

Atomistic Modeling of Quaternized Chitosan Head Groups: Insights into Chemical Stability and Ion Transport for Anion Exchange Membrane Applications

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. DFT Results
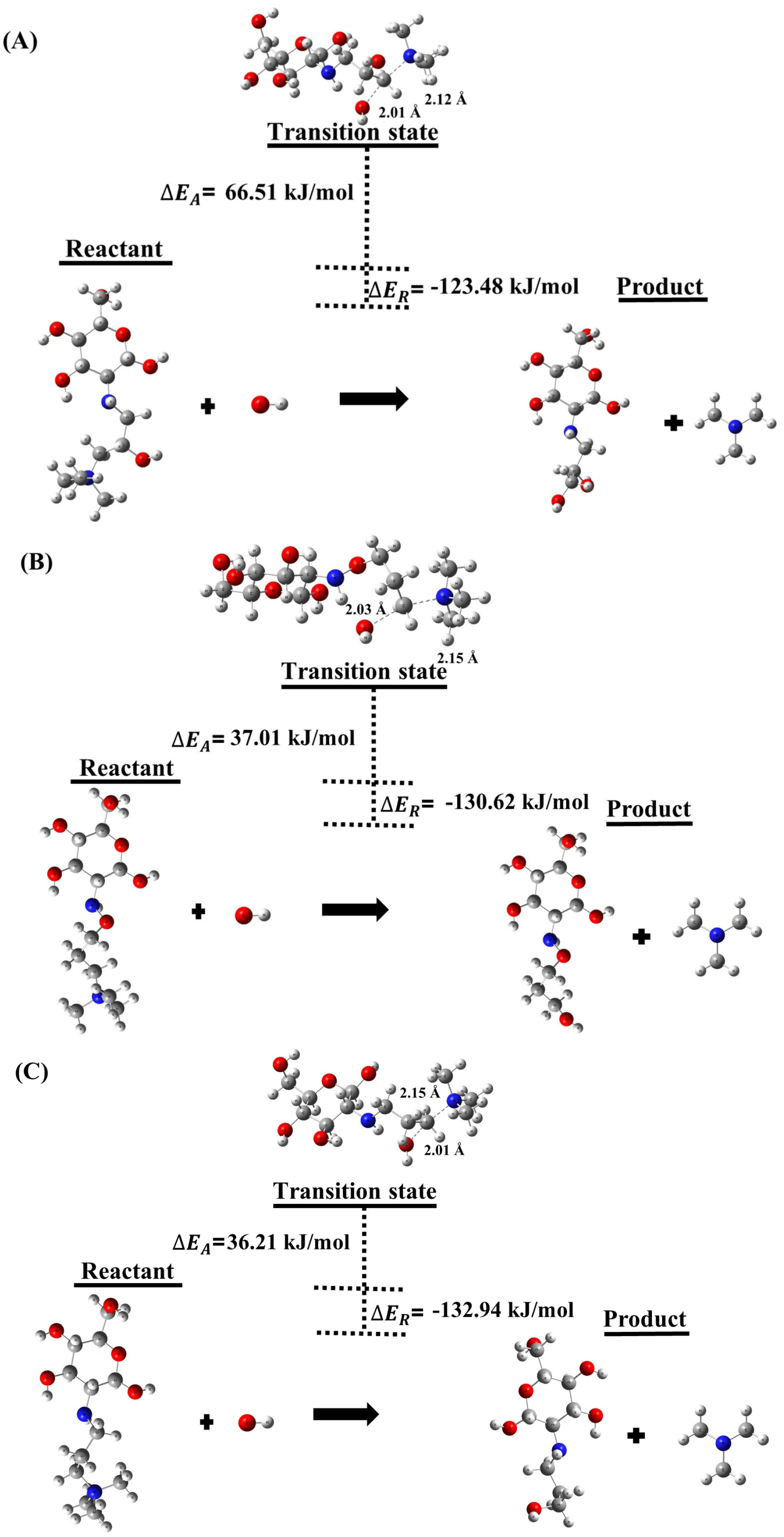
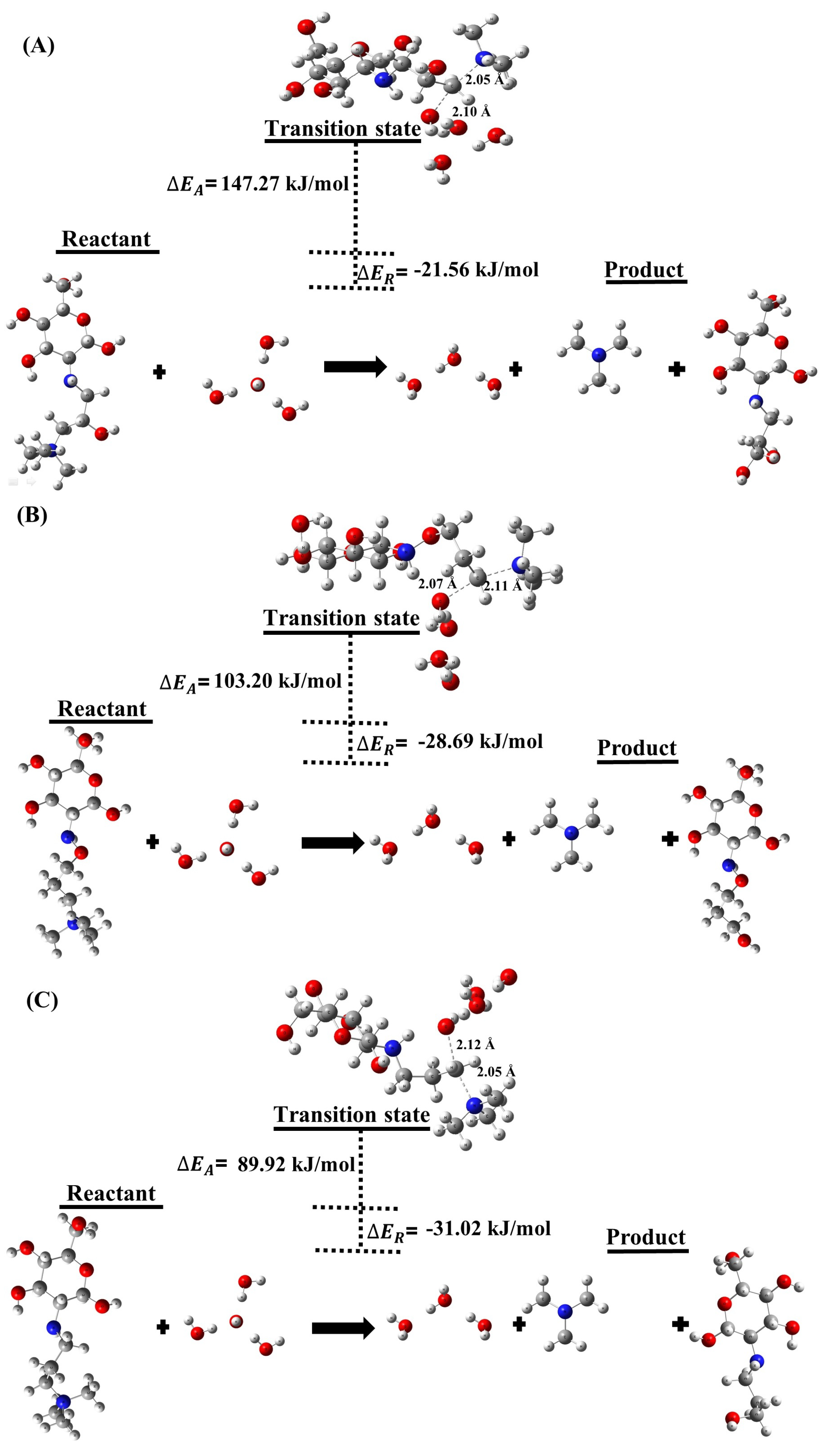
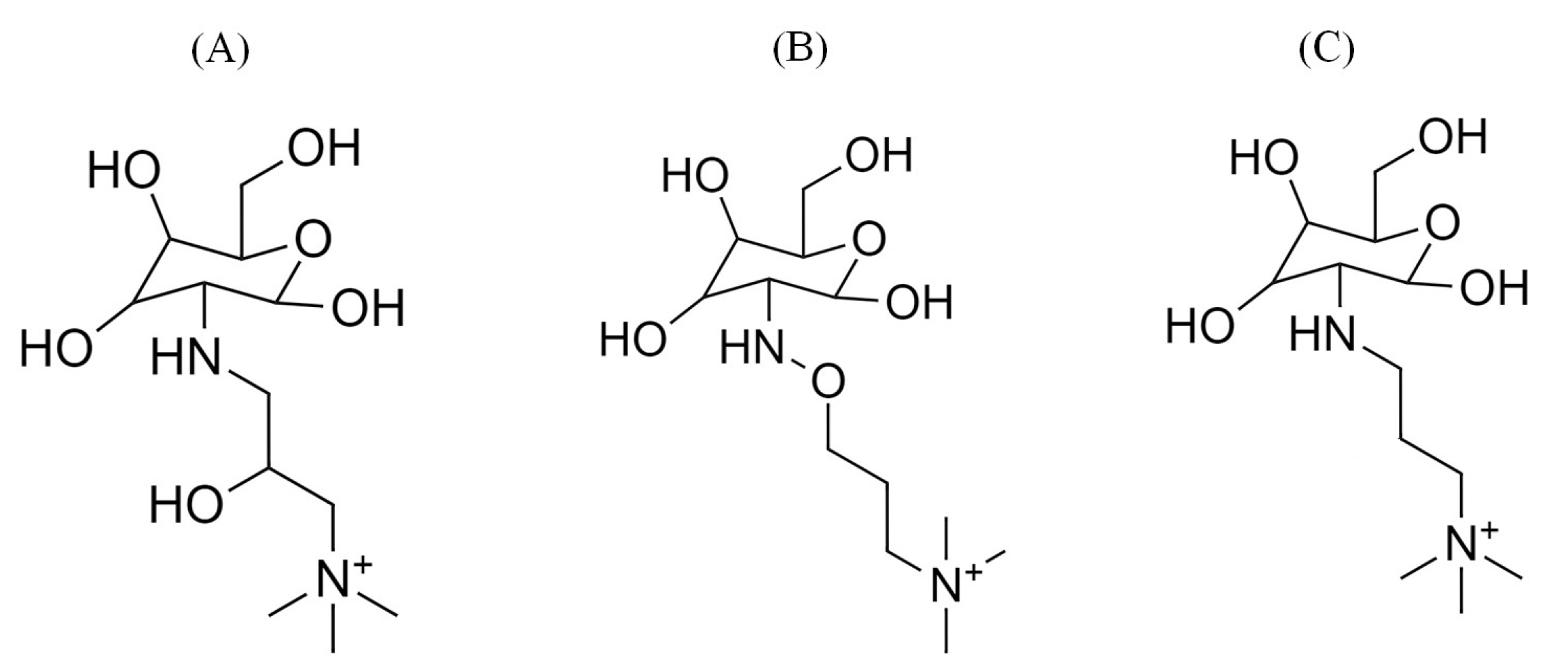
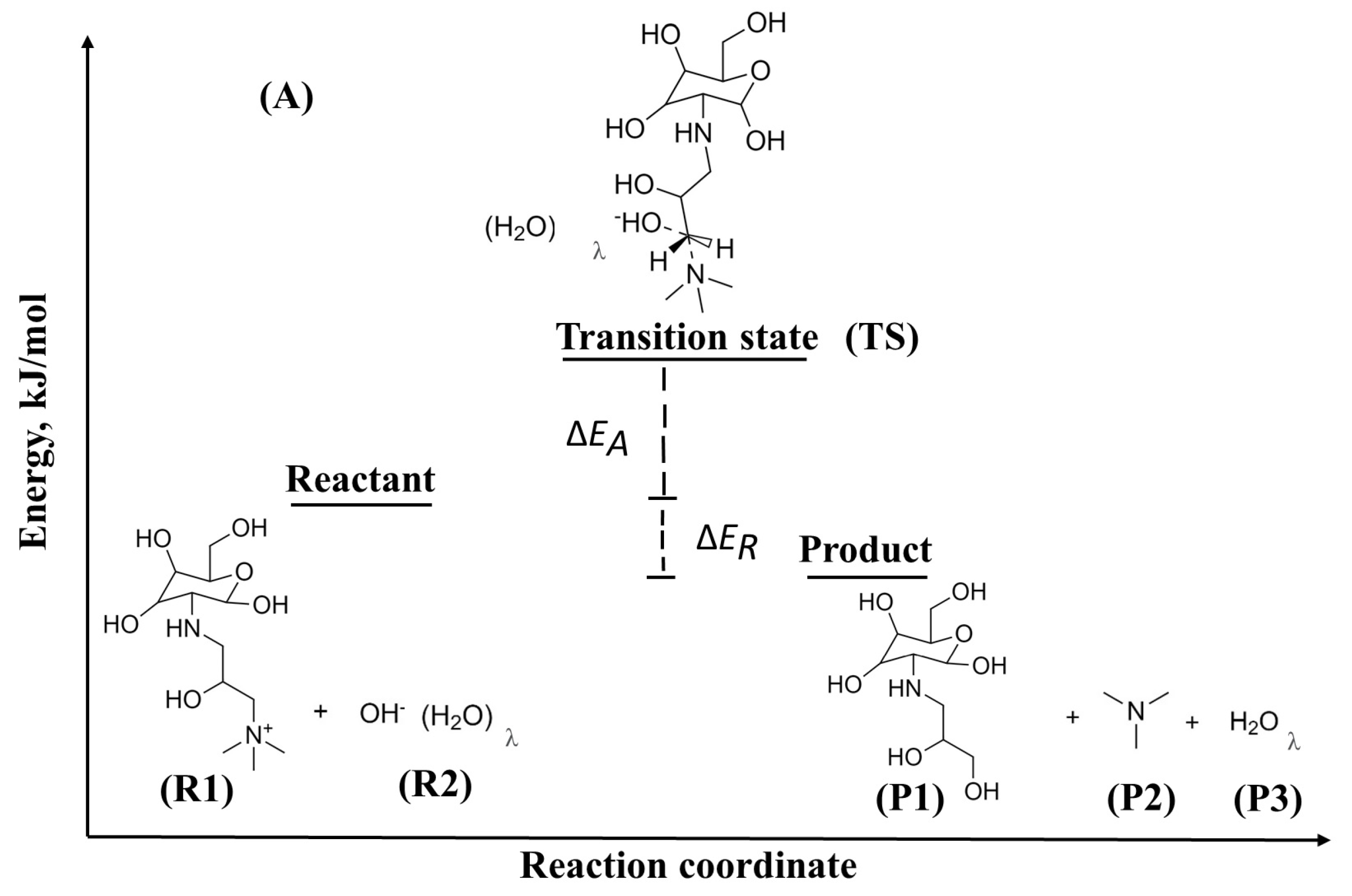
2.1.1. Degradation Reactions
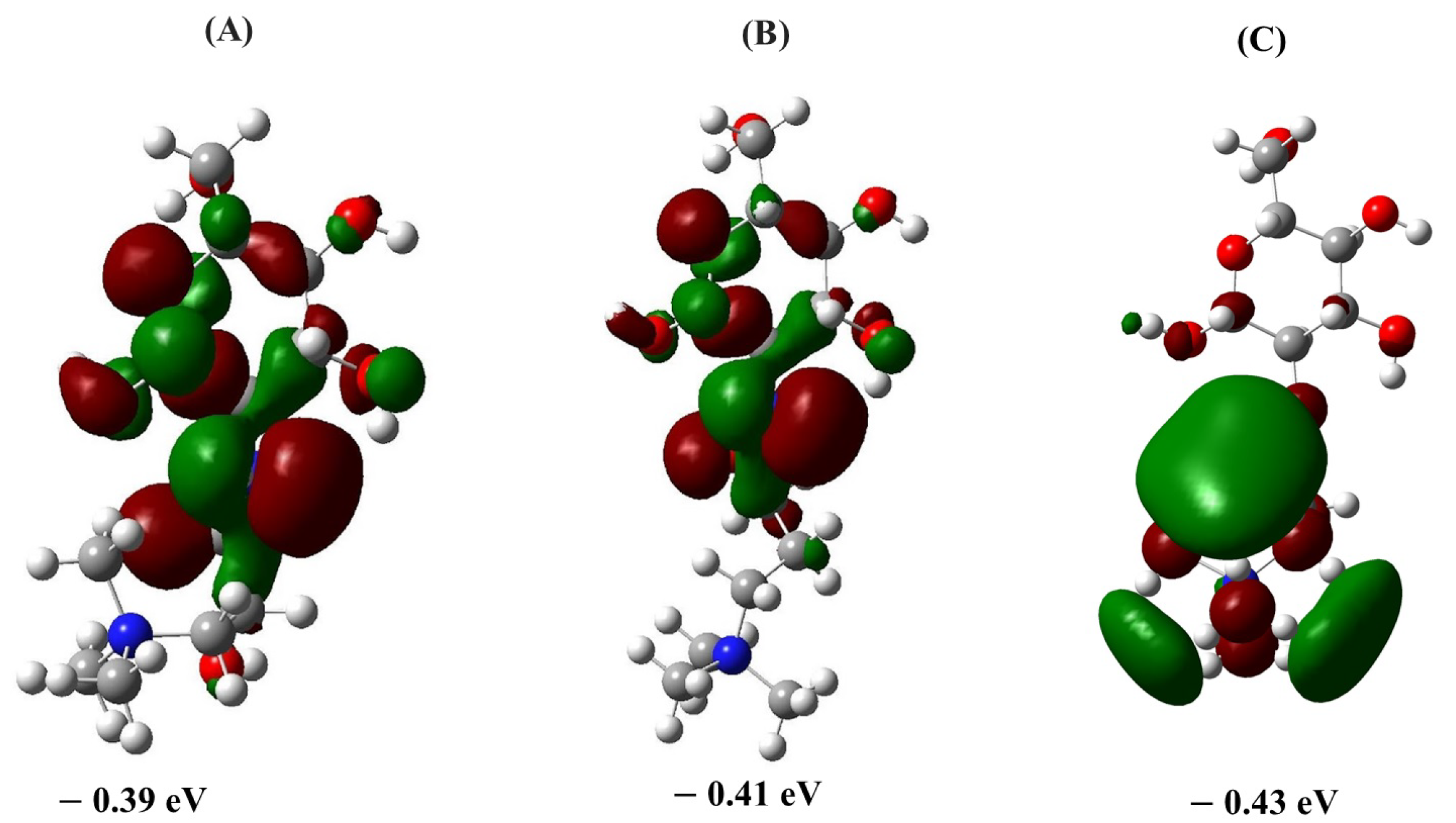
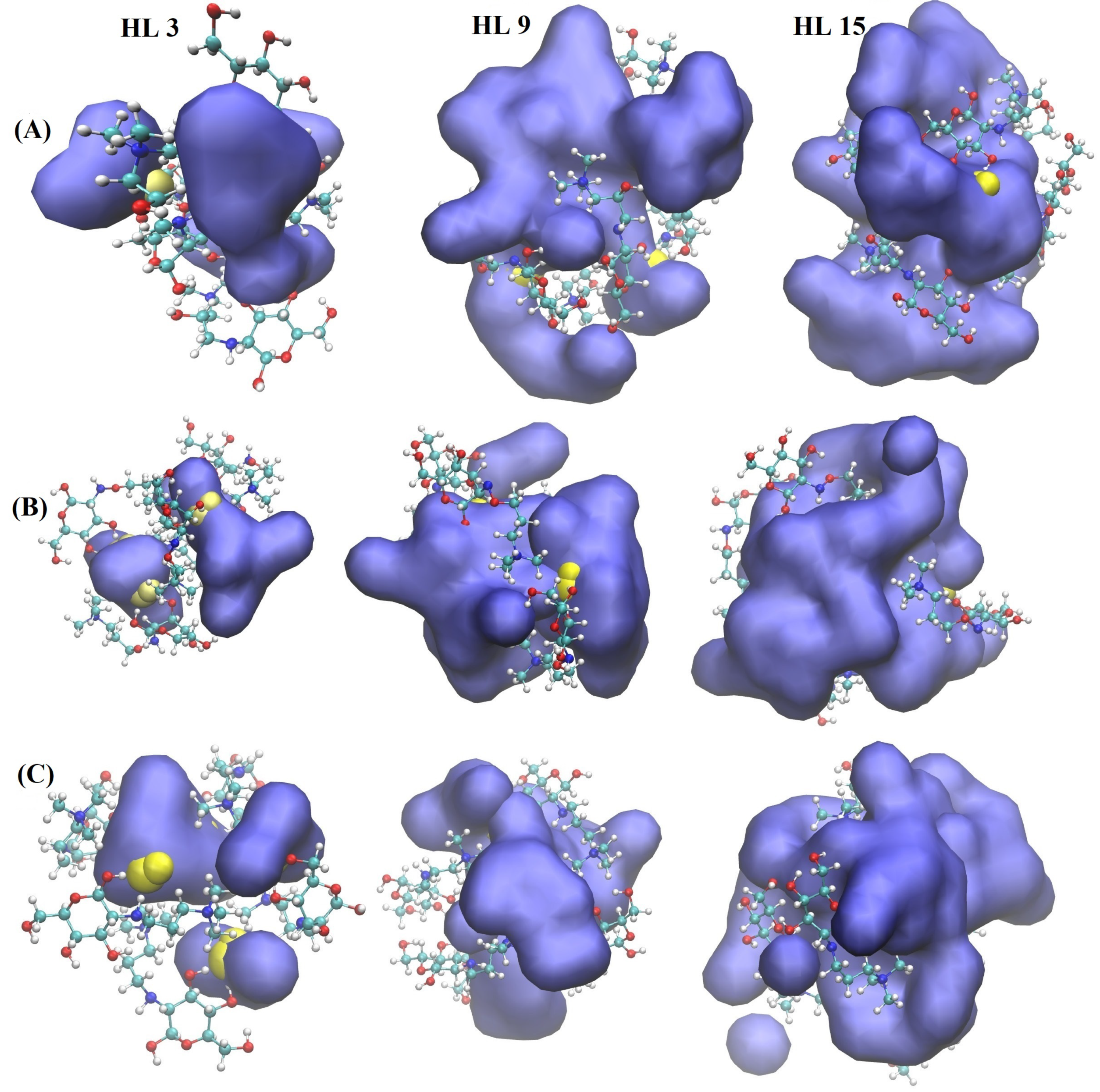
2.1.2. LUMO Distribution and Energies
2.2. Classical All-Atom MD Results
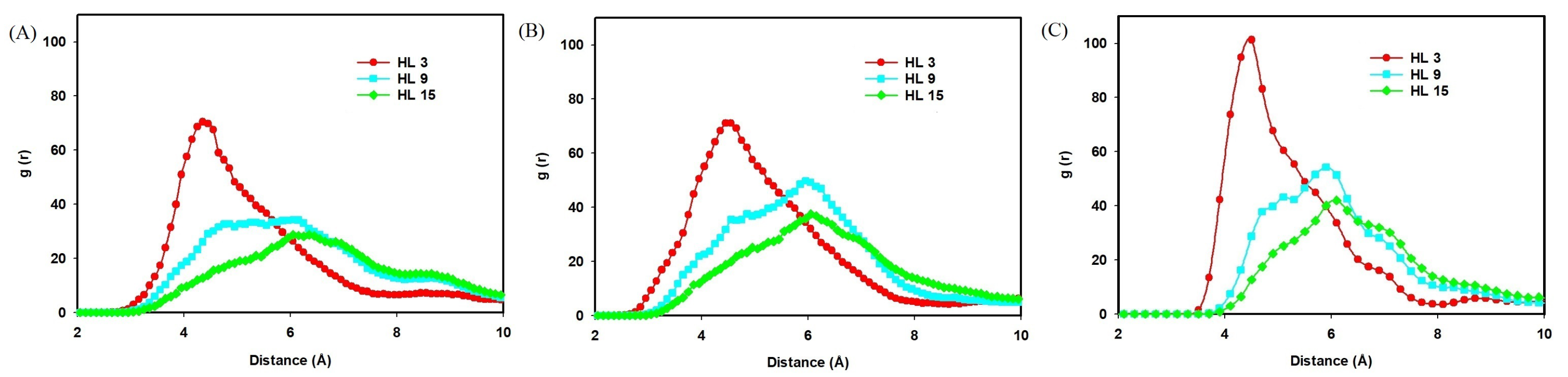
2.2.1. Effect of Hydration Level
2.2.2. Effect of Temperature
3. Methodology
3.1. Computational Models
3.2. DFT Calculations
3.3. Classical All-Atom MD Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AEM | Anion Exchange Membrane |
| AEMFCs | Anion Exchange Membrane Fuel Cells |
| AFCs | Alkaline Fuel Cells |
| BSSE | Basis set superposition error |
| DFT | Density Functional Theory |
| Reaction energy | |
| Activation energy | |
| Binding energy | |
| DMFCs | Direct Methanol Fuel Cells |
| EFCs | Enzymatic Fuel Cells |
| HL | Hydration level |
| Hydroxide ion | |
| LUMO | Lowest unoccupied molecular orbital |
| MCFCs | Molten Carbonate Fuel Cells |
| MD | Molecular dynamics |
| MSD | Mean square displacement |
| Nucleophilic substitution | |
| PAFCs | Phosphoric Acid Fuel Cells |
| PCM | Polarizable continuum model |
| PEMFCs | Proton Exchange Membrane Fuel Cells |
| QCS | Quaternized chitosan |
| QA | Quaternary ammonium |
