
Steric Effects of N-Alkyl Group on the Base-Induced Nitrogen to Carbon Rearrangement of Orthogonally Protected N-Alkyl Arylsulphonamides


Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, A. Vogel’s Textbook of Practical Organic Chemistry, 4th ed.; Furniss, B.S., Hannaford, A.J., Rogers, V., Smith, P.W.G., Tatchell, A.R., Eds.; Longman Scientific and Technical: London, UK, 1978; pp. 1130–1131, 1228–1237. ISBN 0-582-46236-3. [Google Scholar]
- Hinsberg, O. Ueber die Bildung von Säureestern und Säureamiden bei Gegenwart von Wasser und Alkali. Ber. Dtsch. Chem. Ges. 1890, 23, 2962–2965. [Google Scholar] [CrossRef]
- Bel, J.A.L. Sur la dyssymètrie et la creation du pouvoir rotatoire dans les dèrivès alcooliques du chlorure d’ammonium Compt. Rendu 1891, 112, 724–726. [Google Scholar]
- Pope, W.J.; Peachey, S.J. Asymmetric Optically Active Nitrogen Compounds. Dextro- and Laevo-α-benzylphenylallylmethylammonium Iodides and Bromides. J. Chem. Soc. 1899, 75, 1127–1131. [Google Scholar] [CrossRef]
- Pope, W.J.; Read, J. Asymmetric quinquevalent nitrogen compounds of simple molecular constitution. J. Chem. Soc. 1912, 101, 519–529. [Google Scholar] [CrossRef]
- Kocienski, P.J. Protecting Groups, 3rd ed.; Thieme: Stuttgart, Germany, 2000. [Google Scholar]
- Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis, 4th ed.; Wiley: New York, NY, USA, 2007. [Google Scholar]
- Spieβ, P.; Sirvent, A.; Tiefenbrunner, I.; Sargueil, J.; Fernandes, A.J.; Arroyo-Bondia, A.; Meyrelles, R.; Just, D.; Prado-Roller, A.; Shaaban, S.; et al. Nms-Amides: An Amine Protecting Group with Unique Stability and Selectivity. Chem. A Eur. J. 2023, 29, e202301312. [Google Scholar] [CrossRef]
