
Global Reaction Route Mapping of C3H2O: Isomerization Pathways, Dissociation Channels, and Bimolecular Reaction with a Water Molecule


Abstract

1. Introduction
2. Results and Discussion
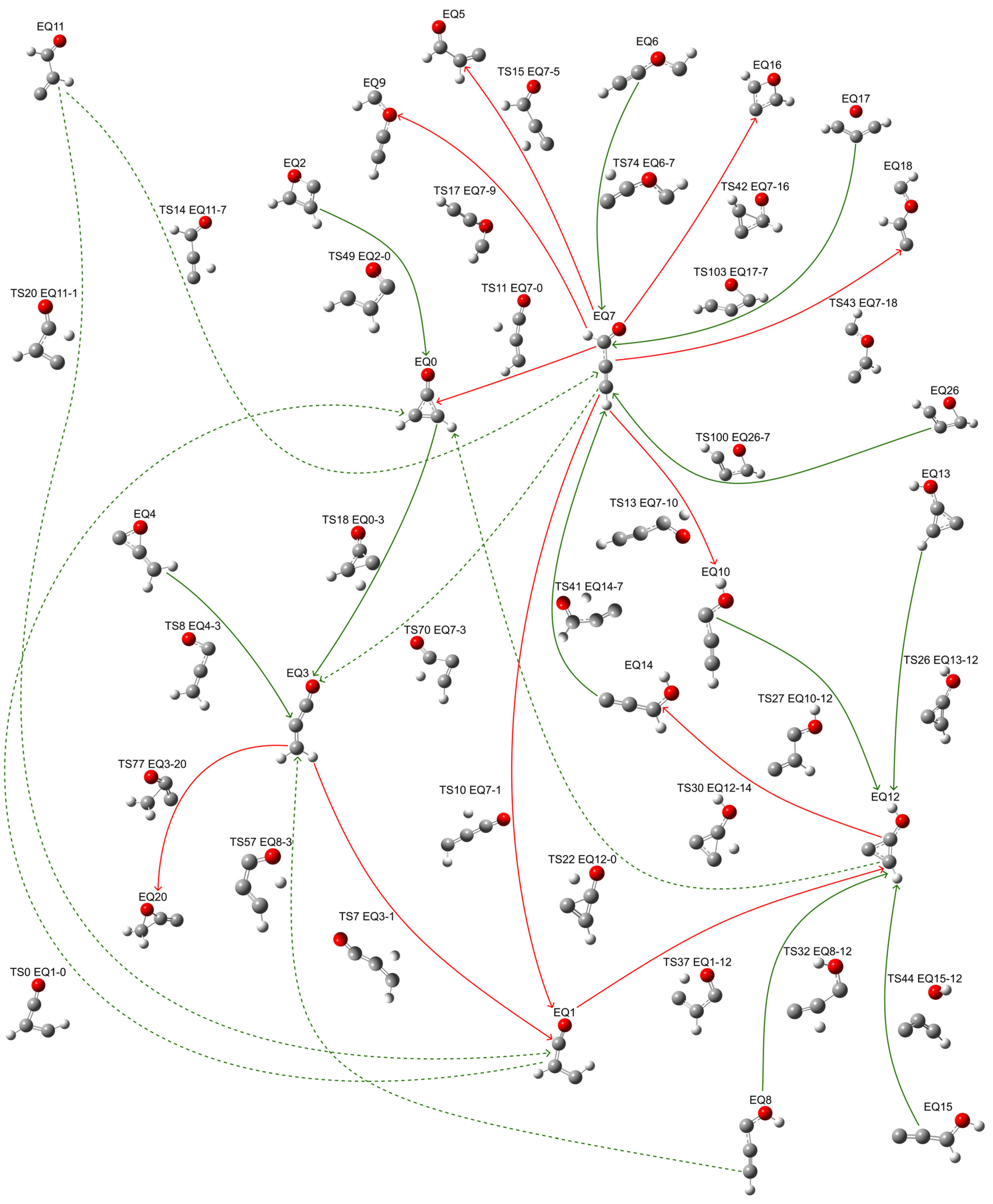
2.1. Isomerization Pathways Without Dissociation (EQx-TSn-EQy)
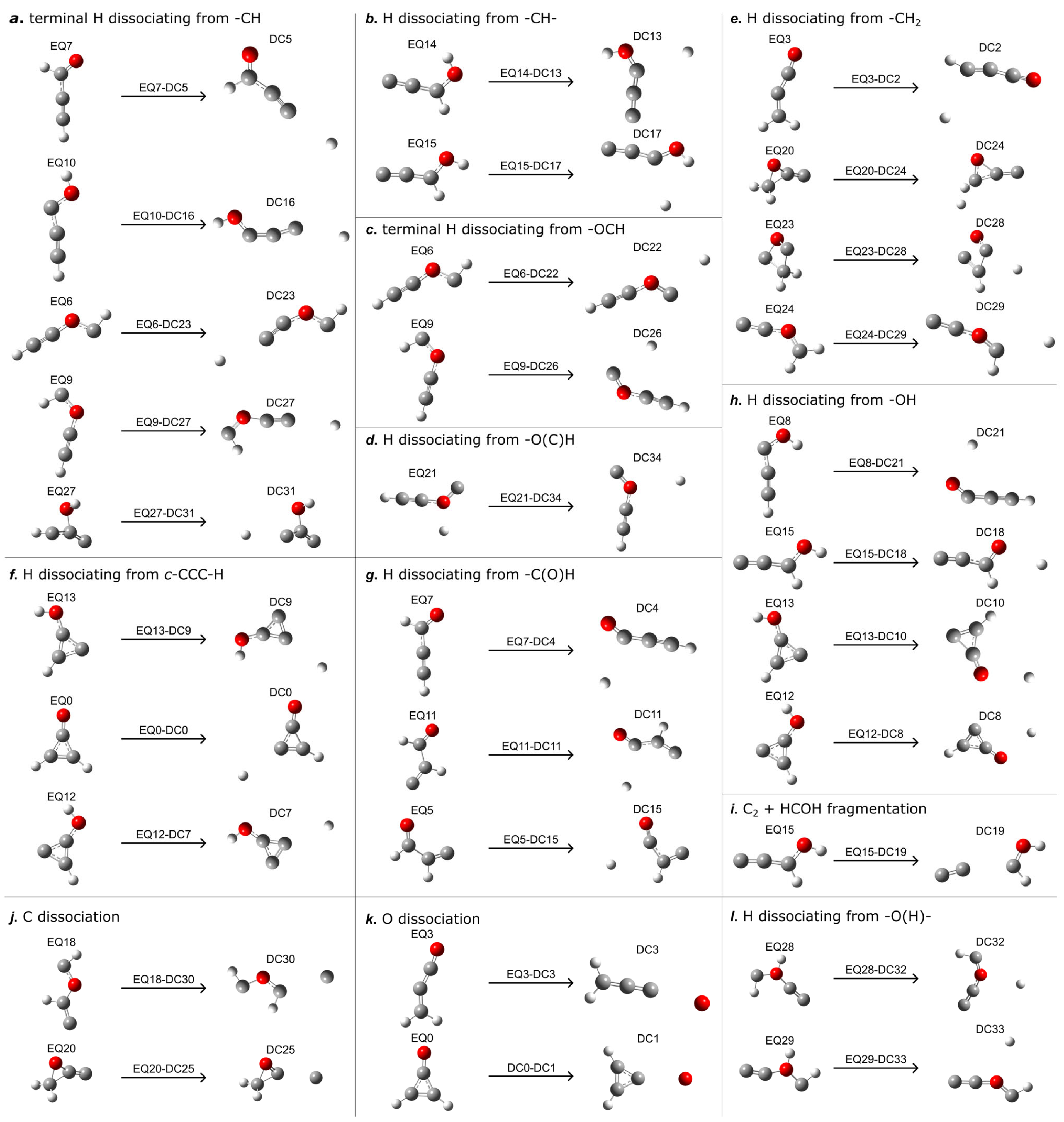
2.2. TS-Mediated Dissociation Pathways (EQa-TSb-DC)
2.3. Direct Dissociation Pathways (EQm-DCn)
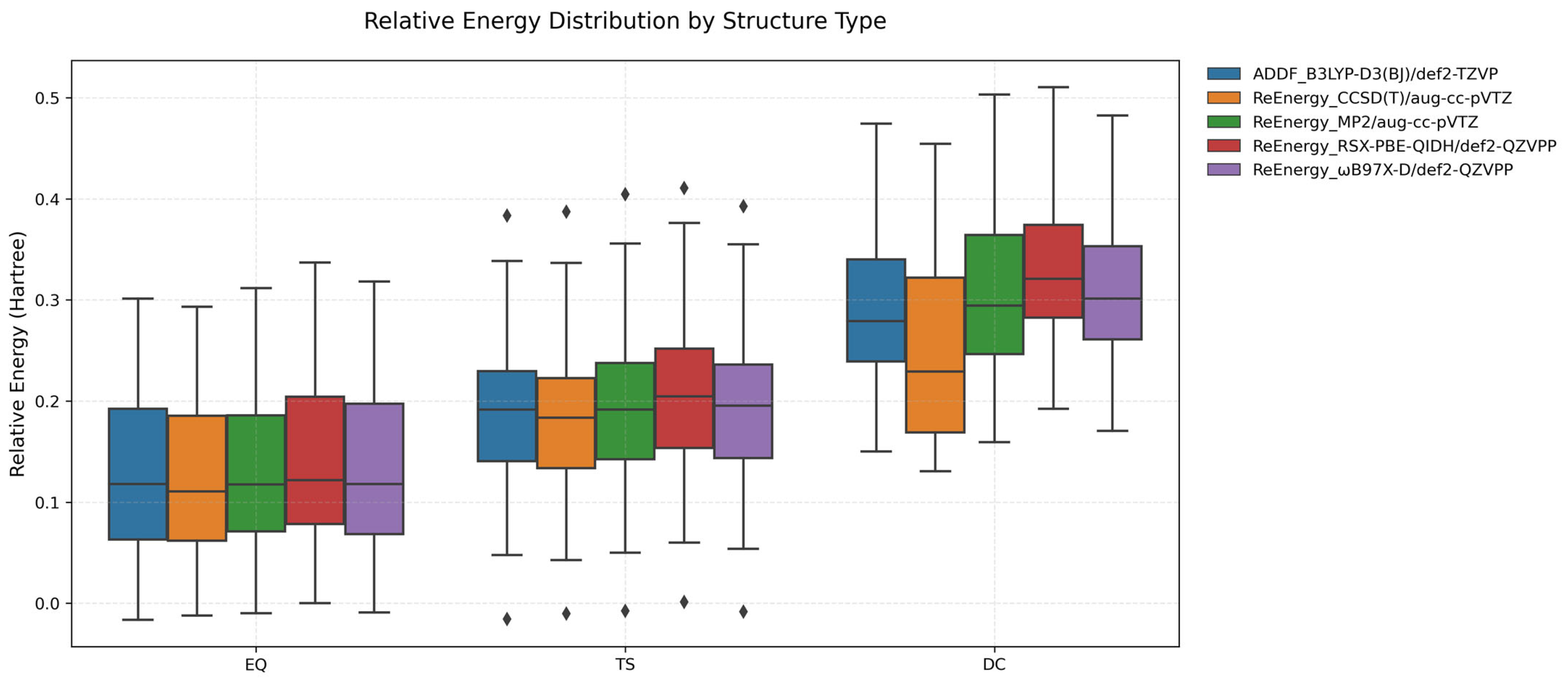
2.4. Benchmarking and Validation of the Exploration Method
2.5. Water-Involved Bimolecular Reactions
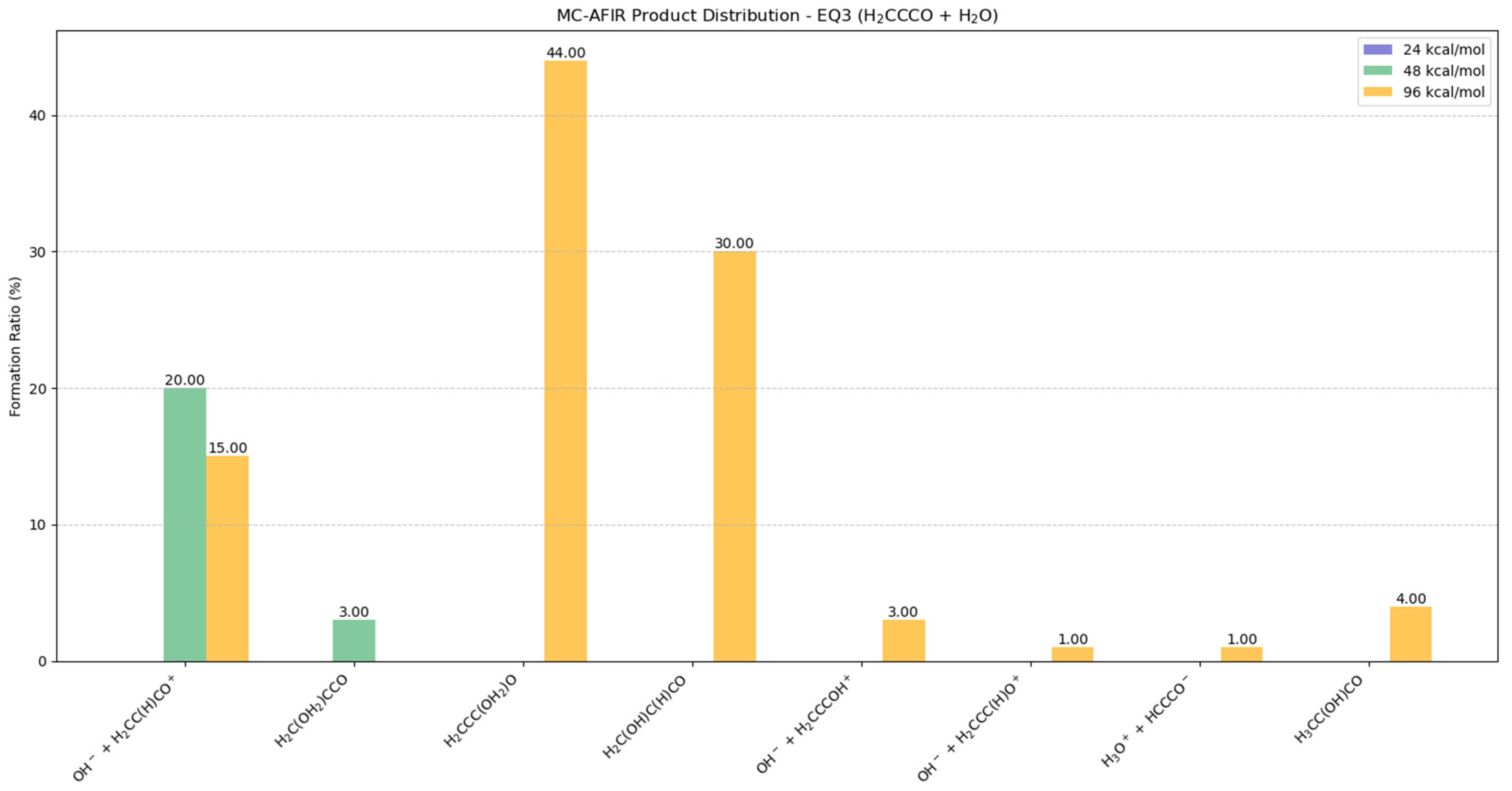
2.5.1. Potential Products from EQ3 (H2CCCO) + H2O
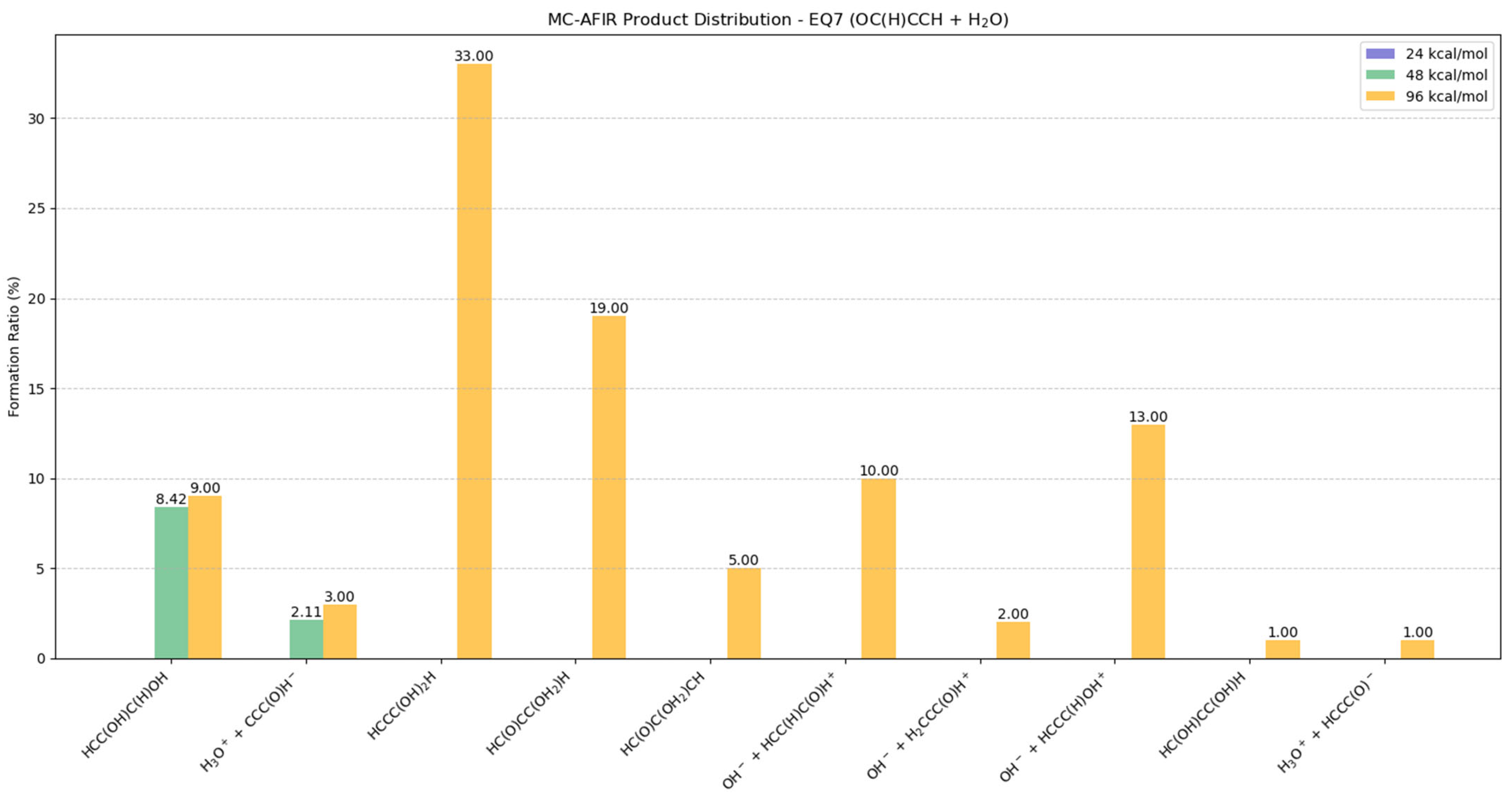
2.5.2. Potential Products from EQ7 (OC(H)CCH) + H2O
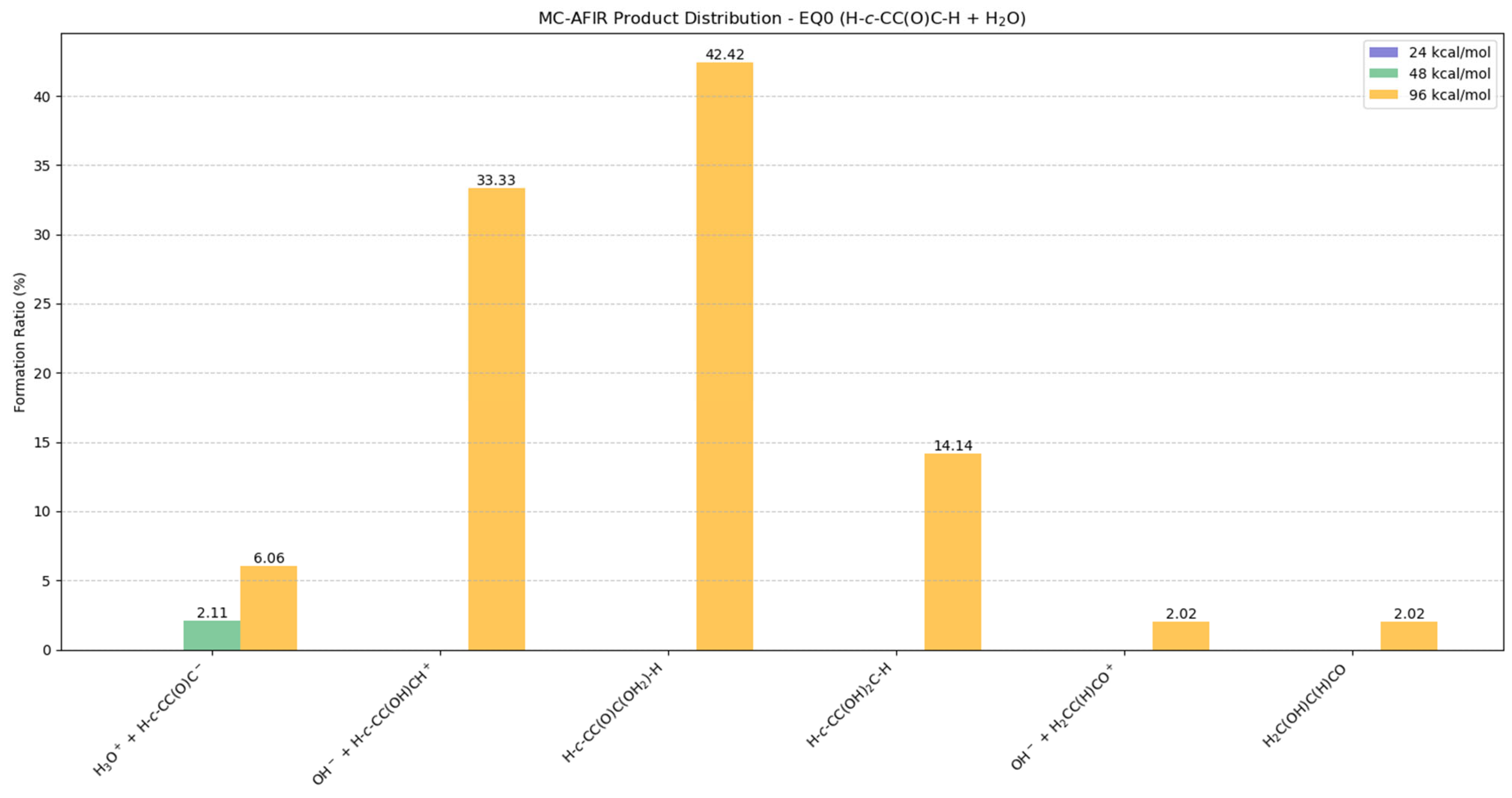
2.5.3. Potential Products from EQ0 (H-c-CC(O)C-H) + H2O
2.5.4. Potential Products from EQ1 (HCC(H)CO) + H2O
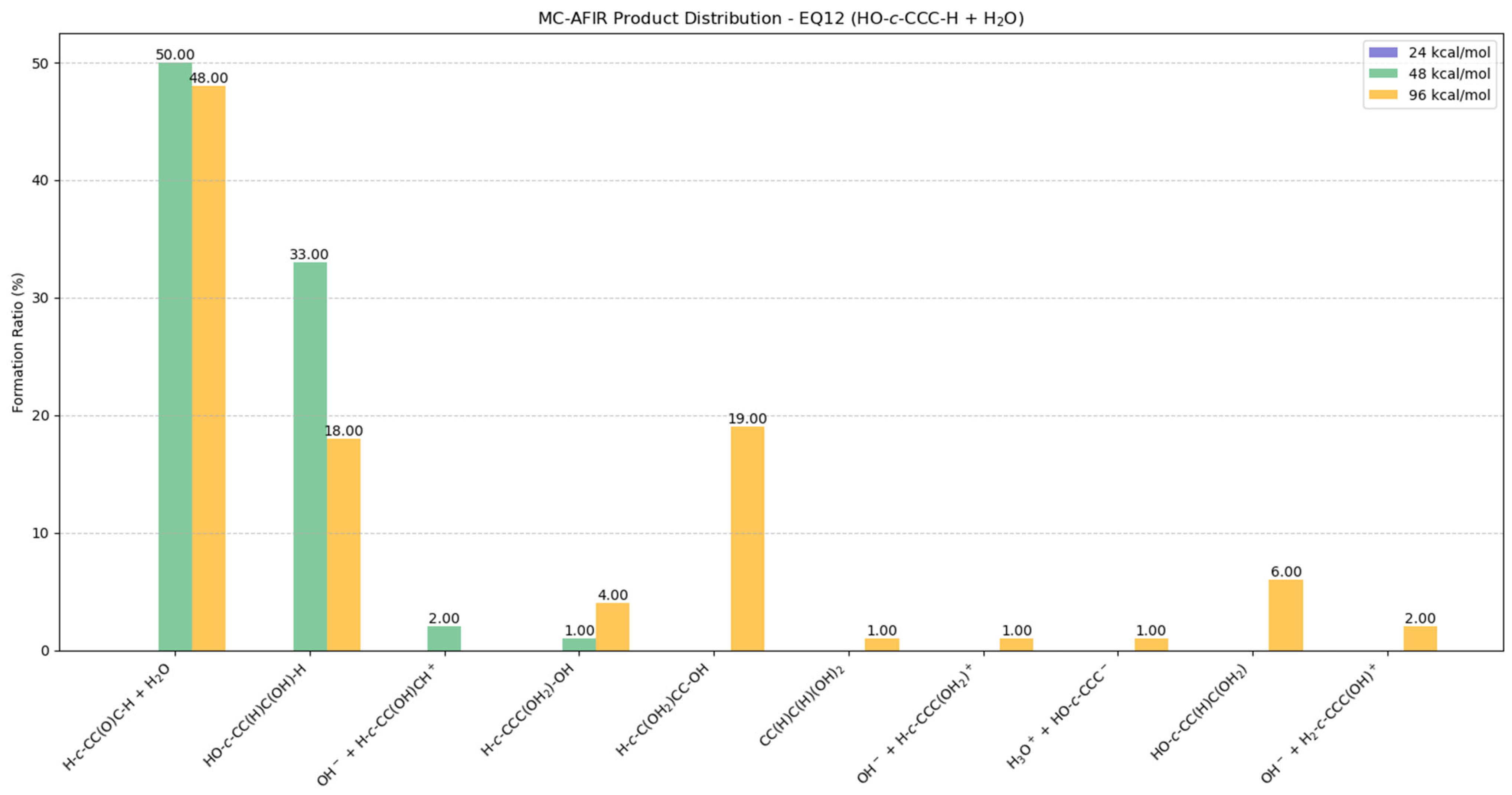
2.5.5. Potential Products from EQ12 (HO-c-CCC-H) + H2O
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zamirri, L.; Ugliengo, P.; Ceccarelli, C.; Rimola, A. Quantum Mechanical Investigations on the Formation of Complex Organic Molecules on Interstellar Ice Mantles: Review and Perspectives. ACS Earth Space Chem. 2019, 3, 1499–1523. [Google Scholar] [CrossRef]
