
Comparing Early Transcriptomic Responses of 18 Soybean (Glycine max) Genotypes to Iron Stress

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Clustering Genotypes into Efficient and Inefficient Classes Based on Phenotypic Data
2.2. Identification of Differentially Expressed Genes in Early Response to IDC Stress
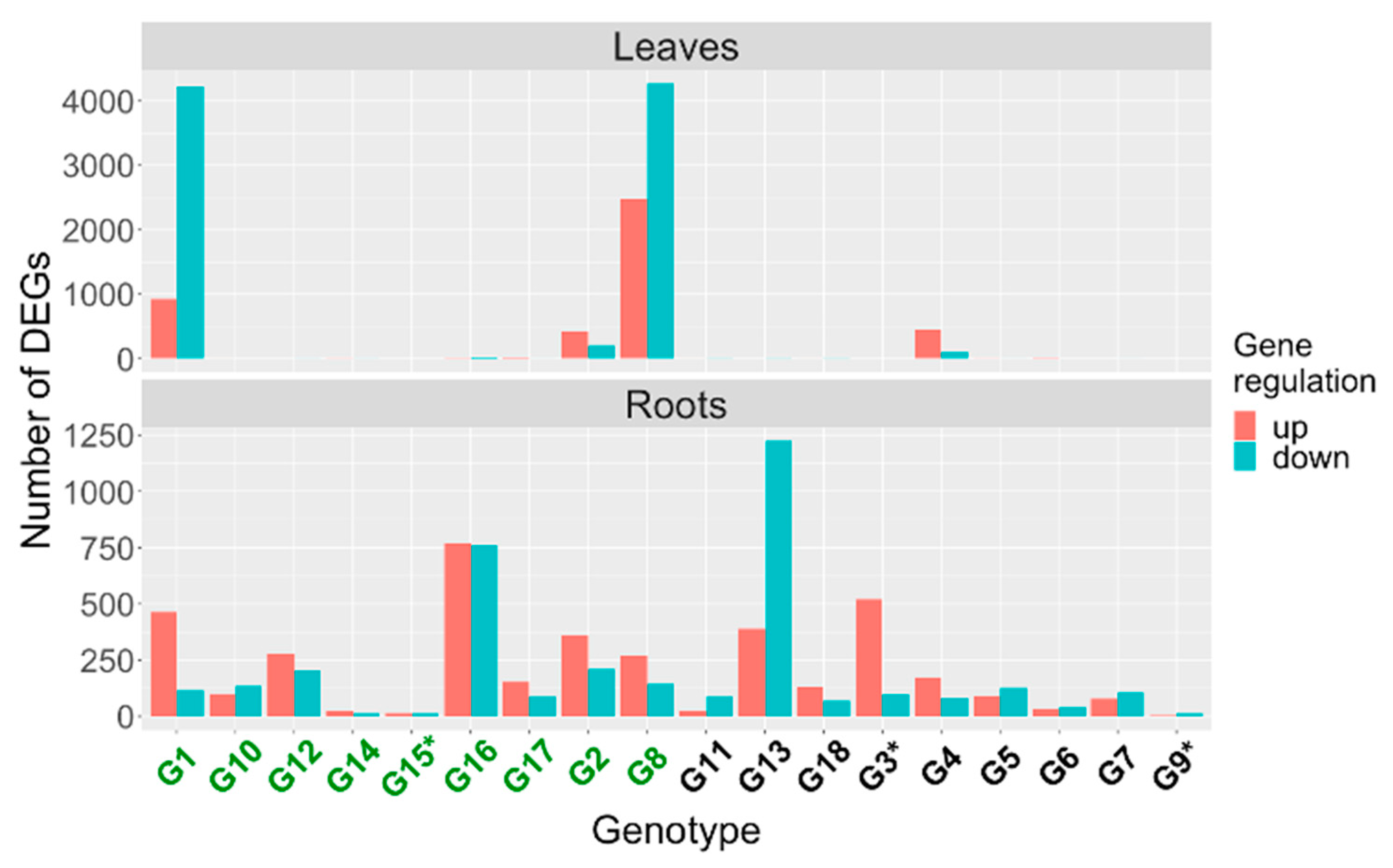
2.3. Comparison of Differentially Expressed Genes between Genotypes
2.4. Comparisons across Genotypes
2.4.1. Differentially Expressed Genes
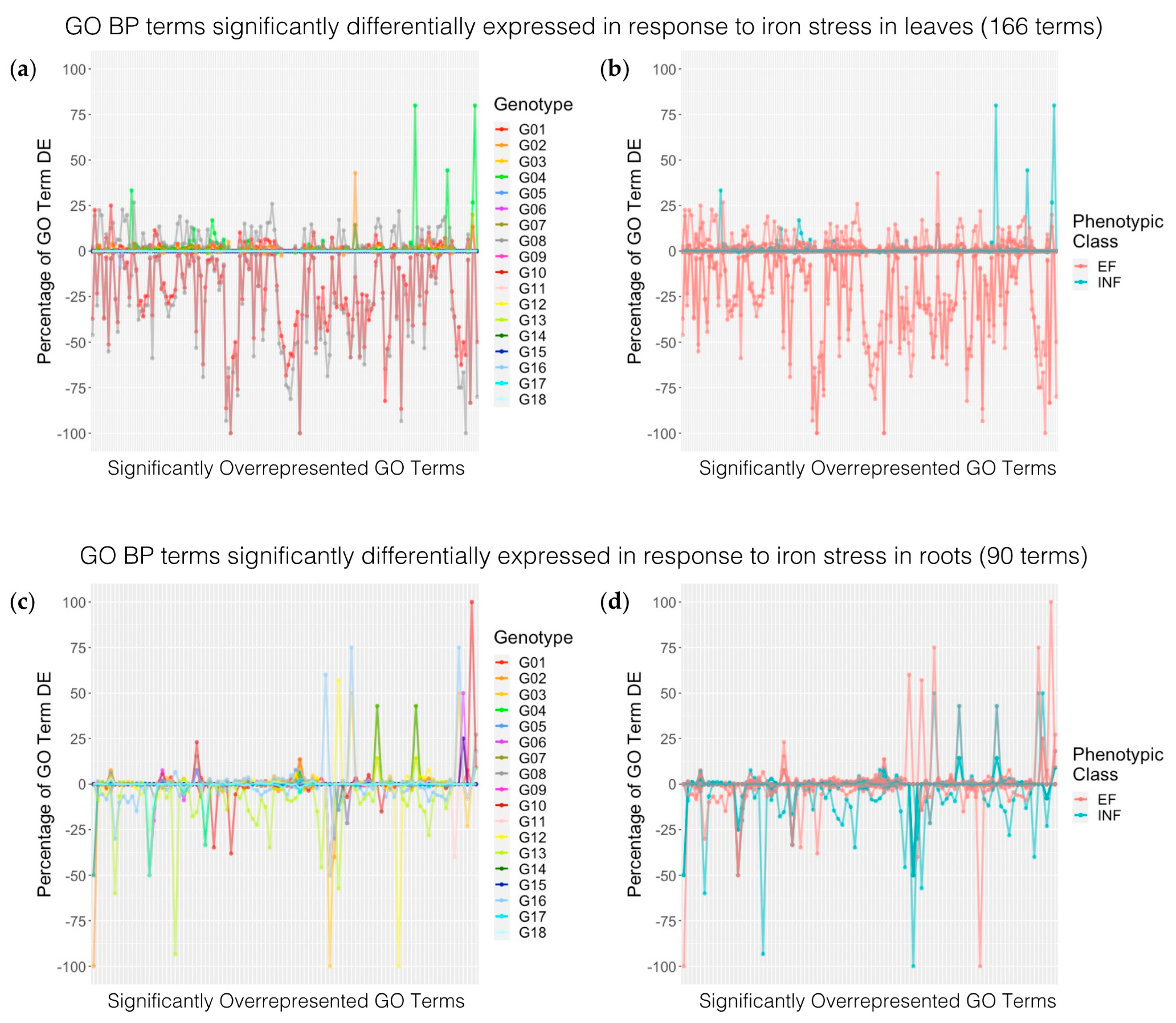
2.4.2. Enriched Biological Process Terms
2.5. Comparing Differentially Expressed Genes between Iron Efficiency Groups
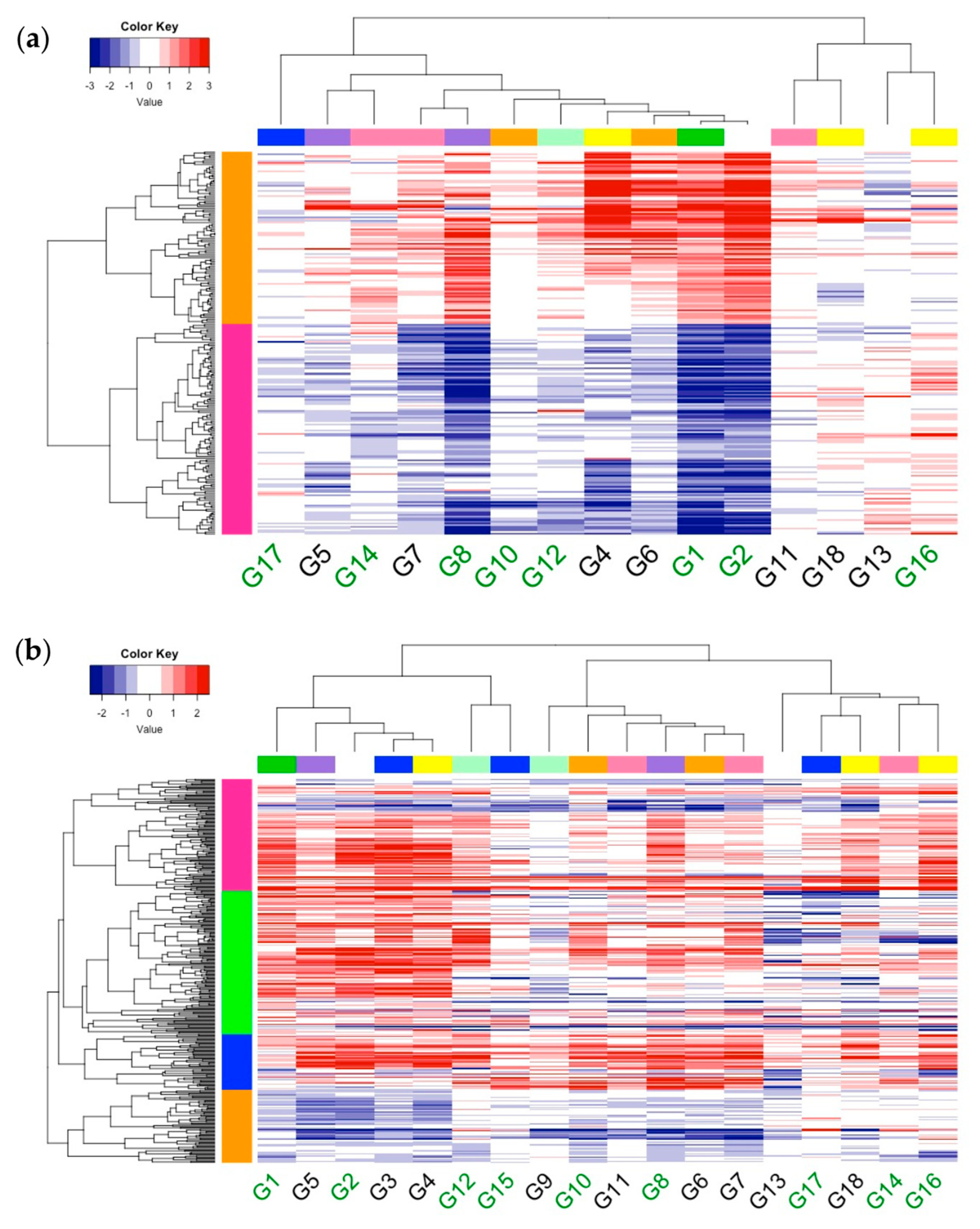
2.6. Characterization of DEG Expression Trends within Biological Processes Terms
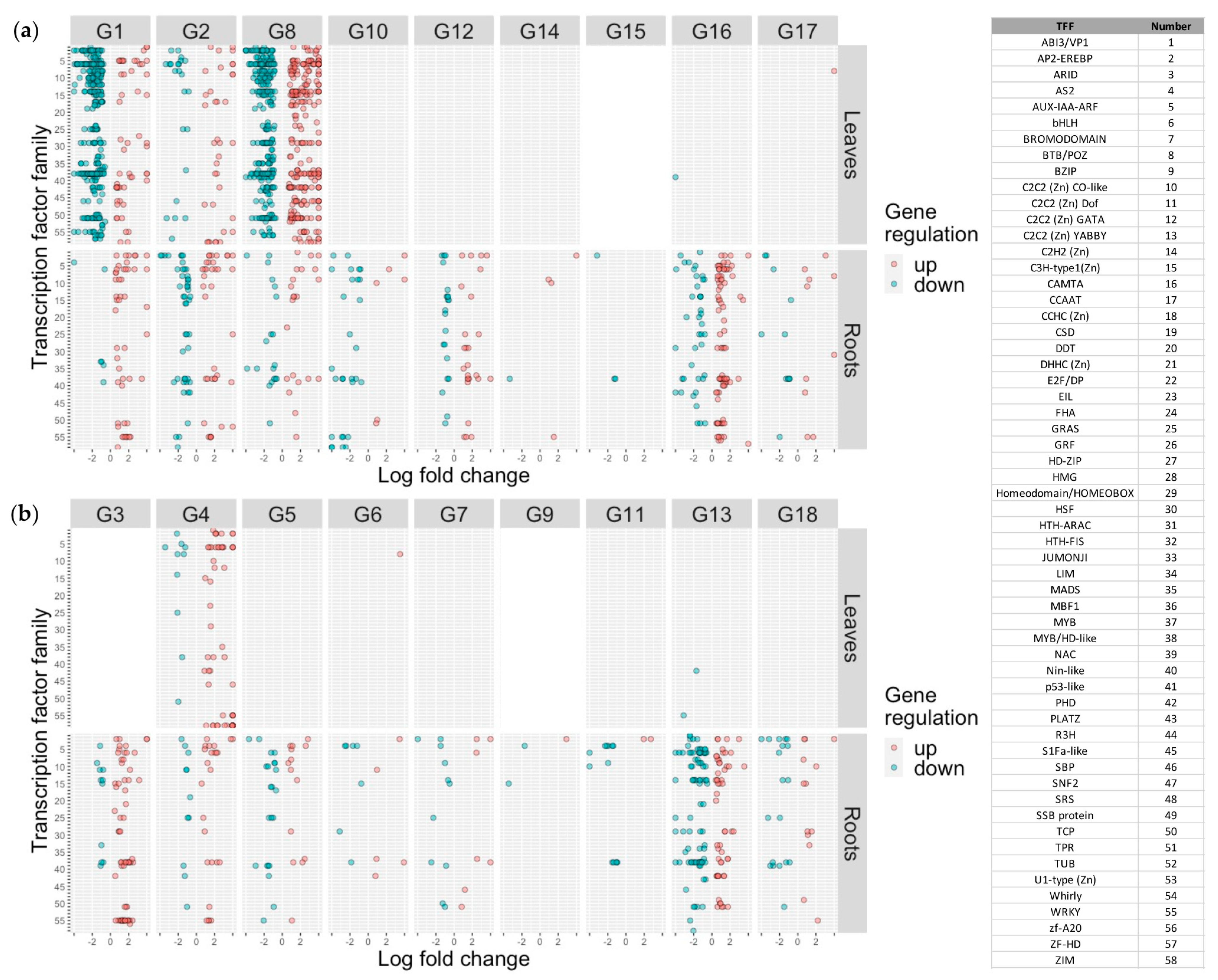
2.7. Characterization of Differentially Expressed Transcription Factors
2.8. Characterization of Differentially Expressed Genes across IDC QTL
2.9. Single Linkage Clustering
3. Discussion
3.1. Soybean Responds Rapidly to Iron Stress
