
Quantitative Nuclear Magnetic Resonance for Small Biological Molecules in Complex Mixtures: Practical Guidelines and Key Considerations for Non-Specialists


Abstract
:1. Introduction
2. Quantification Using NMR, Theoretical Aspects
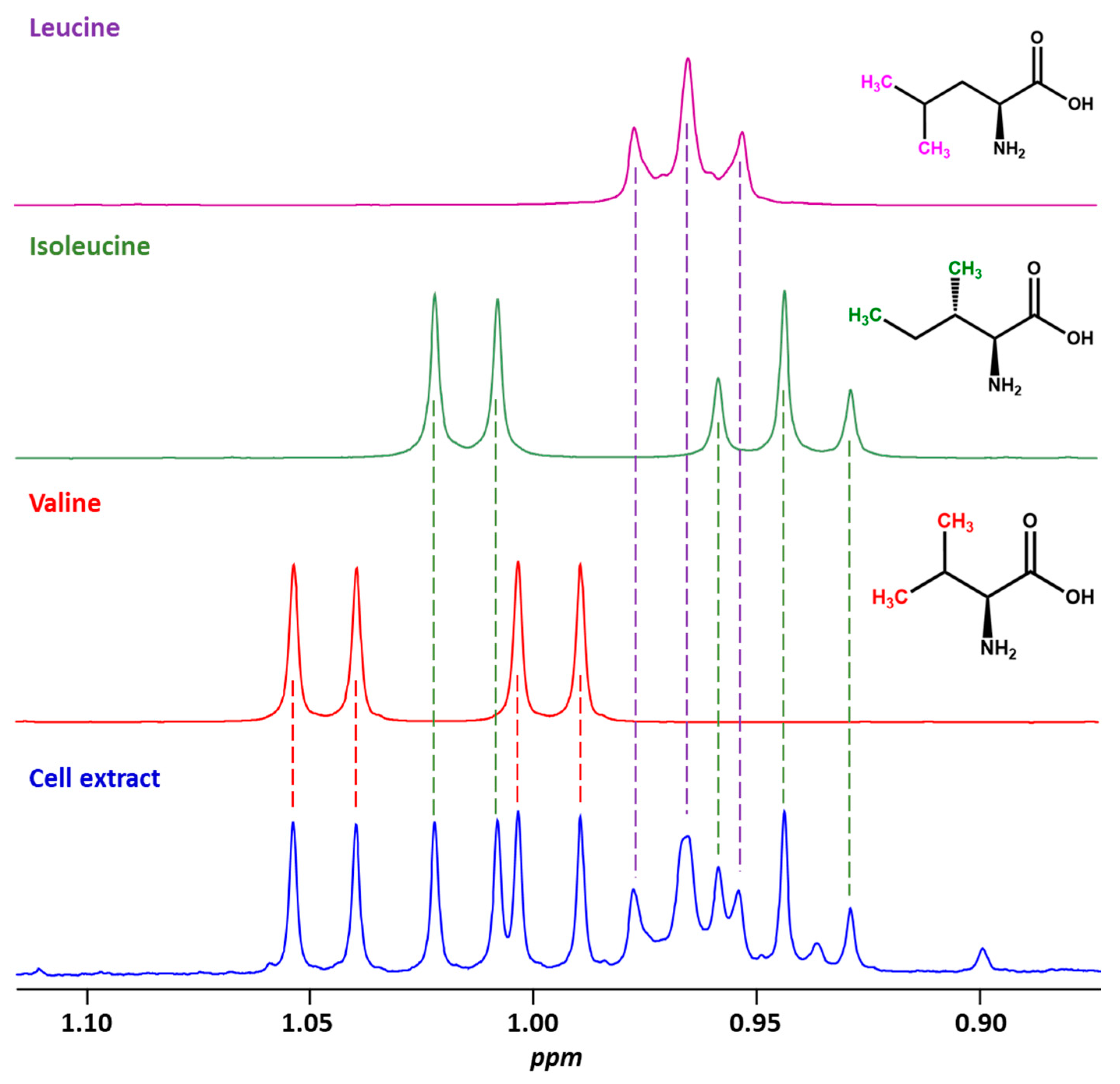
2.1. The Additivity Principle and the Possibility of Studying Mixtures of Small Molecules
2.2. NMR as a Primary Ratio Quantification Method
2.3. NMR Signal Relaxation and Absolute vs. Relative Quantification
3. Applications of Quantitative NMR in Biological and Biomedical Research
4. Practical Aspects
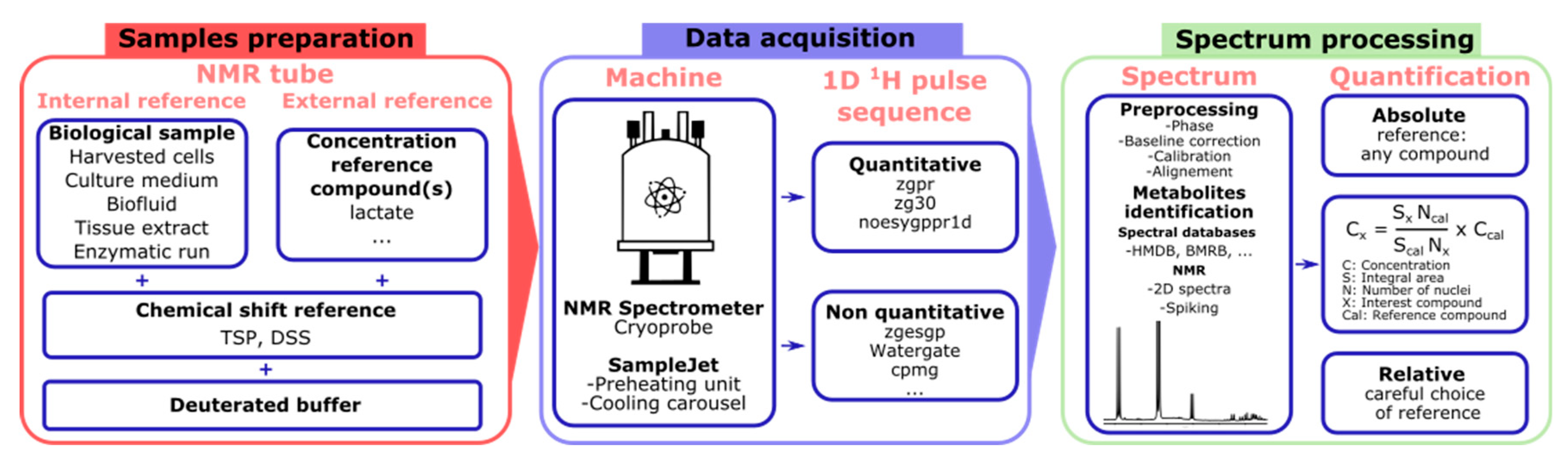
4.1. Sample Preparation
4.2. NMR Data Acquisition
4.2.1. Acquisition Temperature
4.2.2. Which Pulse Sequence?
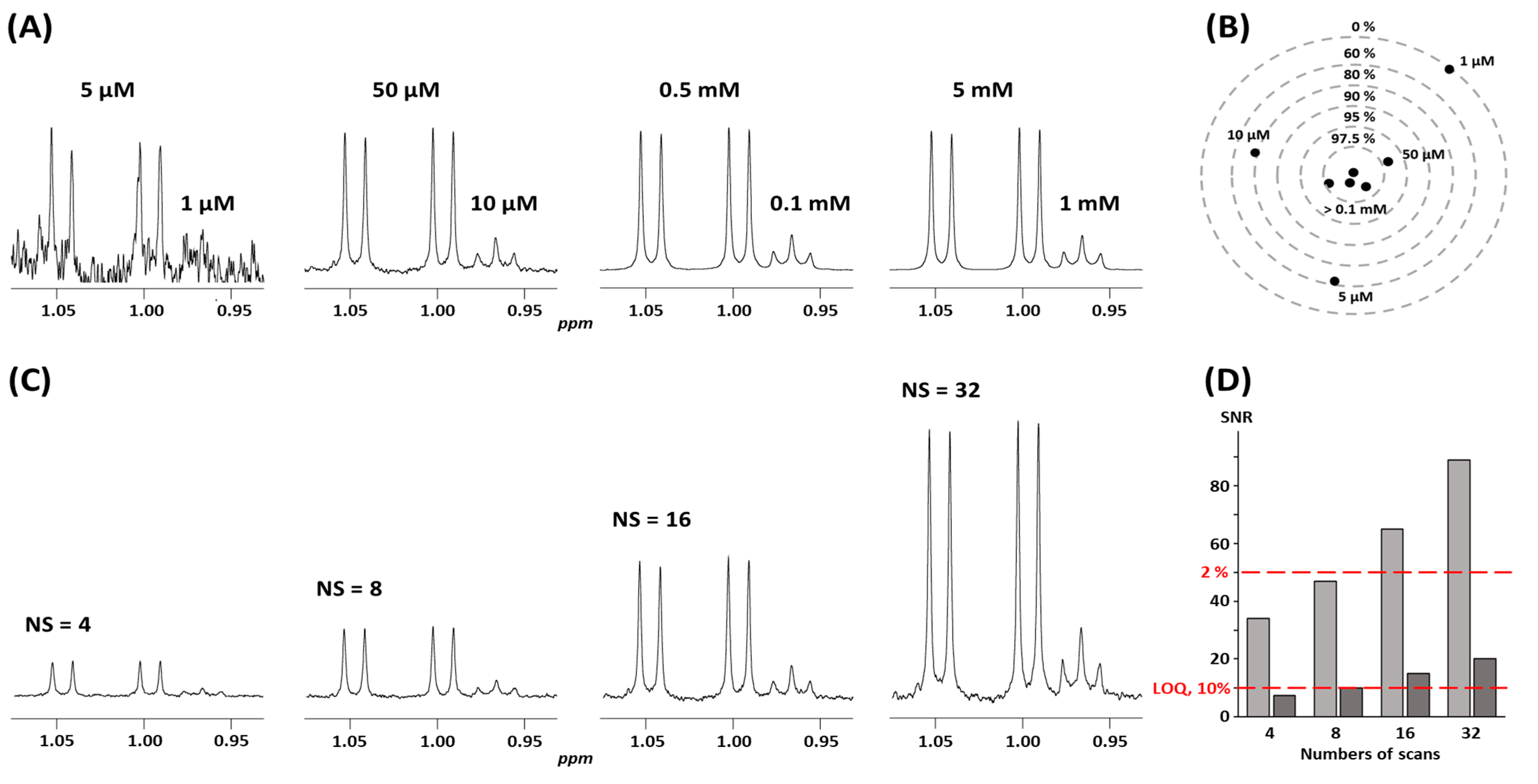
4.2.3. Signal-to-Noise Ratio, Detection Limits, and Accuracy
4.2.4. Experiment Parameters
4.3. Data Exploitation
5. Challenges and Limitations of Quantitative NMR
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vang, J.Y.; Breceda, C.; Her, C.; Krishnan, V.V. Enzyme Kinetics by Real-Time Quantitative NMR (QNMR) Spectroscopy with Progress Curve Analysis. Anal. Biochem. 2022, 658, 114919. [Google Scholar] [CrossRef] [PubMed]
- Løhre, C.; Underhaug, J.; Brusletto, R.; Barth, T. A Workup Protocol Combined with Direct Application of Quantitative Nuclear Magnetic Resonance Spectroscopy of Aqueous Samples from Large-Scale Steam Explosion of Biomass. ACS Omega 2021, 6, 6714–6721. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.-H.; Szczepski, K.; Poulson, B.G.; Chandra, K.; McKay, R.T.; Dhahri, M.; Alahmari, F.; Jaremko, L.; Lachowicz, J.I.; Jaremko, M. NMR as a “Gold Standard” Method in Drug Design and Discovery. Molecules 2020, 25, 4597. [Google Scholar] [CrossRef] [PubMed]
- Crook, A.A.; Powers, R. Quantitative NMR-Based Biomedical Metabolomics: Current Status and Applications. Molecules 2020, 25, 5128. [Google Scholar] [CrossRef]
- Letertre, M.P.M.; Giraudeau, P.; de Tullio, P. Nuclear Magnetic Resonance Spectroscopy in Clinical Metabolomics and Personalized Medicine: Current Challenges and Perspectives. Front. Mol. Biosci. 2021, 8, 698337. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Ingallina, C.; Spano, M.; Di Matteo, G.; Mannina, L. NMR-Based Approaches in the Study of Foods. Molecules 2022, 27, 7906. [Google Scholar] [CrossRef]
- dos Santos Ribeiro, H.S.; Dagnino, D.; Schripsema, J. Rapid and Accurate Verification of Drug Identity, Purity and Quality by 1H-NMR Using Similarity Calculations and Differential NMR. J. Pharm. Biomed. Anal. 2021, 199, 114040. [Google Scholar] [CrossRef]