- McGuire, B.A. 2021 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. Suppl. Ser. 2022, 259, 30. [Google Scholar] [CrossRef]
- Guélin, M.; Cernicharo, J. Organic Molecules in Interstellar Space: Latest Advances. Front. Astron. Space Sci. 2022, 9, 787567. [Google Scholar] [CrossRef]
- Puzzarini, C.; Alessandrini, S.; Bizzocchi, L.; Melosso, M.; Rivilla, V.M. From the Laboratory to the Interstellar Medium: A Strategy to Search for Exotic Molecules in Space. Front. Astron. Space Sci. 2023, 10, 1211784. [Google Scholar] [CrossRef]
- Herbst, E. Unusual Chemical Processes in Interstellar Chemistry: Past and Present. Front. Astron. Space Sci. 2021, 8, 776942. [Google Scholar] [CrossRef]
- Sandford, S.A.; Nuevo, M.; Bera, P.P.; Lee, T.J. Prebiotic Astrochemistry and the Formation of Molecules of Astrobiological Interest in Interstellar Clouds and Protostellar Disks. Chem. Rev. 2020, 120, 4616–4659. [Google Scholar] [CrossRef]
- Herbst, E.; Garrod, R.T. Synthetic Approaches to Complex Organic Molecules in the Cold Interstellar Medium. Front. Astron. Space Sci. 2022, 8, 789428. [Google Scholar] [CrossRef]
