

Symmetry Breaking in the Lowest-Lying Excited-State of CCl4: Valence Shell Spectroscopy in the 5.0–10.8 eV Photon Energy Range

 ,
,  , , , , ,
, , , , ,  and
and
Abstract
:
1. Introduction
2. Structure and Properties of Carbon Tetrachloride
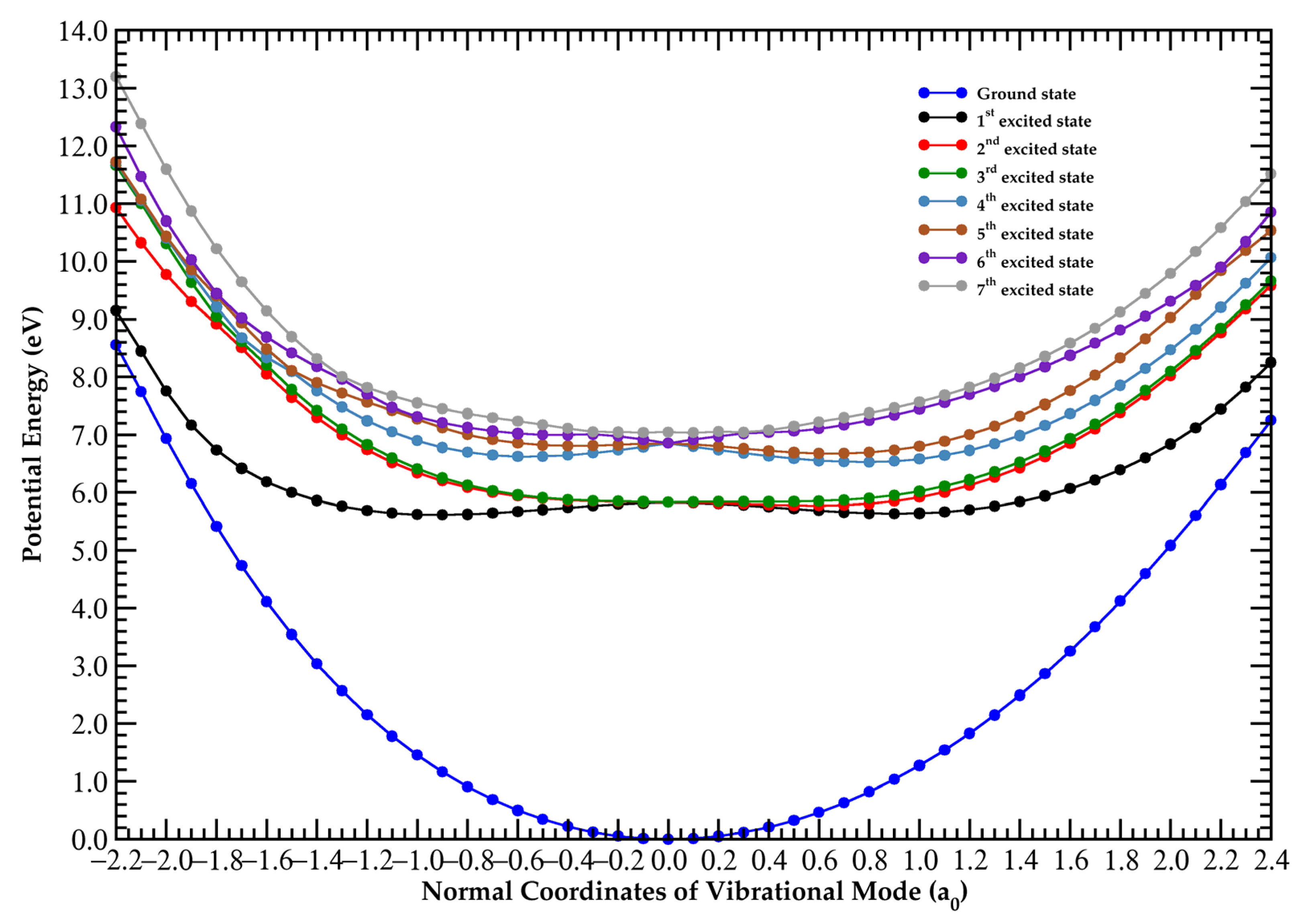
3. Discussion
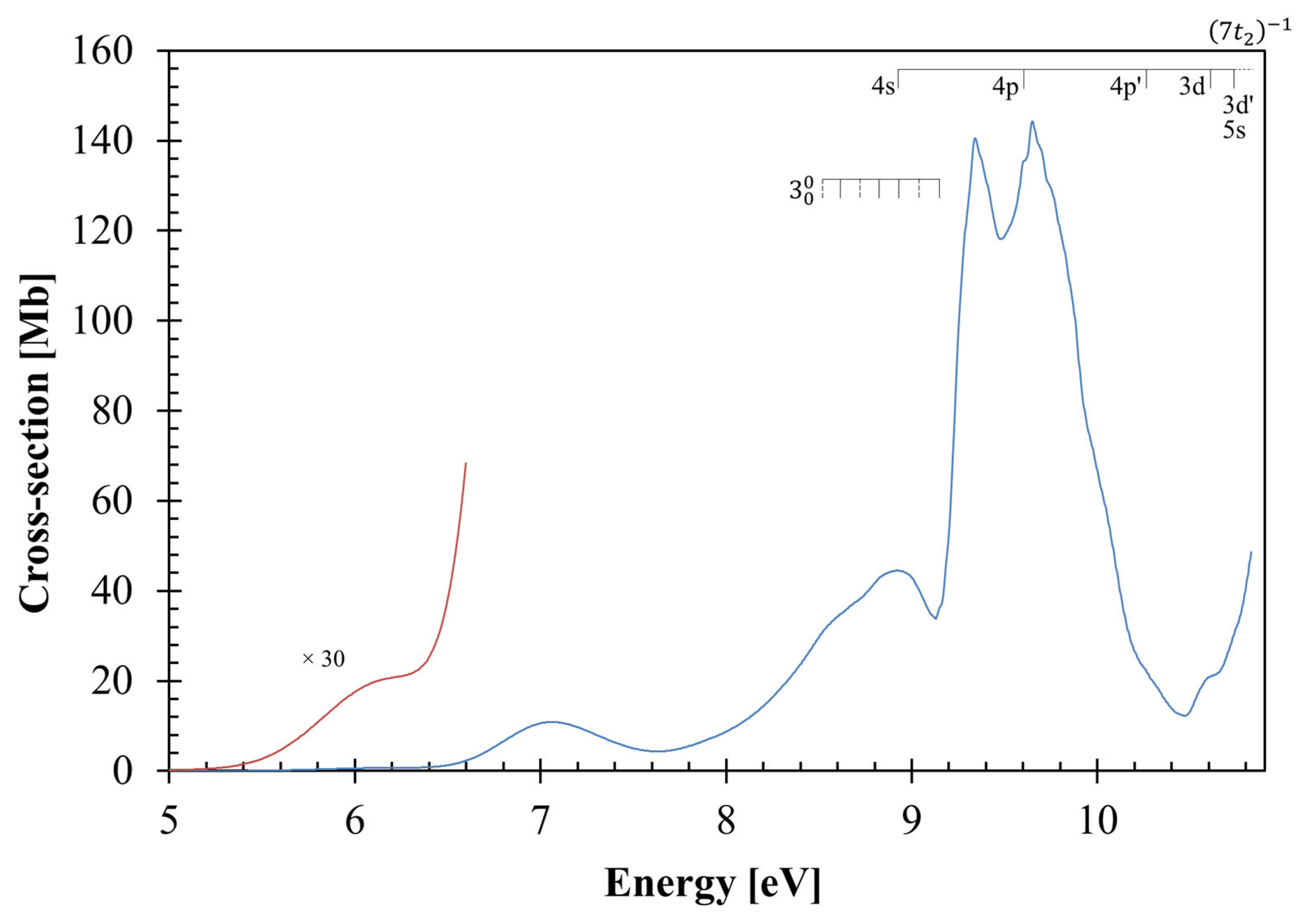
3.1. The 5.0–6.5 eV Photon Energy Range
3.2. The 6.5–9.2 eV Photon Energy Range
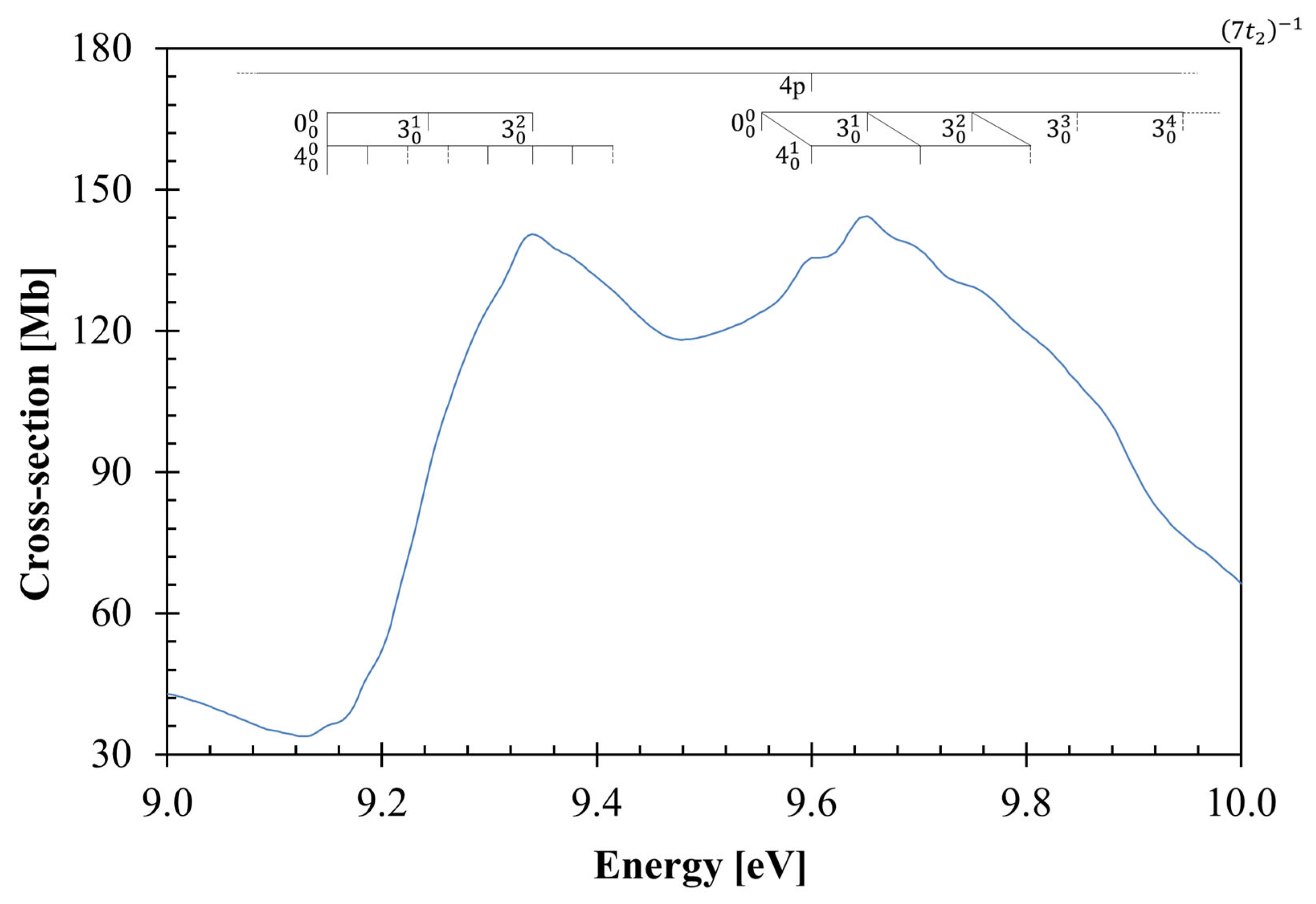
3.3. The 9.2–10.8 eV Photon Energy Range
3.4. Rydberg Transitions
3.5. Absolute Photoabsorption Cross-Sections
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sommerer, T.J.; Kushner, M.J. Monte Carlo-fluid model of chlorine atom production in Cl2, HCl, and CCl4 radio-frequency discharges for plasma etching. J. Vac. Sci. Technol. B 1992, 10, 2179–2187. [Google Scholar] [CrossRef]
- Linn, J.H.; Swartz, W.E., Jr. Spectroscopy Letters: An International Journal for Rapid An XPS Study of the Plasma Etching of Aluminum with CCl4. Spectrosc. Lett. 1985, 18, 335–352. [Google Scholar] [CrossRef]
- Kime, Y.J.; Driscoll, D.C.; Dowben, P.A. The stability of the carbon tetrahalide ions. J. Chem. Soc. Faraday Trans. 1987, 83, 403–410. [Google Scholar] [CrossRef]
- Liang, Q.; Newman, P.A.; Daniel, J.S.; Reimann, S.; Hall, B.D.; Dutton, G.; Kuijpers, L.J.M. Constraining the carbon tetrachloride (CCl4) budget using its global trend and inter-hemispheric gradient. Geophys. Res. Lett. 2014, 41, 5307–5315. [Google Scholar] [CrossRef]
- Sherry, D.; McCulloch, A.; Liang, Q.; Reimann, S.; Newman, P.A. Current sources of carbon tetrachloride (CCl4) in our atmosphere. Environ. Res. Lett. 2018, 13, 024004. [Google Scholar] [CrossRef]
