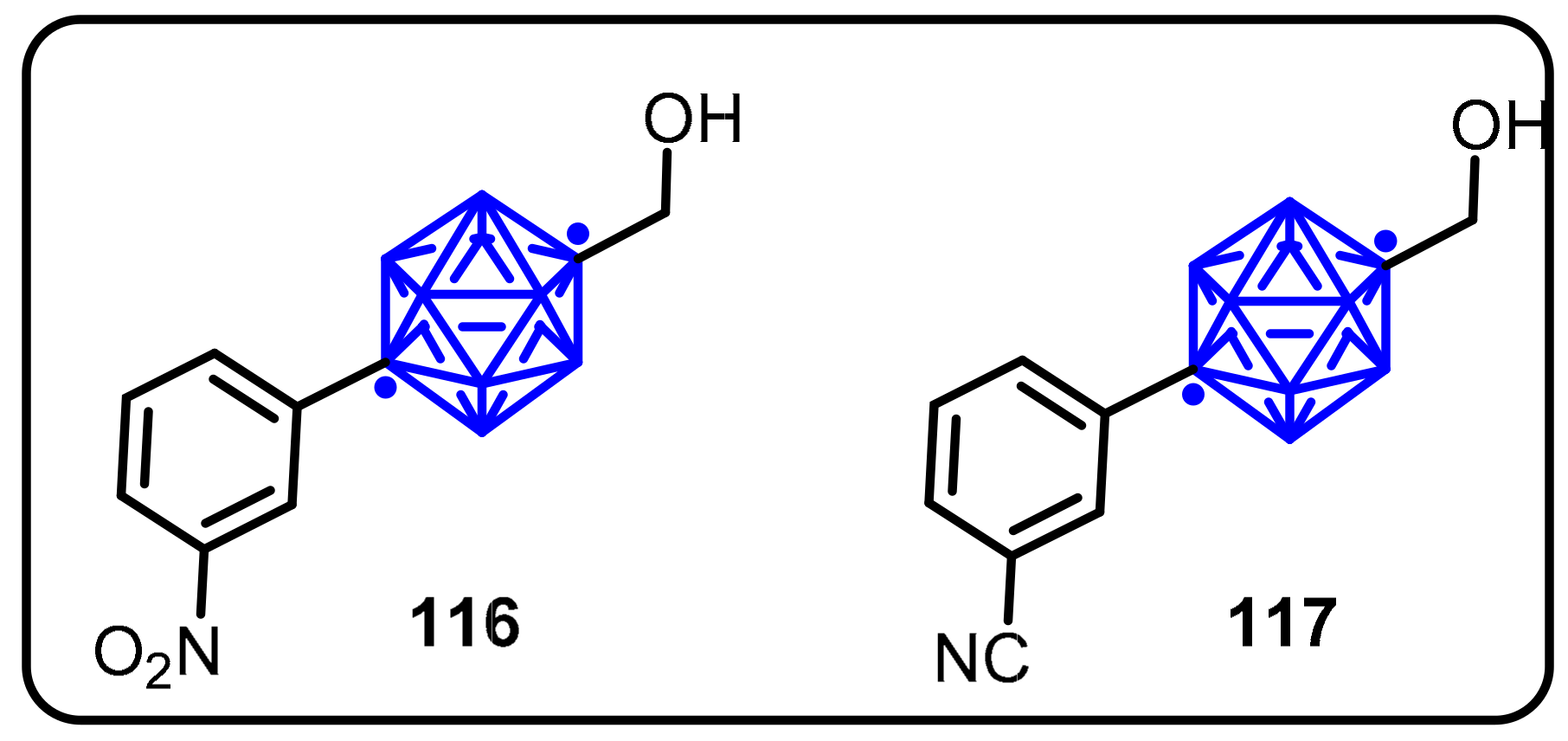
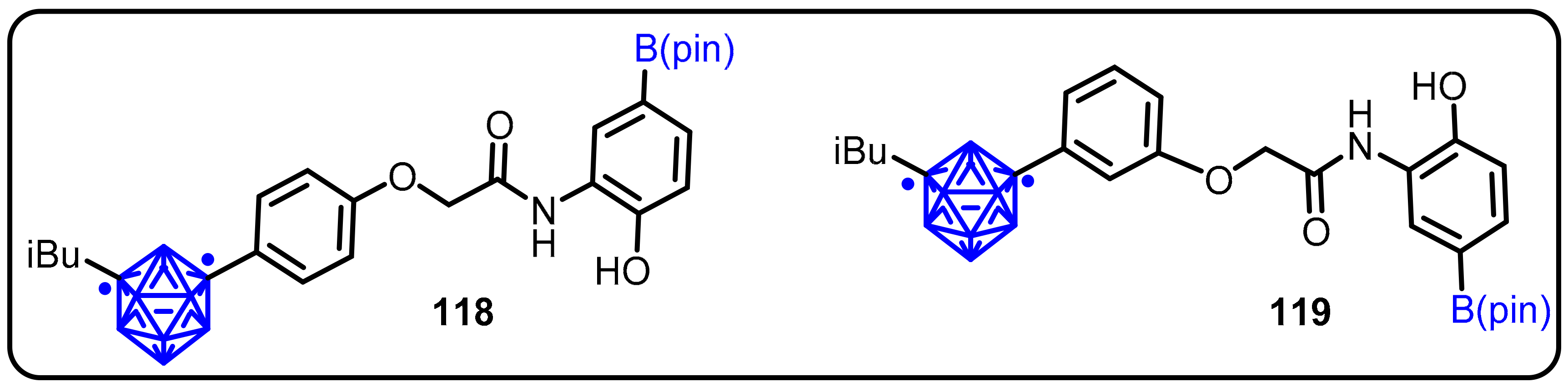
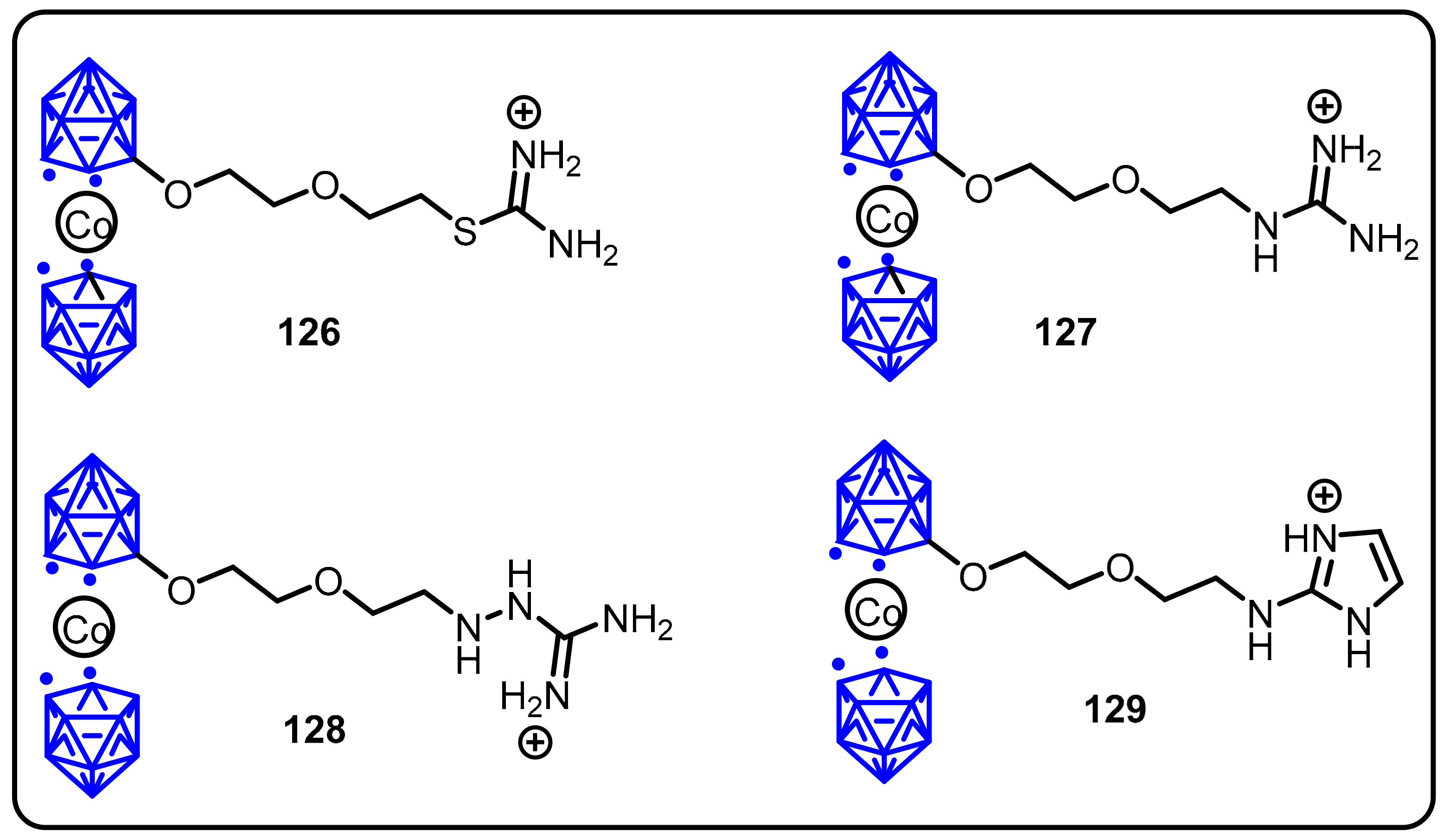
2.3. Carboranes as Drug Motifs
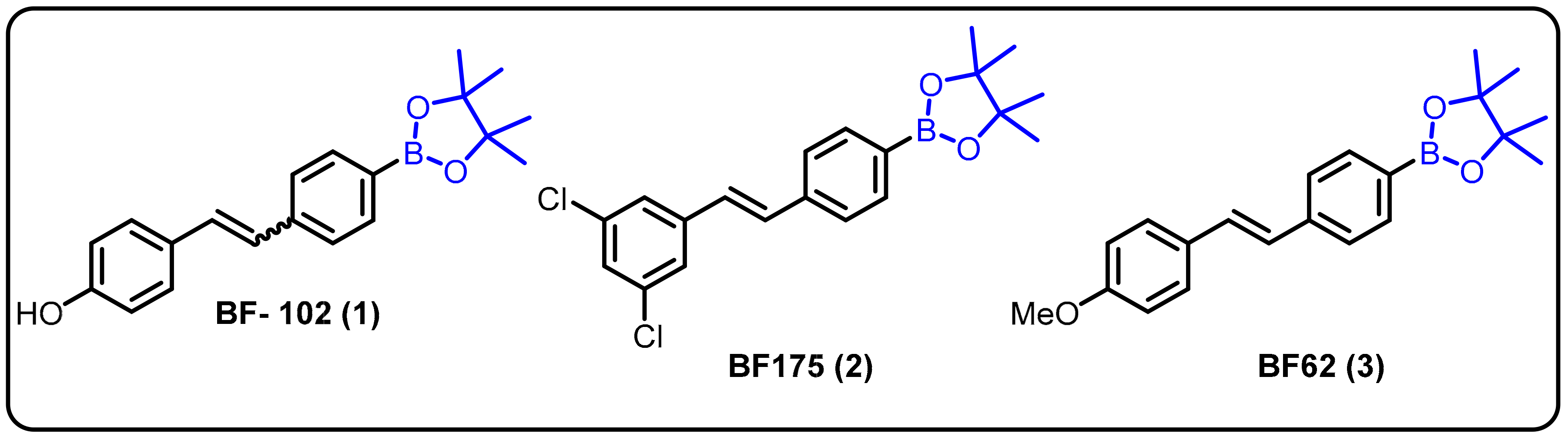
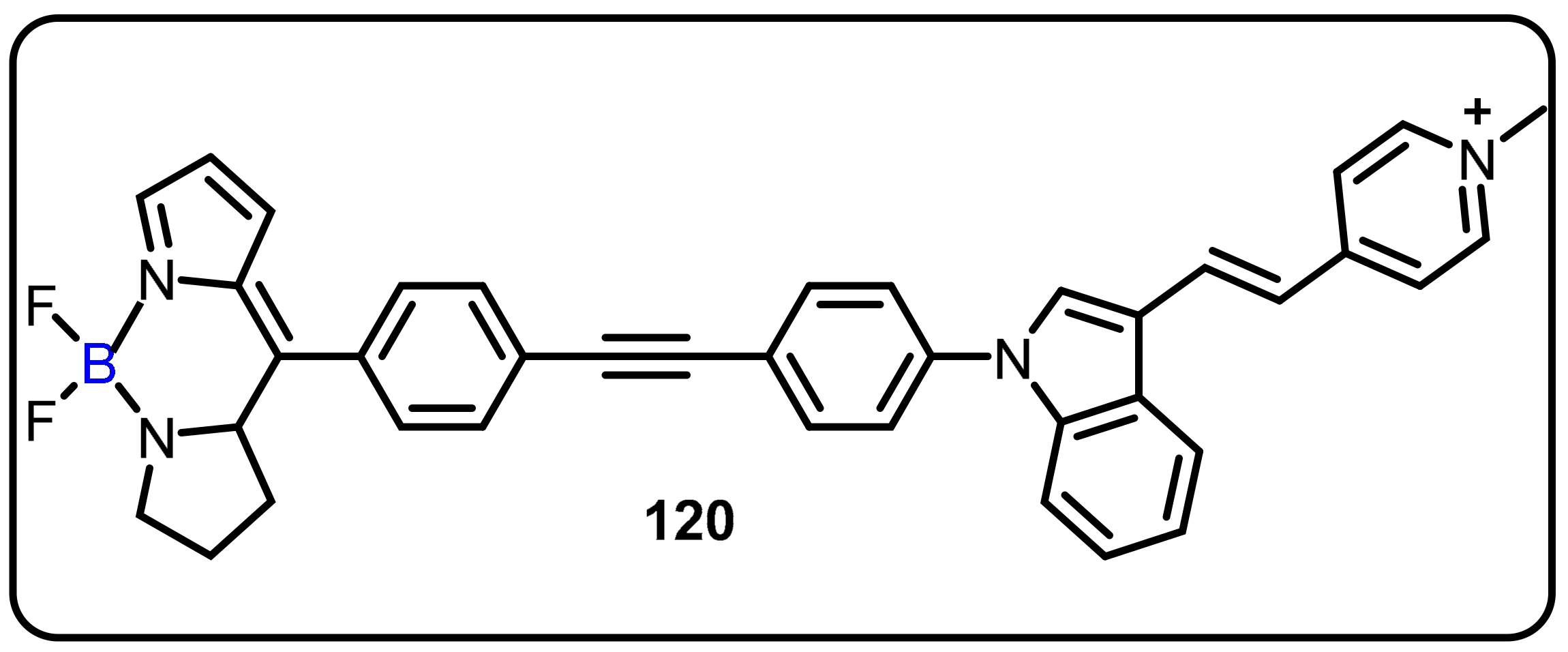
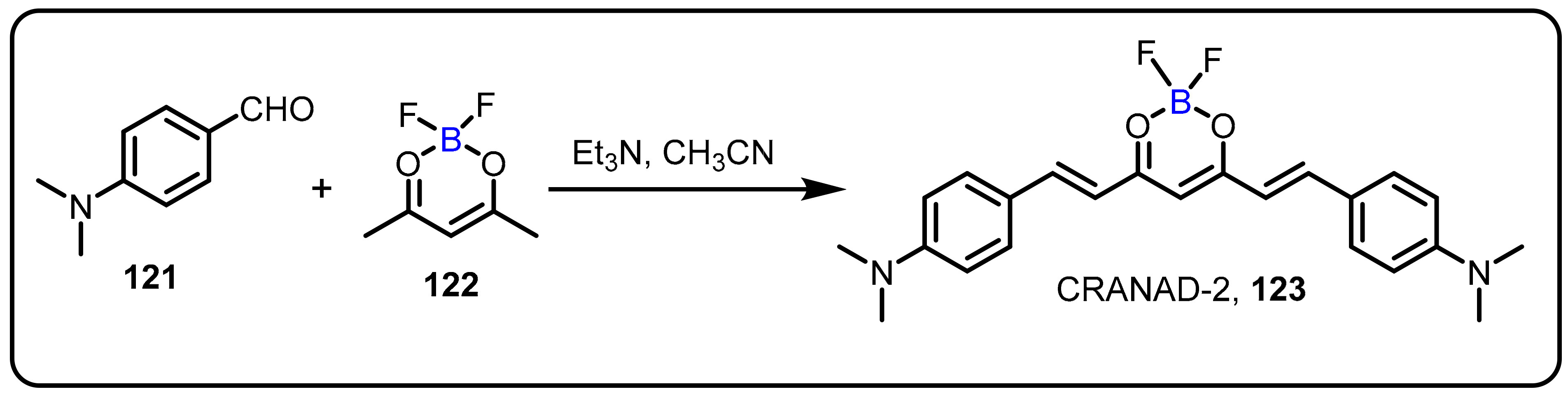
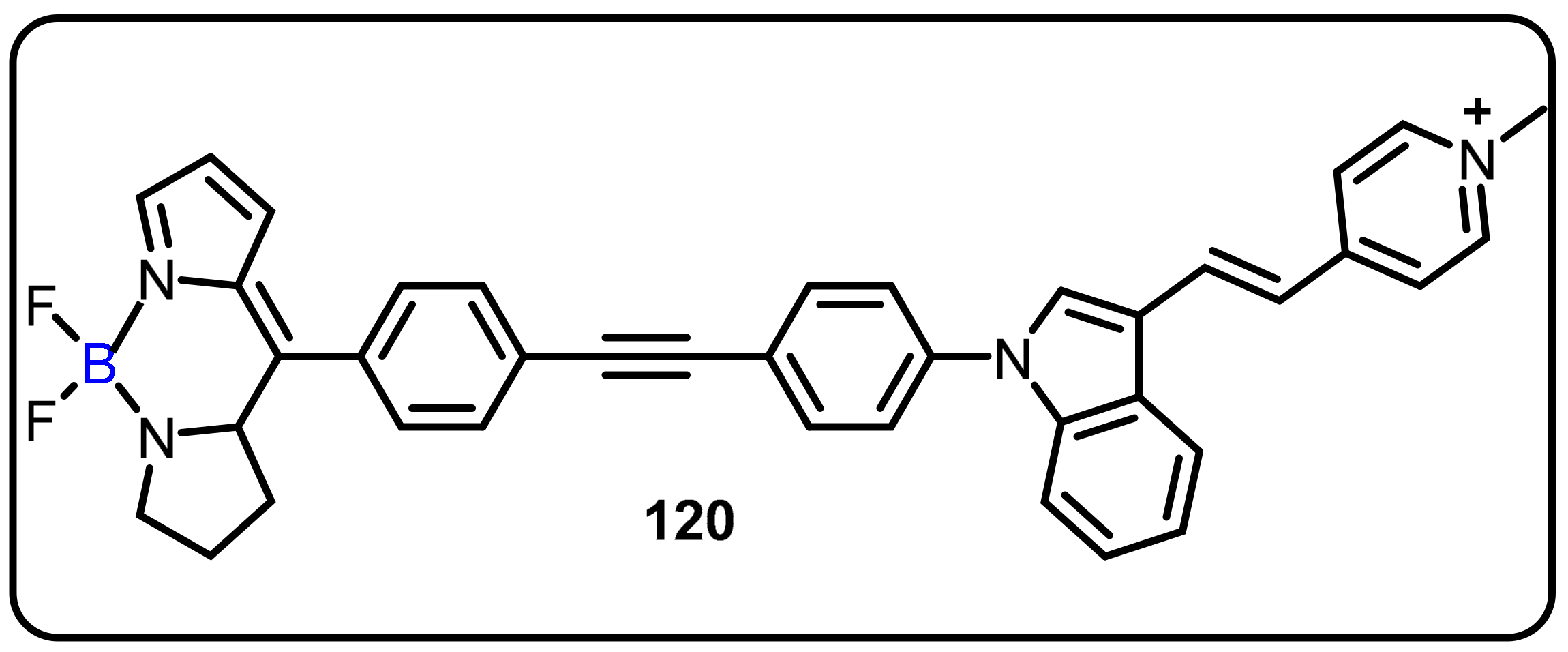
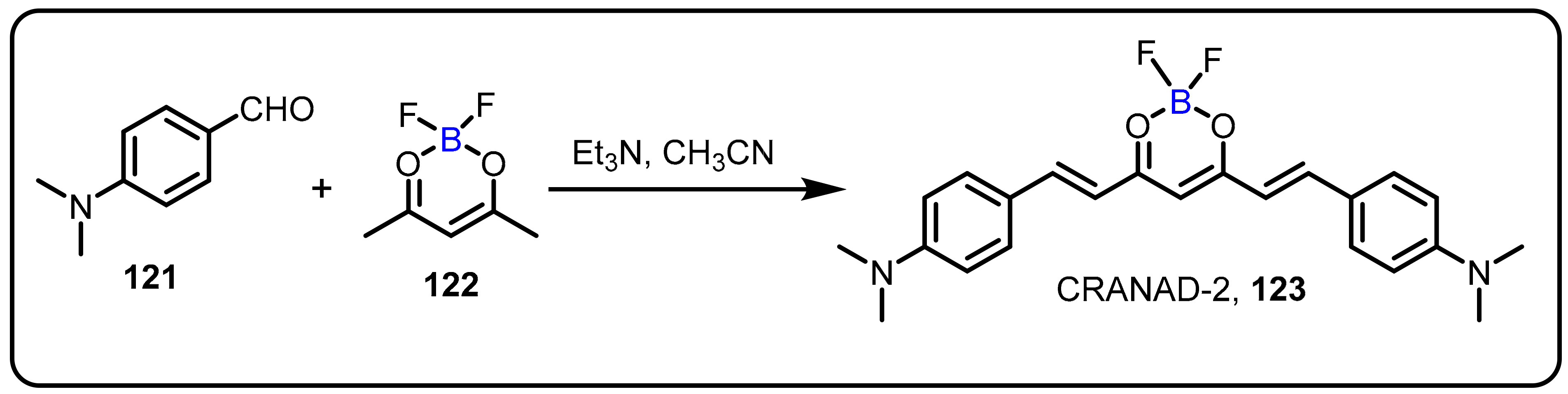
2.4. BODIPY and Other Functionalities
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, S.J.; Ding, C.Z.; Akama, T.; Zhang, Y.-K.; Hernandez, V.; Xia, Y. Therapeutic potential of boron-containing compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslah, H.; Skarbek, C.; Pethe, S.; Labruère, R. Anticancer boron-containing prodrugs responsive to oxidative stress from the tumor microenvironment. Eur. J. Med. Chem. 2020, 207, 112670. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gao, P.; Sun, L.; Kang, D.; Kongsted, J.; Poongavanam, V.; Zhan, P.; Liu, X. Recent developments in the medicinal chemistry of single boron atom-containing compounds. Acta Pharm. Sin. B 2021, 11, 3035–3059. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Thapa, P.; Karki, R.; Schinke, C.; Das, S.; Kambhampati, S.; Banerjee, S.K.; Van Veldhuizen, P.; Verma, A.; Weiss, L.M. Boron chemicals in diagnosis and therapeutics. Future Med. Chem. 2013, 5, 653–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, B.C.; Ojha, D.P.; Das, S.; Evans, T. Boron Compounds in Molecular Imaging. In Boron-Based Compounds: Potential and Emerging Applications in Medicine; John Wiley & Sons: New York, NY, USA, 2018; pp. 205–231. [Google Scholar]
- Hosmane, N.S.; Eagling, R.D. Handbook of Boron Science: With Applications in Organometallics, Catalysis, Materials and Medicine (In 4 Volumes); World Scientific: London, UK, 2018. [Google Scholar]
- Silva, M.P.; Saraiva, L.; Pinto, M.; Sousa, M.E. Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications. Molecules 2020, 25, 4323. [Google Scholar] [CrossRef]
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef]
- Smith, T.P.; Windsor, I.W.; Forest, K.T.; Raines, R.T. Stilbene boronic acids form a covalent bond with human transthyretin and inhibit its aggregation. J. Med. Chem. 2017, 60, 7820–7834. [Google Scholar] [CrossRef] [Green Version]
- Sai, K.K.S.; Das, B.C.; Sattiraju, A.; Almaguel, F.G.; Craft, S.; Mintz, A. Radiolabeling and initial biological evaluation of [18F] KBM-1 for imaging RAR-α receptors in neuroblastoma. Bioorg. Med. Chem. Lett. 2017, 27, 1425–1427. [Google Scholar]
- Zhong, Y.; Wu, Y.; Liu, R.; Li, Z.; Chen, Y.; Evans, T.; Chuang, P.; Das, B.; He, J.C. Novel retinoic acid receptor alpha agonists for treatment of kidney disease. PLoS ONE 2011, 6, e27945. [Google Scholar] [CrossRef] [Green Version]
- Das, B.C.; Tang, X.-Y.; Sanyal, S. Design and synthesis of boron containing 2,4-disubstituted-phthalazin-1 (2H)-one and 3,7-disubstituted-2H-benzo [b] [1,4] oxazine derivatives as potential HGF-mimetic agents. Tetrahedron Lett. 2011, 52, 4292–4294. [Google Scholar] [CrossRef]
- Adamczyk-Wozniak, A.; Borys, K.M.; Sporzynski, A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Tomsho, J.W.; Benkovic, S.J. Boron-containing inhibitors of synthetases. Chem. Soc. Rev. 2011, 40, 4279–4285. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.B.; Yudin, A.K. The versatility of boron in biological target engagement. Nat. Chem. 2017, 9, 731. [Google Scholar] [CrossRef] [PubMed]
- Smoum, R.; Rubinstein, A.; Dembitsky, V.M.; Srebnik, M. Boron containing compounds as protease inhibitors. Chem. Rev. 2012, 112, 4156–4220. [Google Scholar] [CrossRef]
- Touchet, S.; Carreaux, F.; Carboni, B.; Bouillon, A.; Boucher, J.-L. Aminoboronic acids and esters: From synthetic challenges to the discovery of unique classes of enzyme inhibitors. Chem. Soc. Rev. 2011, 40, 3895–3914. [Google Scholar] [CrossRef]