| RDF | Radial distribution function |
| SOFCs | Solid Oxide Fuel Cells |
References
- Dekel, D.R. Review of cell performance in anion exchange membrane fuel cells. J. Power Sources 2018, 375, 158–169. [Google Scholar] [CrossRef]
- Chen, H.; Tao, R.; Bang, K.T.; Shao, M.; Kim, Y. Anion Exchange Membranes for Fuel Cells: State-of-the-Art and Perspectives. Adv. Energy Mater. 2022, 12, 2200934. [Google Scholar] [CrossRef]
- Li, F.; Chan, S.H.; Tu, Z. Recent Development of Anion Exchange Membrane Fuel Cells and Performance Optimization Strategies: A Review. Chem. Rec. 2024, 24, e202300067. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Heras Caballero, A.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef]
- Wan, Y.; Peppley, B.; Creber, K.A.; Bui, V.T. Anion-exchange membranes composed of quaternized-chitosan derivatives for alkaline fuel cells. J. Power Sources 2010, 195, 3785–3793. [Google Scholar] [CrossRef]
- Vijayakumar, V.; Nam, S.Y. A Review of Recent Chitosan Anion Exchange Membranes for Polymer Electrolyte Membrane Fuel Cells. Membranes 2022, 12, 1265. [Google Scholar] [CrossRef]
- Ma, J.; Sahai, Y. Chitosan biopolymer for fuel cell applications. Carbohydr. Polym. 2013, 92, 955–975. [Google Scholar] [CrossRef]
- Myrzakhmetov, B.; Akhmetova, A.; Bissenbay, A.; Karibayev, M.; Pan, X.; Wang, Y.; Bakenov, Z.; Mentbayeva, A. Review: Chitosan-based biopolymers for anion-exchange membrane fuel cell application. R. Soc. Open Sci. 2023, 10, 230843. [Google Scholar] [CrossRef]