- Zhang, W.; Ibuki, T.; Brion, C.E. Absolute dipole differential oscillator strengths for inner shell spectra from high resolution electron energy loss studies of the freon molecules CF4, CF3Cl, CF2Cl2, CFCl3 and CCl4. Chem. Phys. 1992, 160, 435–450. [Google Scholar] [CrossRef]
- Molina, M.J.; Rowland, F.S. Stratospheric sink for chlorofluoromethanes: Chlorine atom-catalysed destruction of ozone. Nature 1974, 249, 810–812. [Google Scholar] [CrossRef]
- Matejcik, S.; Foltin, V.; Stano, M.; Skalny, J.D. Temperature dependencies in dissociative electron attachment to CCl4, CCl2F2, CHCl3 and CHBr3. Int. J. Mass Spectrom. 2003, 223–224, 9–19. [Google Scholar] [CrossRef]
- Oster, T.; Kühn, A.; Illenberger, E. Gas phase negative ion chemistry. Int. J. Mass Spectrom. Ion Proces. 1989, 89, 1–72. [Google Scholar] [CrossRef]
- Carlson, T.A.; Krause, M.O.; Grimm, F.A.; Keller, P.; Taylor, J.W. Angle-resolved photoelectron spectroscopy of CCl4: The Cooper minimum in molecules. J. Chem. Phys. 1982, 77, 5340–5347. [Google Scholar] [CrossRef]
- Causley, G.C.; Russell, B.R. The vacuum ultraviolet absorption spectra of the group IVA tetrachlorides. J. Electron Spectrosc. Relat. Phenom. 1977, 11, 383–397. [Google Scholar] [CrossRef]
- Lee, L.C.; Suto, M. Fluorescence yields from photodissociative excitation of chloromethanes by vacuum ultraviolet radiation. Chem. Phys. 1987, 114, 423–429. [Google Scholar] [CrossRef]
- Russell, B.R.; Edwards, L.O.; Raymonda, J.W. Vacuum Ultraviolet Absorption Spectra of the Chloromethanes. J. Am. Chem. Soc. 1973, 95, 2129–2133. [Google Scholar] [CrossRef]
- Ho, G.H. Absolute photoabsorption cross section of CCl4 in the energy range 6–250 eV. Chem. Phys. 1998, 226, 101–111. [Google Scholar] [CrossRef]