- Vshyvenko, S.; Clapson, M.L.; Suzuki, I.; Hall, D.G. Characterization of the Dynamic Equilibrium between Closed and Open Forms of the Benzoxaborole Pharmacophore. ACS Med. Chem. Lett. 2016, 7, 1097–1101. [Google Scholar] [CrossRef] [Green Version]
- Jemmis, E.D. Overlap control and stability of polyhedral molecules. closo-Carboranes. J. Am. Chem. Soc. 1982, 104, 7017–7020. [Google Scholar] [CrossRef]
- Sachdev, H. MATERIALS SCIENCE. Disclosing boron’s thinnest side. Science 2015, 350, 1468–1469. [Google Scholar] [CrossRef]
- Malouff, T.D.; Seneviratne, D.S.; Ebner, D.K.; Stross, W.C.; Waddle, M.R.; Trifiletti, D.M.; Krishnan, S. Boron neutron capture therapy: A review of clinical applications. Front. Oncol. 2021, 11, 351. [Google Scholar] [CrossRef]
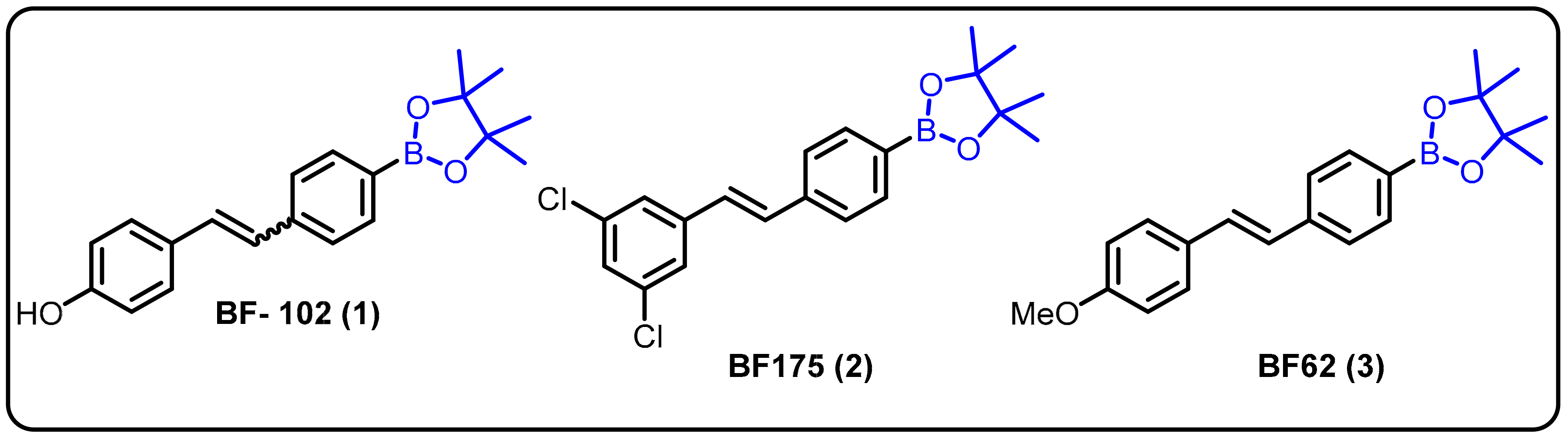
- Das, B.C.; Zhao, X.; Tang, X.-Y.; Yang, F. Design, synthesis and biological study of pinacolyl boronate-substituted stilbenes as novel lipogenic inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5638–5641. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, X.; Zong, H.; Abdulla, A.; Yang, E.S.T.; Wang, Q.; Ji, J.-Y.; Pessin, J.E.; Das, B.C.; Yang, F. Inhibition of SREBP transcriptional activity by aboron-containing compound improves lipid homeostasis in diet-induced obesity. Diabetes 2014, 63, 2464–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, T.T.; Smith, T.P.; Raines, R.T. A Boronic Acid Conjugate of Angiogenin that Shows ROS-Responsive Neuroprotective Activity. Angew. Chem. 2017, 129, 2663–2666. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States Food and Drug Administration approval summary: Bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef] [Green Version]
- Shirley, M. Ixazomib: First global approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.K.; Kirk, C.J. Development of proteasome inhibitors in oncology and autoimmune diseases. Curr. Opin. Drug Discov. Dev. 2008, 11, 616–625. [Google Scholar]
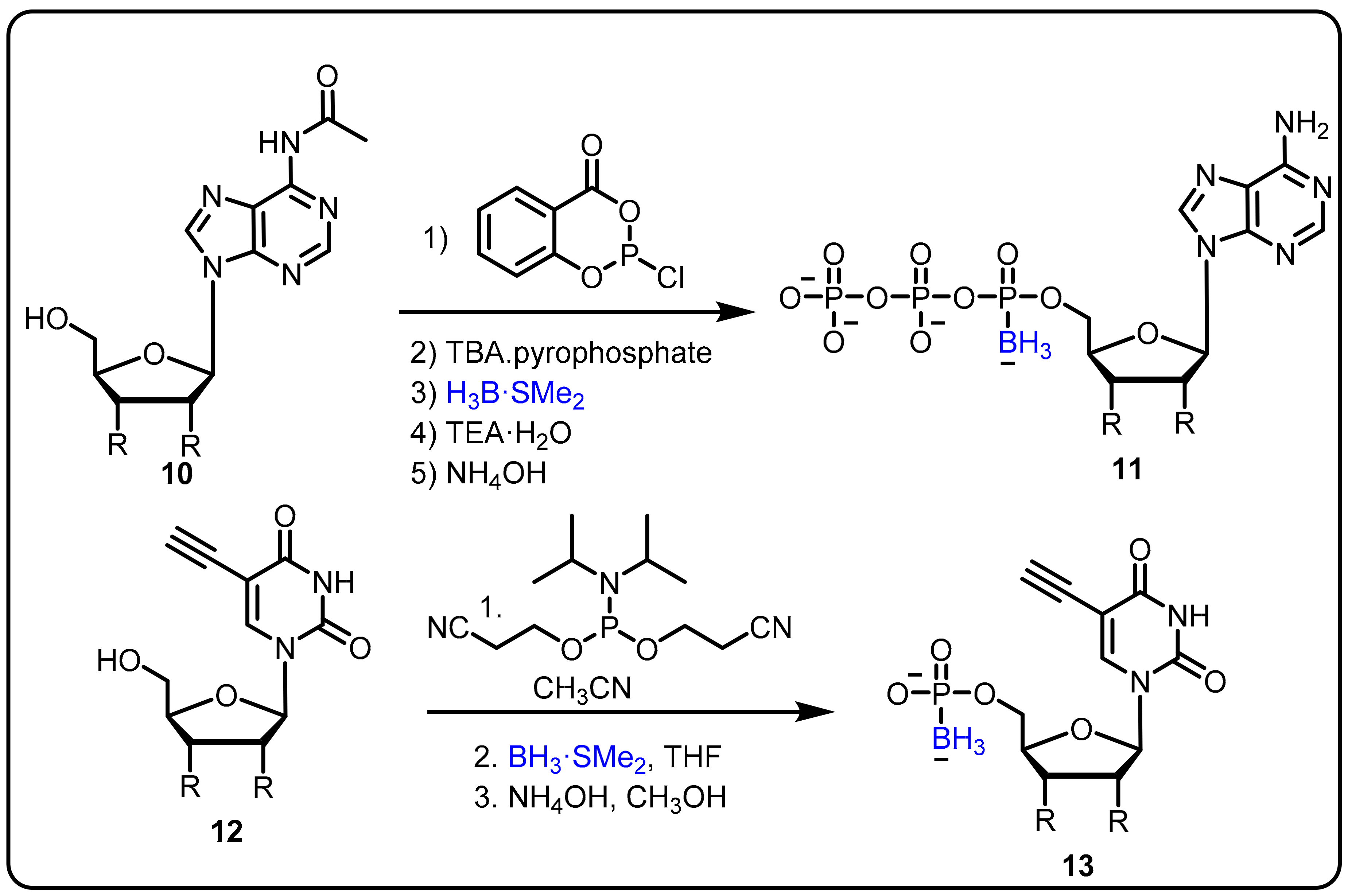
- Khan, S.I.; Dobrikov, M.I.; Shaw, B.R. Synthesis of 5-ethynyl-2′-deoxyuridine-5′-boranomono phosphate as a potential thymidylate synthase inhibitor. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1047–1049. [Google Scholar] [CrossRef]
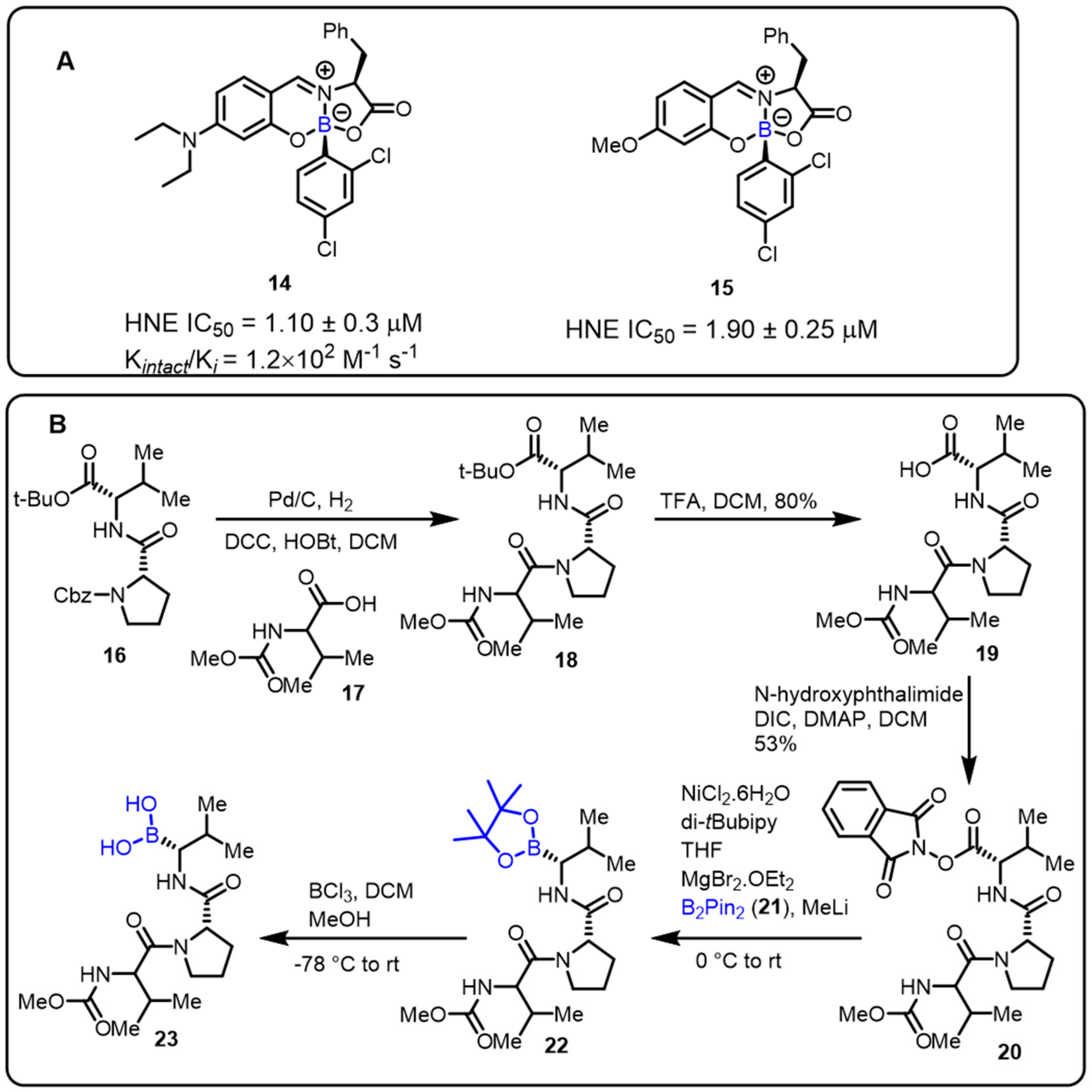
- Montalbano, F.; Cal, P.M.S.D.; Carvalho, M.A.B.R.; Gonçalves, L.M.; Lucas, S.D.; Guedes, R.C.; Veiros, L.F.; Moreira, R.; Gois, P.M.P. Discovery of new heterocycles with activity against human neutrophile elastase based on a boron promoted one-pot assembly reaction. Org. Biomol. Chem. 2013, 11, 4465–4472. [Google Scholar] [CrossRef]
- Moreira, W.; Aziz, D.B.; Dick, T. Boromycin Kills Mycobacterial Persisters without Detectable Resistance. Front. Microbiol. 2016, 7, 199. [Google Scholar] [CrossRef]
- Adamska, A.; Rumijowska-Galewicz, A.; Ruszczynska, A.; Studzińska, M.; Jabłońska, A.; Paradowska, E.; Bulska, E.; Munier-Lehmann, H.; Dziadek, J.; Leśnikowski, Z.J.; et al. Anti-mycobacterial activity of thymine derivatives bearing boron clusters. Eur. J. Med. Chem. 2016, 121, 71–81. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.S.; Bowes, E.G.; Li, H.; Vallée, A.M.; Vogels, C.M.; Decken, A.; Gray, C.A.; Westcott, S.A. Heterocyclic Aminoboron Compounds as Antituberculosis Agents. Heteroat. Chem. 2014, 25, 100–106. [Google Scholar] [CrossRef]
- Maynard, A.; Crosby, R.M.; Ellis, B.; Hamatake, R.; Hong, Z.; Johns, B.A.; Kahler, K.M.; Koble, C.; Leivers, A.; Leivers, M.R.; et al. Discovery of a Potent Boronic Acid Derived Inhibitor of the HCV RNA-Dependent RNA Polymerase. J. Med. Chem. 2014, 57, 1902–1913. [Google Scholar] [CrossRef]