- Maurya, S.; Shin, S.H.; Kim, Y.; Moon, S.H. A review on recent developments of anion exchange membranes for fuel cells and redox flow batteries. Rsc Adv. 2015, 5, 37206–37230. [Google Scholar] [CrossRef]
- Arges, C.G.; Zhang, L. Anion exchange membranes’ evolution toward high hydroxide ion conductivity and alkaline resiliency. ACS Appl. Energy Mater. 2018, 1, 2991–3012. [Google Scholar] [CrossRef]
- Dekel, D.R.; Amar, M.; Willdorf, S.; Kosa, M.; Dhara, S.; Diesendruck, C.E. Effect of water on the stability of quaternary ammonium groups for anion exchange membrane fuel cell applications. Chem. Mater. 2017, 29, 4425–4431. [Google Scholar] [CrossRef]
- Dekel, D.R.; Willdorf, S.; Ash, U.; Amar, M.; Pusara, S.; Dhara, S.; Srebnik, S.; Diesendruck, C.E. The critical relation between chemical stability of cations and water in anion exchange membrane fuel cells environment. J. Power Sources 2018, 375, 351–360. [Google Scholar] [CrossRef]
- Wan, Y.; Peppley, B.; Creber, K.A.; Bui, V.T.; Halliop, E. Quaternized-chitosan membranes for possible applications in alkaline fuel cells. J. Power Sources 2008, 185, 183–187. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Q.L.; Zhang, Q.G.; Zhu, A.M. Synthesis and characterization of cross-linked quaternized poly (vinyl alcohol)/chitosan composite anion exchange membranes for fuel cells. J. Power Sources 2008, 183, 447–453. [Google Scholar] [CrossRef]
- Liao, G.M.; Yang, C.C.; Hu, C.C.; Pai, Y.L.; Lue, S.J. Novel quaternized polyvinyl alcohol/quaternized chitosan nano-composite as an effective hydroxide-conducting electrolyte. J. Membr. Sci. 2015, 485, 17–29. [Google Scholar] [CrossRef]