- Biehl, H.; Boyle, K.J.; Seccombe, D.P.; Smith, D.M.; Tuckett, R.P.; Baumgärtel, H.; Jochims, H.W. Vacuum-UV fluorescence spectroscopy of CCl4, SiCl4 and GeCl4 in the range 9–25 eV. J. Electron Spectrosc. Relat. Phenom. 1998, 97, 89–113. [Google Scholar] [CrossRef]
- Chakraborty, T.; Rai, S.N. Comparative study of infrared and Raman spectra of CCl4 in vapour and condensed phases: Effect of LO-TO splitting resulting from hetero-isotopic TD-TD interactions. Spectrochim. Acta 2006, 65, 406–413. [Google Scholar] [CrossRef]
- Chakraborty, T.; Verma, A.L. Vibrational spectra of CCl4: Isotopic components and hot bands. Part I. Spectrochim. Acta 2002, 58, 1013–1023. [Google Scholar] [CrossRef]
- Wallington, T.J.; Pivesso, B.P.; Lira, A.M.; Anderson, J.E.; Nielsen, C.J.; Andersen, N.H.; Hodnebrog, Ø. CH3Cl, CH2Cl2, CHCl3, and CCl4: Infrared spectra, radiative efficiencies, and global warming potentials. J. Quant. Spectrosc. Radiat. Transf. 2016, 174, 56–64. [Google Scholar] [CrossRef]
- Davis, D.D.; Schmidt, J.F.; Neeley, C.M.; Hanrahan, R.J. Effect of Wavelength in the Gas-Phase Photolysis of Carbon Tetrachloride at 253.7, 184.9, 147.0, and 106.7 nm. J. Phys. Chem. 1975, 79, 11–17. [Google Scholar] [CrossRef]
- Hanf, A.; Läuter, A.; Volpp, H.R. Absolute chlorine atom quantum yield measurements in the UV and VUV gas-phase laser photolysis of CCl4. Chem. Phys. Lett. 2003, 368, 445–451. [Google Scholar] [CrossRef]
- Ibuki, T.; Takahashi, N.; Hiraya, A.; Shobatake, K. CCl2(Ã 1B1) radical formation in VUV photolyses of CCl4 and CBrCl3. J. Chem. Phys. 1986, 85, 5717–5722. [Google Scholar] [CrossRef]
- Tiee, J.J.; Wampler, F.B.; Rice, W.W.; Tiee, J.J.; Wampler, F.B.; Rice, W.W. UV laser photochemistry of CCl4 and CCl3F. J. Chem. Phys. 1980, 72, 2925–2927. [Google Scholar] [CrossRef]