- Ashley, J.D.; Stefanick, J.F.; Schroeder, V.A.; Suckow, M.A.; Kiziltepe, T.; Bilgicer, B. Liposomal Bortezomib Nanoparticles via Boronic Ester Prodrug Formulation for Improved Therapeutic Efficacy in Vivo. J. Med. Chem. 2014, 57, 5282–5292. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Offidani, M.; Vigna, E.; Corvatta, L.; Recchia, A.G.; Morabito, L.; Morabito, F.; Gentili, S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2015, 24, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Gallerani, E.; Zucchetti, M.; Brunelli, D.; Marangon, E.; Noberasco, C.; Hess, D.; Delmonte, A.; Martinelli, G.; Böhm, S.; Driessen, C.; et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer 2013, 49, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Roemmele, R.C.; Christie, M.A. Development and Scale-Up of an Optimized Route to the Peptide Boronic Acid, CEP-18770. Org. Process Res. Dev. 2013, 17, 422–426. [Google Scholar] [CrossRef]
- Satou, Y. Proteasome inhibitor, bortezomib, potently inhibits the growth of adult T-cell leukemia cells both in vivo and in vitro. Leukemia 2004, 18, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Li, D.W.; Yang, L.Y.; Fu, L.; Zhu, X.J.; Wong, W.K.; Jiang, F.L.; Liu, Y. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C.; Zhang, L.; Weigel, L.F.; Schilz, J.; Graf, D.; Bartenschlager, R.; Hilgenfeld, R.; Klein, C.D. Peptide–boronic acid inhibitors of flaviviral proteases: Medicinal chemistry and structural biology. J. Med. Chem. 2017, 60, 511–516. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Ye, L.; Zhou, H.; Waterson, D.; Gamo, F.-J.; Sanz, L.M. Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1, 3-dihydro-1-hydroxy-2, 1-benzoxaboroles. Bioorg. Med. Chem. Lett. 2012, 22, 1299–1307. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Gut, J.; Rosenthal, P.J.; Waterson, D.; Gamo, F.-J.; Angulo-Barturen, I. Synthesis and structure–activity relationships of novel benzoxaboroles as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2011, 21, 644–651. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Waterson, D.; Ge, M.; Li, Z.; Li, L.; Jian, Y. An efficient synthesis for a new class antimalarial agent, 7-(2-carboxyethyl)-1, 3-dihydro-1-hydroxy-2, 1-benzoxaborole. Tetrahedron Lett. 2011, 52, 3909–3911. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Plattner, J.J.; Easom, E.E.; Liu, L.; Retz, D.M.; Ge, M.; Zhou, H.H. Benzoxaborole antimalarial agents. Part 3: Design and syntheses of (carboxy-13C-3, 3-2H2)-labeled and (3-14C)-labeled 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. J. Label. Compd. Radiopharm. 2012, 55, 201–205. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Sanders, V.; Freund, Y.R.; Campo, B.; Rosenthal, P.J.; Bu, W. Benzoxaborole antimalarial agents. Part 4. Discovery of potent 6-(2-(alkoxycarbonyl) pyrazinyl-5-oxy)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. J. Med. Chem. 2015, 58, 5344–5354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Zhou, Y.; Akama, T.; Bu, W.; White, W.H.; Defauw, J.M.; Winkle, J.R.; Balko, T.W. Discovery of an orally bioavailable isoxazoline benzoxaborole (AN8030) as a long acting animal ectoparasiticide. Bioorg. Med. Chem. Lett. 2015, 25, 5589–5593. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, J.; Meng, Q.; Nare, B.; Jacobs, R.T.; Zhou, H. Novel pyrrolobenzoxaboroles: Design, synthesis, and biological evaluation against Trypanosoma brucei. Eur. J. Med. Chem. 2014, 81, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Akama, T.; Dong, C.; Virtucio, C.; Freund, Y.R.; Chen, D.; Orr, M.D.; Jacobs, R.T.; Zhang, Y.-K.; Hernandez, V.; Liu, Y. Discovery and structure–activity relationships of 6-(benzoylamino) benzoxaboroles as orally active anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2013, 23, 5870–5873. [Google Scholar] [CrossRef]
- Bu, W.; Akama, T.; Chanda, S.; Sullivan, D.; Ciaravino, V.; Jarnagin, K.; Freund, Y.; Sanders, V.; Chen, C.-W.; Fan, X.; et al. Early rapid identification of in vivo rat metabolites of AN6414, a novel boron-containing PDE4 inhibitor by QTRAP LC/MS/MS to support drug discovery. J. Pharm. Biomed. Anal. 2012, 70, 344–353. [Google Scholar] [CrossRef]