- Simpson, A.J.; Simpson, M.J.; Soong, R. Environmental Nuclear Magnetic Resonance Spectroscopy: An Overview and a Primer. Anal. Chem. 2018, 90, 628–639. [Google Scholar] [CrossRef]
- Giraudeau, P. Challenges and Perspectives in Quantitative NMR. Magn. Reson. Chem. 2017, 55, 61–69. [Google Scholar] [CrossRef]
- Wishart, D.S.; Cheng, L.L.; Copié, V.; Edison, A.S.; Eghbalnia, H.R.; Hoch, J.C.; Gouveia, G.J.; Pathmasiri, W.; Powers, R.; Schock, T.B.; et al. NMR and Metabolomics—A Roadmap for the Future. Metabolites 2022, 12, 678. [Google Scholar] [CrossRef]
- Alonso-Moreno, P.; Rodriguez, I.; Izquierdo-Garcia, J.L. Benchtop NMR-Based Metabolomics: First Steps for Biomedical Application. Metabolites 2023, 13, 614. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjordahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, Fully-Automated NMR Spectral Profiling for Metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef]
- Hao, J.; Astle, W.; De Iorio, M.; Ebbels, T.M.D. BATMAN—An R Package for the Automated Quantification of Metabolites from Nuclear Magnetic Resonance Spectra Using a Bayesian Model. Bioinformatics 2012, 28, 2088–2090. [Google Scholar] [CrossRef]
- Stavarache, C.; Nicolescu, A.; Duduianu, C.; Ailiesei, G.L.; Balan-Porcăraşu, M.; Cristea, M.; Macsim, A.-M.; Popa, O.; Stavarache, C.; Hîrtopeanu, A.; et al. A Real-Life Reproducibility Assessment for NMR Metabolomics. Diagnostics 2022, 12, 559. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR Spectroscopy. TrAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Hoch, J.C.; Baskaran, K.; Burr, H.; Chin, J.; Eghbalnia, H.R.; Fujiwara, T.; Gryk, M.R.; Iwata, T.; Kojima, C.; Kurisu, G.; et al. Biological Magnetic Resonance Data Bank. Nucleic Acids Res. 2023, 51, D368–D376. [Google Scholar] [CrossRef] [PubMed]
- Beckonert, O.; Keun, H.C.; Ebbels, T.M.D.; Bundy, J.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Metabolic Profiling, Metabolomic and Metabonomic Procedures for NMR Spectroscopy of Urine, Plasma, Serum and Tissue Extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Giraudeau, P. Quantitative NMR Spectroscopy of Complex Mixtures. Chem. Commun. 2023, 59, 6627–6642. [Google Scholar] [CrossRef]
- Quinn, T.J. Primary Methods of Measurement and Primary Standards. Metrologia 1997, 34, 61–65. [Google Scholar] [CrossRef]