- von Niessen, W.; Åsbrink, L.; Bieri, G. 30.4 nm He (II) photoelectron spectra of organic molecules Part VI. Halogeno-compounds (C, H, X; X = Cl, Br, I). J. Electron Spectrosc. Relat. Phenom. 1982, 26, 173–201. [Google Scholar] [CrossRef]
- Dixon, R.N.; Murrell, J.N.; Narayan, B. The photoelectron spectra of the halomethanes. Mol. Phys. 1971, 20, 611–623. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Worley, S.D. Photoelectron Spectra of Molecules. I. Ionization Potentials of Some Organic Molecules and Their Interpretation. J. Chem. Phys. 1969, 50, 654–667. [Google Scholar] [CrossRef]
- Bassett, P.J.; Lloyd, R. Photoelectron Spectra of Halides. Part 1. Tetrafluorides and Tetra- chlorides of Group IVB. J. Chem. Soc. A 1971, 641–645. [Google Scholar] [CrossRef]
- Watanabe, K. Ionization potentials of some molecules. J. Chem. Phys. 1957, 26, 542–547. [Google Scholar] [CrossRef]
- Roszak, S.; Vijayakumar, M.; Balasubramanian, K.; Koski, W.S. A multireference configuration interaction study of photoelectron spectra of carbon tetrahalides. Chem. Phys. Lett. 1993, 208, 225–231. [Google Scholar] [CrossRef]
- Ying, J.F.; Leung, K.T. Absolute transition probability measurement of valence-shell electronic structure of CF4 and CCl4: Bethe surfaces, and dipole-dominated preionization-edge valence and Rydberg states. J. Chem. Phys. 1994, 100, 7120–7134. [Google Scholar] [CrossRef]
- Wang, L.H.; Du, X.J.; Xu, Y.C.; Nie, Z.W.; Wang, D.H.; Wang, S.X.; Zhu, L.F. Generalized Oscillator Strengths for the Valence Shell Excitations in Carbon Tetrachloride Studied by Fast Electron Impact. J. Phys. Chem. A 2022, 126, 453–461. [Google Scholar] [CrossRef]