- Ruan, K.H. Advance in understanding the biosynthesis of prostacyclin and thromboxane A2 in the endoplasmic reticulum membrane via the cyclooxygenase pathway. Mini-Rev. Med. Chem. 2004, 4, 639–647. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crépin, T.; Zhou, H.; Zhang, Y.-K.; Hernandez, V.; Akama, T.; Baker, S.J. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef]
- Hu, Q.-H.; Liu, R.-J.; Fang, Z.-P.; Zhang, J.; Ding, Y.-Y.; Tan, M.; Wang, M.; Pan, W.; Zhou, H.-C.; Wang, E.-D. Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase. Sci. Rep. 2013, 3, 2475. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, V.; Crepin, T.; Palencia, A.; Cusack, S.; Akama, T.; Baker, S.J.; Bu, W.; Feng, L.; Freund, Y.R.; Liu, L.; et al. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob. Agents Chemother. 2013, 57, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanichar, D.; Roppiyakuda, L.; Kosmowska, E.; Faust, M.A.; Tran, K.P.; Chow, F.; Buglo, E.; Groziak, M.P.; Sarina, E.A.; Olmstead, M.M.; et al. Synthesis, characterization, and antibacterial activity of structurally complex 2-acylated 2,3,1-benzodiazaborines and related compounds. Chem. Biodivers. 2014, 11, 1381–1397. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Cao, K.; Zhou, Y.; Alley, M.; Rock, F.; Mohan, M.; Meewan, M.; Baker, S.J.; Lux, S.; Ding, C.Z. Synthesis and SAR of novel benzoxaboroles as a new class of β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2533–2536. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.M.; Gunosewoyo, H.; Barron, M.L.; Boucher, A.; McDonnell, M.; Turner, P.; Morrison, D.E.; Bennett, M.R.; McGregor, I.S.; Rendina, L.M.; et al. The first CNS-active carborane: A novel P2X7 receptor antagonist with antidepressant activity. ACS Chem. Neurosci. 2014, 5, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Synthesis and biological evaluation of novel m-carborane-containing estrogen receptor partial agonists as SERM candidates. Bioorg. Med. Chem. Lett. 2015, 25, 3213–3216. [Google Scholar] [CrossRef]
- Hasabelnaby, S.; Goudah, A.; Agarwal, H.K.; Abd Alla, M.S.; Tjarks, W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur. J. Med. Chem. 2012, 55, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Sekine, R.; Kano, A.; Masuno, H.; Songkram, C.; Kawachi, E.; Hirano, T.; Tanatani, A.; Kagechika, H. Structural development of p-carborane-based potent non-secosteroidal vitamin D analogs. Bioorg. Med. Chem. 2014, 22, 5891–5901. [Google Scholar] [CrossRef]
- Reynolds, R.C.; Campbell, S.R.; Fairchild, R.G.; Kisliuk, R.L.; Micca, P.L.; Queener, S.F.; Riordan, J.M.; Sedwick, W.D.; Waud, W.R.; Leung, A.K.; et al. Novel boron-containing, nonclassical antifolates: Synthesis and preliminary biological and structural evaluation. J. Med. Chem. 2007, 50, 3283–3289. [Google Scholar] [CrossRef]
- Kracke, G.R.; VanGordon, M.R.; Sevryugina, Y.V.; Kueffer, P.J.; Kabytaev, K.; Jalisatgi, S.S.; Hawthorne, M.F. Carborane-derived local anesthetics are isomer dependent. ChemMedChem 2015, 10, 62–67. [Google Scholar] [CrossRef]
- Żołnierczyk, J.; Olejniczak, A.; Mieczkowski, A.; Błoński, J.; Kiliańska, Z.; Robak, T.; Lesnikowski, Z. In vitro antileukemic activity of novel adenosine derivatives bearing boron cluster modification. Bioorg. Med. Chem. 2016, 24, 5076–5087. [Google Scholar] [CrossRef]
- Fujii, S.; Goto, T.; Ohta, K.; Hashimoto, Y.; Suzuki, T.; Ohta, S.; Endo, Y. Potent androgen antagonists based on carborane as a hydrophobic core structure. J. Med. Chem. 2005, 48, 4654–4662. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Azuma, S.; Minegishi, H.; Nakamura, H. Synthesis and biological evaluation of meta-carborane-containing phenoxyacetanilides as inhibitors of hypoxia-inducible factor (HIF)-1 transcriptional activity. J. Organomet. Chem. 2015, 798, 189–195. [Google Scholar] [CrossRef]