- Van Hoof, M.; Mayer, R.J.; Moran, J.; Leboef, D. Triflic Acid-Catalyzed Dehydrative Amination of 2-Arylethanols with Weak N-Nucleophiles in Hexafluoroisopropanol. Angew. Chem. Int. Ed. Engl. 2025, 64, e202417089. [Google Scholar] [CrossRef]
- Spieß, P.; Brześkiewicz, J.; Meyrelles, R.; Just, D.; Maulide, N. Deprotective Functionalization: A Direct Conversion of Nms-Amides to Carboxamides Using Carboxylic Acids. Angew. Chem. Int. Ed. 2024, 63, e202318304. [Google Scholar] [CrossRef]
- Gelmo, P. Über Sulfamide der p-Amidobenzolsulfonsäure. J. Prak. Chem. 1908, 77, 369–382. [Google Scholar] [CrossRef]
- Domagk, G. Ein beitrag zur chemotherapie der bakteriellen infektionen. Dtsch. Med. Wschr. 1935, 61, 250–253. [Google Scholar] [CrossRef]
- Owa, T.; Nagasu, K. Novel sulphonamide derivatives for the treatment of cancer. Expert Opin. Ther. Pat. 2000, 10, 1725–1740. [Google Scholar] [CrossRef]
- Abbate, F.; Casini, A.; Owa, T.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg. Med. Chem. Lett. 2004, 14, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ezabadi, R.R.; Camoutsis, C.; Zoumpoulakis, P.; Geronikaki, A.; Sokovic, M.; Glamocilija, J.; Ciric, A. Sulfonamide-1,2,4-triazole derivatives as antifungal and antibacterial agents: Synthesis, biological evaluation, lipophilicity, and conformational studies. Bioorg. Med. Chem. 2008, 16, 1050–1161. [Google Scholar] [CrossRef]
- Torok, E.; Moran, E.; Cooke, F. Oxford Handbook of Infectious Diseases and Microbiology; Oxford University Press: Oxford, UK, 2009; ISBN 978-0-856925-1. [Google Scholar]
- Sippel, K.H.; Robbins, A.H.; Domsic, J.; Genis, C.; Agbandje-McKenna, M.; McKenna, R. High resolution structure of human carbonic anhydrase ii complexed with acetrazolamide reveals insights into inhibitor drug design. Acta Crystallogr. (Sect. F) 2009, 65, 992–995. [Google Scholar] [CrossRef]