3.2. Diversity of Iron Stress Responses Found within the Soybean Germplasm Collection
3.3. Identifying Targets for Future Analyses
4. Conclusions
5. Materials and Methods
5.1. Phenotypic Clustering
5.2. Plant Materials
5.3. Tissue Collection
5.4. RNA Isolation and Sequencing
5.5. Identification of Differentially Expressed Genes in Response to Iron Stress
5.6. Gene Annotation
5.7. Identification of Overrepresented Gene Ontology (GO) Terms and Transcription Factors
5.8. Single Linkage Clustering
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPM | count per million |
| DEGs | differentially expressed genes |
| EF | iron-efficient |
| FDR | false discovery rate |
| GO | gene ontology |
| GWAS | genome-wide association study |
| IDC | iron deficiency chlorosis |
| INF | iron-inefficient |
| logFC | log2 fold-change |
| NCBI SRA | National Center for Biotechnology Small Reads Archive |
| PI | plant introduction line |
| QTL | quantitative trait loci |
| ROS | reactive oxygen species |
| RNA-seq | RNA sequencing |
| SNP | single nucleotide polymorphism |
| SPAD | soil plant analysis development |
| TAIR10 | The Arabidopsis Information Resource version 10 |
| TF | transcription factor |
| TFF | transcription factor family |
| TMM | trimmed mean of M-values |
References
- Hansen, N.C.; Jolley, V.D.; Naeve, S.L.; Goos, R.J. Iron deficiency of soybean in the North Central U.S. and associated soil properties. Soil Sci. Plant Nutr. 2004, 50, 983–987. [Google Scholar] [CrossRef]
- Brown, J.C.; Weber, C.R.; Caldwell, B.E. Efficient and inefficient use of iron by two soybean genotypes and their isolines. Agron. J. 1967, 59, 459–462. [Google Scholar] [CrossRef]
- Froehlich, D.M.; Fehr, W.R. Agronomic performance of soybeans with differing levels of iron deficiency chlorosis on calcareous soil. Crop Sci. 1981, 21, 438–441. [Google Scholar] [CrossRef]
- Inskeep, W.P.; Bloom, P.R. A comparative study of soil solution chemistry associated with chlorotic and nonchlorotic soybeans in western Minnesota. J. Plant Nutr. 1984, 7, 513–531. [Google Scholar] [CrossRef]