- Limão-Vieira, P.; Horie, M.; Kato, H.; Hoshino, M.; Blanco, F.; García, G.; Buckman, S.J.; Tanaka, H. Differential elastic electron scattering cross sections for CCl4 by 1.5–100 eV energy electron impact. J. Phys. Chem. 2011, 135, 234309. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.R.; Chan, W.F.; Cooper, G.; Brion, C.E. Valence- and inner-shell (Cl 2p, 2s; C 1s) photoabsorption and photoionization of carbon tetrachloride. Absolute oscillator strength (5-400 eV) and dipole-induced breakdown pathways. Chem. Phys. 1994, 181, 147–172. [Google Scholar] [CrossRef]
- Natalense, A.P.P.; Bettega, M.H.F.; Ferreira, L.G.; Lima, M.A.P. Low-energy electron scattering by CF4, CCl4, SiCl4, SiBr4, and SiI4. Phys. Rev. A 1995, 52, R1–R4. [Google Scholar] [CrossRef] [PubMed]
- Varella, M.T.N.; Natalense, A.P.P.; Bettega, M.H.F.; Lima, M.A.P. Low-energy electron scattering by CF4, CCl4, SiCl4, SiBr4, and SiI4. Phys. Rev. A 1999, 60, 3684–3693. [Google Scholar] [CrossRef]
- Azevedo, D.L.; Bettega, M.H.F.; Ferreira, L.G.; Lima, M.A.P. Scattering of low-energy electrons by TiCl4, GeCl4, SiCl4 and CCl4: A comparison of elastic cross sections. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 5467–5478. [Google Scholar] [CrossRef]
- Joucoski, E.; Bettega, M. Elastic scattering of electrons by carbon, silicon, germanium and tin tetrahalides. J. Phys. B At. Mol. Opt. Phys. 2002, 35, 4953–4968. [Google Scholar] [CrossRef]
- Moreira, G.M.; Barbosa, A.S.; Pastega, D.F.; Bettega, M.H.F. Low-energy electron scattering by carbon tetrachloride. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 035202. [Google Scholar] [CrossRef]
- Gaynor, J.D.; Wetterer, A.M.; Cochran, R.M.; Valente, E.J.; Mayer, S.G. Vibrational Spectroscopy of the CCl4 ν1 Mode: Effect of Thermally Populated Vibrational States. J. Chem. Educ. 2015, 92, 1949–1952. [Google Scholar] [CrossRef]