- Ohno, K.; Satoh, H. Exploration on Quantum Chemical Potential Energy Surfaces: Towards the Discovery of New Chemistry. In Theoretical and Computational Chemistry Series; The Royal Society of Chemistry: London, UK, 2022; Volume 23, Chapter 1; pp. 1–16. [Google Scholar] [CrossRef]
- Hollis, J.M.; Remijan, A.J.; Jewell, P.R.; Lovas, F.J. Cyclopropenone (c-H2C3O): A New Interstellar Ring Molecule. Astrophys. J. 2006, 642, 933–939. [Google Scholar] [CrossRef]
- Loison, J.-C.; Agúndez, M.; Marcelino, N.; Wakelam, V.; Hickson, K.M.; Cernicharo, J.; Gerin, M.; Roueff, E.; Guélin, M. The Interstellar Chemistry of H2C3O Isomers. Mon. Not. R. Astron. Soc. 2016, 456, 4101–4110. [Google Scholar] [CrossRef]
- Irvine, W.M.; Brown, R.D.; Cragg, D.M.; Friberg, P.; Godfrey, P.D.; Kaifu, N.; Matthews, H.E.; Ohishi, M.; Suzuki, H.; Takeo, H. A New Interstellar Polyatomic Molecule: Detection of Propynal in the Cold Cloud TMC-1. Astrophys. J. 1988, 335, L89–L93. [Google Scholar] [CrossRef]
- Hollis, J.M.; Jewell, P.R.; Lovas, F.J.; Remijan, A.; Møllendal, H. Green Bank Telescope Detection of New Interstellar Aldehydes: Propenal and Propanal. Astrophys. J. 2004, 610, L21–L24. [Google Scholar] [CrossRef]