- Giraudeau, P.; Tea, I.; Remaud, G.S.; Akoka, S. Reference and Normalization Methods: Essential Tools for the Intercomparison of NMR Spectra. J. Pharm. Biomed. Anal. 2014, 93, 3–16. [Google Scholar] [CrossRef]
- Ogura, T.; Wakayama, M. Coaxial-Tube Quantitative Nuclear Magnetic Resonance for Various Solutions Using SI-Traceable Concentration References. Anal. Chem. 2024, 96, 11657–11665. [Google Scholar] [CrossRef]
- Malz, F. Quantitative NMR in the Solution State NMR. In NMR Spectroscopy in Pharmaceutical Analysis; Elsevier: Amsterdam, The Netherlands, 2008; pp. 43–62. ISBN 9780444531735. [Google Scholar]
- Barding, G.A.; Salditos, R.; Larive, C.K. Quantitative NMR for Bioanalysis and Metabolomics. Anal. Bioanal. Chem. 2012, 404, 1165–1179. [Google Scholar] [CrossRef]
- Jung, Y.S.; Hyeon, J.S.; Hwang, G.S. Software-Assisted Serum Metabolite Quantification Using NMR. Anal. Chim. Acta 2016, 934, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Le Gresley, A.; Fardus, F.; Warren, J. Bias and Uncertainty in Non-Ideal QNMR Analysis. Crit. Rev. Anal. Chem. 2015, 45, 300–310. [Google Scholar] [CrossRef]
- Lanza, I.R.; Zhang, S.; Ward, L.E.; Karakelides, H.; Raftery, D.; Nair, K.S. Quantitative Metabolomics by H-NMR and LC-MS/MS Confirms Altered Metabolic Pathways in Diabetes. PLoS ONE 2010, 5, e10538. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, L.; Massa, G.; Guedens, W.; Louis, E.; Noben, J.-P.; Adriaensens, P. Metabolic Profiling of Type 1 Diabetes Mellitus in Children and Adolescents: A Case–Control Study. Diabetol. Metab. Syndr. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.K.; DeBerardinis, R.J. Applications of Metabolomics to Study Cancer Metabolism. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2018, 1870, 2–14. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.A.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Mutlib, A.E. Application of Quantitative NMR in Drug Metabolism Studies. In Handbook of Metabolic Pathways of Xenobiotics; Wiley: Hoboken, NJ, USA, 2014; pp. 1–31. [Google Scholar]
- 〈761〉 Nuclear Magnetic Resonance Spectroscopy; United States Pharmacopeia and National Formulary; UPS: Rockville, MD, USA, 2023. [CrossRef]
- European Directorate for the Quality of Medicines & HealthCare, Council of Europe. European Pharmacopoeia 2023, 11th ed.; European Directorate for the Quality of Medicines & HealthCare, Council of Europe: Strasbourg, France, 2023. [Google Scholar]