- Ran, C.; Xu, X.; Raymond, S.B.; Ferrara, B.J.; Neal, K.; Bacskai, B.J.; Medarova, Z.; Moore, A. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near-infrared probes for in vivo detection of amyloid-beta deposits. J. Am. Chem. Soc. 2009, 131, 15257–15261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Zheng, C.; Zhang, L.; Da, C.; Zhao, K. 2-Aminoethoxydiphenyl borate administration into the nostril alleviates murine allergic rhinitis. Am. J. Otolaryngol. 2011, 32, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.; Cao, G.L.; Tomczuk, B.; Suzdak, P.D.; Cross, A.S.; Shapiro, P.; Rosen, G.M. Effect of the mammalian arginase inhibitor 2(S)-amino-6-boronohexanoic acid on Bacillus anthracis arginase. Curr. Microbiol. 2012, 64, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Tapia, V.S.; Swanton, T.; White, C.S.; Beswick, J.A.; Brough, D.; Freeman, S. Design, synthesis and evaluation of oxazaborine inhibitors of the NLRP3 inflammasome. ChemMedChem 2018, 13, 312. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Verges, B. New insight into the pathophysiology of lipid abnormalities in type 2 diabetes. Diabetes Metab. 2005, 31, 429–439. [Google Scholar] [CrossRef]
- Girard, J.; Lafontan, M. Impact of visceral adipose tissue on liver metabolism and insulin resistance. Part II: Visceral adipose tissue production and liver metabolism. Diabetes Metab. 2008, 34, 439–445. [Google Scholar] [CrossRef]
- Lanier, M.; Cole, D.C.; Istratiy, Y.; Klein, M.G.; Schwartz, P.A.; Tjhen, R.; Jennings, A.; Hixon, M.S. Repurposing Suzuki coupling reagents as a directed fragment library targeting serine hydrolases and related enzymes. J. Med. Chem. 2017, 60, 5209–5215. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Nishimasu, H.; Ishitani, R.; Kojima, H.; Nureki, O.; Aoki, J.; Nagano, T. Screening and X-ray crystal structure-based optimization of autotaxin (ENPP2) inhibitors, using a newly developed fluorescence probe. ACS Chem. Biol. 2013, 8, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Henneberg, F.; Mata, R.A.; Tittmann, K.; Schneider, T.R.; Stark, H.; Bourenkov, G.; Chari, A. The inhibition mechanism of human 20S proteasomes enables next-generation inhibitor design. Science 2016, 353, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E. Antiviral drugs: Current state of the art. J. Clin. Virol. 2001, 22, 73–89. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; Mian, A.M.; So, A.G.; Scott, W.A. A mechanism of AZT resistance: An increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 1999, 4, 35–43. [Google Scholar] [CrossRef]
- Barth, R.F.; Soloway, A.H.; Fairchild, R.G.; Brugger, R.M. Boron neutron capture therapy for cancer. Realities and prospects. Cancer 1992, 70, 2995–3007. [Google Scholar] [PubMed]
- Alvarez, K.; Deval, J.; Selmi, B.; Barral, K.; Boretto, J.; Guerreiro, C.; Mulard, L.; Sarfati, R.; Canard, B. Borano-nucleotides: New analogues to circumvent HIV-1 RT-mediated nucleoside drug-resistance. Nucleosides Nucleotides Nucleic Acids 2005, 24, 419–422. [Google Scholar] [CrossRef]
- Cheek, M.A.; Dobrikov, M.I.; Wennefors, C.K.; Xu, Z.; Hashmi, S.N.; Shen, X.; Shaw, B.R. Synthesis and Properties of (α-P-Borano)-Nucleoside 5′-Triphosphate Analogues as Potential Antiviral Agents; Nucleic Acids Symposium Series; Oxford University Press: New York, NY, USA, 2008; pp. 81–82. [Google Scholar]
- Li, C.; Wang, J.; Barton, L.M.; Yu, S.; Tian, M.; Peters, D.S.; Kumar, M.; Yu, A.W.; Johnson, K.A.; Chatterjee, A.K.; et al. Decarboxylative borylation. Science 2017, 356, eaam7355. [Google Scholar] [CrossRef] [Green Version]
- Torssell, K.; Larsson, E. Darstellung langkettiger Alkylborsauren. Acta Chem. Scand. 1957, 11, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Zhdankin, V.V.; Persichini, P., III; Zhang, L.; Fix, S.; Kiprof, P. Synthesis and structure of benzoboroxoles: Novel organoboron heterocycles. Tetrahedron Lett. 1999, 40, 6705–6708. [Google Scholar] [CrossRef]