- Mirian, M.; Zarghi, A.; Sadeghhi, S.; Tabaraki, P.; Tavallaee, M.; Dadrass, O.; Sadeghi-aliabadi, H. Synthesis and cytotoxic evaluation of some novel sulfonamide derivatives against a few human cancer cells. Iran. J. Pharm. Res. 2011, 10, 741–748. [Google Scholar] [CrossRef]
- Boechat, N.; Pinheiro, L.C.S.; Santos-Filho, O.A.; Silva, I. Design and Synthesis of New N-(5-Trifluoromethyl)-1H-1,2,4-triazol-3-yl Benzenesulfonamides as Possible Antimalarial Prototypes. Molecules 2011, 16, 8083–8097. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Said, H.M.; Hagemann, C.; Carta, F.; Katzer, A.; Polat, B.; Staab, A.; Scozzafava, A.; Anacker, J.; Vince, G.H.; Flentje, M.; et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including acetazolamide in human glioblastoma. Bioorg. Med. Chem. 2013, 21, 3949–3957. [Google Scholar] [CrossRef]
- Shah, S.S.A.; Rivera, G.; Ashfaq, M. Recent Advances in Medicinal Chemistry of Sulfonamides. Rational Design as Anti-Tumoral, Anti-Bacterial and Anti-Inflammatory Agents. Mini. Rev. Med. Chem. 2013, 13, 70–86. [Google Scholar] [CrossRef]
- Lyden, P.; Pereira, B.; Chen, B.; Zhao, L.; Lamb, J.; Lei, I.-F.; Rajput, P. Direct Thrombin Inhibitor Argatroban Reduces Stroke Damage in 2 Different Models. Stroke 2014, 45, 896–899. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; El-Gazzar, M.G.; Zahran, S.S. Synthesis, anticancer and radiosensitizing evaluation of some novel sulfonamide derivatives. Eur. J. Med. Chem. 2015, 92, 682–692. [Google Scholar] [CrossRef]