- Lamichhane, S.; Yde, C.C.; Schmedes, M.S.; Jensen, H.M.; Meier, S.; Bertram, H.C. Strategy for Nuclear-Magnetic-Resonance-Based Metabolomics of Human Feces. Anal. Chem. 2015, 87, 5930–5937. [Google Scholar] [CrossRef]
- Bervoets, L.; Ippel, J.H.; Smolinska, A.; van Best, N.; Savelkoul, P.H.M.; Mommers, M.A.H.; Penders, J. Practical and Robust NMR-Based Metabolic Phenotyping of Gut Health in Early Life. J. Proteome Res. 2021, 20, 5079–5087. [Google Scholar] [CrossRef]
- Wu, H.; Southam, A.D.; Hines, A.; Viant, M.R. High-Throughput Tissue Extraction Protocol for NMR- and MS-Based Metabolomics. Anal. Biochem. 2008, 372, 204–212. [Google Scholar] [CrossRef]
- Cruz, T.; Gleizes, M.; Balayssac, S.; Mornet, E.; Marsal, G.; Millán, J.L.; Malet-Martino, M.; Nowak, L.G.; Gilard, V.; Fonta, C. Identification of Altered Brain Metabolites Associated with TNAP Activity in a Mouse Model of Hypophosphatasia Using Untargeted NMR-based Metabolomics Analysis. J. Neurochem. 2017, 140, 919–940. [Google Scholar] [CrossRef] [PubMed]
- Mili, M.; Panthu, B.; Madec, A.M.; Berger, M.A.; Rautureau, G.J.P.; Elena-Herrmann, B. Fast and Ergonomic Extraction of Adherent Mammalian Cells for NMR-Based Metabolomics Studies. Anal. Bioanal. Chem. 2020, 412, 5453–5463. [Google Scholar] [CrossRef]
- Ghini, V.; Abuja, P.M.; Polasek, O.; Kozera, L.; Laiho, P.; Anton, G.; Zins, M.; Klovins, J.; Metspalu, A.; Wichmann, H.-E.; et al. Impact of the Pre-Examination Phase on Multicenter Metabolomic Studies. New Biotechnol. 2022, 68, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Saude, E.J.; Sykes, B.D. Urine Stability for Metabolomic Studies: Effects of Preparation and Storage. Metabolomics 2007, 3, 19–27. [Google Scholar] [CrossRef]
- Kelly, A.E.; Ou, H.D.; Withers, R.; Dötsch, V. Low-Conductivity Buffers for High-Sensitivity NMR Measurements. J. Am. Chem. Soc. 2002, 124, 12013–12019. [Google Scholar] [CrossRef]
- Saude, E.J.; Slupsky, C.M.; Sykes, B.D. Optimization of NMR Analysis of Biological Fluids for Quantitative Accuracy. Metabolomics 2006, 2, 113–123. [Google Scholar] [CrossRef]
- Mak, J.Y.W. Determination of Sample Concentrations by PULCON NMR Spectroscopy. Aust. J. Chem. 2021, 75, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, G. Sample Collection and Preparation of Biofluids and Extracts for NMR Spectroscopy. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1277, pp. 15–28. [Google Scholar]
