Abstract
Licorice (Glycyrrhiza uralensis Fisch), a significant traditional Chinese herbal medicine, has been extensively utilized in China to treat various ailments. Natural bioactive coumarins, glycycoumarin, glycyrin, and 3-O-methylglycyrol, were isolated from licorice, and they exhibited various pharmacological properties. In this report, we have accomplished the total synthesis of glycycoumarin, glycyrin, and 3-O-methylglycyrol in 5–7 linear steps from commercially available 2,4,6-trihydroxybenzaldehyde with yields of 12.3–21.2%. Additionally, their anti-inflammatory activities were studied and compared. Glycycoumarin, glycyrin, and 3-O-methylglycyrol exhibited different levels of anti-inflammatory activities, with glycyrin being the most potent. Mechanistic studies indicated that glycyrin exerted its anti-inflammatory properties by inhibiting the activation of TNF-α, IL-6, and IL-1β, making it a potential anti-inflammatory lead compound for further optimization and discovery of new agents.
1. Introduction
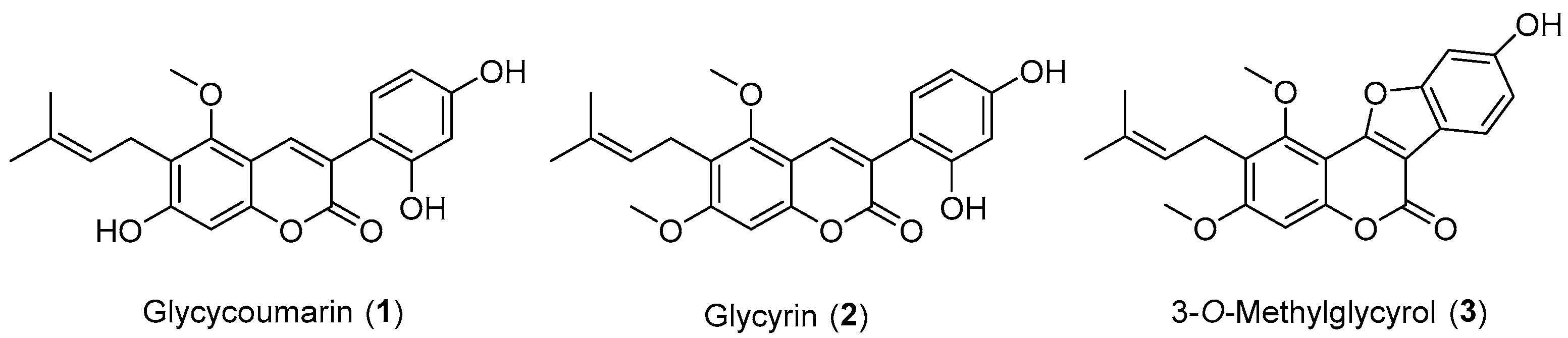
Licorice (Glycyrrhiza uralensis Fisch), a significant traditional Chinese herbal medicine listed in the pharmacopoeia of the People’s Republic of China, has been extensively utilized in China to treat various ailments, including spleen and stomach disorders, palpitations, shortness of breath, coughs, influenza infection, and liver disease [1,2]. Licorice is also recognized for producing a variety of bioactive natural products, such as glycosides, flavonoids, and coumarins [3,4]. Glycycoumarin (Figure 1, compound 1) is a naturally occurring coumarin that was initially extracted from licorice by Zhu et al. [5]. It has exhibited several beneficial pharmacological properties, including anti-inflammatory, antioxidant, and hepatoprotective effects [6]. Among these properties, its anti-inflammatory activity is particularly noteworthy. Glycycoumarin (1) has been shown to inhibit the production of nitric oxide (NO), interleukin-6 (IL-6), and prostaglandin E2 (PGE2) in LPS-induced macrophages [7]. Glycycoumarin (1) ameliorates alcohol-induced hepatic injury by activating Nrf2 and autophagy [8]. Furthermore, glycycoumarin (1) protects mice against acetaminophen-induced liver injury primarily by activating sustained autophagy [9].

Figure 1.
Structures of natural coumarins 1–3.