- Zelder, F.; Sonnay, M.; Prieto, L. Antivitamins for Medicinal Applications. ChemBioChem 2015, 16, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Chinthakindi, P.K.; Naicker, T.; Thota, N.; Govender, T.; Kruger, H.; Arvidsson, P.L. Sulfonimidamides in Medicinal and Agricultural Chemistry. Angew. Int. Ed. Engl. 2017, 56, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Stanczak, A. Cinnoline Scaffold—A Molecular Heart of Medicinal Chemistry? Molecules 2019, 24, 2271. [Google Scholar] [CrossRef] [PubMed]
- Jafarinejad, S.; Martin, W.H.C.; Ras, B.A.; Isreb, M.; Jacob, B.; Aziz, A.; Adoul, Z.; Lagnado, R.; Bowen, R.D.; Najafzadeh, M. The anticancer/cytotoxic effect of a novel gallic acid derivative in non-small cell lung carcinoma A549 cells and peripheral blood mononuclear cells from healthy individuals and lung cancer patients. BioFactors 2024, 50, 201–213. [Google Scholar] [CrossRef]
- Koyuncu, I.; Temiz, E.; Güler, E.M.; Durgun, M.; Yuksekdag, O.; Giovannuzzi, S.; Supuran, C.T. Effective Anticancer Potential of a New Sulfonamide as a Carbonic Anhydrase IX Inhibitor Against Aggressive Tumors. ChemMedChem 2024, 19, e202300680. [Google Scholar] [CrossRef]
- Yerrabelly, J.R.; Bommagani, M.B.; Yerrabelly, H.; Mullaguri, S.C.; Kancha, R.K. Synthesis and anticancer activity of cinnoline sulphonamides and 4-heteroyclic derivatives: Cross-coupling approach. J. Heterocycl. Chem. 2024, 61, 958–970. [Google Scholar] [CrossRef]
- Siliveri, S.; Gudishetty, L.P.; Vaddiraju, N.; Kavali, J. Design, Synthesis, In Silico Molecular Docking, ADMET Studies, and Biological Evaluation of Novel Substituted Diphenyl Pyrazole Incorporated 1,2,3-Triazolyl Benzene Sulfonamides. Russ. J. Bioorg. Chem. 2024, 50, 1119–1132. [Google Scholar] [CrossRef]
- Snieckus, V. Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev. 1990, 90, 879–933. [Google Scholar] [CrossRef]