- Rautureau, G.J.P.; Morio, B.; Guibert, S.; Lefevre, C.; Perrier, J.; Alves, A.; Chauvin, M.A.; Pinteur, C.; Monet, M.A.; Godet, M.; et al. Dietary Obesity in Mice Is Associated with Lipid Deposition and Metabolic Shifts in the Lungs Sharing Features with the Liver. Sci. Rep. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Ameta, K.; Gupta, A.; Kumar, S.; Sethi, R.; Kumar, D.; Mahdi, A.A. Essential Hypertension: A Filtered Serum Based Metabolomics Study. Sci. Rep. 2017, 7, 2153. [Google Scholar] [CrossRef]
- Hong, Y.; Coen, M.; Rhode, C.M.; Reily, M.D.; Robertson, D.G.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Chemical Shift Calibration of 1 H MAS NMR Liver Tissue Spectra Exemplified Using a Study of Glycine Protection of Galactosamine Toxicity. Magn. Reson. Chem. 2009, 47, S47–S53. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, J.R.; Ebeling, P.R.; Gooley, P.R.; McConville, M.J. A Sample Preparation Protocol for 1H Nuclear Magnetic Resonance Studies of Water-Soluble Metabolites in Blood and Urine. Anal. Biochem. 2010, 398, 263–265. [Google Scholar] [CrossRef]
- Weber, M.; Hellriegel, C.; Rueck, A.; Wuethrich, J.; Jenks, P. Using High-Performance 1H NMR (HP-QNMR®) for the Certification of Organic Reference Materials under Accreditation Guidelines—Describing the Overall Process with Focus on Homogeneity and Stability Assessment. J. Pharm. Biomed. Anal. 2014, 93, 102–110. [Google Scholar] [CrossRef]
- Giraudeau, P.; Silvestre, V.; Akoka, S. Optimizing Water Suppression for Quantitative NMR-Based Metabolomics: A Tutorial Review. Metabolomics 2015, 11, 1041–1055. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Cassani, J.; Probert, F.; Palace, J.; Claridge, T.D.W.; Botana, A.; Kenwright, A.M. Reliable, High-Quality Suppression of NMR Signals Arising from Water and Macromolecules: Application to Bio-Fluid Analysis. Analyst 2019, 144, 7270–7277. [Google Scholar] [CrossRef] [PubMed]
- Mckay, R.T. How the 1D-NOESY Suppresses Solvent Signal in Metabonomics NMR Spectroscopy: An Examination of the Pulse Sequence Components and Evolution. Concepts Magn. Reson. Part A 2011, 38A, 197–220. [Google Scholar] [CrossRef]
- Amiel, A.; Tremblay-Franco, M.; Gautier, R.; Ducheix, S.; Montagner, A.; Polizzi, A.; Debrauwer, L.; Guillou, H.; Bertrand-Michel, J.; Canlet, C. Proton NMR Enables the Absolute Quantification of Aqueous Metabolites and Lipid Classes in Unique Mouse Liver Samples. Metabolites 2019, 10, 9. [Google Scholar] [CrossRef]
- Torsten Schoenberger Guideline for QNMR Analysis. 2019. Available online: https://fr.scribd.com/document/548437169/QNMR-Guideline-Version001 (accessed on 26 March 2025).