- Karibayev, M.; Myrzakhmetov, B.; Kalybekkyzy, S.; Wang, Y.; Mentbayeva, A. Binding and Degradation Reaction of Hydroxide Ions with Several Quaternary Ammonium Head Groups of Anion Exchange Membranes Investigated by the DFT Method. Molecules 2022, 27, 2686. [Google Scholar] [CrossRef]
- Zhang, N.; Huo, J.; Yang, B.; Ruan, X.; Zhang, X.; Bao, J.; Qi, W.; He, G. Understanding of imidazolium group hydration and polymer structure for hydroxide anion conduction in hydrated imidazolium-g-PPO membrane by molecular dynamics simulations. Chem. Eng. Sci. 2018, 192, 1167–1176. [Google Scholar] [CrossRef]
- Dubey, V.; Maiti, A.; Daschakraborty, S. Predicting the solvation structure and vehicular diffusion of hydroxide ion in an anion exchange membrane using nonreactive molecular dynamics simulation. Chem. Phys. Lett. 2020, 755, 137802. [Google Scholar] [CrossRef]
- Luo, X.; Liu, H.; Bae, C.; Tuckerman, M.E.; Hickner, M.A.; Paddison, S.J. Mesoscale Simulations of Quaternary Ammonium-Tethered Triblock Copolymers: Effects of the Degree of Functionalization and Styrene Content. J. Phys. Chem. C 2020, 124, 16315–16323. [Google Scholar] [CrossRef]
- Munoz-Santiburcio, D.; Marx, D. Confinement-controlled aqueous chemistry within nanometric slit pores: Focus review. Chem. Rev. 2021, 121, 6293–6320. [Google Scholar] [CrossRef]
- Zelovich, T.; Tuckerman, M.E. OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics. Membranes 2021, 11, 355. [Google Scholar] [CrossRef]