- Rushwinga, E.; Touray, H.K.; Bowen, R.D.; Gallagher, R.T.; Martin, W.H.C. Novel nitrogen to carbon rearrangement forming nicotinic acid sulfonamides. Tetrahedron Lett. 2013, 54, 4726–4728. [Google Scholar] [CrossRef]
- Watanabe, H.; Gay, R.; Hauser, C.R. Ortho metalation of N-substituted benzenesulfonamides by excess N-butyllithium. Condensation with carbonyl compounds. Cyclizations. J. Org. Chem. 1968, 33, 900–903. [Google Scholar] [CrossRef]
- Lombardino, J.G. Preparation of substituted 1,2-benzoisothiazolin-3-one 1,1-dioxides (o-benzoic sulfimides). J. Org. Chem. 1971, 36, 1843–1845. [Google Scholar] [CrossRef]
- Court, J.J.; Hlasta, D.J. Ortho versus adjacent-benzylic directed lithiations of substituted N,N-diethylbenzamides. Tetrahedron Lett. 1996, 37, 1335–1338. [Google Scholar] [CrossRef]
- Metallinos, C.; Nerdinger, S.; Snieckus, V. N-Cumyl Benzamide, Sulfonamide, and Aryl O-Carbamate Directed Metalation Groups. Mild Hydrolytic Lability for Facile Manipulation of Directed Ortho Metalation Derived Aromatics. Org. Lett. 1999, 1, 1183–1186. [Google Scholar] [CrossRef]
- MacNeil, S.L.; Familoni, O.B.; Snieckus, V. Selective Ortho and Benzylic Functionalization of Secondary and Tertiary p-Tolylsulfonamides. Ipso-Bromo Desilylation and Suzuki Cross-Coupling Reactions. J. Org. Chem. 2001, 66, 3663–3670. [Google Scholar] [CrossRef]
- Saidykhan, A.; Bowen, R.D.; Gallagher, R.T.; Martin, W.H.C. Intramolecular N-C rearrangements involving sulfonamide protecting groups. Tetrahedron Lett. 2015, 56, 66–68. [Google Scholar] [CrossRef]