- Watanabe, N.; Takahashi, M. Theoretical Study of Valence Shell Excitation by Electron Impact in CCl4. J. Phys. Chem. A 2023, 127, 1866–1873. [Google Scholar] [CrossRef]
- Regeta, K.; Kumar, S.; Cunha, T.; Mendes, M.; Lozano, A.I.; Pereira, P.J.S.; García, G.; Moutinho, A.M.C.; Bacchus-Montabonel, M.C.; Limão-Vieira, P. Combined Experimental and Theoretical Studies on Electron Transfer in Potassium Collisions with CCl4. J. Phys. Chem. A 2020, 124, 3220–3227. [Google Scholar] [CrossRef]
- Mondal, T.; Varandas, A.J.C. The Jahn-Teller effect in the triply degenerate electronic state of methane radical cation. J. Chem. Phys. 2011, 135, 174304. [Google Scholar] [CrossRef] [PubMed]
- Carlon, N.R.; Papanastasiou, D.K.; Fleming, E.L.; Jackman, C.H.; Newman, P.A.; Burkholder, J.B. UV absorption cross sections of nitrous oxide (N2O) and carbon tetrachloride (CCl4) between 210 and 350 K and the atmospheric implications. Atmos. Chem. Phys. 2010, 10, 6137–6149. [Google Scholar] [CrossRef]
- Eden, S.; Limão-Vieira, P.; Hoffmann, S.V.; Mason, N.J. VUV photoabsorption in CF3X (X = Cl, Br, I) fluoro-alkanes. Chem. Phys. 2006, 323, 313–333. [Google Scholar] [CrossRef]
- Palmer, M.H.; Ridley, T.; Hoffmann, S.V.; Jones, N.C.; Coreno, M.; De Simone, M.; Grazioli, C.; Biczysko, M.; Baiardi, A.; Limão-Vieira, P. Interpretation of the vacuum ultraviolet photoabsorption spectrum of iodobenzene by ab initio computations. J. Chem. Phys. 2015, 142, 134302. [Google Scholar] [CrossRef] [PubMed]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E. Time-dependent density-functional theory for molecules and molecular solids. J. Mol. Struct.-Theochem 2009, 914, 3–18. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef]