- Arcus, V.L.; Main, L.; Nicholson, B.K. ortho-Directed electrophilic boronation of a benzyl ketone: The preparation, X-ray crystal structure, and some reactions of 4-ethyl-1-hydroxy-3-(4-hydroxyphenyl)-2-oxa-1-boranaphthalene. J. Organomet. Chem. 1993, 460, 139–147. [Google Scholar] [CrossRef]
- Sporzyński, A.; Lewandowski, M.; Rogowska, P.; Cyrański, M.K. 1, 3-Dihydro-1-hydroxy-3-morpholin-4-yl-2, 1-benzoxaborole: Product of the reaction of o-formylphenylboronic acid with morpholine. Appl. Organomet. Chem. 2005, 19, 1202–1203. [Google Scholar] [CrossRef]
- Ding, D.; Zhao, Y.; Meng, Q.; Xie, D.; Nare, B.; Chen, D.; Bacchi, C.J.; Yarlett, N.; Zhang, Y.-K.; Hernandez, V. Discovery of novel benzoxaborole-based potent antitrypanosomal agents. ACS Med. Chem. Lett. 2010, 1, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Meng, Q.; Gao, G.; Zhao, Y.; Wang, Q.; Nare, B.; Jacobs, R.; Rock, F.; Alley, M.R.; Plattner, J.J. Design, synthesis, and structure−Activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J. Med. Chem. 2011, 54, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wang, Q.; Zhang, F.; Wang, Z.; Bowling, T.; Nare, B.; Jacobs, R.T.; Zhang, J.; Ding, D.; Liu, Y. Chalcone–benzoxaborole hybrid molecules as potent antitrypanosomal agents. J. Med. Chem. 2012, 55, 3553–3557. [Google Scholar] [CrossRef]
- Alley, M.R.; Baker, S.J.; Beutner, K.R.; Plattner, J. Recent progress on the topical therapy of onychomycosis. Expert Opin. Investig. Drugs 2007, 16, 157–167. [Google Scholar] [CrossRef]
- Baker, S.J.; Zhang, Y.-K.; Akama, T.; Lau, A.; Zhou, H.; Hernandez, V.; Mao, W.; Alley, M.; Sanders, V.; Plattner, J.J. Discovery of a new boron-containing antifungal agent, 5-fluoro-1, 3-dihydro-1-hydroxy-2, 1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 2006, 49, 4447–4450. [Google Scholar] [CrossRef]
- Wardlaw, T.; Salama, P.; Johansson, E.W.; Mason, E. Pneumonia: The leading killer of children. Lancet 2006, 368, 1048–1050. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Hooper, D.C. Hospital-acquired infections due to gram-negative bacteria. N. Engl. J. Med. 2010, 362, 1804–1813. [Google Scholar] [CrossRef]
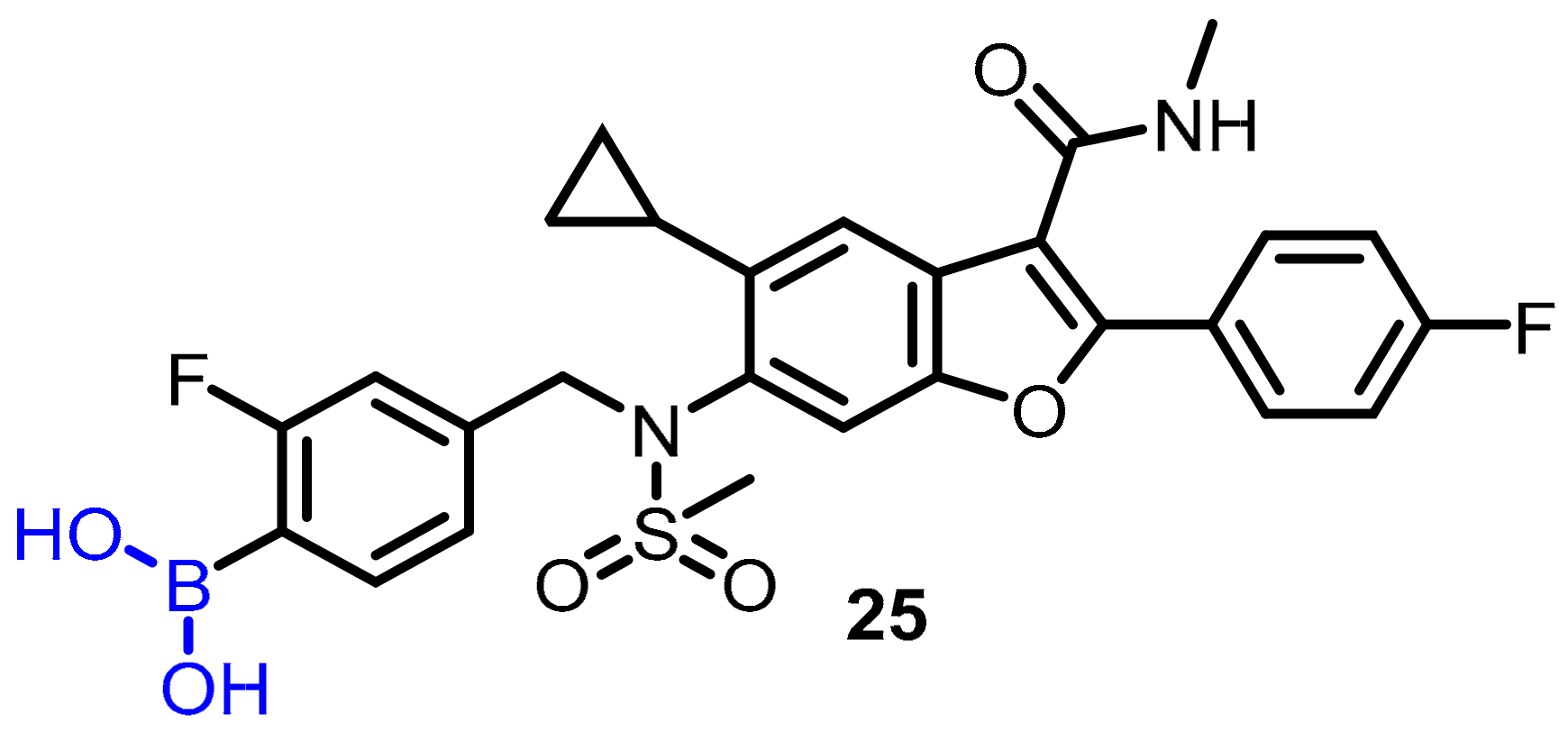
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class a Serine Carbapenemases; ACS Publications: Washington, DC, USA, 2015. [Google Scholar]
- Soloway, A.; Wright, R.; Messer, J. Evaluation of boron compounds for use in neutron capture therapy of brain tumors. I. Animal investigations. J. Pharmacol. Exptl. Therap. 1961, 134, 117–122. [Google Scholar]
- Sivaev, I.B.; Bregadze, V.V. Polyhedral boranes for medical applications: Current status and perspectives. Eur. J. Inorg. Chem. 2009, 2009, 1433–1450. [Google Scholar] [CrossRef]
- Leukart, O.; Caviezel, M.; Eberle, A.; Escher, E.; Tun-Kyi, A.; Schwyzer, R. L-O-Carboranylalanine, a boron analogue of phenylalanine. Helv. Chim. Acta 1976, 59, 2184–2187. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Grebennikov, A.V.; L’Vov, A.I. Synthesis of some nitrogenous barene derivatives. Bull. Acad. Sci. USSR Div. Chem. Sci. 1970, 19, 97–102. [Google Scholar] [CrossRef]
- Kahl, S.B.; Li, J. Synthesis and Characterization of a Boronated Metallophthalocyanine for Boron Neutron Capture Therapy. Inorg. Chem. 1996, 35, 3878–3880. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1–3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Scholz, M.; Bensdorf, K.; Gust, R.; Hey-Hawkins, E. Asborin: The Carbaborane Analogue of Aspirin. ChemMedChem 2009, 4, 746–748. [Google Scholar] [CrossRef]