- Saidykhan, A.; Ebert, J.; Ally, H.; Gallagher, R.T.; Martin, W.H.C.; Bowen, R.D. The scope and regioselectivity of intramolecular N-C rearrangements of orthogonally protected sulfonamides, including cyclization to saccharin derivatives. Tetrahedron Lett. 2017, 58, 3089–3091. [Google Scholar] [CrossRef]
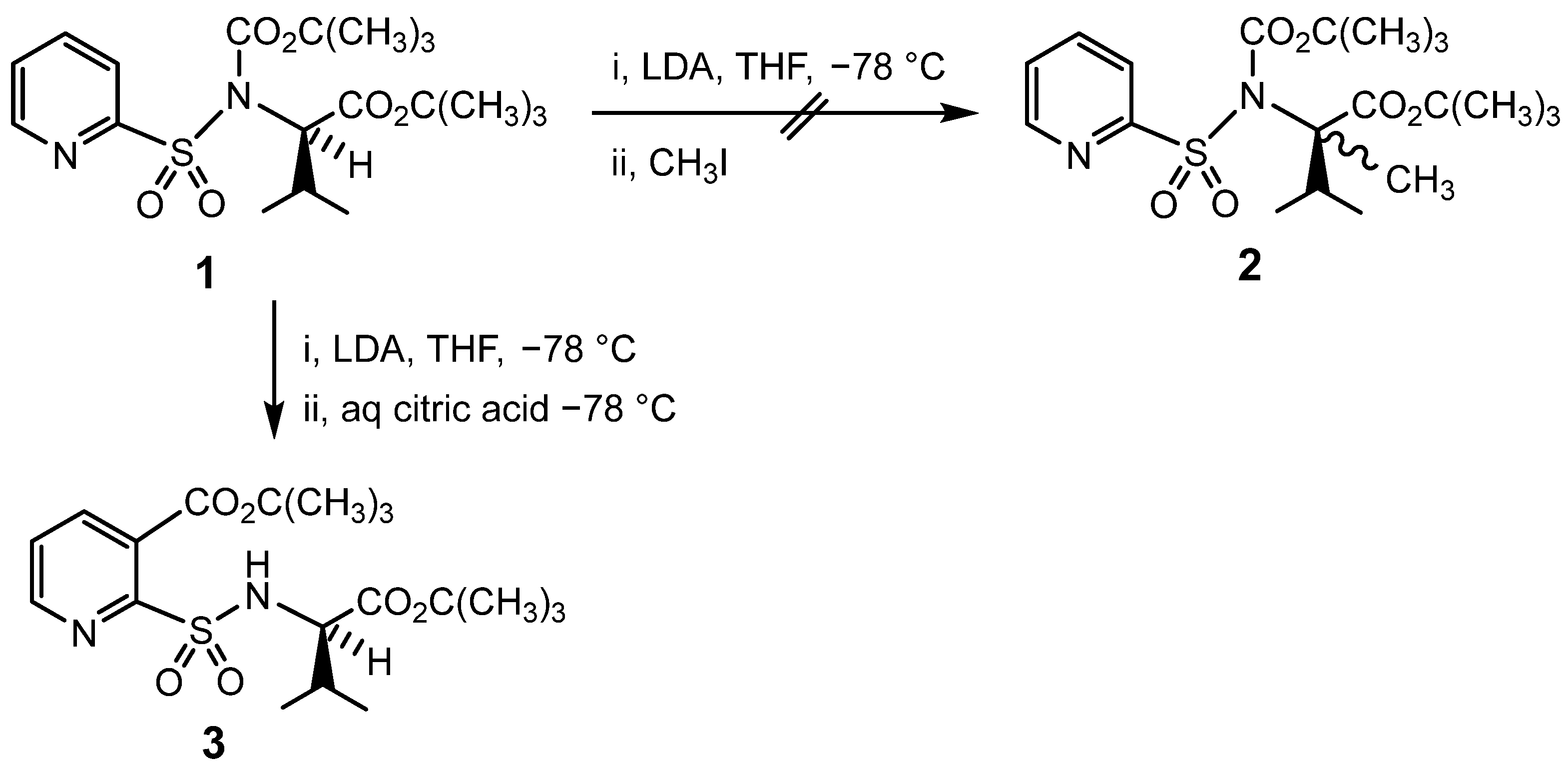
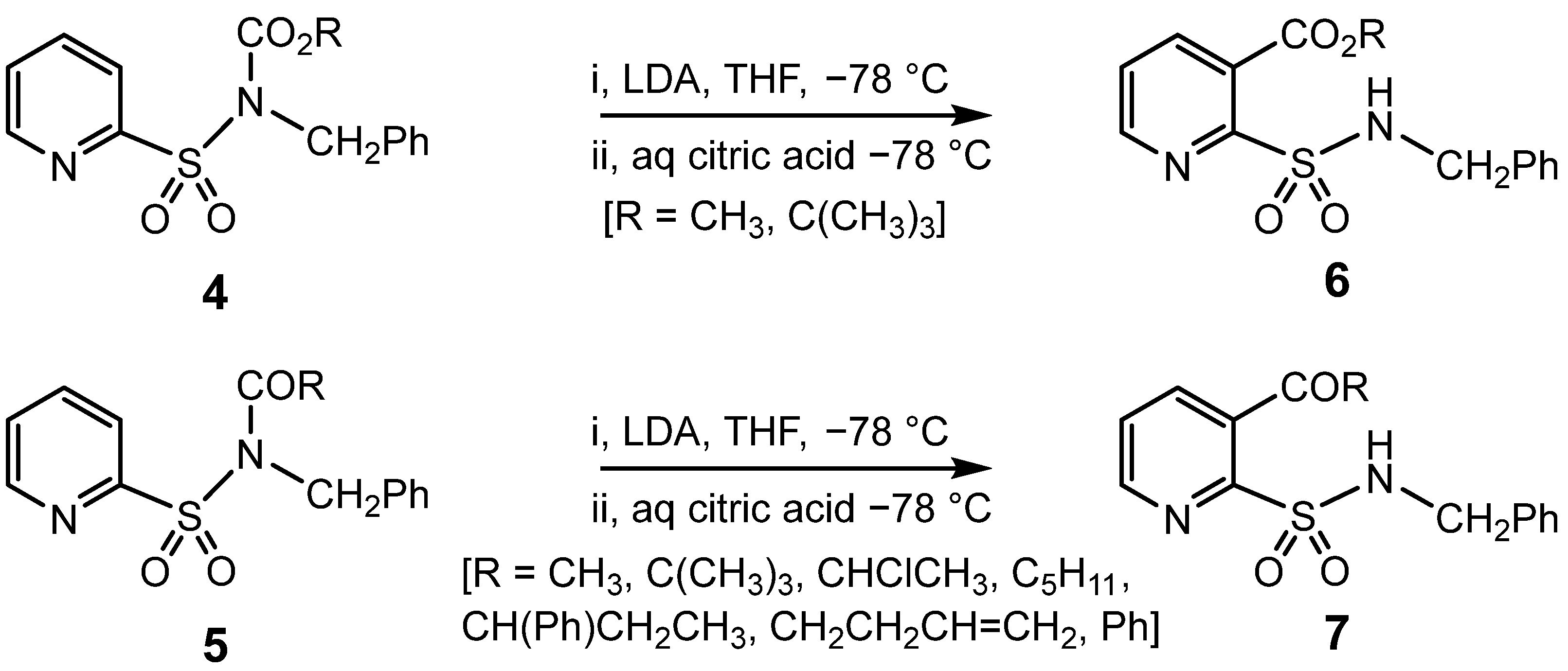

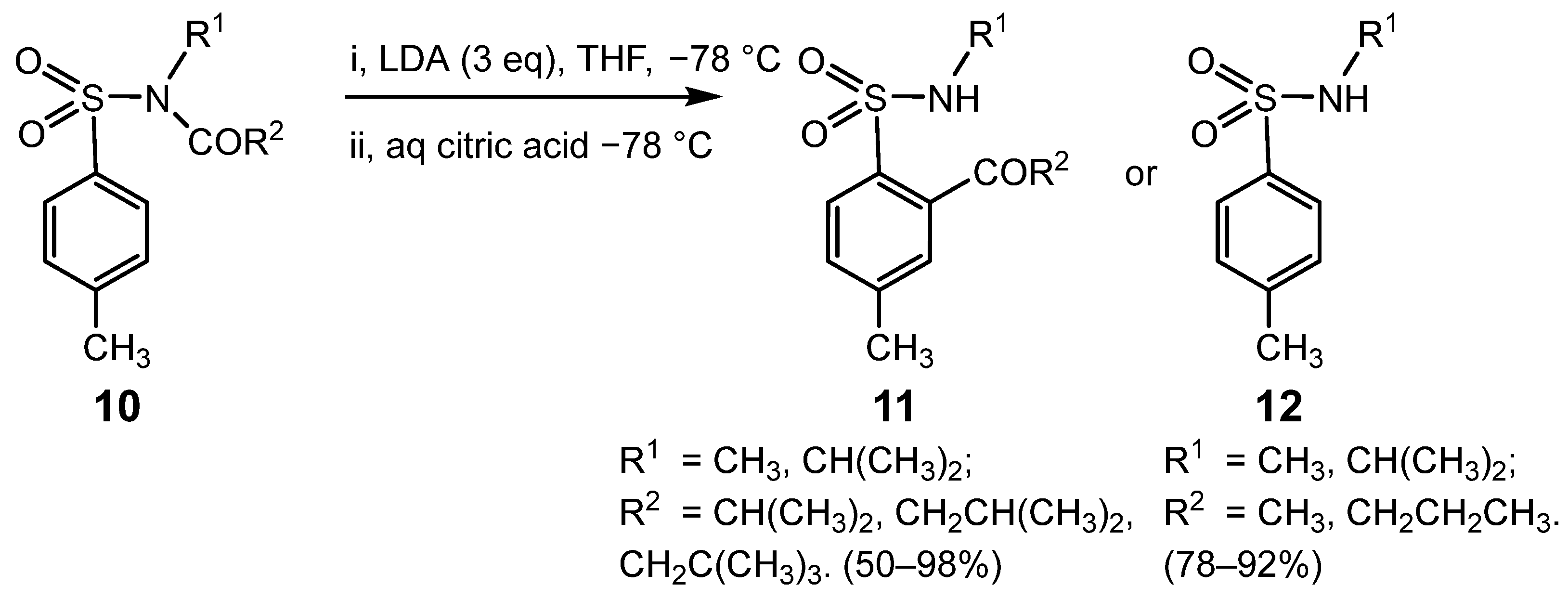
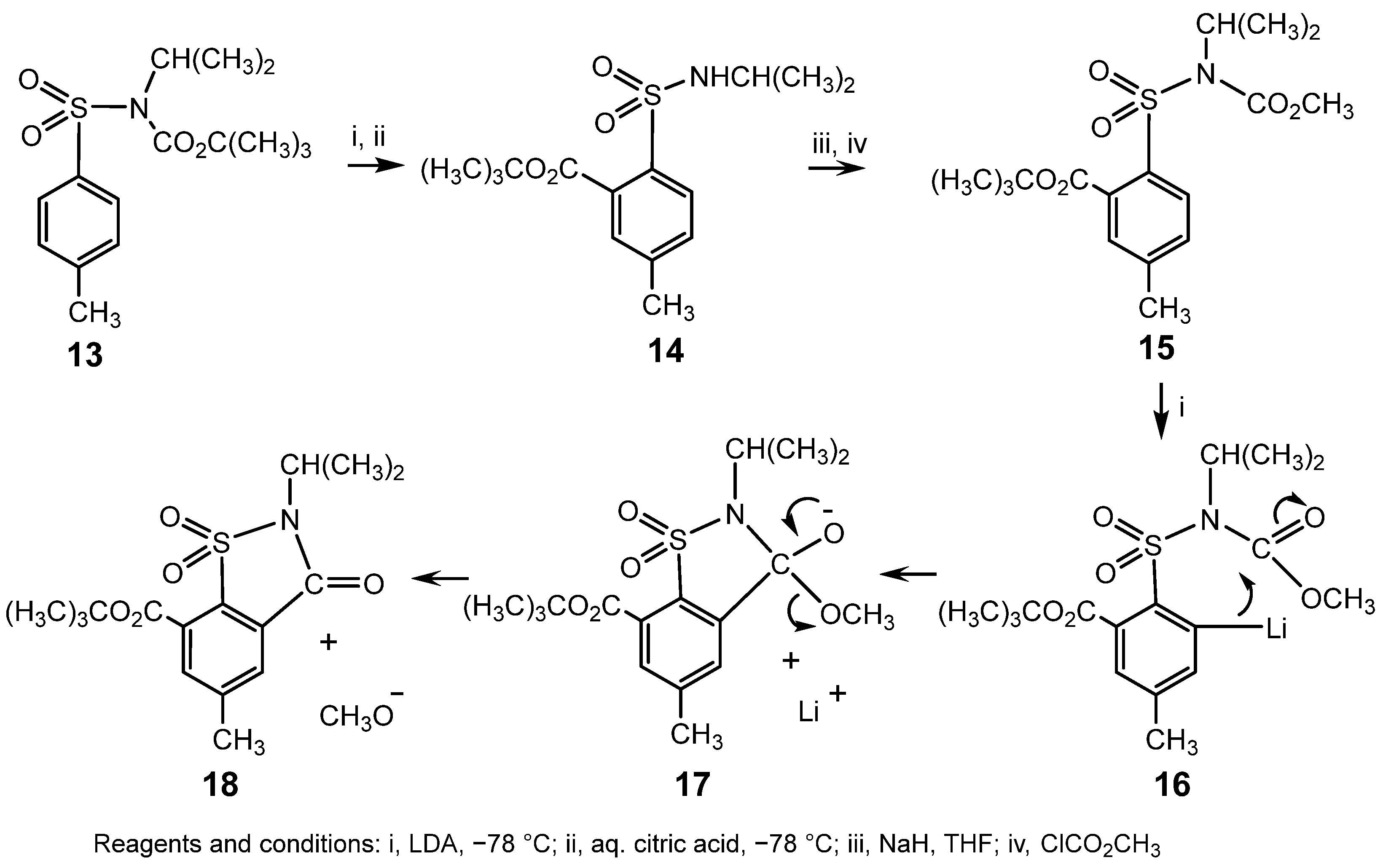
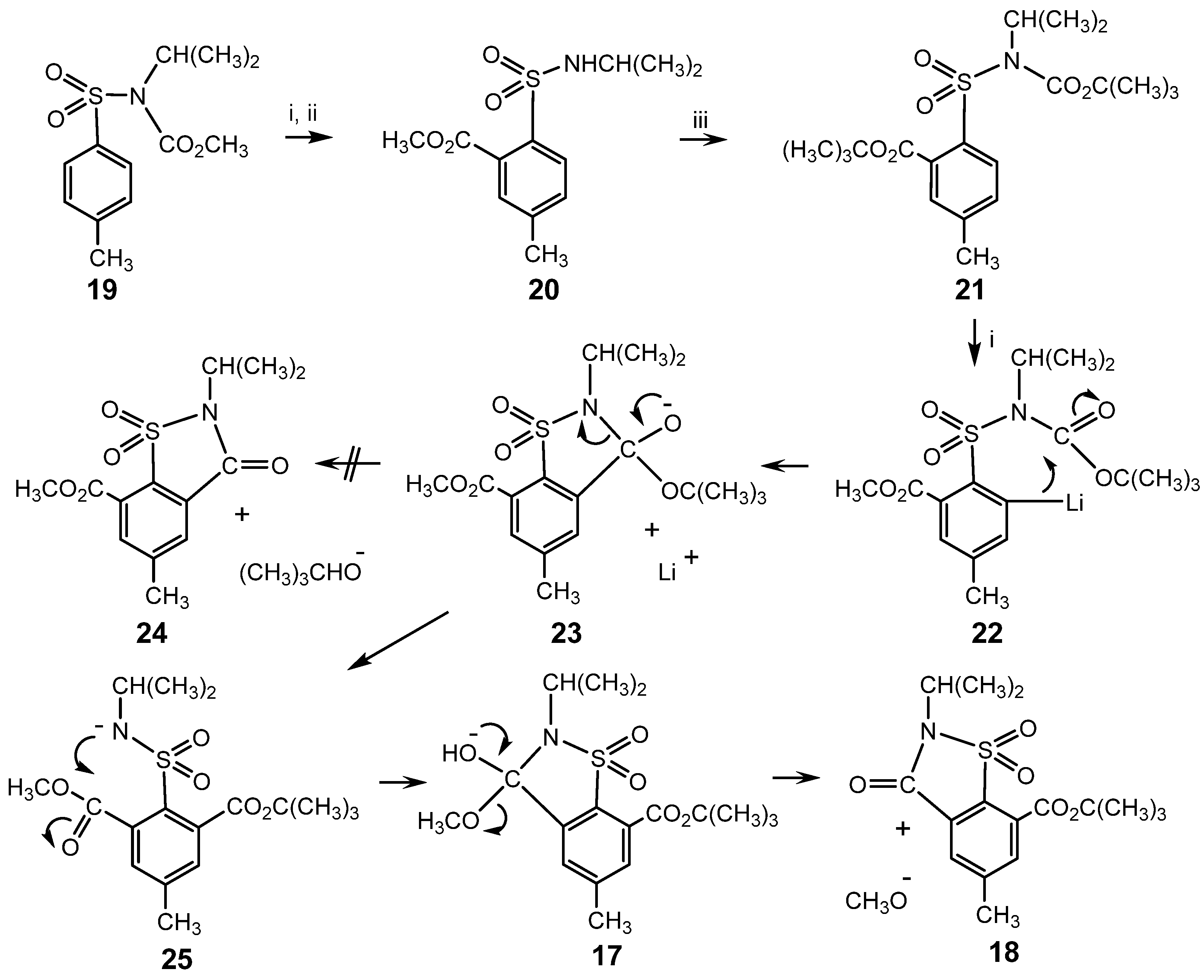

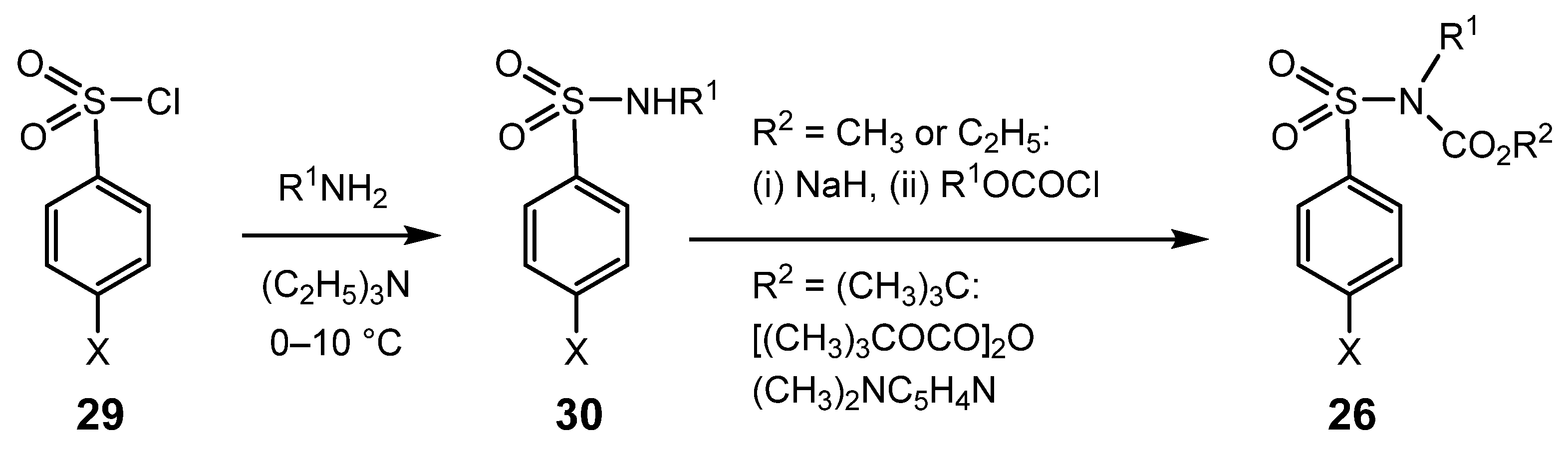
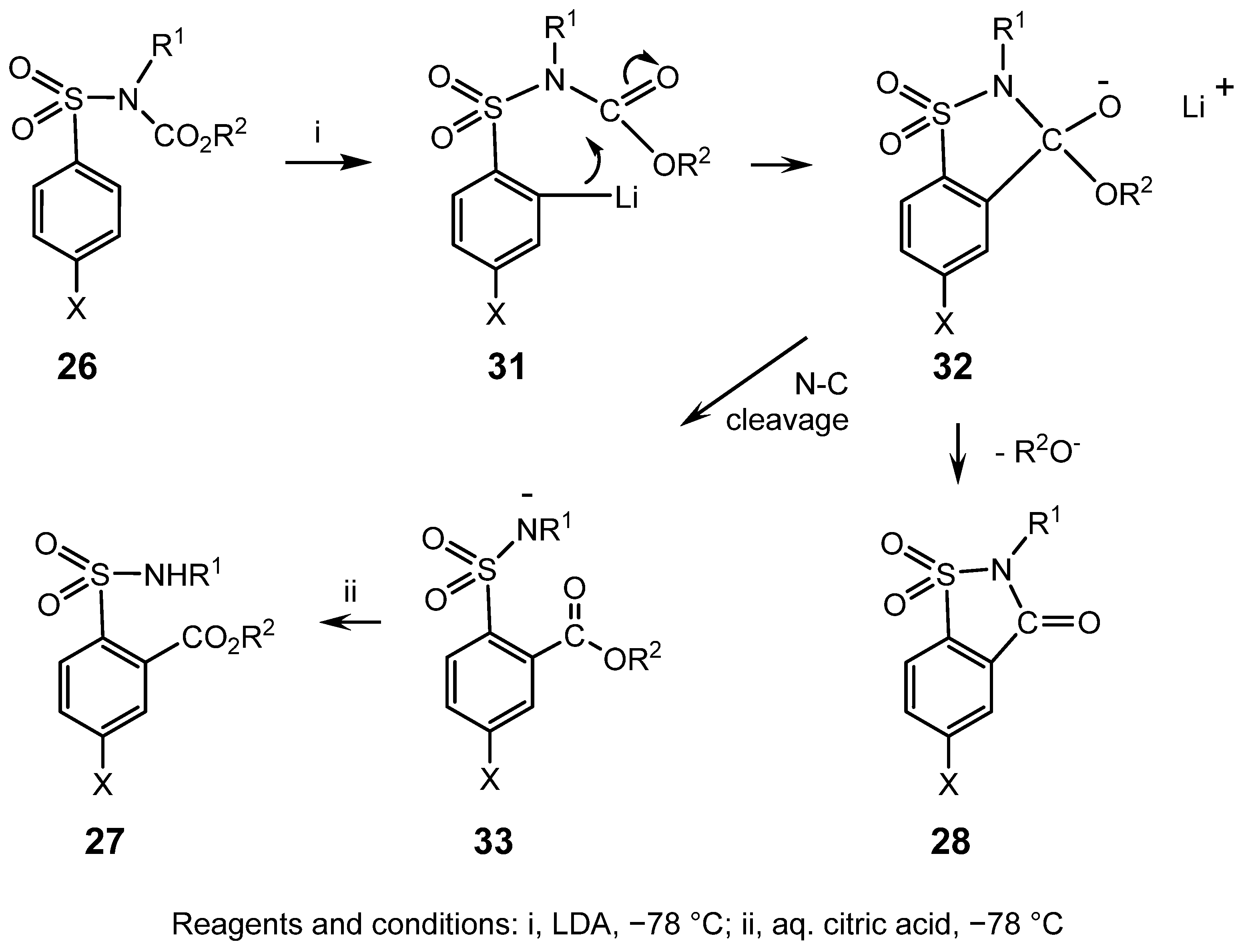

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Products | ||||
|---|---|---|---|---|---|
| Relative Yield (%) a | Ratio a | ||||
| Compound | R1 | R2 | 27 | 28 | 27:28 |
| 26ai | CH3 | CH3 | 48 | 52 | 1:1.1 |
| 26aii | CH3 | C2H5 | 67 | 33 | 2:1 |
| 26aiii | CH3 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26bi | C2H5 | CH3 | 80 | 20 | 4:1 |
| 26bii | C2H5 | C2H5 | 95 | 5 | 19:1 |
| 26biii | C2H5 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26ci | CH2CH2CH3 | CH3 | 95 | ~5 | ~20:1 |
| 26cii | CH2CH2CH3 | C2H5 | 98 | 2 | ~50:1 |
| 26ciii | CH2CH2CH3 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26di | CH(CH3)2 | CH3 | ~98 | ~2 | ~50:1 |
| 26dii | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26diii | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26ei | CH2CH(CH3)2 | CH3 | ≳98 | ≳1 | ~98:1 |
| 26eii | CH2CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26eiii | CH2CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| Substrate | Products | ||||
|---|---|---|---|---|---|
| Relative Yield (%) a | Ratio a | ||||
| Compound | R1 | R2 | 27 | 28 | 27:28 |
| 26fi | CH3 | CH3 | 90 | 10 | 9:1 |
| 26fii | CH3 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26gi | CH(CH3)2 | CH3 | ≳99 b | ≲1 | ≳99:1 |