- Turner, B.E. A Molecular Line Survey of Sagittarius B2 and Orion-KL from 70 to 115 GHz. II. Analysis of the Data. Astrophys. J. Suppl. Ser. 1991, 76, 617. [Google Scholar] [CrossRef]
- Ohishi, M.; Kaifu, N. Chemical and Physical Evolution of Dark Clouds: Molecular Spectral Line Survey toward TMC-1. Faraday Discuss. 1998, 109, 205–216. [Google Scholar] [CrossRef]
- Karton, A.; Talbi, D. Pinning the Most Stable HxCyOz Isomers in Space by Means of High-Level Theoretical Procedures. Chem. Phys. 2014, 436, 22–28. [Google Scholar] [CrossRef]
- Loomis, R.A.; McGuire, B.A.; Shingledecker, C.; Johnson, C.H.; Blair, S.; Robertson, A.; Remijan, A.J. Investigating the Minimum Energy Principle in Searches for New Molecular Species—The Case of H2C3O Isomers. Astrophys. J. 2015, 799, 34. [Google Scholar] [CrossRef]
- Song, X.; Wang, M.; Yang, C.; Liu, Y.; Ma, S.; Ma, X.; Pang, W. The Molecular Structure and Spectroscopic Properties of C3H2O and Its Isomers: An Ab Initio Study. Spectrochim. Acta A 2022, 265, 120388. [Google Scholar] [CrossRef]
- Jacobs, C.A.; Brahms, J.C.; Dailey, W.P.; Beran, K.; Harmony, M.D. Synthesis, Microwave Spectrum, and Ab Initio Calculations for Difluorocyclopropenone. J. Am. Chem. Soc. 1992, 114, 115–121. [Google Scholar] [CrossRef]