- Mulder, F.A.A.; Tenori, L.; Licari, C.; Luchinat, C. Practical Considerations for Rapid and Quantitative NMR-Based Metabolomics. J. Magn. Reson. 2023, 352, 107462. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Bruderer, S.; Paruzzo, F.; Fischetti, G.; Toscano, G.; Graf, D.; Fey, M.; Henrici, A.; Ziebart, V.; Heitmann, B.; et al. Deconvolution of 1D NMR Spectra: A Deep Learning-Based Approach. J. Magn. Reson. 2023, 347, 107357. [Google Scholar] [CrossRef]
- Izquierdo-García, J.L.; Villa, P.; Kyriazis, A.; del Puerto-Nevado, L.; Pérez-Rial, S.; Rodriguez, I.; Hernandez, N.; Ruiz-Cabello, J. Descriptive Review of Current NMR-Based Metabolomic Data Analysis Packages. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 59, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.; Deborde, C.; Lefebvre, M.; Maucourt, M.; Moing, A. NMRProcFlow: A Graphical and Interactive Tool Dedicated to 1D Spectra Processing for NMR-Based Metabolomics. Metabolomics 2017, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Viant, M.R. Two-Dimensional J-Resolved NMR Spectroscopy: Review of a Key Methodology in the Metabolomics Toolbox. Phytochem. Anal. 2010, 21, 22–32. [Google Scholar] [CrossRef]
- Marchand, J.; Martineau, E.; Guitton, Y.; Dervilly-Pinel, G.; Giraudeau, P. Multidimensional NMR Approaches towards Highly Resolved, Sensitive and High-Throughput Quantitative Metabolomics. Curr. Opin. Biotechnol. 2017, 43, 49–55. [Google Scholar] [CrossRef]
- Mauve, C.; Khlifi, S.; Gilard, F.; Mouille, G.; Farjon, J. Sensitive, Highly Resolved, and Quantitative 1 H–13 C NMR Data in One Go for Tracking Metabolites in Vegetal Extracts. Chem. Commun. 2016, 52, 6142–6145. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.J.; Loening, N.M. QQ-HSQC: A Quick, Quantitative Heteronuclear Correlation Experiment for NMR Spectroscopy. Magn. Reson. Chem. 2007, 45, 937–941. [Google Scholar] [CrossRef]
- Giraudeau, P. Quantitative 2D Liquid-state NMR. Magn. Reson. Chem. 2014, 52, 259–272. [Google Scholar] [CrossRef]
- Abhyankar, N.; Szalai, V. Challenges and Advances in the Application of Dynamic Nuclear Polarization to Liquid-State NMR Spectroscopy. J. Phys. Chem. B 2021, 125, 5171–5190. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Shanaiah, N.; Raftery, D. Isotope Enhanced Approaches in Metabolomics. Adv Exp Med Biol. 2012, 992, 147–164. [Google Scholar]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Giraudeau, P. NMR-Based Metabolomics and Fluxomics: Developments and Future Prospects. Analyst 2020, 145, 2457–2472. [Google Scholar] [CrossRef]
- Srivastava, S.; Uddin, M. Recent Trends in Metabolomics, Machine Learning and Artificial Intelligence. In Artificial Intelligence in Biomedical and Modern Healthcare Informatics; Elsevier: Amsterdam, The Netherlands, 2025; pp. 91–100. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Specification | Comments |
|---|---|---|
| Biological sample | Biofluids: urine, plasma, saliva, cells supernatant | For mice, urine and serum volumes might reach more than 50 µL, while for humans, volumes might be higher. Thus, dilution in buffers, preferentially deuterated, is often needed to complete the total volume needed in an NMR tube [44]. |
| Cells lysate | The resuspension of metabolites extracted from 2 to 10 million cells typically yields a satisfactory signal-to-noise ratio [38]. | |
| Tissues | 20 to 150 mg of tissues are commonly used for NMR acquisition. Metabolites are extracted and resuspended in deuterated buffers [45]. When quantifying metabolites in tissue samples, additional considerations are necessary to ensure accuracy and reproducibility. Tissue weighing precision, water content variability, and metabolite extraction efficiencies can introduce variability in concentration measurements and should be carefully controlled [18,36]. Additionally, repeatability assessments and extraction validation should be performed to ensure quantitative reliability across different samples and biological replicates. Comprehensive discussions on optimized tissue preparation protocols for qNMR can be found in previously established guidelines [37,39]. | |
| Total volume | 200 µL or 550 µL | The sample size depends on the characteristics of the NMR probe used. Most often, 5 mm probes can accommodate 3 mm tubes with appropriate spinners or adapters. To minimize the solvent contribution to spectra, more concentrated biological material in a 3 mm tube is preferred, rather than diluting samples to fit into 5 mm tubes. NMR is non-destructive: samples can be used for other purposes after NMR analysis. |