- Zelovich, T.; Long, Z.; Hickner, M.; Paddison, S.J.; Bae, C.; Tuckerman, M.E. Ab initio molecular dynamics study of hydroxide diffusion mechanisms in nanoconfined structural mimics of anion exchange membranes. J. Phys. Chem. C 2019, 123, 4638–4653. [Google Scholar] [CrossRef]
- Wan, Y.; Creber, K.A.; Peppley, B.; Bui, V.T. Ionic conductivity of chitosan membranes. Polymer 2003, 44, 1057–1065. [Google Scholar] [CrossRef]
- Muller-Plathe, F. Permeation of polymers—a computational approach. Acta Polym. 1994, 45, 259–293. [Google Scholar] [CrossRef]
- Wan, Y.; Creber, K.A.; Peppley, B.; Bui, V.T. Synthesis, characterization and ionic conductive properties of phosphorylated chitosan membranes. Macromol. Chem. Phys. 2003, 204, 850–858. [Google Scholar] [CrossRef]
- Akhmetova, A.; Myrzakhmetov, B.; Wang, Y.; Bakenov, Z.; Mentbayeva, A. Development of Quaternized Chitosan Integrated with Nanofibrous Polyacrylonitrile Mat as an Anion-Exchange Membrane. ACS Omega 2022, 7, 45371–45380. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Creber, K.A.; Peppley, B.; Bui, V.T. Ionic conductivity and related properties of crosslinked chitosan membranes. J. Appl. Polym. Sci. 2003, 89, 306–317. [Google Scholar] [CrossRef]
- Tomasino, E.; Mukherjee, B.; Ataollahi, N.; Scardi, P. Water Uptake in an Anion Exchange Membrane Based on Polyamine: A First-Principles Study. J. Phys. Chem. B 2022, 126, 7418–7428. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Wang, Y.; Zhang, Y.; Zhang, W.; You, W. Alkaline-Stable Anion-Exchange Membranes with Barium [2.2. 2] Cryptate Cations: The Importance of High Binding Constants. Angew. Chem. 2023, 135, e202217742. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Gill, P.M.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke—Lee—Yang—Parr (B—LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef]