- Yang, J.; McCann, K.; Laane, J. Vibrational Frequencies and Structure of Cyclopropenone from Ab Initio Calculations. J. Mol. Struct. 2004, 695, 339–343. [Google Scholar] [CrossRef]
- Şahin, S.; Bleda, E.A.; Altun, Z.; Trindle, C. Computational Characterization of Isomeric C4H2O Systems: Thermochemistry, Vibrational Frequencies, and Optical Spectra for Butatrienone, Ethynyl Ketene, Butadiynol, and Triafulvenone. Int. J. Quantum Chem. 2016, 116, 444–451. [Google Scholar] [CrossRef]
- Ahmadvand, S.; Zaari, R.R.; Varganov, S.A. Spin-Forbidden and Spin-Allowed Cyclopropenone (c-H2C3O) Formation in Interstellar Medium. Astrophys. J. 2014, 795, 173. [Google Scholar] [CrossRef]
- Petrie, S. Can Gas-Phase Radical/Neutral Reactions Account for Observed Abundances of Propynal within Dense Interstellar Clouds? Astrophys. J. 1995, 454, L165–L168. [Google Scholar] [CrossRef]
- Nhlabatsi, Z.P.; Bhasi, P.; Sitha, S. Possible Interstellar Formation of Glycine from the Reaction of CH2=NH, CO, and H2O: Catalysis by Extra Water Molecules through the Hydrogen Relay Transport. Phys. Chem. Chem. Phys. 2016, 18, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Rimola, A.; Sodupe, M.; Ugliengo, P. Computational Study of Interstellar Glycine Formation Occurring at Radical Surfaces of Water-Ice Dust Particles. Astrophys. J. 2012, 754, 24. [Google Scholar] [CrossRef]
- Ferrero, S.; Ceccarelli, C.; Ugliengo, P.; Sodupe, M.; Rimola, A. Formation of Interstellar Complex Organic Molecules on Water-Rich Ices Triggered by Atomic Carbon Freezing. Astrophys. J. 2024, 960, 22. [Google Scholar] [CrossRef]
- Ibrahim, M.; Guillemin, J.-C.; Chaquin, P.; Markovits, A.; Krim, L. The Significant Role of Water in Reactions Occurring on the Surface of Interstellar Ice Grains: Hydrogenation of Pure Ketene H2C=C=O Ice versus Hydrogenation of Mixed H2C=C=O/H2O Ice at 10 K. Phys. Chem. Chem. Phys. 2024, 26, 4200–4207. [Google Scholar] [CrossRef]
- Benson, R.C.; Flygare, W.H.; Oda, M.; Breslow, R. Microwave Spectrum, Substitutional Structure, and Stark and Zeeman Effects in Cyclopropenone. J. Am. Chem. Soc. 1973, 95, 2772–2777. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Sumiya, Y.; Takagi, M.; Suzuki, K.; Hatanaka, M.; Osada, Y.; Taketsugu, T.; Morokuma, K.; Ohno, K. GRRM17. Available online: http://iqce.jp/GRRM/index_e.shtml (accessed on 7 January 2025).