As illustrated in Figure 1, the natural coumarins glycyrin (2) [10] and 3-O-methylglycyrol (3) were also isolated from licorice [11]. They possess chemical structures closely related to glycycoumarin (1). Glycyrin (2) has displayed therapeutic activities against metabolic syndrome [12] and exhibited antimicrobial and antiviral properties [13]. However, no data on anti-inflammatory activity for either compound 2 or 3 have been reported in the literature. Given the promising anti-inflammatory activity associated with the structurally similar natural coumarin 1, obtaining robust and easy access to these natural products is of significant interest. However, the isolation of compounds 1–3 from licorice plants has been achieved with low yield. Therefore, establishing a synthetic method to obtain these compounds for further pharmacological investigation is highly desirable.
To the best of our knowledge, the first total synthesis of compound 1 was recently reported by Song et al. [14], while the syntheses of natural coumarins 2 and 3 remain undescribed. Our ongoing interest in discovering new anti-inflammatory agents from natural products prompted us to develop a reliable and efficient synthetic route for natural compounds 1–3 [15,16,17]. Herein, we present systematic studies toward the syntheses of natural compounds 1–3, with compounds 2 and 3 being synthesized for the first time. Additionally, their anti-inflammatory activities were studied and compared.
2. Results and Discussion
2.1. Synthesis of Natural Coumarins 1–3
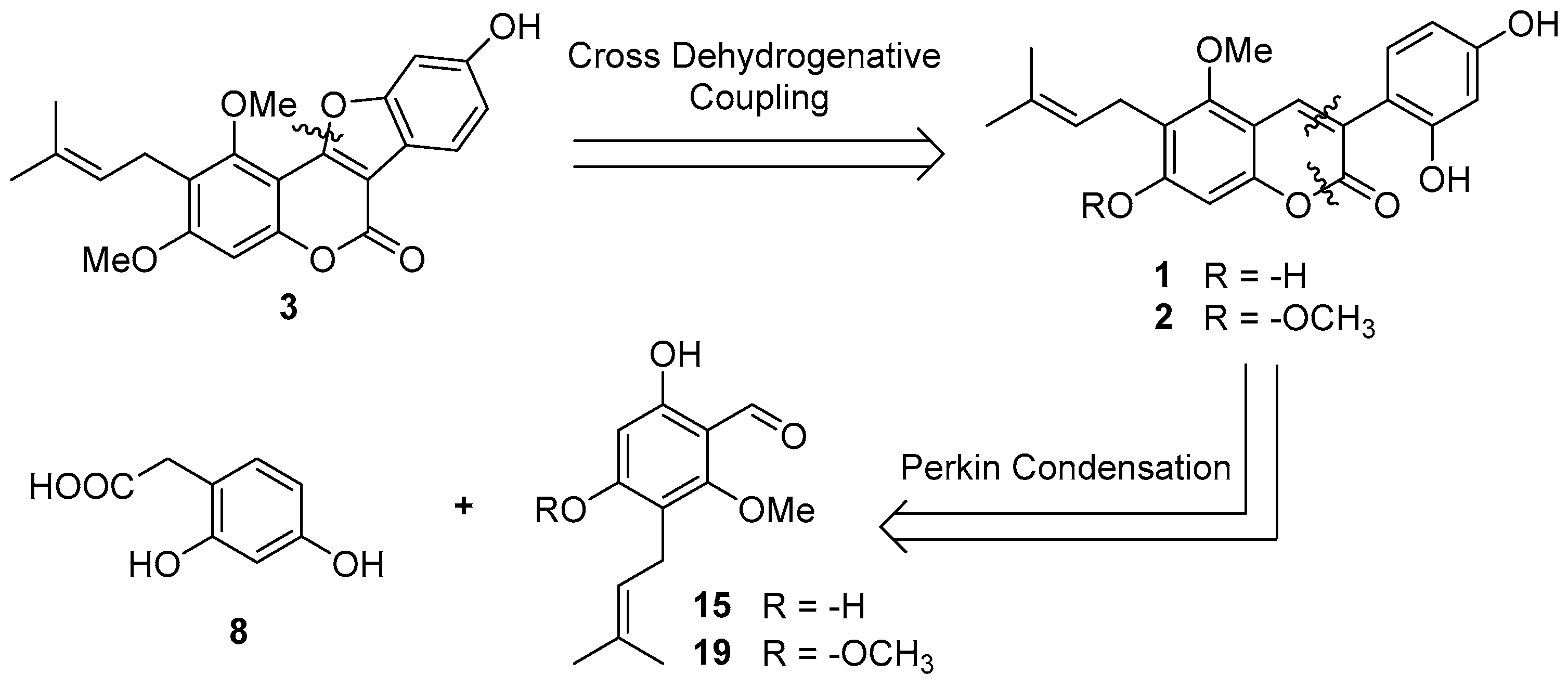
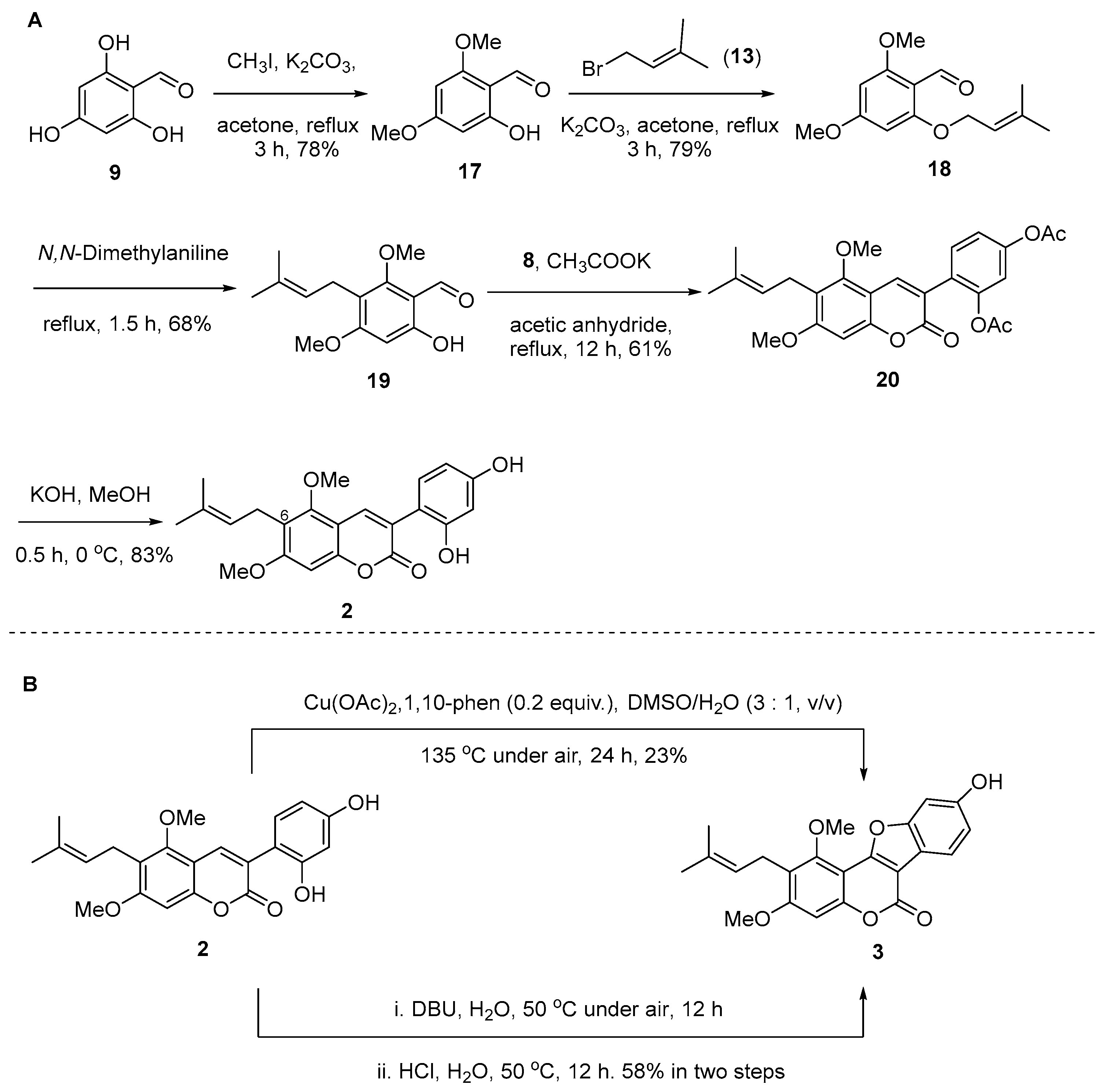
Scheme 1 outlines the retrosynthetic methodology aimed at synthesizing the desired compounds 1–3. We envisaged that compound 3 could be generated through an intramolecular cross dehydrogenative C-O coupling reaction of compound 2, and both target compounds 1 and 2 could be readily obtained through Perkin condensation between 2-(2,4-dihydroxyphenyl)acetic acid (8) and the corresponding 2-hydroxybenzaldehydes (15 or 19).

Scheme 1.
Retrosynthetic analysis of compounds 1–3.
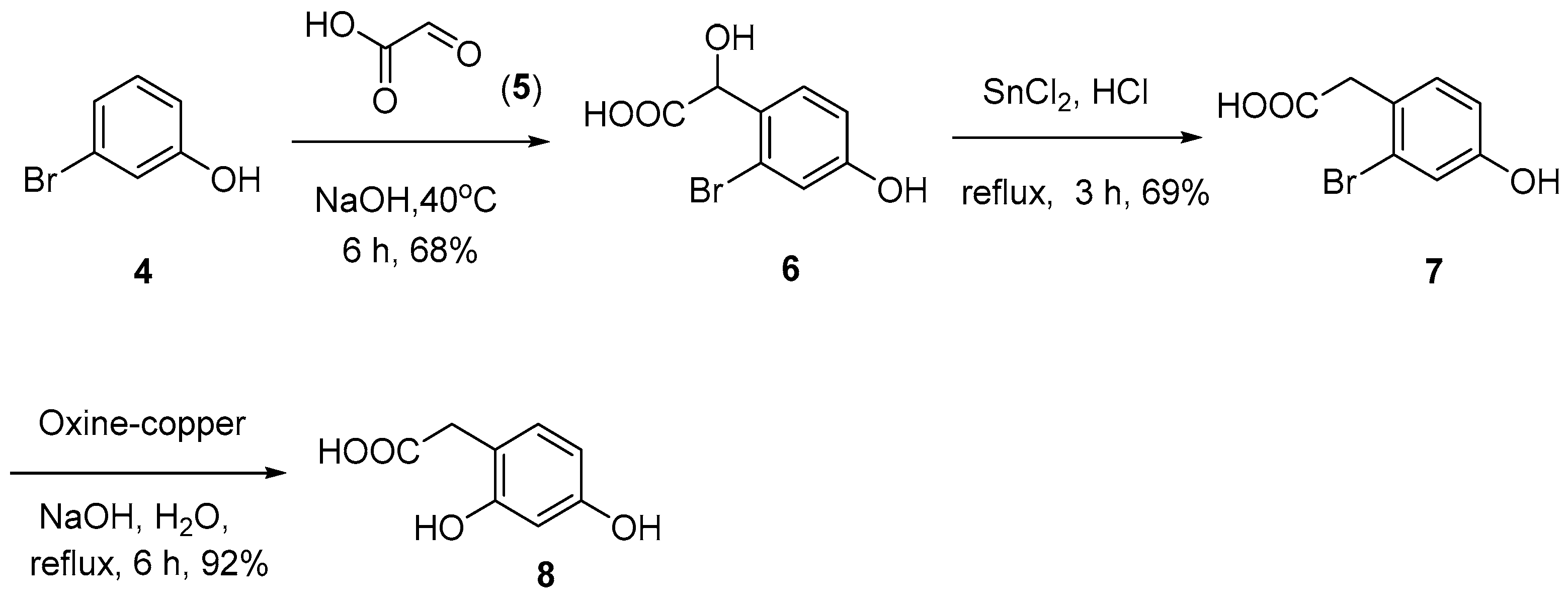
As shown in Scheme 2, the synthesis of key intermediate 8 commenced with readily available 3-bromophenol (4). Treatment of compound 4 with glyoxylic acid (5) under basic conditions yielded compound 6 in 68%. Subsequently, the α-OH group of compound 6 was reduced using SnCl2/HCl to give 2-bromo-4-hydroxyphenylacetic acid (7), which was then hydroxylated in the presence of oxine-copper/NaOH to generate the desired intermediate, 2-(2,4-dihydroxyphenyl)acetic acid (8), with a 92% yield [18].

Scheme 2.
Synthesis of compound 8.