- De Castro, E.; Jorge, F. Accurate universal Gaussian basis set for all atoms of the periodic table. Chem. Phys. 1998, 108, 5225–5229. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. G16_C01. Gaussian 16, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2016; Volume 248. [Google Scholar]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolym. Orig. Res. Biomol. 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Han, K.W.; Ko, K.H.; Abu-Hakmeh, K.; Bae, C.; Sohn, Y.J.; Jang, S.S. Molecular dynamics simulation study of a polysulfone-based anion exchange membrane in comparison with the proton exchange membrane. J. Phys. Chem. C 2014, 118, 12577–12587. [Google Scholar] [CrossRef]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The nose–hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Berendsen, H.J.C. Transport properties computed by linear response through weak coupling to a bath. In Computer Simulation in Materials Science: Interatomic Potentials, Simulation Techniques and Applications; Springer: Dordrecht, The Netherlands, 1991; pp. 139–155. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Karibayev, M.; Bekeshov, D.; Myrzakhmetov, B.; Kalybekkyzy, S.; Wang, Y.; Bakenov, Z.; Mentbayeva, A. Effect of Hydration on the Intermolecular Interaction of Various Quaternary Ammonium Based Head Groups with Hydroxide Ion of Anion Exchange Membrane Studied at the Molecular Level. Eurasian-Chem.-Technol. J. 2023, 25, 89–102. [Google Scholar] [CrossRef]