- Reddy, M.V.; Mallireddigari, M.R.; Pallela, V.R.; Venkatapuram, P.; Boominathan, R.; Bell, S.C.; Reddy, E.P. Design, synthesis, and biological evaluation of (E)- and (Z)-styryl-2-acetoxyphenyl sulfides and sulfones as cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. 2005, 13, 1715–1723. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Watley, R.L.; Awuah, S.G.; Bio, M.; Cantu, R.; Gobeze, H.B.; Nesterov, V.N.; Das, S.K.; D’Souza, F.; You, Y. Dual Functioning Thieno-Pyrrole Fused BODIPY Dyes for NIR Optical Imaging and Photodynamic Therapy: Singlet Oxygen Generation without Heavy Halogen Atom Assistance. Chem. Asian J. 2015, 10, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.D.; Kim, N.N.; Traish, A.M.; Christianson, D.W. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat. Struct. Biol. 1999, 6, 1043–1047. [Google Scholar] [PubMed]
- Baggio, R.; Elbaum, D.; Kanyo, Z.F.; Carroll, P.J.; Cavalli, R.C.; Ash, D.E.; Christianson, D.W. Inhibition of Mn2+2-Arginase by Borate Leads to the Design of a Transition State Analogue Inhibitor, 2(S)-Amino-6-boronohexanoic Acid. J. Am. Chem. Soc. 1997, 119, 8107–8108. [Google Scholar] [CrossRef]
- Kanyo, Z.F.; Scolnick, L.R.; Ash, D.E.; Christianson, D.W. Structure of a unique binuclear manganese cluster in arginase. Nature 1996, 383, 554–557. [Google Scholar] [CrossRef]
- Kettner, C.A.; Shenvi, A.B. Inhibition of the serine proteases leukocyte elastase, pancreatic elastase, cathepsin G, and chymotrypsin by peptide boronic acids. J. Biol. Chem. 1984, 259, 15106–15114. [Google Scholar] [CrossRef]
- Shenvi, A.B. alpha-Aminoboronic acid derivatives: Effective inhibitors of aminopeptidases. Biochemistry 1986, 25, 1286–1291. [Google Scholar] [CrossRef]
- Kaplánek, R.; Martásek, P.; Grüner, B.; Panda, S.; Rak, J.; Masters, B.S.; Král, V.; Roman, L.J. Nitric oxide synthases activation and inhibition by metallacarborane-cluster-based isoform-specific affectors. J. Med. Chem. 2012, 55, 9541–9548. [Google Scholar] [CrossRef] [Green Version]
- Simerska, P.; Suksamran, T.; Ziora, Z.M.; De Labastida Rivera, L.; Engwerda, C.; Toth, I. Ovalbumin lipid core peptide vaccines and their CD4+ and CD8+ T cell responses. Vaccine 2014, 32, 4743–4750. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; De Veerman, M.; Coyle, A.J.; Gutierrez-Ramos, J.C.; Thielemans, K.; Pauwels, R.A. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J. Clin. Investig. 2000, 106, 551–559. [Google Scholar] [CrossRef]
- Watzl, B.; Abrahamse, S.L.; Treptow-van Lishaut, S.; Neudecker, C.; Hänsch, G.M.; Rechkemmer, G.; Pool-Zobel, B.L. Enhancement of ovalbumin-induced antibody production and mucosal mast cell response by mercury. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 1999, 37, 627–637. [Google Scholar] [CrossRef]
- Parnes, J.R. Molecular biology and function of CD4 and CD8. Adv. Immunol. 1989, 44, 265–311. [Google Scholar] [PubMed]
- O’Rourke, A.M.; Mescher, M.F. The roles of CD8 in cytotoxic T lymphocyte function. Immunol. Today 1993, 14, 177–183. [Google Scholar] [CrossRef]
- Routray, I.; Ali, S. Boron Induces Lymphocyte Proliferation and Modulates the Priming Effects of Lipopolysaccharide on Macrophages. PLoS ONE 2016, 11, e0150607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, A.G.; Rivers-Auty, J.; Daniels, M.J.; White, C.S.; Schwalbe, C.H.; Schilling, T.; Hammadi, H.; Jaiyong, P.; Spencer, N.G.; England, H. Boron-based inhibitors of the NLRP3 inflammasome. Cell Chem. Biol. 2017, 24, 1321–1335. [Google Scholar] [CrossRef] [Green Version]
- Yinghuai, Z.; Hosmane, N.S. Applications and perspectives of boron-enriched nanocomposites in cancer therapy. Future Med. Chem. 2013, 5, 705–714. [Google Scholar] [CrossRef]
- Pizzorno, L. Nothing Boring About Boron. Integr. Med. 2015, 14, 35–48. [Google Scholar]
- Soriano-Ursúa, M.A.; Das, B.C.; Trujillo-Ferrara, J.G. Boron-containing compounds: Chemico-biological properties and expanding medicinal potential in prevention, diagnosis and therapy. Expert Opin. Ther. Pat. 2014, 24, 485–500. [Google Scholar] [CrossRef]

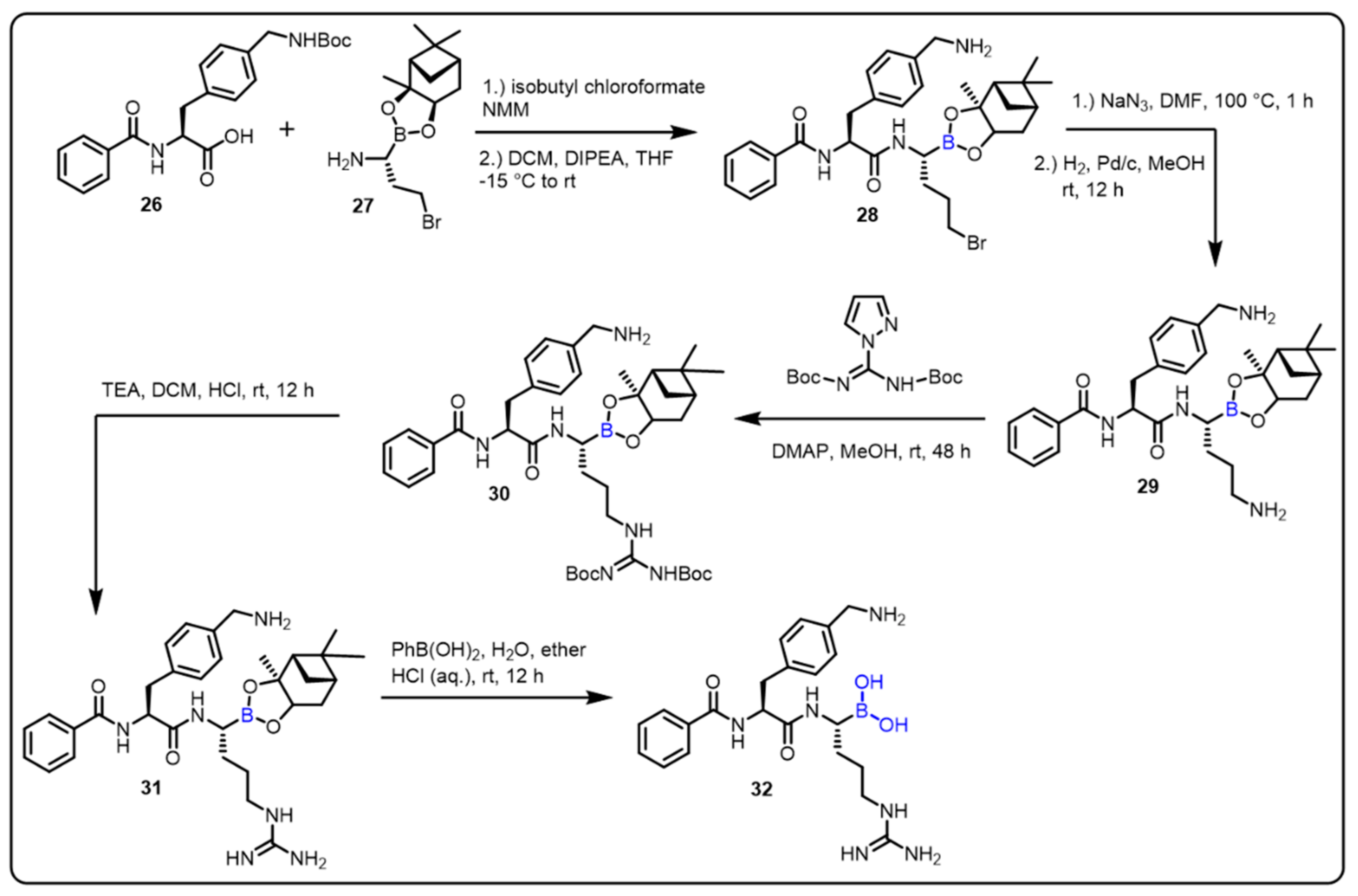
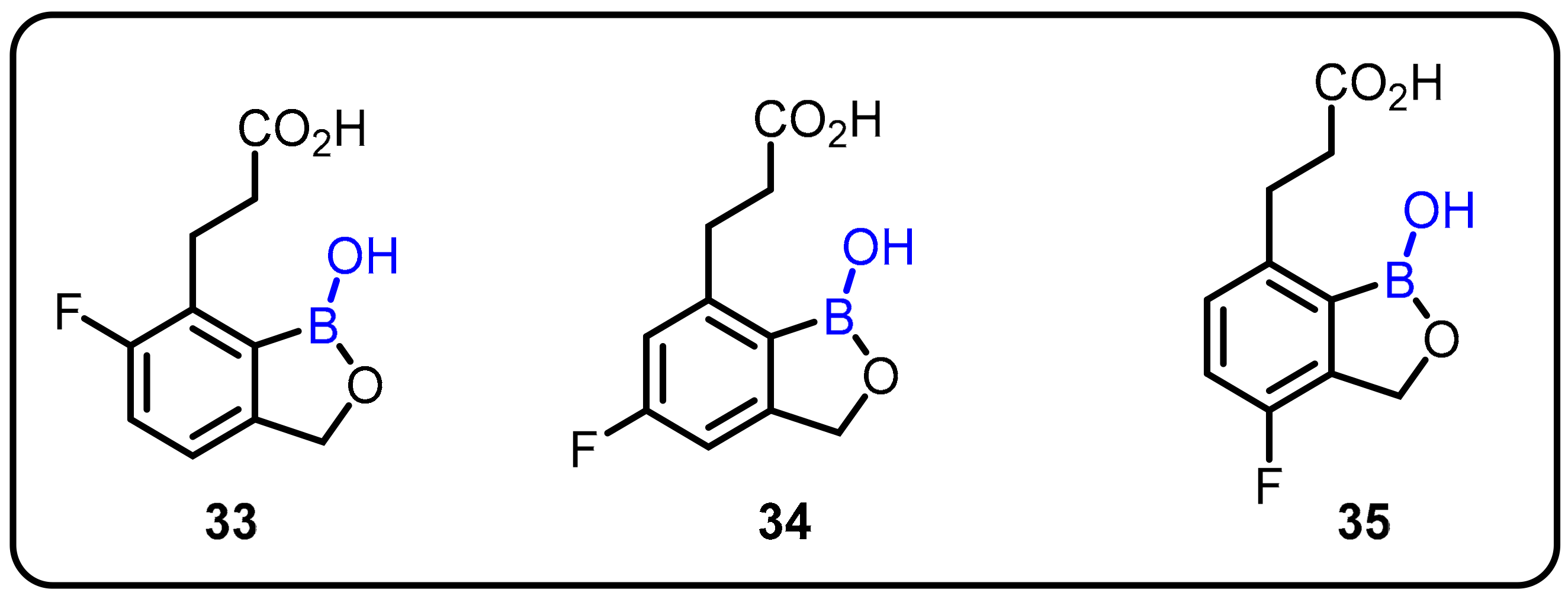
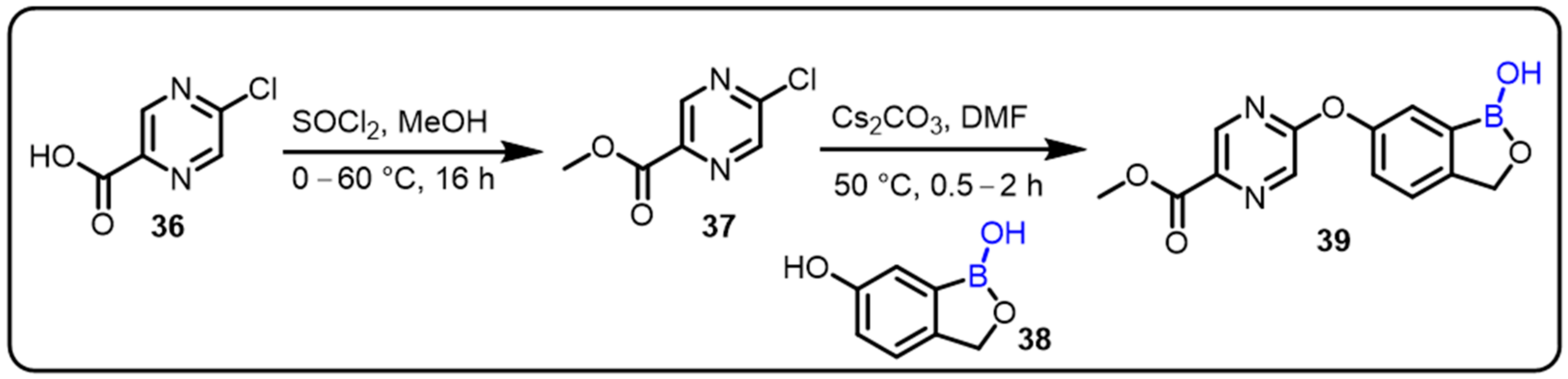
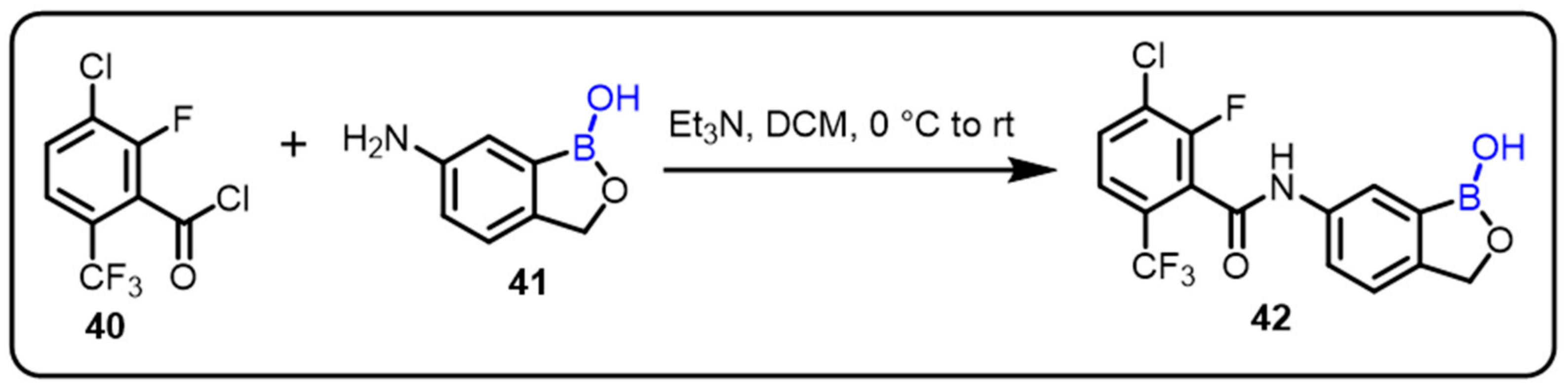