| Small-molecule concentration range | 10 µM to 50 mM | Higher concentrations allow for faster acquisition and better signal-to-noise ratios, but mixtures often contain components spanning several orders of magnitude in concentration. In metabolomics, for example, certain metabolites may be present at sub-µM levels, while others exceed 10 mM. This large dynamic range poses a challenge to quantification, as high-concentration signals may obscure weaker ones and saturate the receiver. Pulse sequences and processing strategies should be adapted accordingly to preserve quantitative accuracy across the entire concentration range. Pre-fractionation or targeted profiling can help to address this issue in complex samples. |
| Protein content | As low as possible | A lower protein content is preferable. Acquisition schemes, such as those inserted into the CPMG pulse sequence, can reduce the impact of the protein broad spectral background at the cost of lowered sensitivity and accuracy. Samples can be filtered using low-molecular-weight cutoffs to remove proteins. Low protein concentrations and catalytic amounts are usually not an issue [46]. |
| Additives | Flexible, with the same 10 µM to 50 mM concentration limit | Caution should be taken with protonated molecules that may have peaks overlaying important signals, such as buffers and common additives, or contaminants, such as DMSO, glycerol, methanol, etc. |
| Chemical shift reference | 10 µM to 1 mM | TSP or DSS are the most common. Some metabolites from the sample, such as glucose or alanine, can also be used as references [47]. |
| Concentration reference | See below | High-quality concentration standards should be used for accurate quantification. Concentration references can be internal (added directly to the sample or placed inside a capillary immersed in the sample) or external (in another NMR tube). Lactate solutions are often used as they are available commercially at standardized concentrations. TSP or DSS can be used as concentration references, but they tend to bind proteins in biological samples, such as serum or urine, which alters their linewidth. In this situation, a strategy based on diluting untreated samples in deuterated solvents can be used to precipitate proteins and recover metabolites quantitated relative to standard reference compounds, such as DSS [48]. Alternative standards, such as Certified Reference Materials [49], are widely used in the pharmaceutical field to ensure compliance with regulatory guidelines [49]. |
| Sample quality | Homogeneous | Samples can be centrifuged or filtered if necessary. Be cautious of glycerol contamination from 0.5 µm filters, as well as acetate or formate originating from various lab consumables. |
| Solvent | Deuterated solvents (e.g., D2O, CDCl3) | The final NMR solvent must contain at least 5 to 10% deuterium for the spectrometer to compensate the magnetic field drift over the time of the NMR experiment. Typically, 5–10% D2O is added to aqueous samples. Cellular and tissue extracts often separate into polar and non-polar fractions. Dried samples from polar fractions can be resuspended in 100% D2O buffered solutions. Hydrophobic phases resulting from metabolite extractions can be dissolved in organic solvents such as CDCl3. |
| pH and buffers | Adjust to the required pH | As pH influences chemical shifts, adequate buffers should be used to minimize pH variations between samples; proton-less buffers such as phosphate are most commonly used in the range from 20 to 200 mM. Lower concentrations of salts are preferred, as low-conductivity buffers favor higher NMR sensitivity [41]. |
| Sample storage | Store under appropriate conditions | Store biological samples at low temperatures (−80 °C or in liquid nitrogen) to prevent metabolite evolution or degradation. Avoid repeated freeze–thaw cycles. NMR samples can be kept at a low temperature (4 °C) before acquisition. |
| Acquisition temperature | 4 °C to 90 °C | Higher temperatures allow for better NMR signal sensitivity. Temperatures of 30–37 °C are most common in metabolomics and enzymology. Low temperatures should only be used in proven cases of instability. |
| Bruker Pulse Sequence or Tools | Considered Quantitative Provided That a Sufficiently Long Recycle Delay Is Used | Description, Applications |