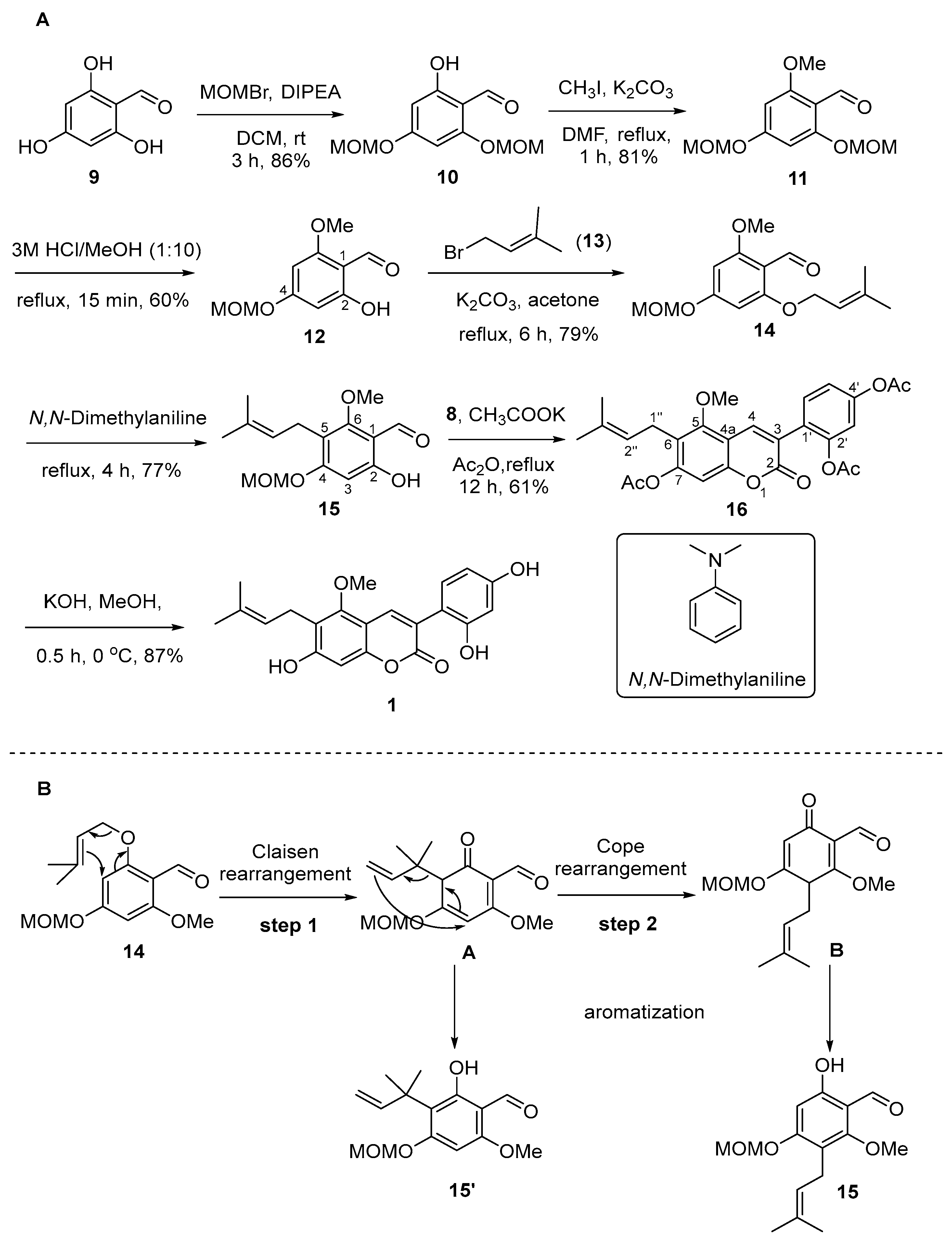
Meanwhile, the 2- and 4-phenol groups of 2,4,6-trihydroxybenzaldehyde (9) were selectively protected, yielding compound 10 (66%) through treatment with bromo(methoxy)methane (MOMBr) in the presence of diisopropylethylamine (DIPEA) (Scheme 3A) [19]. We anticipated that the selective O-methylation after the coumarin scaffold formation would be challenging. Hence, the methoxyl group was introduced early, resulting in compound 11 (81%). Due to the hydrogen bond between the 2-phenol group and the formyl group (-CHO), the MOM group at the 2-phenol position can be selectively deprotected using a 3M HCl solution in MeOH (1:10), resulting in compound 12 with a 60% yield. Prenylation of compound 12 furnished prenyl ether 14 with a yield of 79% [20]. Compound 14 was then subjected to a Claisen/Cope rearrangement to give the 5-prenylbenzaldehyde 15 in N,N-dimethylaniline as a solvent [20]. Upon heating prenyl ether 14 in N,N-dimethylaniline at reflux for 1 h, we found that two products, 15 and 15′ (Scheme 3B), could be isolated [21]. The formation of the byproduct 15′ has been rationalized by assuming a dearomatized intermediate A that results from a Claisen rearrangement (step 1), and from intermediate A, the byproduct 15′ is formed through aromatization. Intermediate A may undergo a second [3,3]-sigmatropic rearrangement (step 2, Cope-rearrangement) to cyclohexadienone intermediate B, from which the desired para-product 15 is formed after aromatization. After understanding the reaction mechanism, we extended the reaction time to 5 h, successfully obtaining product 15 with a yield of 77% without the byproduct 15′.

Scheme 3.
(A) Synthesis of glycycoumarin (1). (B) Mechanism of Claisen/Cope rearrangement.
With intermediates 2-(2,4-dihydroxyphenyl)acetic acid (8) and 5-prenylbenzaldehyde 15 in our hands, these two fragments were then coupled under Perkin condensation conditions, using CH3COOK in refluxing acetic anhydride (Ac2O), to give acetylated glycycoumarin (16) with a 61% yield [18,22,23,24]. Under these reaction conditions, the MOM protecting group at the C-7 position was concurrently transformed into the acetyl (-Ac) group. Global deacetylation with KOH in MeOH at 0 °C afforded the target compound glycycoumarin (1) with an 87% yield [23]. The NMR data of the synthetic sample were consistent with reported data [25] for natural glycycoumarin (1) (Table 1).

Table 1.
Comparison of the 13C NMR Data of synthesized compound 1 with literature data [25].
Following the successful synthesis of glycycoumarin (1), the syntheses of coumarins 2 and 3 were then pursued (Scheme 4). The prenylbenzaldehyde fragment of compound 2 was prepared from commercially available 2,4,6-trihydroxybenzaldehyde (9), which underwent a selective methylation to give 2-hydroxybenzaldehyde 17. Compound 17 was prenylated, and the resulting prenyl ether 18 was treated with N,N-dimethylaniline at 195 °C to furnish 19 via a Claisen/Cope rearrangement. Subsequently, Perkin condensation of prenylbenzaldehyde 19 and 2-(2,4-dihydroxyphenyl)acetic acid (8) gave acetylated coumarin 20, which was deacetylated to give target molecule 2 with an 83% yield. The synthetic compound 2 was characterized by 1H NMR and 13C NMR, and the data were identical with those of the natural product (Table 2) [3].

Scheme 4.
(A) Synthesis of glycyrin (2). (B) Synthesis of 3-O-methylglycyrol (3).
Table 2.
Comparison of the 13C NMR Data of synthesized compounds 2 and 3 with literature data [3,26].
As shown in Scheme 4B, treatment of compound 2 with 0.2 equivalents of Cu(OAc)2 and 1,10-phen in DMSO/H2O (v/v, 3:1) at 135 °C generated product 3 in a low yield (23%) [22], which was difficult to isolate. We speculated that the isopentenyl group at the C-6 position was unstable under these harsh conditions, leading to the production of numerous by-products. Therefore, a 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU)-promoted intramolecular dehydrogenation/oxa-Michael reaction was used to successfully obtain 3 in a moderate yield (58%) under milder conditions (DBU/H2O at 50 °C and then HCl at 50 °C) [18]. The NMR data of the synthetic compound 3 were consistent with the data reported for natural 3-O-methylglycyrol (3) (Table 2) [26].