- Nhung, L.T.T.; Kim, I.Y.; Yoon, Y.S. Quaternized chitosan-based anion exchange membrane composited with quaternized poly (vinylbenzyl chloride)/polysulfone blend. Polymers 2020, 12, 2714. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Chuang, F.S.; Tsen, W.C.; Kuo, T.W. Quaternized chitosan/functionalized carbon nanotubes composite anion exchange membranes. J. Appl. Polym. Sci. 2019, 136, 47778. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QCS | BSSE | ||
|---|---|---|---|
| (A) | −123.48 | 15.12 | 66.51 |
| (B) | −130.62 | 16.11 | 37.01 |
| (C) | −132.94 | 14.72 | 36.21 |
| QCS | BSSE | ||
|---|---|---|---|
| (A) | −21.56 | 11.47 | 147.27 |
| (B) | −28.69 | 8.38 | 103.20 |
| (C) | −31.02 | 8.47 | 89.92 |
| D, , (SE) | HL Values | |||
|---|---|---|---|---|
| 3 | 9 | 15 | ||
| (A) | 0.011 (0.004) | 0.017 (0.005) | 0.027 (0.003) | |
| (B) | 0.011 (0.003) | 0.016 (0.006) | 0.026 (0.002) | |
| (C) | 0.011 (0.001) | 0.015 (0.002) | 0.026 (0.002) | |
| D, , (SE) | HL Values | |||
|---|---|---|---|---|
| 3 | 9 | 15 | ||
| (A) | 0.20 (0.07) | 0.40 (0.06) | 0.46 (0.04) | |
| (B) | 0.18 (0.04) | 0.39 (0.05) | 0.43 (0.06) | |
| (C) | 0.13 (0.03) | 0.39 (0.02) | 0.42 (0.05) | |
| D, , (SE) | Temperature Values (K) | |||
|---|---|---|---|---|
| 298 | 330 | 350 | ||
| (A) | 0.011 (0.004) | 0.023 (0.003) | 0.027 (0.003) | |
| (B) | 0.011 (0.003) | 0.023 (0.003) | 0.026 (0.003) | |