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic Exploration of the Mechanism of Chemical Reactions: The Global Reaction Route Mapping (GRRM) Strategy by the ADDF and AFIR Methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ohno, K.; Kishimoto, N.; Iwamoto, T.; Satoh, H. Global Exploration of Isomers and Isomerization Channels on the Quantum Chemical Potential Energy Surface of H3CNO3. J. Comput. Chem. 2017, 38, 669–687. [Google Scholar] [CrossRef]
- Ohno, K.; Maeda, S. A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy Surface. Chem. Phys. Lett. 2004, 384, 277–282. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K. Global Mapping of Equilibrium and Transition Structures on Potential Energy Surfaces by the Scaled Hypersphere Search Method: Applications to ab Initio Surfaces of Formaldehyde and Propyne Molecules. J. Phys. Chem. A 2005, 109, 5742–5753. [Google Scholar] [CrossRef]
- Ohno, K.; Maeda, S. Global Reaction Route Mapping on Potential Energy Surfaces of Formaldehyde, Formic Acid, and Their Metal-Substituted Analogues. J. Phys. Chem. A 2006, 110, 8933–8941. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Taketsugu, T.; Morokuma, K.; Ohno, K. Anharmonic Downward Distortion Following for Automated Exploration of Quantum Chemical Potential Energy Surfaces. Bull. Chem. Soc. Jpn. 2014, 87, 1315–1334. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Brémond, É.; Savarese, M.; Pérez-Jiménez, Á.J.; Sancho-García, J.C.; Adamo, C. Range-Separated Double-Hybrid Functional from Nonempirical Constraints. J. Chem. Theory Comput. 2018, 14, 4052–4062. [Google Scholar] [CrossRef]
- Brémond, É.; Sancho-García, J.C.; Pérez-Jiménez, Á.J.; Adamo, C. Communication: Double-Hybrid Functionals from Adiabatic-Connection: The QIDH Model. J. Chem. Phys. 2014, 141, 031101. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A Fifth-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Eyring, H. The Activated Complex and the Absolute Rate of Chemical Reactions. Chem. Rev. 1935, 17, 65–77. [Google Scholar] [CrossRef]
- Wigner, E. On the Quantum Correction for Thermodynamic Equilibrium. Phys. Rev. 1932, 40, 749–759. [Google Scholar] [CrossRef]
- Eckart, C. The Penetration of a Potential Barrier by Electrons. Phys. Rev. 1930, 35, 1303–1309. [Google Scholar] [CrossRef]
- Bao, J.L.; Truhlar, D.G. Variational Transition State Theory: Theoretical Framework and Recent Developments. Chem. Soc. Rev. 2017, 46, 7548–7596. [Google Scholar] [CrossRef]
- Peters, P.S.; Duflot, D.; Faure, A.; Kahane, C.; Ceccarelli, C.; Wiesenfeld, L.; Toubin, C. Theoretical Investigation of the Isomerization of trans-HCOH to H2CO: An Example of a Water-Catalyzed Reaction. J. Phys. Chem. A 2011, 115, 8983–8989. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Saita, K.; Suzuki, K.; Ichino, T.; Sumiya, Y.; Sugiyama, K.; Ono, Y. Implementation and Performance of the Artificial Force Induced Reaction Method in the GRRM17 Program. J. Comput. Chem. 2018, 39, 233–250. [Google Scholar] [CrossRef]
- Maeda, S.; Morokuma, K. A Systematic Method for Locating Transition Structures of A + B → X Type Reactions. J. Chem. Phys. 2010, 132, 241102. [Google Scholar] [CrossRef]
- Maeda, S.; Morokuma, K. Finding Reaction Pathways of Type A + B → X: Toward Systematic Prediction of Reaction Mechanisms. J. Chem. Theory Comput. 2011, 7, 2335–2345. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | EQx | TSn x-y | Energy Barrier | EQy | Imaginary Frequency (cm−1) | Pathways | EQx | TSn x-y | Energy Barrier | EQy | Imaginary Frequency (cm−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EQ1-TS0-EQ0 | 21.46 | 30.07 | 8.61 | 0 | −815.12 | EQ12-TS30-EQ14 | 33.98 | 95.91 | 61.93 | 39.36 | −958.30 |
| EQ3-TS7-EQ1 | −10.56 | 66.38 | 76.94 | 21.46 | −1678.08 | EQ8-TS32-EQ12 | 43.38 | 90.90 | 47.52 | 33.98 | −387.95 |
| EQ4-TS8-EQ3 | 58.21 | 73.90 | 15.69 | −10.56 | −1001.41 | EQ1-TS37-EQ12 | 21.46 | 119.47 | 98.01 | 33.98 | −1710.32 |
| EQ7-TS10-EQ1 | −4.91 | 55.65 | 60.56 | 21.46 | −1102.08 | EQ14-TS41-EQ7 | 39.36 | 73.11 | 33.75 | −4.91 | −1738.97 |
| EQ7-TS11-EQ0 | −4.91 | 52.42 | 57.33 | 0 | −1239.08 | EQ7-TS42-EQ16 | −4.91 | 71.71 | 76.62 | 70.75 | −467.34 |
| EQ7-TS13-EQ10 | −4.91 | 72.76 | 77.67 | 40.96 | −2092.46 | EQ7-TS43-EQ18 | −4.91 | 120.95 | 125.86 | 117.86 | −91.47 |
| EQ11-TS14-EQ7 | 36.44 | 37.85 | 1.41 | −4.91 | −615.25 | EQ15-TS44-EQ12 | 55.21 | 152.72 | 97.51 | 33.98 | −758.24 |
| EQ7-TS15-EQ5 | −4.91 | 39.25 | 44.16 | 38.93 | −496.21 | EQ2-TS49-EQ0 | 57.93 | 87.45 | 29.52 | 0 | −506.28 |