2.2. Anti-Inflammatory Activity of Natural Coumarins 1–3
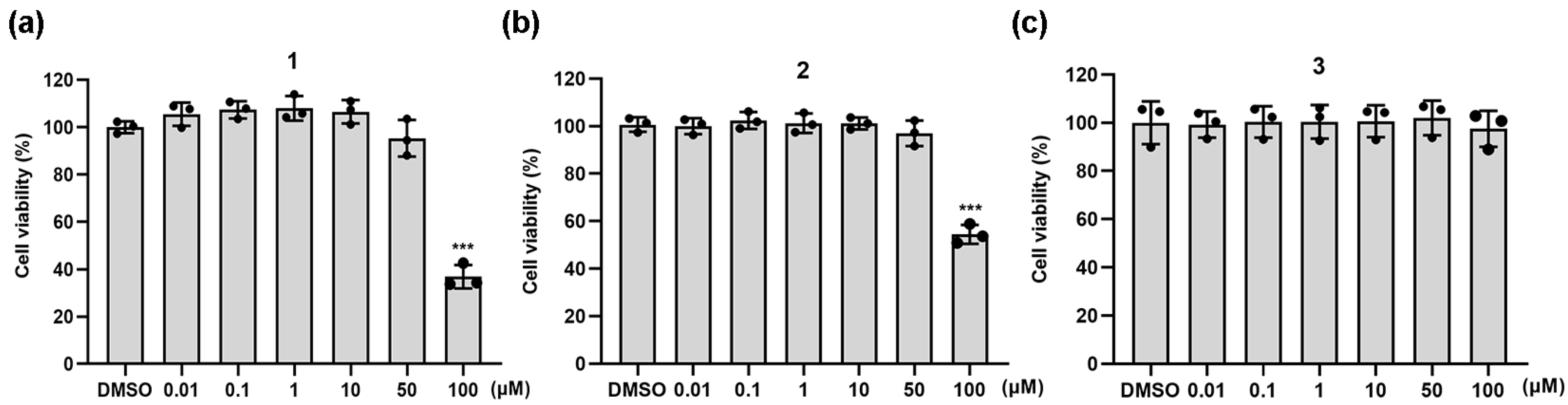
Since nitric oxide (NO), a vital gas signaling molecule, is a unique transmitter of acute or chronic inflammation [27], the anti-inflammatory activities and cytotoxic effects of compounds 1–3 were evaluated by monitoring cell viability and NO inhibition in LPS-induced RAW264.7 cells, respectively. The effects of compounds 1–3 on cell viability were first evaluated using a CCK8 assay. RAW264.7 cells were exposed to different doses of the tested compound for 24 h. Meanwhile, 0.1% DMSO was used as the control. Significant cytotoxic effects were observed on the growth of RAW264.7 cells incubated with 1 and 2 at a dose of 100 μM (Figure 2). Cell survival was restored to that of the normal control group at reduced doses of 0.01, 0.1, 1, 10, 25, and 50 μM. The same concentration ranges were also used in cell-based studies to determine their anti-inflammatory activities.

Figure 2.
CCK-8 detected the viability of RAW264.7 cells after treatment with compounds 1 (a), 2 (b) and 3 (c) for 24 h. Graphic data were run in triplicate and are presented as the mean ± SD (compared with DMSO, *** p < 0.001).
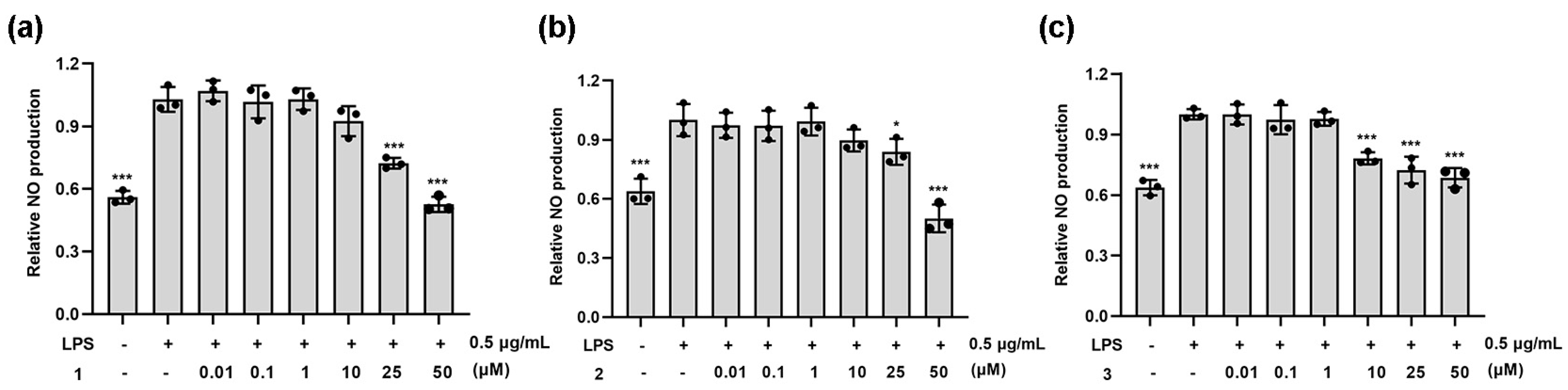
To determine the anti-inflammatory activity of compounds 1–3, the mouse macrophage-like cell line RAW264.7 was used. Upon treatment with LPS (0.5 μg/mL), RAW264.7 cells produced an increased amount of NO. After treatment with different concentrations of coumarins 1–3, NO secretion was strongly inhibited (Figure 3). Compound 1 exhibited greater activity in inhibiting NO secretion at a dose of 25 μM and 50 μM, and the concentration of 50 μM with the inhibition rate exceeding 50%. Compound 2 showed strong inhibition of NO secretion only at 50 μM. Compound 3 inhibited NO secretion in a concentration-dependent manner.

Figure 3.
Effects of the tested compounds 1 (a), 2 (b) and 3 (c) on LPS-stimulated NO production in RAW264.7 cells; 0.1% DMSO was used as the control. Graphic data were run in triplicate and are presented as the mean ± SD (compared with LPS, * p < 0.05, *** p < 0.001).
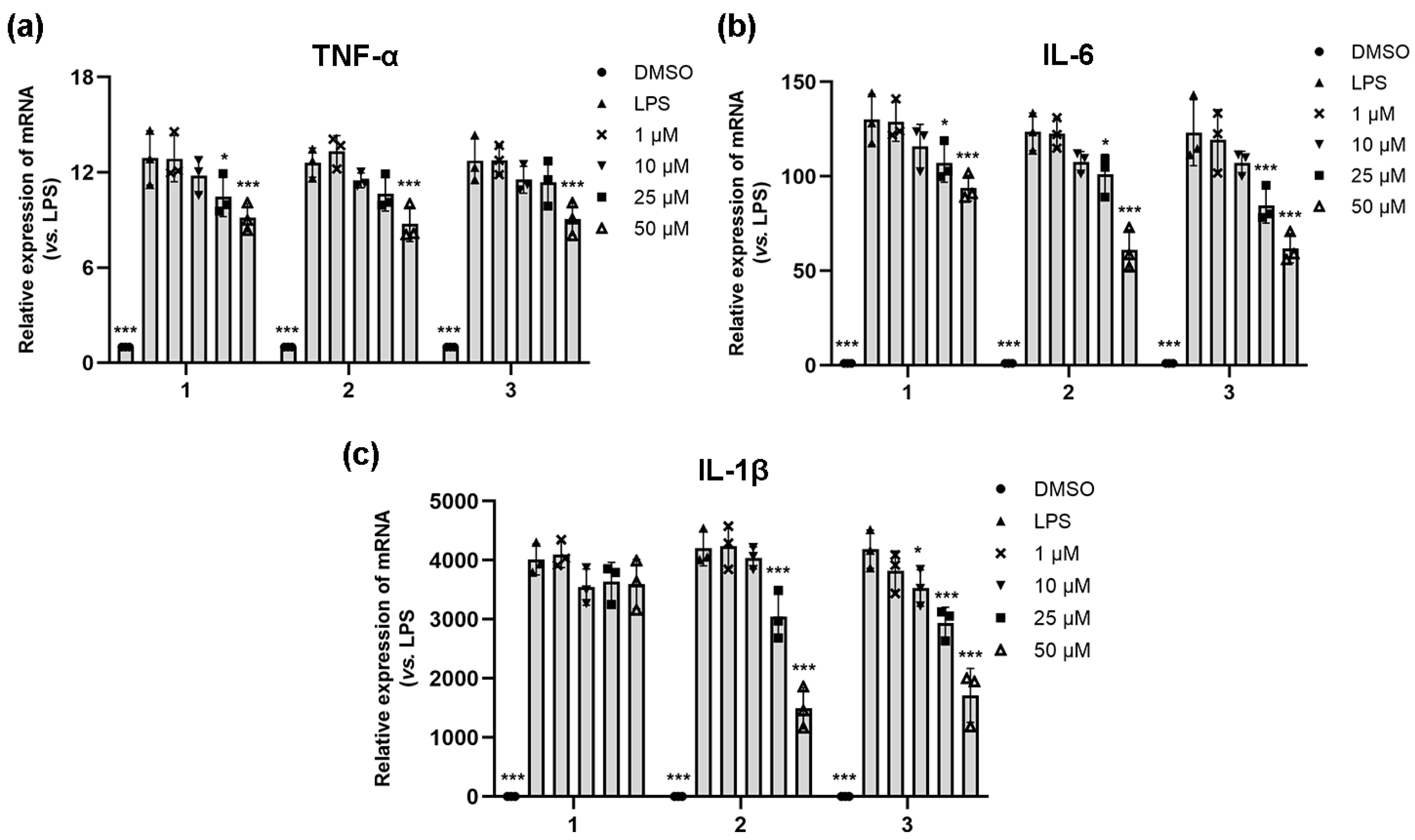
To confirm the function of compounds 1–3 in the LPS-induced production of pro-inflammatory cytokines, the expression levels of three critical pro-inflammatory cytokines, TNF-α, IL-6, and IL-1β mRNA, were analyzed by qRT-PCR, respectively [28]. As shown in Figure 4, LPS significantly induced the mRNA expressions of TNF-α, IL-6, and IL-1β compared with the normal control. Compound 1 demonstrated significant inhibition of IL-6 and TNF-α expression at concentrations of 25 and 50 μM but had no effect on IL-1β. Compound 2 exhibited significant inhibition of IL-6 and IL-1β expression at concentrations of 25 and 50 μM. It also significantly inhibited TNF-α at 50 μM. Compound 3 exhibited concentration-dependent inhibition of IL-6 and IL-1β, and it inhibited TNF-α only at a concentration of 50 μM.