- Morino, Y.; Nakamura, Y.; Iijima, T. Mean square amplitudes and force constants of tetrahedral molecules. I. Carbon tetrachloride and germanium tetrachloride. J. Chem. Phys. 1960, 32, 643–652. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Tetrachloride, CCl4 | E (eV) Expt. a |
Cross-Section (Mb) | ||||
|---|---|---|---|---|---|---|
|
State (C2v) |
State (Td) | E (eV) | fL | Dominant Excitations | ||
| 1A1 | 1A1 | |||||
| 2 1A1 | 1 1T2 | 6.853 | 0.0202 | (87%) | ||
| 1 1B2 | (87%) | 7.06(1) | 10.87 | |||
| 1 1B1 | (87%) | |||||
| 3 1B2 | 3 1T2 | 8.468 | 0.1254 | (42%) | ||
| 3 1B1 | (6%) | 8.92(9) | 44.48 | |||
| 4 1A1 | (42%) | |||||
| 4 1B2 | 4 1T2 | 9.047 | 0.0358 | (4%) | ||
| 4 1B1 | (4%) | 9.343 | 140.41 | |||
| 5 1A1 | (4%) | |||||
| 5 1B1 | 5 1T2 | 9.339 | 0.2174 | (6%) | ||
| 5 1B2 | (6%) | 9.652 | 144.31 | |||
| 6 1A1 | (8%) | |||||
| 7 1A1 | 6 1T2 | 9.584 | 0.0157 | (28%) | ||
| 6 1B2 | (28%) | 10.26(4) | 22.16 | |||
| 6 1B1 | (28%) | |||||
| 8 1A1 | 7 1T2 | 10.344 | 0.0155 | (14%) | ||
| 7 1B1 | (11%) | |||||
| 7 1B2 | (11%) | |||||
| Td | C1 | |||
|---|---|---|---|---|
| Assignment | Energy | Assignment | ||
| 8.52(1)(s,w) | – | – | – | |
| 8.63(4)(w) | 0.113 | – | – | |
| 8.73(4)(w) | 0.100 | – | – | |
| 8.81(8)(s) | 0.084 | – | – | |
| 8.92(9)(b) | 0.111 | – | ||
| 9.03(7)(s,w) | 0.108 | – | ||
| 9.14(7)(s,b) | 0.110 | – | ||
| 9.14(7)(s,b) | – | – | – | |
| 9.19(1)(s) | – | 0.044 | – | |
| 9.22(5)(s,w) | – | 0.034 | – | |
| 9.24(2)(s,w) | 0.095 | – | – | |
| 9.26(3)(s,w) | – | 0.038 | – | |
| 9.30(1)(s,w) | – | 0.038 | – | |
| 9.343 | 0.101 | 0.042 | – | |
| 9.37(9)(b) | – | 0.036 | – | |
| 9.41(1)(s,w) | – | 0.032 | – | |
| 9.55(2)(s,w) | – | – | – | |
| 9.60(4)(b) | – | 0.052 | ||
| 9.652 | 0.100 | – | ||
| 9.70(1)(b) | 0.101 | 0.052 | ||
| 9.75(1)(b,w) | 0.099 | – | ||
| 9.80(5)(s,w) | 0.104 | 0.054 | ||