| 26gii | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26giii | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26hi | CH2CH(CH3)2 | CH3 | ~98 | 2 | ~50:1 |
| 26hii | CH2CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26hiii | CH2CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| Substrate | Products | |||||
|---|---|---|---|---|---|---|
| Relative Yield (%) a | Ratio a | |||||
| Compound | X | R1 | R2 | 27 | 28 | 27:28 |
| 26ji | F | CH3 | CH3 | 95 | 5 | 17:1 |
| 26jii | F | CH3 | C(CH3)3 | 99 | 1 | 99:1 |
| 26jiii | F | CH(CH3)2 | CH3 | ~99 | ~1 | ~95:1 |
| 26jiv | F | CH(CH3)2 | C2H5 | 99 | 1 | 99:1 |
| 26jv | F | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26ki | Cl | CH3 | CH3 | 94 | 6 | 14:1 |
| 26kii | Cl | CH3 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26kiii | Cl | CH(CH3)2 | CH3 | ~99 | ~1 | 98:1 |
| 26kiv | Cl | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26kv | Cl | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26li | Br | CH3 | CH3 | 90 | 10 | 9:1 |
| 26lii | Br | CH3 | C(CH3)3 | ~99 | ~1 | ~99:1 |
| 26liii | Br | CH(CH3)2 | CH3 | ~99 | ~1 | ~99:1 |
| 26liv | Br | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26lv | Br | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26mi | CH3O | CH3 | CH3 | 88 | 12 | 7:1 |
| 26mii | CH3O | CH3 | C(CH3)3 | ~99 | ~1 | ~99:1 |
| 26miii | CH3O | CH(CH3)2 | CH3 | ~99 | ~1 | ~99:1 |
| 26miv | CH3O | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26mv | CH3O | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26ni | CF3 | CH3 | CH3 | 90 | 10 | 9:1 |
| 26nii | CF3 | CH3 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26niii | CF3 | CH(CH3)2 | CH3 | ~99 | ~1 | ~99:1 |