|---|---|---|
| Zgpr | Yes | The standard 1D proton NMR pulse sequence with water presaturation is commonly used for the routine analysis of small molecules in metabolomics. However, signals close to the presaturation frequency may experience a loss of intensity. |
| Zgpr30 | Yes | A variant of zgpr with a 30° flip angle that allows for reducing the relaxation delay (d1). Useful when faster data acquisition is needed, but at the cost of a lessened sensitivity. PULCON method cannot be used with this sequence. |
| Noesygppr1d | Yes | NOESY sequence with presaturation to suppress water. Routine spectrum in metabolomics. |
| Cpmgpr1d | No | Includes a CPMG sequence to suppress broad signals from macromolecules. Suitable for samples that display altered baseline due to the background contribution of lipids or proteins. |
| zgesgp | No | 1D 1H excitation sculpting (ES) with gradients. Uses shaped pulses and gradients to selectively excite and suppress specific signals. Effective water suppression and improved detection of overlapping signals. |
| Diffusion-edited NMR (ledbp2s1d) | No | Suppresses signals from small molecules by exploiting differences in diffusion rates. Useful for complex mixtures and identifying macromolecules. |
| Most 1D NMR sequences with pulsed field gradients (PFG) | No (except in some cases) | PFG techniques are used to improve solvent suppression and reduce artifacts. However, in many applications (e.g., diffusion-editing or selective excitation), PFG elements can interfere with signal intensity or introduce relaxation-based biases, precluding quantitative use. Notably, some PFG-containing sequences, such as noesygppr1d, can be quantitative under appropriate acquisition conditions. |
| Conditions of NMR Acquisition | Common Pulse Sequence | Repetition Time | Chemical reference | Exploitation of Data, Quantification and Comments |
|---|---|---|---|---|
| Quantitative conditions | Short (zgpr, zgpr30, noesygppr1d) | At least 5 × T1 (e.g., 5–60 s depending on compound). T1 should be measured using inversion–recovery or saturation–recovery methods. | A single-concentration reference compound (internal, spiked, or external) can be used to determine the concentration of any other compound | Absolute quantification by integration and Equation (1). For pulses of less than 90°. the repetition time can be shortened (e.g., 3 T1 for a 30° pulse with the zgpr30 pulse sequence). |
| Non quantitative conditions | Any sequence, short or longer (cpmgpr1D) | Below 5 T1: 2–4 s between experiments | One standard for each quantified compound (spiking experiment or external reference) | Relative quantification by integration and absolute quantification by direct comparison with the same molecule at a known standard Possible in the absence of standard: determination of variation by comparison of same peak integrals between a set of spectra. |
| Chenomx | noesygppr1D, cpmgpr1d | 4 s | One compound (TSP at known concentration) | Strict adherence to Chenomx acquisition parameters recommendations. Dedicated software and peak-fitting tool. Limited library of biomolecules. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drevet Mulard, E.; Gilard, V.; Balayssac, S.; Rautureau, G.J.P. Quantitative Nuclear Magnetic Resonance for Small Biological Molecules in Complex Mixtures: Practical Guidelines and Key Considerations for Non-Specialists. Molecules 2025, 30, 1838. https://doi.org/10.3390/molecules30081838
Drevet Mulard E, Gilard V, Balayssac S, Rautureau GJP. Quantitative Nuclear Magnetic Resonance for Small Biological Molecules in Complex Mixtures: Practical Guidelines and Key Considerations for Non-Specialists. Molecules. 2025; 30(8):1838. https://doi.org/10.3390/molecules30081838
Chicago/Turabian StyleDrevet Mulard, Eva, Véronique Gilard, Stéphane Balayssac, and Gilles J. P. Rautureau. 2025. "Quantitative Nuclear Magnetic Resonance for Small Biological Molecules in Complex Mixtures: Practical Guidelines and Key Considerations for Non-Specialists" Molecules 30, no. 8: 1838. https://doi.org/10.3390/molecules30081838
APA StyleDrevet Mulard, E., Gilard, V., Balayssac, S., & Rautureau, G. J. P. (2025). Quantitative Nuclear Magnetic Resonance for Small Biological Molecules in Complex Mixtures: Practical Guidelines and Key Considerations for Non-Specialists. Molecules, 30(8), 1838. https://doi.org/10.3390/molecules30081838