| 9.84(4)(s,w) | 0.093 | – | ||
| 9.94(7)(s) | 0.103 | – | ||
| 0.102 | 0.042 | |||
| En | δ | Assignment |
|---|---|---|
| (ns ← 7t2) | ||
| 8.92(9)(b) | 1.98 | 4s |
| 10.74(9)(s,w) | 2.00 | 5s |
| (np ← 7t2) | ||
| 9.60(4)(s) | 1.74 | 4p |
| (np’ ← 7t2) | ||
| 10.26(4)(s,w) | 1.39 | 4p’ |
| (nd ← 7t2) | ||
| 10.61(5)(s) | 0.13 | 3d |
| (nd’ ← 7t2) | ||
| 10.74(9)(s,w) | 0.00 | 3d’ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalagnol, L.V.S.; Kumar, S.; Barbosa, A.S.; Akther, U.S.; Jones, N.C.; Hoffmann, S.V.; Bettega, M.H.F.; Limão-Vieira, P. Symmetry Breaking in the Lowest-Lying Excited-State of CCl4: Valence Shell Spectroscopy in the 5.0–10.8 eV Photon Energy Range. Molecules 2024, 29, 5619. https://doi.org/10.3390/molecules29235619
Dalagnol LVS, Kumar S, Barbosa AS, Akther US, Jones NC, Hoffmann SV, Bettega MHF, Limão-Vieira P. Symmetry Breaking in the Lowest-Lying Excited-State of CCl4: Valence Shell Spectroscopy in the 5.0–10.8 eV Photon Energy Range. Molecules. 2024; 29(23):5619. https://doi.org/10.3390/molecules29235619
Chicago/Turabian StyleDalagnol, Luiz V. S., Sarvesh Kumar, Alessandra S. Barbosa, Umma S. Akther, Nykola C. Jones, Søren V. Hoffmann, Márcio H. F. Bettega, and Paulo Limão-Vieira. 2024. "Symmetry Breaking in the Lowest-Lying Excited-State of CCl4: Valence Shell Spectroscopy in the 5.0–10.8 eV Photon Energy Range" Molecules 29, no. 23: 5619. https://doi.org/10.3390/molecules29235619
APA StyleDalagnol, L. V. S., Kumar, S., Barbosa, A. S., Akther, U. S., Jones, N. C., Hoffmann, S. V., Bettega, M. H. F., & Limão-Vieira, P. (2024). Symmetry Breaking in the Lowest-Lying Excited-State of CCl4: Valence Shell Spectroscopy in the 5.0–10.8 eV Photon Energy Range. Molecules, 29(23), 5619. https://doi.org/10.3390/molecules29235619