Figure 4.
The effects of the compounds 1–3 on the expression of TNF-a (a), IL-6 (b), and IL-1β (c) detected by qRT-PCR. LPS-stimulated (0.5 μg/mL) RAW264.7 cells were treated with 1, 10, 25, and 50 μM of compounds 1–3 for 24 h, and 0.1% DMSO was used as the control. Graphic data were run in triplicate and are presented as the mean ± SD (compared with LPS, * p < 0.05, *** p < 0.001).
3. Materials and Methods
3.1. General Experimental Procedures
Melting points were recorded on a Büchi B-545 melting point apparatus (Sigma-Aldrich, St. Louis, MO, USA). 1H NMR, 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer (Billerica, MA, USA) or a JEOL Eclips-600 pectrometer (Akishima, Japan), and tetramethylsilane (TMS) was used as the internal reference. The HR-MS spectra were recorded by Thermo QExactive (Thermo Scientific, Waltham, MA, USA) and Agilent 6545 LC/QTOF mass spectrometers (Santa Clara, CA, USA). Column chromatography was performed on silica gel (100–200 mesh). Reagents were purchased from commercial sources and used as received, unless mentioned otherwise. The solvents were of analytical grade.
3.2. Synthesis and Characterization of the Compounds
3.2.1. 2-(2-Bromo-4-hydroxyphenyl)-2-hydroxyacetic Acid (6)
3-Bromophenol (4, 8.65 g, 50.00 mmol, 1.0 equiv.) was added to a three-necked round-bottom flask. Once the reaction temperature reached 40 °C, a 50% aqueous solution of glyoxylic acid (5, 7.2 mL, 65.0 mmol, 1.3 equiv.) and an 8% aqueous NaOH (37.5 mL, 75.00 mmol, 1.5 equiv.) solution were simultaneously added slowly. The mixture was stirred for 6 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature and acidified to pH 1–2 using 3 M HCl (60 mL). The aqueous solution was washed with toluene (3 × 50 mL) to remove 3-bromophenol, and the product was extracted using EtOAc (3 × 50 mL). The organic layer was separated, washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo to yield compound 6 (8.40 g, 34.00 mmol, 68%) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.24 (d, J = 8.4 Hz, 1H), 6.97 (s, 1H), 6.77 (d, d, J = 8.4 Hz, 1H), 5.13 (s, 1H). The spectroscopic data correspond to reported values [22].
3.2.2. 2-(2-Bromo-4-hydroxyphenyl)acetic Acid (7)
A solution of compound 6 (5.00 g, 20.24 mmol, 1.0 equiv.), SnCl2·2H2O (5.02 g, 22.26 mmol, 1.1 equiv.), and concentrated HCl (10.0 mL) was added to a round-bottom flask, and the mixture was stirred at 80 °C for 3 h. After the reaction was complete (monitored by TLC), H2O (20 mL) was added, and the mixture was heated to reflux until a clear solution was obtained. The resulting mixture was cooled to room temperature, whereupon compound 7 recrystallized to afford a white solid (3.23 g, 13.97 mmol, 69% yield). mp: 172–174 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.12 (d, J = 8.3 Hz, 1H), 6.99 (d, J = 2.4 Hz, 1H), 6.71 (dd, J = 8.3, 2.4 Hz, 1H), 3.45 (s, 2H). The spectroscopic data correspond to reported values [22].
3.2.3. 2-(2,4-Dihydroxyphenyl)acetic Acid (8)
Under a N2 atmosphere, a solution of compound 7 (2.31 g, 10.00 mmol, 1.0 equiv.), bis(8-quinolinolato)copper(II) (0.34 g, 1.00 mmol, 0.1 equiv.), NaOH (4.00 g, 100.00 mmol, 10 equiv.), and H2O (40 mL) was added to a round-bottom flask. The mixture was stirred at 110 °C for 6 h. After the reaction was complete (monitored by TLC), the resulting mixture was cooled to room temperature. The solid was filtered off, and the aqueous solution was acidified to pH 1-2 using 3 M HCl (40 mL). The product was extracted using EtOAc (3 × 50 mL). The organic layer was separated, washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo to yield compound 8 (1.55 g, 9.20 mmol, 92%) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.24 (d, J = 2.4 Hz, 1H), 6.12 (dd, J = 8.1, 2.4 Hz, 1H), 3.29 (s, 2H). The spectroscopic data correspond to reported values [22].
3.2.4. 2-Hydroxy-4,6-bis(methoxymethoxy)benzaldehyde (10)
Under a N2 atmosphere, a solution of 2,4,6-trihydroxybenzaldehyde (9, 5.00 g, 32.44 mmol, 1.0 equiv.) and DIPEA (22.60 mL, 129.76 mmol, 4.0 equiv.) in CH2Cl2 (200 mL) was cooled to 0 °C, and MOMBr (5.30 mL, 64.88 mmol, 2.0 equiv.) was added. The resulting mixture was stirred at room temperature for 3 h. After the reaction was complete (monitored by TLC), H2O (100 mL) was added, and the mixture was extracted with CH2Cl2 (3 × 50 mL). The organic layer and extracts were combined, dried, and evaporated to give a red oil, which was chromatographed on silica gel (petroleum ether/EtOAc = 50/1) to yield compound 10 (6.76 g, 27.90 mmol, 86%) as a white solid. mp: 67–68 °C. 1H NMR (400 MHz, CDCl3) δ 12.31 (s, 1H), 10.19 (s, 1H), 6.28 (d, J = 2.1 Hz, 1H), 6.25 (d, J = 2.1 Hz, 1H), 5.26 (s, 2H), 5.20 (s, 2H), 3.53 (s, 3H), 3.50 (s, 3H). The spectroscopic data correspond to reported values [29].
3.2.5. 2-Methoxy-4,6-bis(methoxymethoxy)benzaldehyde (11)
Under a N2 atmosphere, a solution of compound 10 (3.50 g, 14.45 mmol, 1.0 equiv.), K2CO3 (9.99 g, 72.25 mmol, 5.0 equiv.), and CH3I (2.25 mL, 36.13 mmol, 2.5 equiv.) in anhydrous DMF (50 mL) was stirred at 153 °C for 1 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature, quenched with water (20 mL), and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 20/1) to yield compound 11 (3.00 g, 11.70 mmol, 81% yield) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.40 (s, 1H), 6.46 (d, J = 1.8 Hz, 1H), 6.31 (d, J = 1.8 Hz, 1H), 5.25 (s, 2H), 5.22 (s, 2H), 3.89 (s, 3H), 3.51 (s, 3H), 3.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.8, 163.7, 163.5, 161.7, 110.2, 95.4, 94.9, 94.2, 93.3, 56.6, 56.5, 56.1. HRMS (ESI) calculated for C12H17O6+ [M + H]+ 257.1020, found 257.1014.
3.2.6. 2-Hydroxy-6-methoxy-4-(methoxymethoxy)benzaldehyde (12)
A solution of compound 11 (3.00 g, 11.71 mmol) and 3 M HCl (27 mL) in anhydrous MeOH (270 mL) was stirred at 66 °C for 15 min. After the reaction was complete (monitored by TLC), the mixture was poured into ice-cold water, and the resulting solution was adjusted to pH 7.0 with saturated NaHCO3 (100 mL) and extracted three times with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 50/1) to yield compound 12 (1.49 g, 7.03 mmol, 60%) as a white solid. mp: 63–65 °C; 1H NMR (400 MHz, CDCl3) δ 12.38 (s, 1H), 10.14 (s, 1H), 6.19 (d, J = 1.9 Hz, 1H), 6.04 (d, J = 1.9 Hz, 1H), 5.21 (s, 2H), 3.88 (s, 3H), 3.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.1, 165.9, 165.7, 163.8, 106.6, 95.7, 94.1, 91.2, 56.5, 55.8. HRMS (ESI) calculated for C10H13O5+ [M + H]+ 213.0758, found 213.0752.