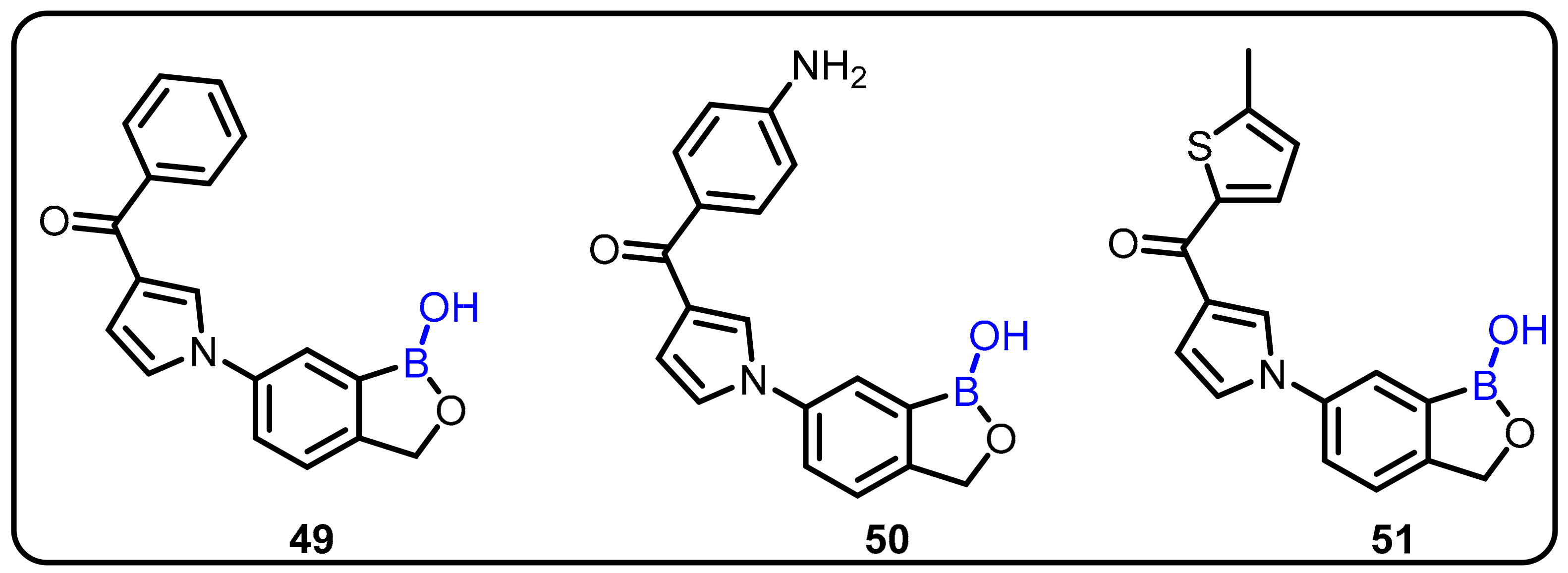
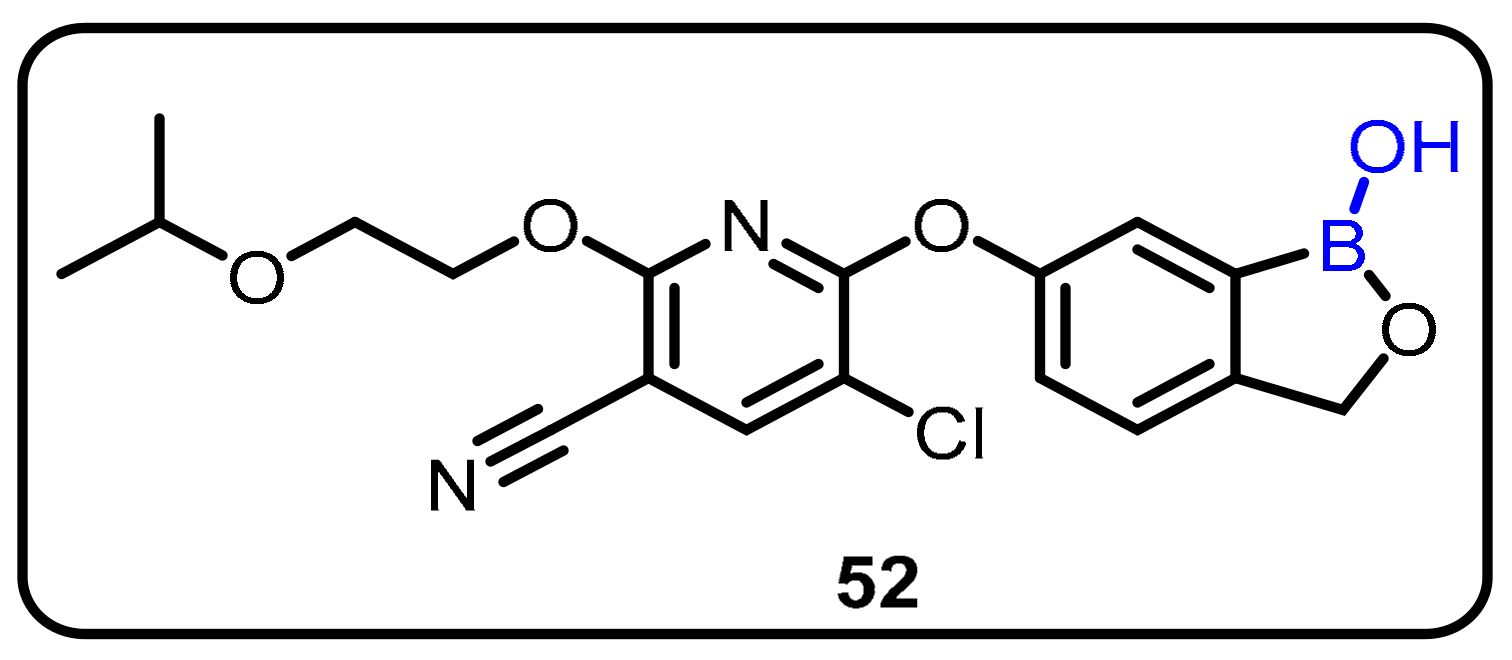

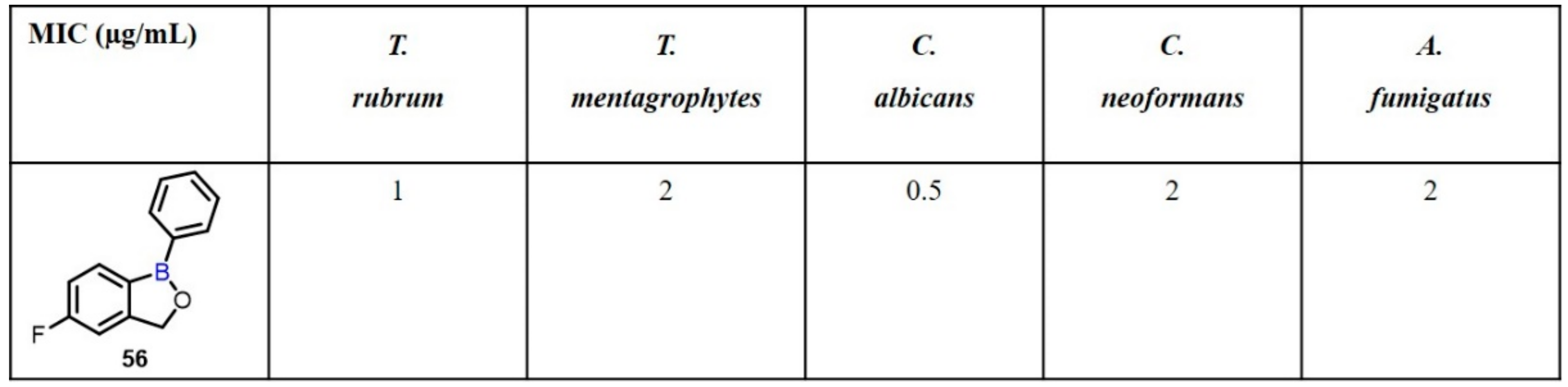
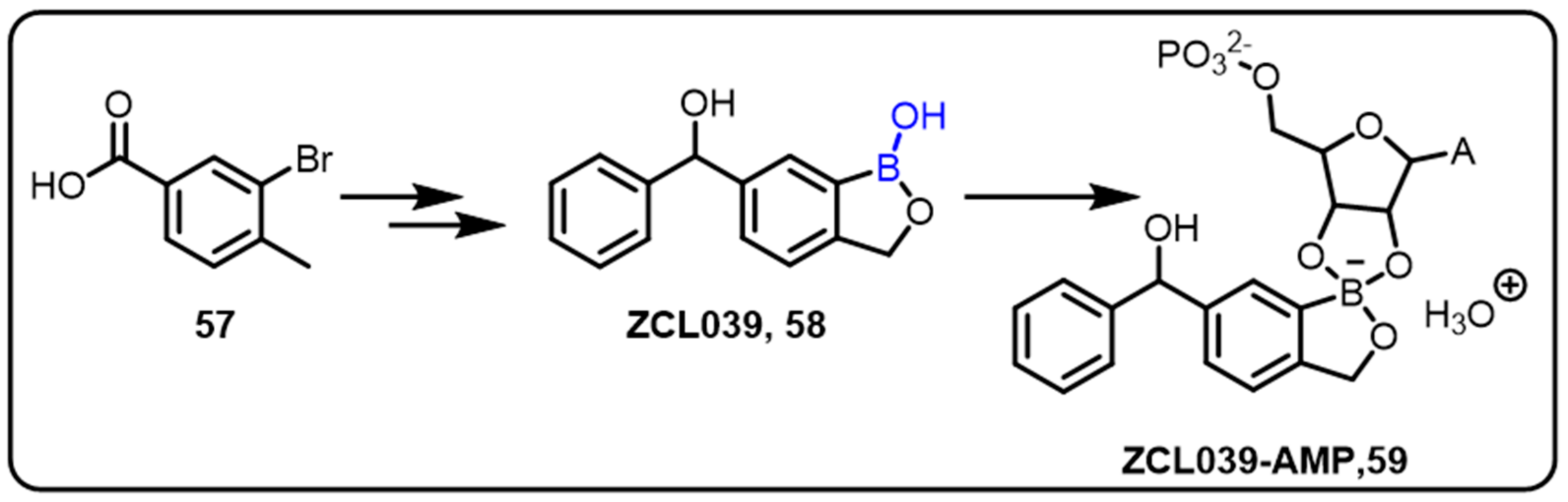
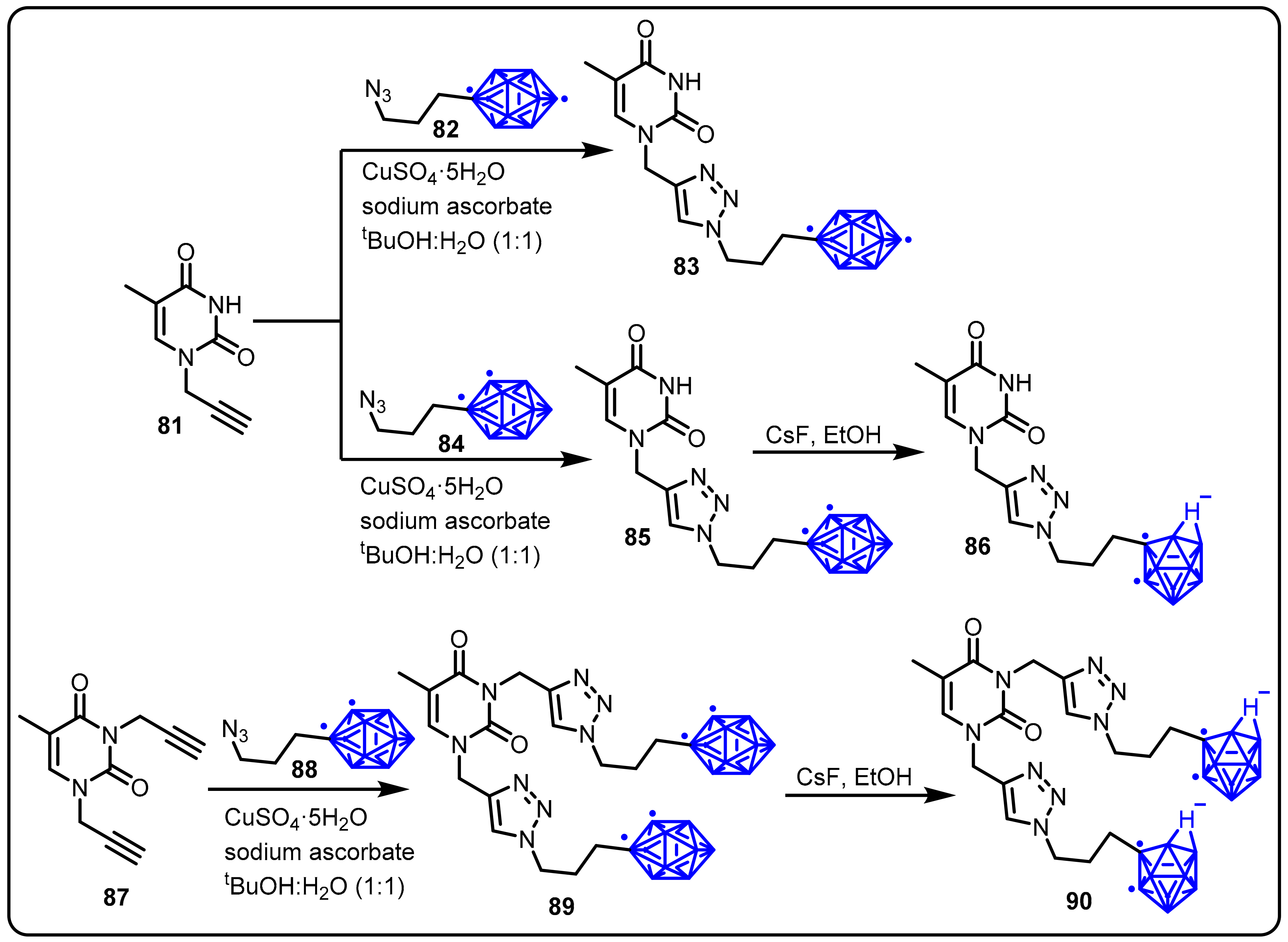
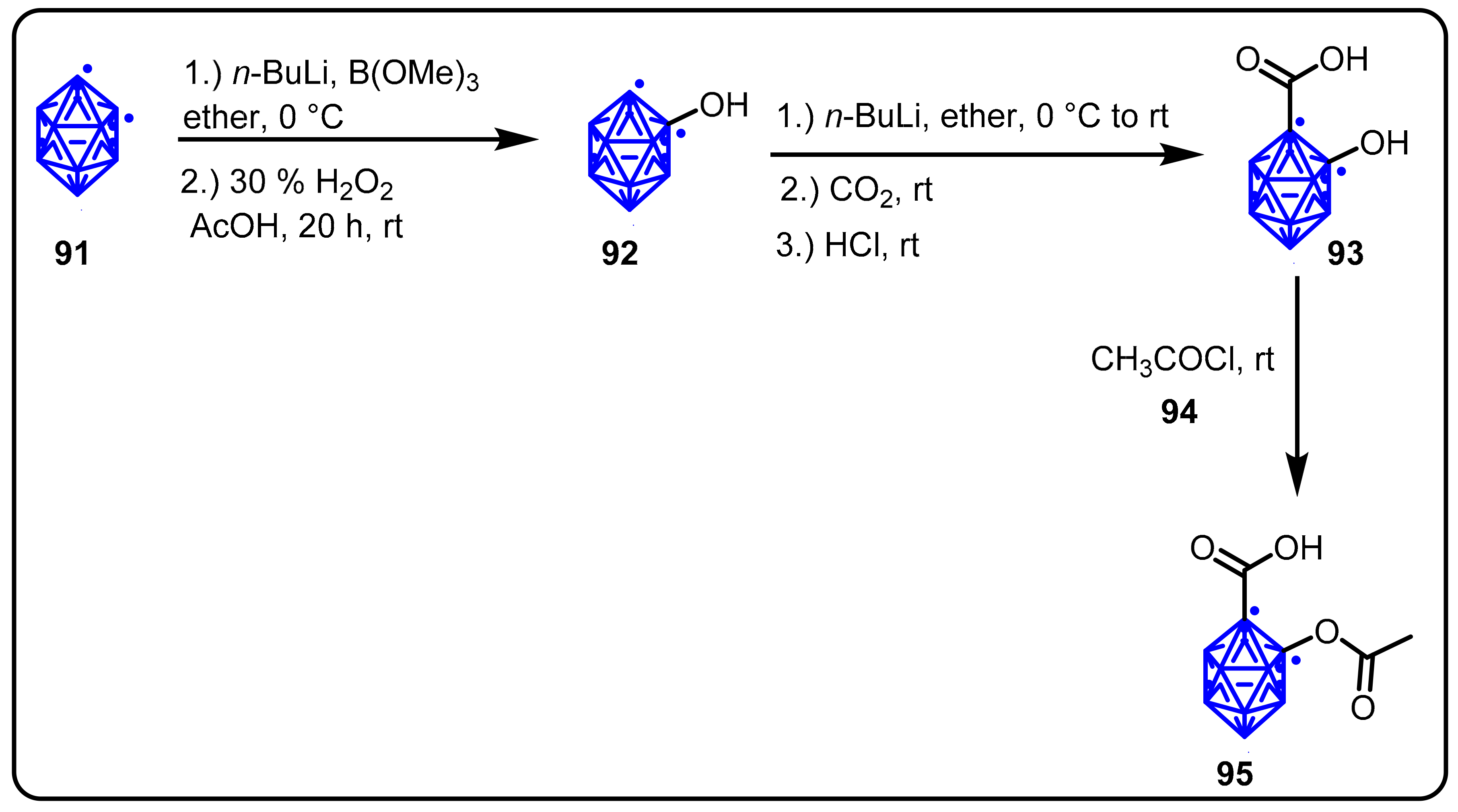

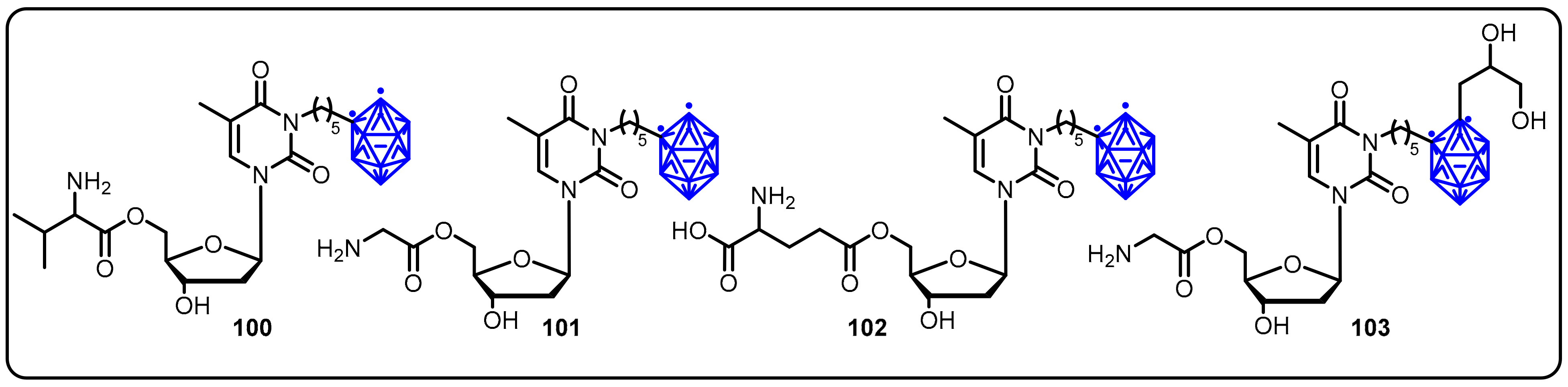



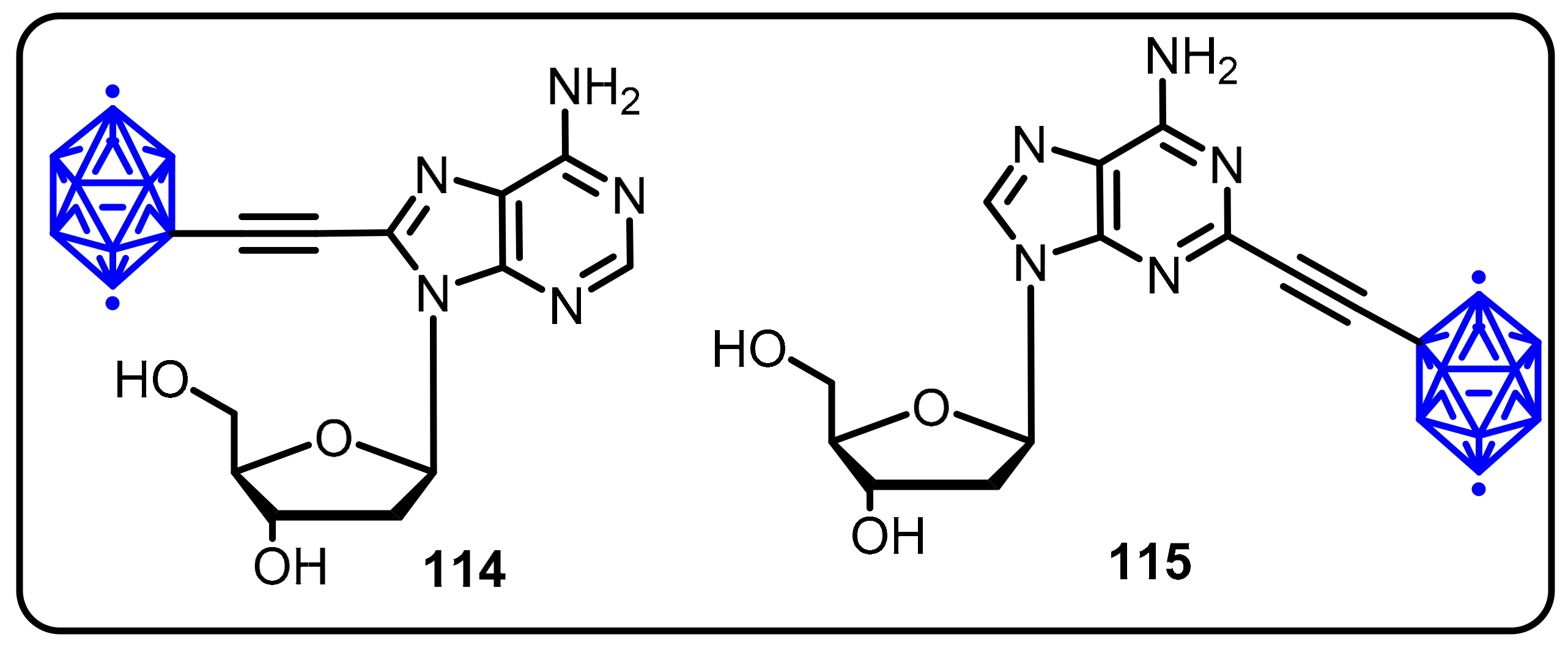



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Boronic Acid/Esters/Aminoboronic Acids | Benzoxaborole Derivatives | Carboranes | BODIPYand Other Boron Reagents | Ref | |
|---|---|---|---|---|---|
| Lipogenic inhibitors (heart) | 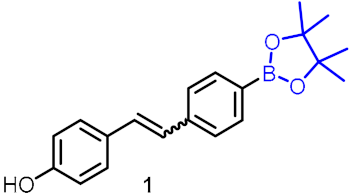 | [22] | |||
| Improves lipid homeostasis |  | [23] | |||
| Amyotrophic lateral sclerosis |  | [24] | |||
| Proteasome inhibitor | Bortezomib (7) Ixazomib (8) Delanzomib (9) | [25,26,27] | |||
| Inhibitors of HIV reverse transcriptase (RT) | 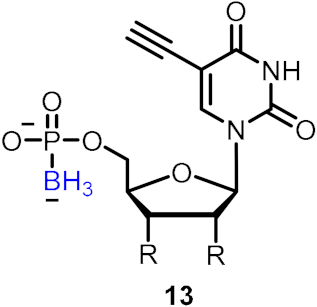 | [28] | |||
| Inhibitors of human neutrophil elastase (HNE) | 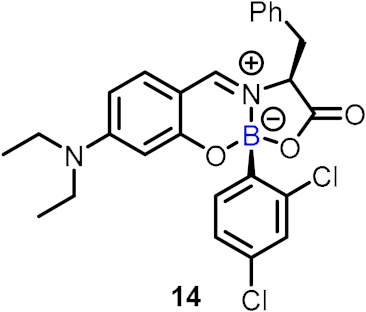 | [29] | |||
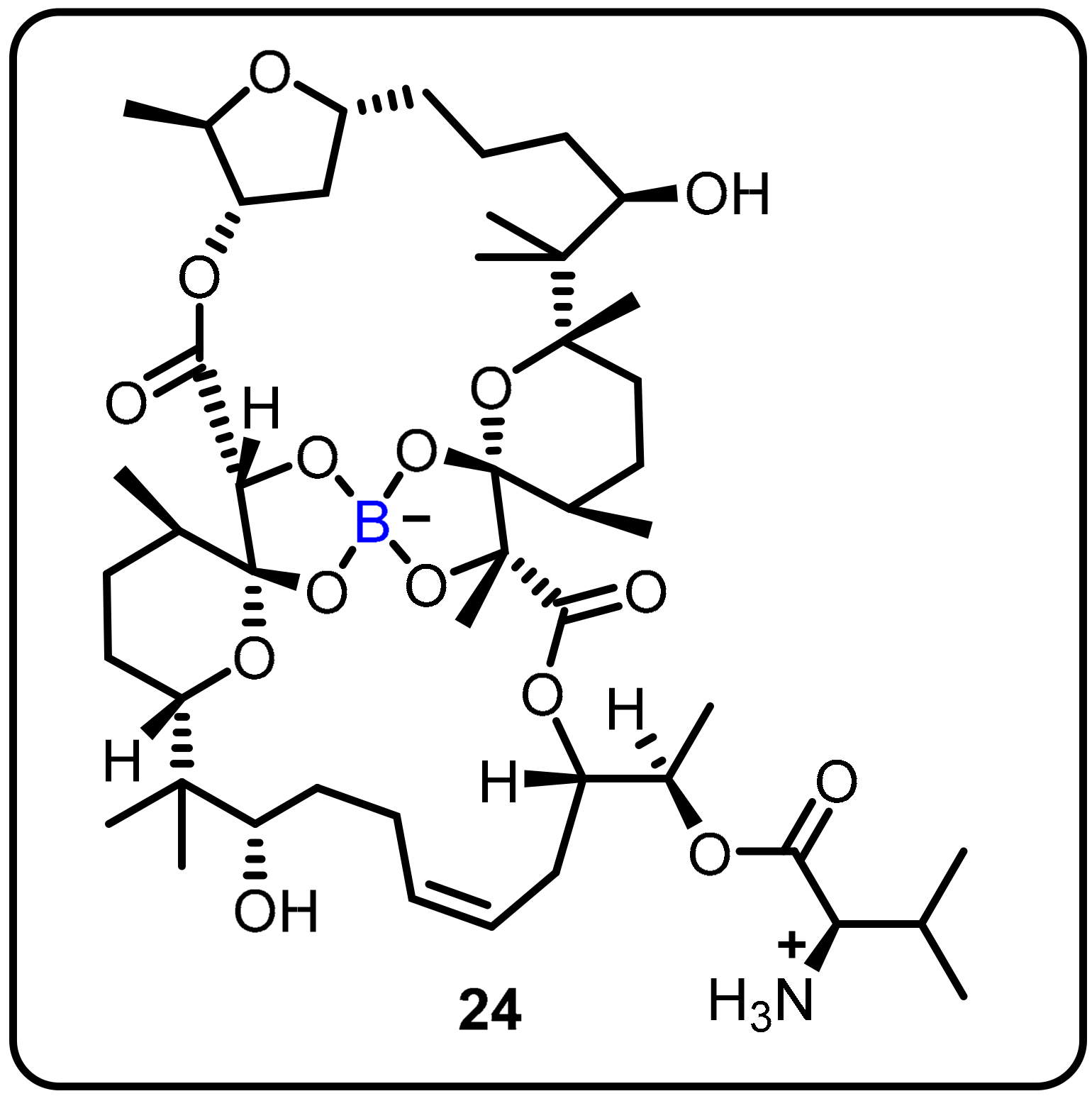
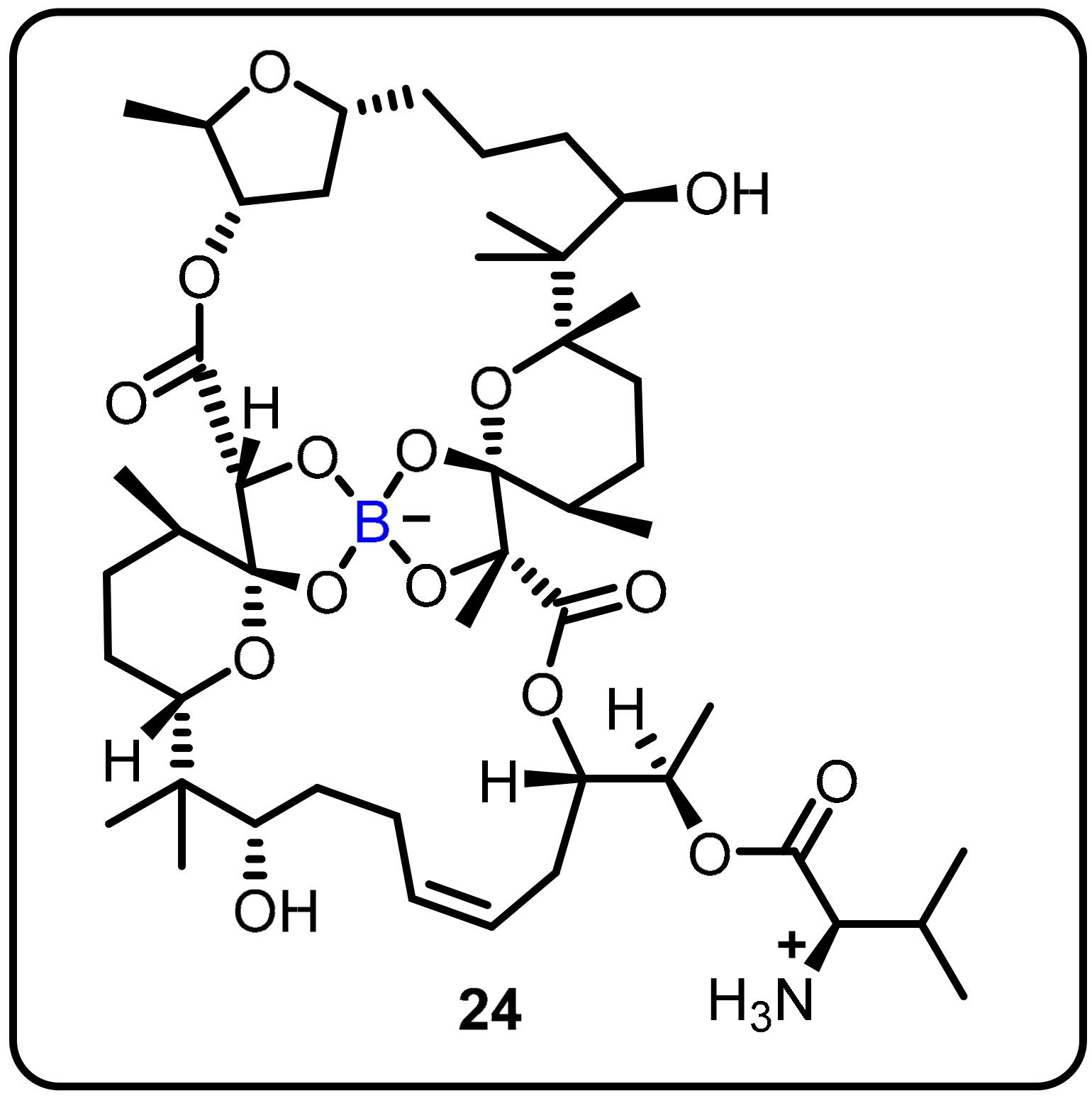
| Anti-mycobacterial agents | Boromycin (24) | 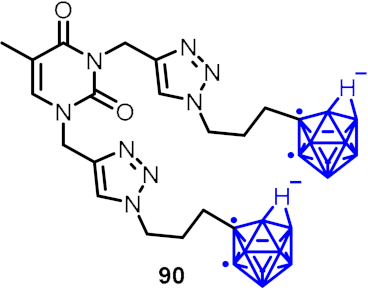 |  | [30,31,32] | |
| Inhibitor of HCV |  | [33] | |||
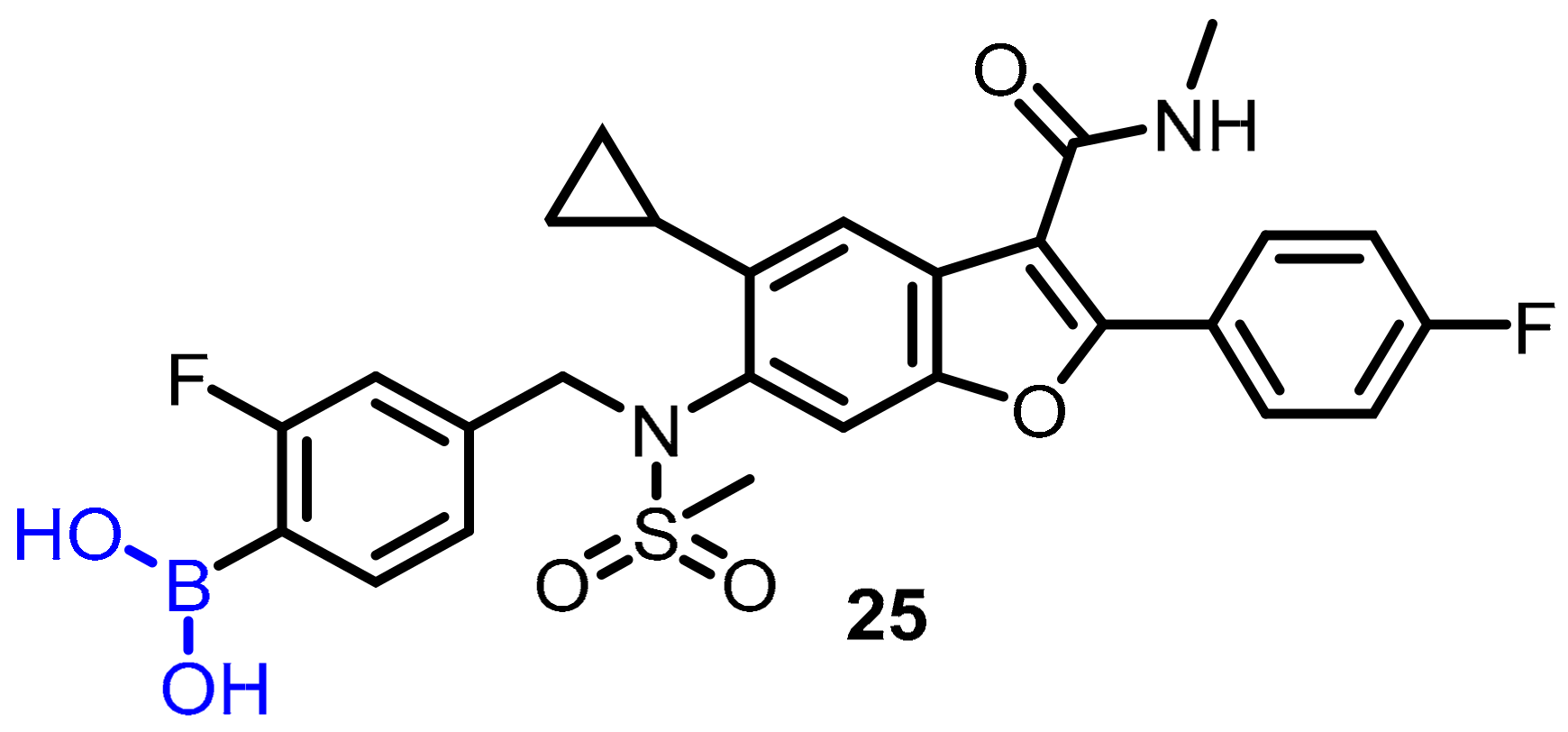
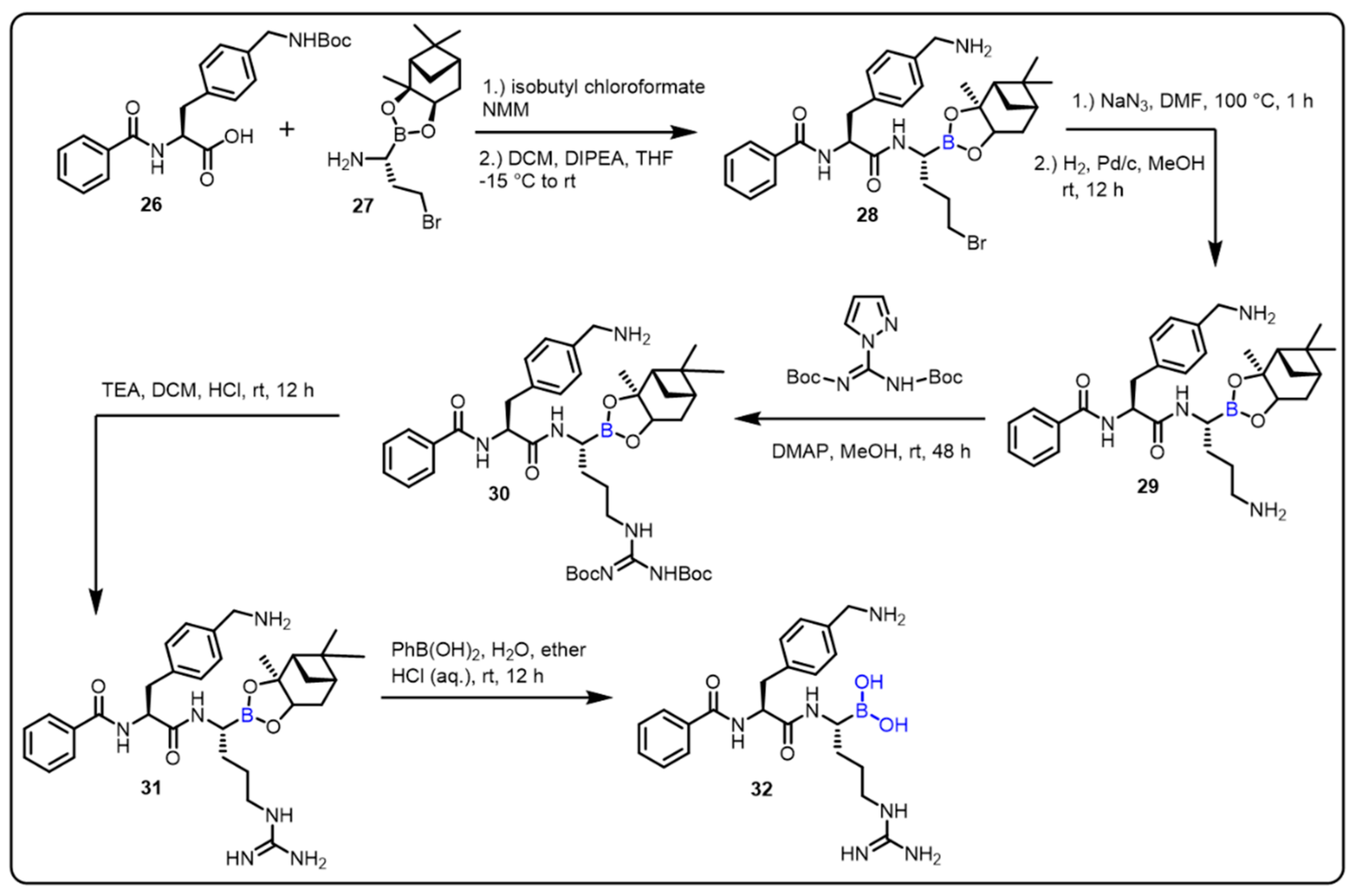
| Anticancer agent | 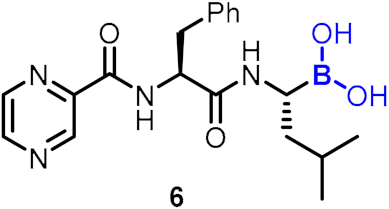 | 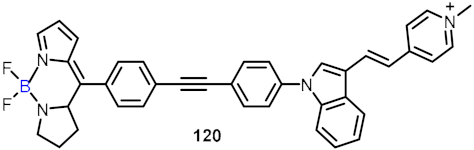 | [34,35,36,37,38,39] | ||
| Protease inhibitor | 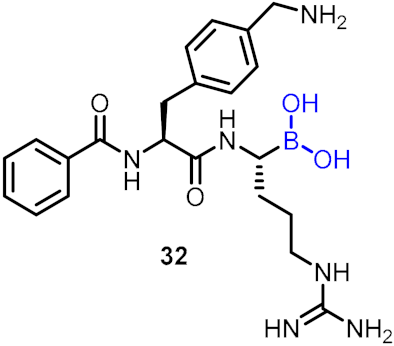 | [40] | |||
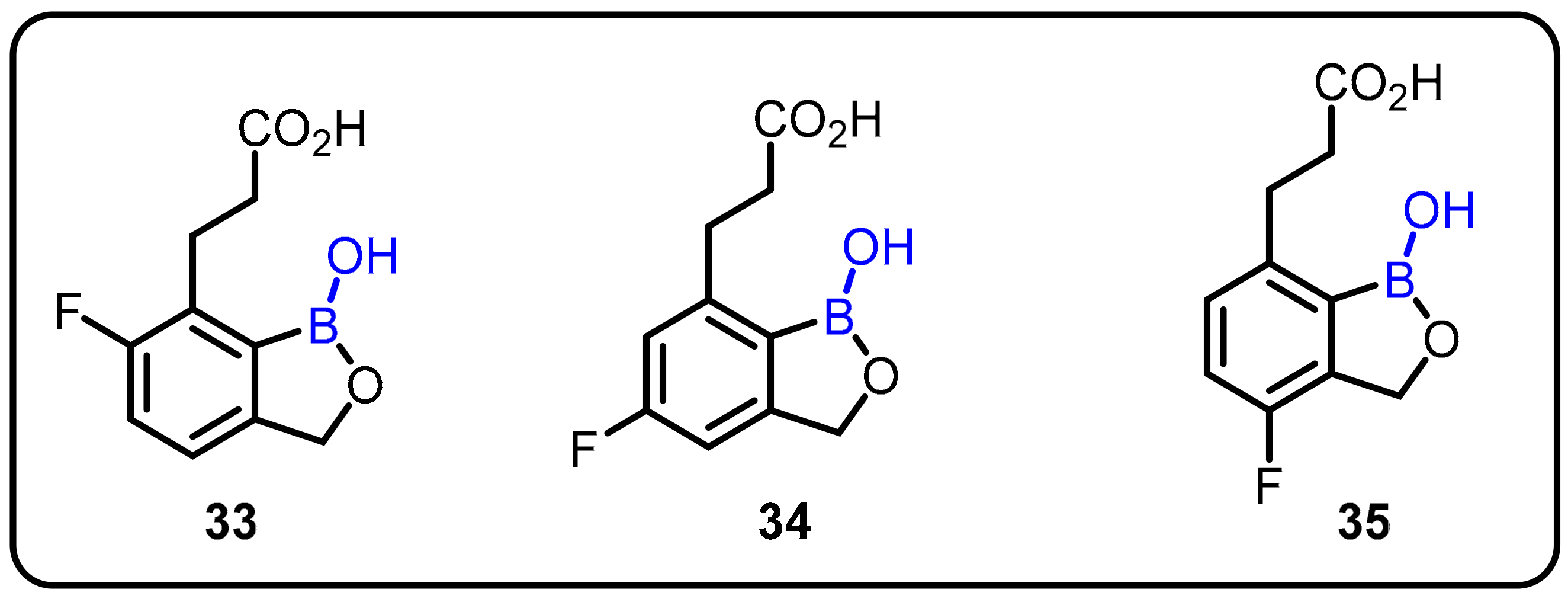
| Antimalarial agents |  | [41,42,43,44,45] | |||
| Animal ectoparasiticide |  | [46] | |||
| Anti-trypanosomal agents | 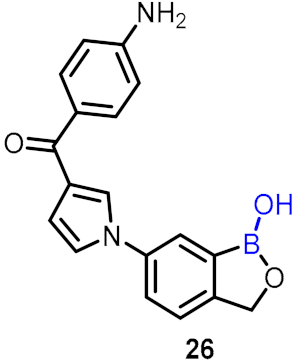 | [47] | |||
| Anti-inflammatory diseases | 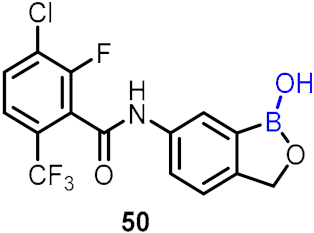 |  | [48,49,50] | ||
| Antifungal drug |  | [51] | |||
| Anti-pneumococcal agent (lung) | 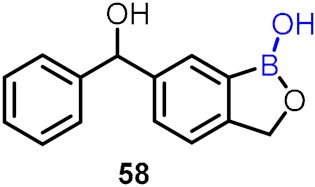 | [52] | |||
| Antibacterial activity | 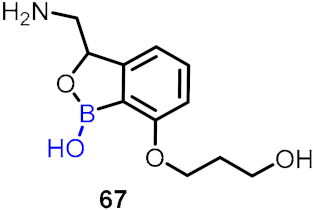 |  | [53,54] | ||
| β-lactamase inhibitors |  | [55] | |||
| Antidepressant activity | 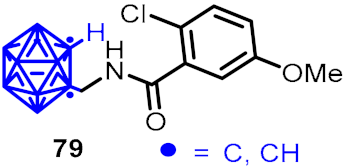 | [56] | |||
| Estrogen receptor partial agonist candidates | 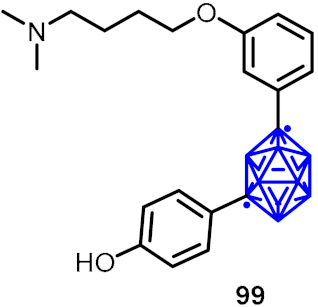 | [57] | |||
| Brain antitumor agents |  | [58] | |||
| Non-secosteroidal vitamin D receptor | 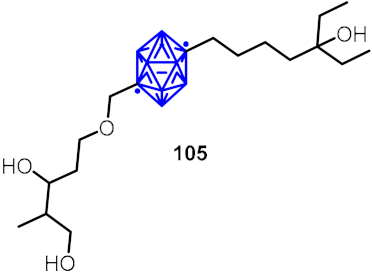 | [59] | |||
| Antifolate analog |  | [60] | |||
| Anesthetics |  | [61] | |||
| Antileukemic activity |  | [62] | |||
| Androgen receptor antagonists |  | [63] | |||
| Hypoxia-inducible factor (HIF-1α) inhibitor | 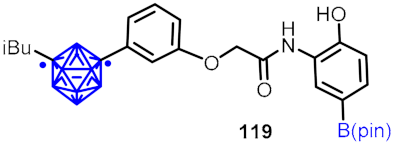 | [64] | |||
| Amyloid-β detection | 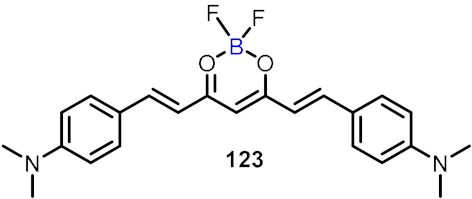 | [65] | |||
| Antiallergic activity |  | [66] | |||
| Anthrax |  | [67] | |||
| NLRP3 inhibitor |  | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y.; Saito, M.; Weiss, L.M.; Almaguel, F.; Hosmane, N.S.; et al. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules 2022, 27, 2615. https://doi.org/10.3390/molecules27092615
Das BC, Nandwana NK, Das S, Nandwana V, Shareef MA, Das Y, Saito M, Weiss LM, Almaguel F, Hosmane NS, et al. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules. 2022; 27(9):2615. https://doi.org/10.3390/molecules27092615
Chicago/Turabian StyleDas, Bhaskar C., Nitesh K. Nandwana, Sasmita Das, Varsha Nandwana, Mohammed Adil Shareef, Yogarupa Das, Mariko Saito, Louis M. Weiss, Frankis Almaguel, Narayan S. Hosmane, and et al. 2022. "Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective" Molecules 27, no. 9: 2615. https://doi.org/10.3390/molecules27092615
APA StyleDas, B. C., Nandwana, N. K., Das, S., Nandwana, V., Shareef, M. A., Das, Y., Saito, M., Weiss, L. M., Almaguel, F., Hosmane, N. S., & Evans, T. (2022). Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules, 27(9), 2615. https://doi.org/10.3390/molecules27092615