| 26niv | CF3 | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26oi | CN | CH3 | CH3 | 93 | 7 | 13:1 |
| 26oii | CN | CH3 | C(CH3)3 | ~99 | ~1 | 99:1 |
| 26oiii | CN | CH(CH3)2 | CH3 | ≳99 b | ≲1 | ≳99:1 |
| 26oiv | CN | CH(CH3)2 | C2H5 | ≳99 b | ≲1 | ≳99:1 |
| 26ov | CN | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
| 26pi | C(CH3)3 | CH3 | CH3 | 92 | 8 | 11:1 |
| 26pii | C(CH3)3 | CH3 | C(CH3)3 | ~99 | ~1 | ~99:1 |
| 26piii | C(CH3)3 | CH(CH3)2 | CH3 | ≳99 b | ≲1 | ≳99:1 |
| 26piv | C(CH3)3 | CH(CH3)2 | C(CH3)3 | ≳99 b | ≲1 | ≳99:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saidykhan, A.; Ebert, J.; Fenwick, N.W.; Martin, W.H.C.; Bowen, R.D. Steric Effects of N-Alkyl Group on the Base-Induced Nitrogen to Carbon Rearrangement of Orthogonally Protected N-Alkyl Arylsulphonamides. Molecules 2025, 30, 1823. https://doi.org/10.3390/molecules30081823
Saidykhan A, Ebert J, Fenwick NW, Martin WHC, Bowen RD. Steric Effects of N-Alkyl Group on the Base-Induced Nitrogen to Carbon Rearrangement of Orthogonally Protected N-Alkyl Arylsulphonamides. Molecules. 2025; 30(8):1823. https://doi.org/10.3390/molecules30081823
Chicago/Turabian StyleSaidykhan, Amie, Jenessa Ebert, Nathan W. Fenwick, William H. C. Martin, and Richard D. Bowen. 2025. "Steric Effects of N-Alkyl Group on the Base-Induced Nitrogen to Carbon Rearrangement of Orthogonally Protected N-Alkyl Arylsulphonamides" Molecules 30, no. 8: 1823. https://doi.org/10.3390/molecules30081823
APA StyleSaidykhan, A., Ebert, J., Fenwick, N. W., Martin, W. H. C., & Bowen, R. D. (2025). Steric Effects of N-Alkyl Group on the Base-Induced Nitrogen to Carbon Rearrangement of Orthogonally Protected N-Alkyl Arylsulphonamides. Molecules, 30(8), 1823. https://doi.org/10.3390/molecules30081823