| (C) | 0.011 (0.001) | 0.016 (0.002) | 0.026 (0.003) | |
| D, (SE) | Temperature Values (K) | |||
|---|---|---|---|---|
| 298 | 330 | 350 | ||
| (A) | 0.20 (0.07) | 1.54 (0.04) | 2.86 (0.25) | |
| (B) | 0.18 (0.04) | 1.38 (0.06) | 1.92 (0.20) | |
| (C) | 0.13 (0.03) | 0.44 (0.05) | 0.84 (0.09) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karibayev, M.; Myrzakhmetov, B.; Bekeshov, D.; Wang, Y.; Mentbayeva, A. Atomistic Modeling of Quaternized Chitosan Head Groups: Insights into Chemical Stability and Ion Transport for Anion Exchange Membrane Applications. Molecules 2024, 29, 3175. https://doi.org/10.3390/molecules29133175
Karibayev M, Myrzakhmetov B, Bekeshov D, Wang Y, Mentbayeva A. Atomistic Modeling of Quaternized Chitosan Head Groups: Insights into Chemical Stability and Ion Transport for Anion Exchange Membrane Applications. Molecules. 2024; 29(13):3175. https://doi.org/10.3390/molecules29133175
Chicago/Turabian StyleKaribayev, Mirat, Bauyrzhan Myrzakhmetov, Dias Bekeshov, Yanwei Wang, and Almagul Mentbayeva. 2024. "Atomistic Modeling of Quaternized Chitosan Head Groups: Insights into Chemical Stability and Ion Transport for Anion Exchange Membrane Applications" Molecules 29, no. 13: 3175. https://doi.org/10.3390/molecules29133175
APA StyleKaribayev, M., Myrzakhmetov, B., Bekeshov, D., Wang, Y., & Mentbayeva, A. (2024). Atomistic Modeling of Quaternized Chitosan Head Groups: Insights into Chemical Stability and Ion Transport for Anion Exchange Membrane Applications. Molecules, 29(13), 3175. https://doi.org/10.3390/molecules29133175