| EQ7-TS17-EQ9 | −4.91 | 99.23 | 104.14 | 77.33 | −404.69 | EQ8-TS57-EQ3 | 43.38 | 87.86 | 44.48 | −10.56 | −1668.23 |
| EQ0-TS18-EQ3 | 0 | 75.58 | 75.58 | −10.56 | −420.51 | EQ7-TS70-EQ3 | −4.91 | 74.30 | 79.21 | −10.56 | −989.34 |
| EQ11-TS20-EQ1 | 36.44 | 52.19 | 15.75 | 21.46 | −659.98 | EQ6-TS74-EQ7 | 75.25 | 120.98 | 45.73 | −4.91 | −311.80 |
| EQ12-TS22-EQ0 | 33.98 | 84.14 | 50.16 | 0 | −2047.35 | EQ3-TS77-EQ20 | −10.56 | 86.05 | 96.61 | 77.45 | −504.66 |
| EQ13-TS26-EQ12 | 36.34 | 44.67 | 8.33 | 33.98 | −605.25 | EQ26-TS100-EQ7 | 118.01 | 119.07 | 1.06 | −4.91 | −783.66 |
| EQ10-TS27-EQ12 | 40.96 | 85.99 | 45.03 | 33.98 | −305.30 | EQ17-TS103-EQ7 | 118.16 | 119.73 | 1.57 | −4.91 | −383.34 |
| Pathways | EQa | TSb a-DC | Energy Barrier | Imaginary Frequency (cm−1) | Dissociation Process |
|---|---|---|---|---|---|
| EQ1-TS2-DC | 21.46 | 27.77 | 6.31 | −565.83 | HCC(H)CO → CO + HCCH |
| EQ3-TS9-DC | −10.56 | 70.21 | 80.77 | −1320.69 | H2CCCO → H+ + HCCCO− |
| EQ0-TS71-DC | 0 | 36.09 | 36.09 | −673.89 | H-c-CC(O)C-H → CO + HCCH |
| EQ7-TS82-DC | −4.91 | 104.78 | 109.69 | −2028.17 | OC(H)CCH → H2·OCCC |
| EQ2-TS1-DC | 57.93 | 83.64 | 25.71 | −865.52 | H-c-CCOC-H → HCC(H)CO |
| EQ4-TS4-DC | 58.21 | 95.03 | 36.82 | −770.26 | H2C-c-CCO → H2CCOC |
| EQ4-TS63-DC | 58.21 | 139.28 | 81.07 | −649.19 | H2C-c-CCO → C-c-CCO + H2 |
| EQ14-TS47-DC | 39.36 | 123.04 | 83.68 | −1446.83 | HOC(H)CC → OH− + CCCH+ |
| EQ15-TS56-DC | 55.21 | 114.48 | 59.27 | −2265.85 | HOC(H)CC → H2·OCCC |
| EQ8-TS59-DC | 43.38 | 91.10 | 47.72 | −2345.41 | HOCCCH → COH− + HCC+ |
| EQ20-TS64-DC | 77.45 | 113.69 | 36.24 | −661.85 | H2-c-COC-C → OCCCH2 |
| EQ23-TS65-DC | 93.09 | 94.85 | 1.76 | −404.27 | H2-c-CCOC → H2CCOC |
| EQ23-TS90-DC | 93.09 | 166.52 | 73.43 | −604.55 | H2-c-CCOC → CCCO + H2 |
| EQ9-TS84-DC | 77.33 | 127.66 | 50.33 | −1475.36 | HCOCCH → H+ + COCCH− |
| EQ9-TS85-DC | 77.33 | 128.06 | 50.73 | −1053.52 | HCOCCH → H+ + COCCH− |
| EQ9-TS104-DC | 77.33 | 137.06 | 59.73 | −1205.31 | HCOCCH → H+ + COCCH− |
| EQ9-TS87-DC | 77.33 | 123.11 | 45.78 | −172.39 | HCOCCH → H+ + CCOCH− |
| EQ18-TS96-DC | 117.86 | 141.88 | 24.02 | −452.57 | HCOC(H)C → CC(H)OCH |
| EQ19-TS114-DC | 144.21 | 184.23 | 40.02 | −1220.97 | HOC(C)CH → H2·OC(C)C |
| EQ21-TS123-DC | 180.83 | 181.72 | 0.89 | −592.26 | COCCH− + H+ → COCCH− + H+ |
| Pathways | EQm | DCn | Energy Barrier |
|---|---|---|---|
| EQ0-DC0 | 0 | 110.63 | 110.63 |
| EQ0-DC1 | 0 | 180.71 | 180.71 |
| EQ3-DC2 | −10.56 | 93.36 | 103.92 |
| EQ3-DC3 | −10.56 | 199.99 | 210.55 |
| EQ7-DC4 | −4.91 | 88.10 | 93.01 |
| EQ7-DC5 | −4.91 | 145.64 | 150.55 |
| EQ12-DC7 | 33.98 | 155.25 | 121.27 |
| EQ12-DC8 | 33.98 | 123.85 | 89.87 |
| EQ13-DC9 | 36.34 | 153.12 | 116.78 |
| EQ13-DC10 | 36.34 | 128.06 | 91.72 |
| EQ11-DC11 | 36.44 | 140.95 | 104.51 |
| EQ14-DC13 | 39.36 | 157.52 | 118.16 |
| EQ5-DC15 | 38.93 | 133.75 | 94.82 |
| EQ10-DC16 | 40.96 | 169.29 | 128.33 |
| EQ15-DC17 | 55.21 | 147.34 | 92.13 |
| EQ15-DC18 | 55.21 | 159.54 | 104.33 |
| EQ15-DC19 | 55.21 | 197.12 | 141.91 |
| EQ8-DC21 | 43.38 | 104.85 | 61.47 |
| EQ6-DC22 | 75.25 | 167.64 | 92.39 |
| EQ6-DC23 | 75.25 | 224.37 | 149.12 |
| EQ20-DC24 | 77.45 | 196.67 | 119.22 |
| EQ20-DC25 | 77.45 | 218.81 | 141.36 |
| EQ9-DC26 | 77.33 | 166.05 | 88.72 |
| EQ9-DC27 | 77.33 | 224.29 | 146.96 |
| EQ23-DC28 | 93.09 | 180.46 | 87.37 |
| EQ24-DC29 | 95.42 | 224.61 | 129.19 |
| EQ18-DC30 | 117.86 | 291.26 | 173.40 |
| EQ27-DC31 | 153.10 | 237.04 | 83.94 |
| EQ28-DC32 | 178.18 | 242.97 | 64.79 |
| EQ29-DC33 | 200.29 | 235.39 | 35.10 |
| EQ21-DC34 | 180.83 | 194.06 | 13.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Kishimoto, N. Global Reaction Route Mapping of C3H2O: Isomerization Pathways, Dissociation Channels, and Bimolecular Reaction with a Water Molecule. Molecules 2025, 30, 1829. https://doi.org/10.3390/molecules30081829
Zhang D, Kishimoto N. Global Reaction Route Mapping of C3H2O: Isomerization Pathways, Dissociation Channels, and Bimolecular Reaction with a Water Molecule. Molecules. 2025; 30(8):1829. https://doi.org/10.3390/molecules30081829
Chicago/Turabian StyleZhang, Dapeng, and Naoki Kishimoto. 2025. "Global Reaction Route Mapping of C3H2O: Isomerization Pathways, Dissociation Channels, and Bimolecular Reaction with a Water Molecule" Molecules 30, no. 8: 1829. https://doi.org/10.3390/molecules30081829
APA StyleZhang, D., & Kishimoto, N. (2025). Global Reaction Route Mapping of C3H2O: Isomerization Pathways, Dissociation Channels, and Bimolecular Reaction with a Water Molecule. Molecules, 30(8), 1829. https://doi.org/10.3390/molecules30081829