3.2.7. 2-Methoxy-4-(methoxymethoxy)-6-((3-methylbut-2-en-1-yl)oxy)benzaldehyde (14)
Under a N2 atmosphere, to a solution of 12 (1.70 g, 8.01 mmol, 1.0 equiv.) and K2CO3 (2.21 g, 16.02 mmol, 2.0 equiv.) in acetone (20 mL) was added 13 (1.41 mL, 12.02 mmol, 1.5 equiv.) and refluxed for 6 h. After the reaction was completed (detected by TLC). The mixture was cooled to room temperature, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 20/1) to give 14 (1.77 g, 6.33 mmol, 79%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 10.39 (s, 1H), 6.26 (d, J = 1.9 Hz, 1H), 6.23 (d, J = 1.9 Hz, 1H), 5.48 (t, J = 6.6 Hz, 1H), 5.22 (s, 2H), 4.60 (d, J = 6.6 Hz, 2H), 3.88 (s, 3H), 3.51 (s, 3H), 1.80 (s, 3H), 1.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.0, 163.8, 163.7, 163.4, 138.4, 119.0, 109.8, 94.2, 93.3, 92.2, 66.0, 56.4, 56.0, 25.8, 18.3.HRMS (ESI) calculated for C15H21O5+ [M + H]+ 281.1384, found 281.1378.
3.2.8. 6-Hydroxy-2-methoxy-4-(methoxymethoxy)-3-(3-methylbut-2-en-1-yl)benzaldehyde (15)
Under a N2 atmosphere, a solution of compound 14 (1.00 g, 3.57 mmol) in N,N-dimethylaniline (5 mL) was refluxed for 4 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature, and the resulting solution was adjusted to pH 7.0 with 3 M HCl (5 mL) and extracted three times with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 50/1) to give 15 (0.77 g, 2.75 mmol, 77%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 11.99 (s, 1H), 10.07 (s, 1H), 6.45 (s, 1H), 5.25 (s, 2H), 5.20–5.11 (m, 1H), 3.87 (s, 3H), 3.49 (s, 3H), 3.29 (d, J = 6.9 Hz, 2H), 1.79 (s, 3H), 1.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.9, 163.7, 163.6, 162.0, 131.8, 122.6, 115.9, 109.5, 98.13, 94.0, 64.4, 56.4, 25.7, 22.3, 17.8.HRMS (ESI) calculated for C15H21O5+ [M + H]+ 281.1384, found 281.1377.
3.2.9. 4-(7-Acetoxy-5-methoxy-6-(3-methylbut-2-en-1-yl)-2-oxo-2H-chromen-3-yl)-1,3-phenylene Diacetate (16)
Under a N2 atmosphere, a solution of compound 15 (0.28 g, 1.00 mmol, 1.0 equiv.), compound 8 (0.84 g, 5.00 mmol, 5.0 equiv.), and anhydrous CH3COOK (0.18 g, 1.80 mmol, 1.8 equiv.) in Ac2O (1.2 mL) was refluxed for 12 h. After the reaction was complete (monitored by TLC), the mixture was poured into ice-cold water, and the resulting solution was adjusted to pH 7.0 with saturated NaHCO3 (5 mL) and extracted three times with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 10/1) to yield compound 16 (0.3 g, 0.61 mmol, 61%) as a white solid. mp: 71–73 °C; 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.47 (d, J = 9.1 Hz, 1H), 7.14-7.09 (m, 2H), 6.95 (s, 1H), 5.08 (t, J = 6.6 Hz, 1H), 3.87 (s, 3H), 3.35 (d, J = 6.6 Hz, 2H), 2.35 (s, 3H), 2.34 (s, 3H), 2.21 (s, 3H), 1.79 (s, 3H), 1.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.0, 168.8, 168.5, 159.3, 156.0, 152.8, 152.5, 151.3, 149.0, 137.4, 132.9, 131.4, 125.5, 124.4, 123.9, 121.4, 119.3, 117.0, 112.1, 107.5, 63.5, 25.6, 23.5, 21.1, 21.0, 20.9, 18.0. HRMS (ESI) calculated for C27H27O9+ [M + H]+ 495.1650, found 495.1642.
3.2.10. Glycycoumarin (1)
A solution of compound 16 (0.20 g, 0.40 mmol, 1.0 equiv.) in MeOH (5 mL) was treated with KOH (0.07 g, 1.20 mmol, 3 equiv.). The mixture was stirred at 0 °C for 0.5 h. After the reaction was complete (monitored by TLC), the mixture was poured into ice-cold water. The resulting solution was adjusted to pH 7.0 with 3 M HCl (5 mL) and extracted with EtOAc (3 × 20 mL). The organic layer was combined, washed with saturated aqueous NaCl solution, and dried with anhydrous Na2SO4. The solvents were then removed in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 5/1) to yield compound 1 (0.13 g, 0.35 mmol, 87%) as a yellow solid. mp: 234–236 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 9.40 (s, 2H), 7.82 (s, 1H,), 7.11 (d, J = 8.3 Hz, 1H), 6.61 (s, 1H), 6.37 (d, J = 2.3 Hz, 1H), 6.27 (dd, J = 8.3, 2.3 Hz, 1H), 5.16 (t, J = 6.9 Hz), 3.77 (s, 3H), 3.27 (d, J = 6.9 Hz, 2H), 1.74 (s, 3H), 1.64 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 160.0, 159.2, 158.3, 156.0, 155.2, 152.9, 136.4, 131.5, 130.7, 122.6, 120.3, 118.3, 113.4, 106.2, 106.1, 102.6, 97.9, 62.8, 25.4, 22.3, 17.7.
3.2.11. 2-Hydroxy-4,6-dimethoxybenzaldehyde (17)
Under a N2 atmosphere, a suspension of 2,4,6-trihydroxybenzaldehyde (9, 5.00 g, 32.44 mmol, 1.0 equiv.), CH3I (4.45 mL, 71.40 mmol, 2.2 equiv.), and K2CO3 (8.97 g, 64.89 mmol, 2.0 equiv.) in acetone (50 mL) was stirred at 50 °C for 3 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature, quenched with water (50 mL), and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 20/1) to yield compound 17 (4.61 g, 25.30 mmol, 78%) as a yellow solid. mp: 67–69 °C; 1H NMR (400 MHz, CDCl3) δ 12.53 (s, 1H), 10.10 (s, 1H), 6.02 (d, J = 2.1 Hz, 1H), 5.92 (d, J = 2.1 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H). The spectroscopic data correspond to reported values [30].
3.2.12. 2,4-Dimethoxy-6-((3-methylbut-2-en-1-yl)oxy)benzaldehyde (18)
Under a N2 atmosphere, a solution of compound 17 (1.83 g, 10.00 mmol) and K2CO3 (2.76 g, 20.00 mmol, 2.0 equiv.) in acetone (20 mL) was treated with compound 13 (1.76 mL, 15.00 mmol, 1.5 equiv.) and refluxed for 3 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 10/1) to yield compound 18 (1.98 g, 7.90 mmol, 79%) as white solid. mp: 62–63 °C; 1H NMR (400 MHz, CDCl3) δ 10.37 (s, 1H), 6.09 (s, 1H), 6.07 (s, 1H), 5.48 (t, J = 6.1 Hz), 4.60 (d, J = 6.1 Hz), 3.88 (s, 3H), 3.87 (s, 3H), 1.79 (s, 3H), 1.75 (s, 3H). The spectroscopic data correspond to reported values [31].
3.2.13. 6-Hydroxy-2,4-dimethoxy-3-(3-methylbut-2-en-1-yl)benzaldehyde (19)
Under a N2 atmosphere, a solution of compound 18 (1.00 g, 4.00 mmol) in N,N-dimethylaniline (5 mL) was refluxed for 1.5 h. After the reaction was complete (monitored by TLC), the mixture was cooled to room temperature, and the resulting solution was adjusted to pH 7.0 with 3 M HCl (5 mL) and extracted three times with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 50/1) to yield compound 19 (0.68 g, 2.72 mmol, 68%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 12.14 (s, 1H), 10.04 (s, 1H), 6.24 (s, 1H), 5.14 (t, J = 7.0 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.26 (d, J = 7.0 Hz, 2H), 1.78 (s, 3H), 1.70 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 192.7, 166.4, 164.1, 161.7, 131.8, 122.5, 115.5, 108.8, 95.5, 64.3, 56.0, 25.7, 22.1, 17.8. HRMS (ESI) calculated for C14H19O4+ [M + H]+ 251.1278, found 251.1272.
3.2.14. 4-(5,7-Dimethoxy-6-(3-methylbut-2-en-1-yl)-2-oxo-2H-chromen-3-yl)-1,3-phenylene Diacetate (20)
Under a N2 atmosphere, a solution of 19 (0.25 g, 1.00 mmol, 1.0 equiv.), 8 (0.84 g, 5.00 mmol, 5 equiv.), and anhydrous CH3COOK (0.18 g, 1.80 mmol, 1.8 equiv.) in Ac2O (1.2 mL) was refluxed for 12 h. After the reaction was completed (detected by TLC), the mixture was poured into ice-cold water and the obtained solution was adjusted to pH = 7.0 with saturated NaHCO3 (5 mL) and extracted three times with EtOAc (3 × 50 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 10/1) to give 20 (0.28 g, 0.61 mmol, 61%) as yellow solid. mp: 160–161 °C; 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.48 (dd, J = 8.0, 0.8 Hz, 1H), 7.13-7.07 (m, 2H), 6.69 (s, 1H), 5.16 (t, J = 6.9 Hz, 1H), 3.92 (s, 3H), 3.85 (s, 3H), 3.39 (d, J = 6.9 Hz, 2H), 2.34 (s, 3H), 2.20 (s, 3H), 1.80 (s, 3H), 1.71 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.0, 168.6, 161.9, 160.0, 155.4, 154.5, 151.0, 148.9, 138.3, 132.2, 131.5, 126.0, 122.1, 120.6, 120.4, 119.2, 116.9, 107.4, 95.1, 63.2, 56.2, 25.8, 22.8, 21.2, 21.0, 17.9. HRMS (ESI) calculated for C26H27O8+ [M + H]+ 467.1701, found 467.1691. The spectroscopic data correspond to reported values [10].
3.2.15. Glycyrin (2)
A solution of compound 20 (0.12 g, 0.26 mmol, 1.0 equiv.) in MeOH (5 mL) was treated with KOH (0.03 g, 0.52 mmol, 2 equiv.). The mixture was stirred at 0 °C for 0.5 h. After the reaction was complete (monitored by TLC), the mixture was poured into ice-cold water. The resulting solution was adjusted to pH 7.0 with 3 M HCl (2 mL) and extracted with EtOAc (3 × 20 mL). The organic layers were combined, washed with saturated aqueous NaCl solution, and dried with anhydrous Na2SO4. The solvents were then removed in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 5/1) to yield compound 2 (0.083 g, 0.217 mmol, 83%) as a yellow solid. mp: 203–205 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.44 (s, 1H), 7.86 (s, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.89 (s, 1H), 6.38 (d, J = 2.3 Hz, 1H), 6.28 (dd, J = 8.4, 2.3 Hz, 1H), 5.11 (t, J = 7.0 Hz, 1H), 3.89 (s, 3H), 3.78 (s, 3H), 3.29 (d, J = 7.0 Hz, 2H), 1.74 (s, 3H), 1.64 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 160.9, 160.4, 159.0, 156.6, 155.0, 153.8, 136.6, 132.1, 131.6, 122.8, 121.9, 119.6, 113.7, 107.5, 106.7, 103.2, 95.7, 63.4, 56.9, 25.9, 22.7, 18.2.
3.2.16. 3-O-Methylglycyrol (3)
A solution of compound 2 (0.10 g, 0.26 mmol, 1.0 equiv.) in H2O (2 mL) was treated with DBU (0.18 mL, 1.30 mmol, 5 equiv.). The mixture was stirred at 50 °C in a water bath for 12 h. After the reaction was complete (monitored by TLC), the mixture was acidified to pH 1.0 with 3 M HCl (5 mL) and stirred for an additional 12 h. The mixture was then extracted with EtOAc (3 × 15 mL). The organic layers were combined, washed with saturated aqueous NaCl solution, and dried with anhydrous Na2SO4. The solvents were then removed in vacuo. The crude material was purified by flash chromatography (PE/EtOAc = 5/1) to yield compound 3 (0.06 g, 0.15 mmol, 58%). mp: 255–257 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1H), 7.04 (s, 1H), 6.97 (dd, J = 8.4, 2.0 Hz, 1H), 5.14 (t, J = 7.2 Hz, 1H), 3.91 (s, 6H), 3.33 (d, J = 7.2 Hz, 2H), 1.76 (s, 3H), 1.64 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 161.0, 158.3, 157.9, 157.6, 156.7, 153.8, 153.6, 131.8, 122.6, 121.1, 121.0, 114.6 (2 × C), 103.4, 101.3, 99.0, 96.9, 63.0, 57.0, 25.9, 22.4, 18.2.
3.2.17. Cell Culture
The RAW264.7 cell line was obtained from Procell (Wuhan, China), authenticated by short tandem repeat profiling (STR), and examined for mycoplasma contamination. RAW264.7 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Hyclone, UT, USA), which was supplemented with 10% fetal bovine serum (FBS; Anweisci, Shanghai, China), and maintained at 37 °C and 5% CO2.
3.2.18. Cell Viability Assay
RAW264.7 cells were seeded into 96-well plates (1 × 103 cells/well) overnight and treated with 0.1% DMSO (negative control) and different concentration of test compounds. After 24 h, 10% (v/v) CCK-8 (Beyotime, Shanghai, China) was added to each well and incubated for 1 h. Fluorescence intensities were measured by using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm.
3.2.19. NO Determination
RAW264.7 cells were seeded in 96-well plate at a density of 1 × 105 cells/well. The cells were treated with different concentrations of the test compounds; after 1 h, the cells were stimulated with LPS (0.5 μg/mL) and then incubated for 24 h. The nitrite accumulated in the culture medium was measured as an indicator of NO production based on a diazotisation reaction using the Griess reagent system as previously described [32].
3.2.20. Real-Time Quantitative PCR
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was using to extract total cellular RNA according to the manufacturer’s instructions. RNA concentration was determined by examining the absorbance at 260 nm using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific). Total RNA (2 μg) was reverse-transcribed using TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (Cat No: AT341-01, TransGen Biotech, Beijing, China). Equal amounts of cDNA were subjected to RT-qPCR with TransStart® Top Green qPCR SuperMix (Cat No: AQ131-02, TransGen Biotech, Beijing, China) using a Chromo4 detection system (Bio-Rad, California, USA). GAPDH served as reference genes to eliminate differences in the number of cells. Quantitative analyses were performed using the threshold cycle number (Ct), where the signal was detected above the background and was in the exponential phase. Relative RNA expression was analyzed by 2−△△C(t), and DMSO was used as a control. The sequences of primers used are showed in Table 3.
Table 3.
Primers used in quantitative RT-PCR.
4. Conclusions
In summary, the total synthesis of glycycoumarin (1), glycyrin (2), and 3-O-methylglycyrol (3) has been accomplished in 5–7 steps from commercially available 2,4,6-trihydroxybenzaldehyde (9) with yields of 13.5%, 21.2%, and 12.3%, respectively. Notably, glycyrin (2) and 3-O-methylglycyrol (3) were synthesized for the first time. Our synthetic strategy features a Perkin condensation to establish the 3-phenyl-2H-chromen-2-one framework and a Claisen/Cope rearrangement to introduce the isopentene group to the coumarin core. Furthermore, the anti-inflammatory potencies of the synthetic natural products 1–3 were investigated using various in vitro systems, including the inhibition of NO production in LPS-induced RAW264.7 cells and the inhibition of three critical pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β). Among compounds 1–3, the anti-inflammatory activities of glycyrin (2) and 3-O-methylglycyrol (3) were first reported. Generally, compounds 1–3 exhibited different levels of anti-inflammatory activities, with compound 2 being the most potent. Mechanistic studies indicated that compound 2 exerted its anti-inflammatory property by inhibiting the activation of TNF-α, IL-6, and IL-1β. Hence, compound 2 could be a potential anti-inflammatory lead compound for further optimization and discovery of new agents.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29163942/s1. 1H NMR and 13C NMR spectra for the synthesized compounds.
Author Contributions
Y.H. and H.D. conceived and designed this research; T.P. synthesized compounds 1 and 2; B.L. synthesized compound 3; X.Y. evaluated the anti-inflammatory activity of 1–3; N.W. Writing—original draft; X.W. Writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 22307007, 82003619; and the Sichuan Provincial Science and Technology Foundation, grant numbers 2023NSFSC0609, 2023NSFSC1837, 2024NSFSC0995.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Asl, M.N.; Hosseinzadeh, H. Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-S.; Kang, S.-M.; Kim, Y.-J.; Lee, I.-J. Exploring the dietary and therapeutic potential of licorice (Glycyrrhiza uralensis Fisch.) sprouts. J. Ethnopharmacol. 2024, 328, 118101. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, Z.; Song, W.; Wang, Y.; Liang, W.; Li, K.; Tang, S.; Wang, Q.; Qiao, X.; Zhou, D.; et al. Bioactive constituents of Glycyrrhiza uralensis (licorice): Discovery of the effective components of a traditional herbal medicine. J. Nat. Prod. 2016, 79, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Annapurna, K.; Narsaiah, A.V. Total synthesis of natural alkaloids Schwarzinicines A-D. Tetrahedron 2024, 155, 133909. [Google Scholar] [CrossRef]
- Zhu, D.-Y.; Song, G.-Q.; Jian, F.-X.; Chang, X.-R.; Guo, W.-B. Studies on chemical constituents of Glycyrrhiza uralensis Fisch. Acta Chim. Sin. 1984, 42, 1080–1084. [Google Scholar]
- Zang, Y. Pharmacological activities of coumarin compounds in licorice: A review. Nat. Prod. Commun. 2020, 15, 1–17. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Li, Y.; Zheng, Y.; Li, P. Antioxidant and anti-inflammatory activities of six flavonoids separated from licorice. Food. Chem. 2013, 141, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yin, S.; Huo, Y.; Liang, M.; Fan, L.; Ye, M.; Hu, H. Glycycoumarin ameliorates alcohol-induced hepatotoxicity via activation of Nrf2 and autophagy. Free Radic. Biol. Med. 2015, 89, 135–146. [Google Scholar] [CrossRef]
- Yan, M.; Ye, L.; Yin, S.; Lu, X.; Liu, X.; Lu, S.; Cui, J.; Fan, L.; Kaplowitz, N.; Hu, H. Glycycoumarin protects mice against acetaminophen-induced liver injury predominantly via activating sustained autophagy. Brit. J. Pharmacol. 2018, 175, 3747–3757. [Google Scholar] [CrossRef]
- Kinoshita, T.; Saitoh, T.; Shibata, S. A new 3-Arylcoumarin from licorice root. Chem. Pharm. Bull. 1978, 26, 135–140. [Google Scholar] [CrossRef]
- Shiozawa, T.; Urata, S.; Kinoshita, T.; Saitoh, T. Revised structures of glycyrol and isoglycyrol, constituents of the root of Glycyrrhiza uralensis. Chem. Pharm. Bull. 1989, 39, 2239–2240. [Google Scholar] [CrossRef]
- Kuroda, M.; Mimaki, Y.; Sashida, Y.; Mae, T.; Kishida, H.; Nishiyama, T.; Tsukagawa, M.; Konishi, E.; Takahashi, K.; Kawada, T.; et al. Phenolics with PPAR-gamma ligand-binding activity obtained from licorice (Glycyrrhiza uralensis roots) and ameliorative effects of glycyrin on genetically diabetic KK-A(y) mice. Bioorg. Med. Chem. Lett. 2003, 13, 4267–4272. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kikuzaki, H.; Fukuda, S.; Nakatani, N. Antibacterial compounds of licorice against upper airway respiratory tract pathogens. J. Nutr. Sci. Vitaminol. 2001, 47, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xu, Y.; Duan, X.; Qin, X.; Huang, L.; Zhang, L. Method for Preparing 7,3(2′,4′)-trihydroxy-5-methoxy-6-isopentenyl coumarin. CN 116903570A, 20 October 2023. [Google Scholar]
- Dong, H.; Wu, M.; Wang, Y.; Du, W.; He, Y.; Shi, Z. Total syntheses and anti-inflammatory activities of syringin and its natural analogues. J. Nat. Prod. 2021, 84, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wu, M.; Li, Y.; Lu, L.; Qin, J.; He, Y.; Shi, Z. Total syntheses and anti-inflammatory evaluations of pongamosides A−C, natural furanoflavonoid glucosides from fruit of Pongamia pinnata (L.) Pierre. J. Nat. Prod. 2022, 85, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Long, B.; Feng, C.; Yang, T.; Jiang, X.; He, Y.; Dong, H. Total syntheses of pongaflavone and its natural analogues. J. Asian Nat. Prod. Res. 2023, 25, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Jiang, Y.; Song, X.; Lu, G.; Zhang, Q.; Du, Z.; Chan, A.S.C.; Zou, Y. Synthesis of phenolic coumestans via a sequential Dehydrogenation/Oxa-Michael addition reaction of 2′,4′-dihydroxyl-3-arylcoumarins. J. Org. Chem. 2022, 87, 5785–5794. [Google Scholar] [CrossRef]
- Dong, H.; Liao, L.; Long, B.; Che, Y.; Peng, T.; He, Y.; Mei, L.; Xu, B. Total synthesis and antibacterial evaluation of lupinifolin and its natural analogues. J. Nat. Prod. 2024, 87, 1044–1058. [Google Scholar] [CrossRef]
- Dong, H.; Che, Y.; Zhu, X.; Zhong, Y.; Lin, J.; Wang, J.; Du, W.; Song, T. Total syntheses and antibacterial studies of natural isoflavones: Scandenone, osajin, and 6,8-diprenylgenistein. Molecules 2024, 29, 2574. [Google Scholar] [CrossRef]
- Nchiozem-Ngnitedem, V.; Sperlich, E.; Matieta, V.Y.; Kuete, J.R.N.; Kuete, V.; Omer, E.A.; Efferth, T.; Schmidt, B. Synthesis and bioactivity of isoflavones from Ficus carica and some non-natural analogues. J. Nat. Prod. 2023, 86, 1520–1528. [Google Scholar] [CrossRef]
- Song, X.; Luo, X.; Sheng, J.; Li, J.; Zhu, Z.; Du, Z.; Miao, H.; Yan, M.; Li, M.; Zou, Y. Copper-catalyzed intramolecular cross dehydrogenative coupling approach to coumestans from 2′-hydroxyl-3-arylcoumarins. Rsc. Adv. 2019, 9, 17391–17398. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Xu, T.; Zhang, E.; Zhang, X.; Wei, W.; Zou, Y. Synthesis of coumestrol and aureol. J. Nat. Prod. 2016, 79, 2749–2753. [Google Scholar] [CrossRef] [PubMed]
- Robledo-O’Ryan, N.; Matos, M.J.; Vazquez-Rodriguez, S.; Santana, L.; Uriarte, E.; Moncada-Basualto, M.; Mura, F.; Lapier, M.; Maya, J.D.; Olea-Azar, C. Synthesis, antioxidant and antichagasic properties of a selected series of hydroxy-3-arylcoumarins. Bioorgan. Med. Chem. 2017, 25, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Gou, S.; Liu, J.; He, M.; Qiang, Y.; Ni, J. Quantification and bio-assay of α-glucosidase inhibitors from the roots of Glycyrrhiza uralensis Fisch. Nat. Prod. Res. 2016, 30, 2130–2134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, D.; Gao, Z.; Kan, H.; Qiu, F.; Chen, L.; Li, H. Licocoumarone induces BxPC-3 pancreatic adenocarcinoma cell death by inhibiting DYRK1A. Chem-Biol. Interact. 2020, 316, 108913. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-L.; Raja, A.; Hong, B.-C.; Lee, O.-H. Organocatalytic Enantioselective Michael–Acetalization–Reduction–Nef Reaction for a One-Pot Entry to the Functionalized Aflatoxin System. Total Synthesis of (−)-Dihydroaflatoxin D2 and (−)-and (+)-Microminutinin. Org. Lett. 2017, 19, 3494–3497. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Riemer, M. Synthesis of allyl- and prenylcoumarins via microwave-promoted tandem claisen rearrangement/wittig olefination. Synthesis 2016, 48, 141–149. [Google Scholar] [CrossRef]
- Mali, R.S.; Joshi, P.P.; Sandhua, P.K.; Manekar-Tilve, A. Efficient syntheses of 6-prenylcoumarins and linear pyranocoumarins: Total synthesis of suberosin, toddaculin, O-methylapigravin (O-methylbrosiperin), O-methylbalsamiferone, dihydroxanthyletin, xanthyletin and luvangetin. J. Chem. Soc. Perkin Trans. 2002, 1, 371–376. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Broderick, M.; Fein, H. Measurement of nitric oxide production in biological systems by using griess reaction assay. Sensors 2003, 3, 276–284. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).