- Hansen, N.C.; Schmitt, M.A.; Andersen, J.E.; Strock, J.S. Iron deficiency of soybean in the upper midwest and associated soil properties. Agron. J. 2003, 95, 1595–1601. [Google Scholar] [CrossRef]
- Weiss, M.G. Inheritance and physiology of efficiency in iron utilization in soybeans. Genetics 1943, 28, 253–268. [Google Scholar] [CrossRef]
- Cianzio, S.R.; Fehr, W.R. Genetic control of iron deficiency chlorosis in soybeans. Iowa State J. Res. 1980, 54, 367–375. [Google Scholar]
- Cianzio, S.R.; Fehr, W.R. Variation in the inheritance of resistance to iron deficiency chlorosis in soybeans. Crop Sci. 1982, 5, 433–434. [Google Scholar] [CrossRef]
- Lin, S.; Cianzio, S.; Shoemaker, R. Mapping genetic loci for iron deficiency chlorosis in soybean. Mol. Breed. 1997, 3, 219–229. [Google Scholar] [CrossRef]
- Mamidi, S.; Chikara, S.; Goos, R.J.; Hyten, D.L.; Annam, D.; Moghaddam, S.M.; Lee, R.K.; Cregan, P.B.; McClean, P.E. Genome-wide association analysis identifies candidate genes associated with iron deficiency chlorosis in soybean. Plant Genome 2011, 4, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Mamidi, S.; Lee, R.K.; Goos, J.R.; McClean, P.E. Genome-wide association studies identifies seven major regions responsible for iron deficiency chlorosis in soybean (Glycine max). PLoS ONE 2014, 9, e107469. [Google Scholar] [CrossRef]
- Assefa, T.; Zhang, J.; Moran Lauter, A.N.; Singh, A.; O’Rourke, J.A.; Graham, M.A.; Singh, A.K. Deconstructing the genetic architecture of iron deficiency chlorosis in soybean using genome-wide approaches. BMC Plant Biol. 2020, 20, 42. [Google Scholar] [CrossRef]
- Diers, B.W.; Cianzio, S.R.; Shoemaker, R.C. Possible identification of quantitative trait loci affecting iron efficiency in soybean. J. Plant Nutr. 1992, 15, 2127–2136. [Google Scholar] [CrossRef]
- Severin, A.J.; Peiffer, G.A.; Xu, W.W.; Hyten, D.L.; Bucciarelli, B.; O’Rourke, J.A.; Bolon, Y.-T.; Grant, D.; Farmer, A.D.; May, G.D.; et al. An integrative approach to genomic introgression mapping. Plant Physiol. 2010, 154, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, G.A.; King, K.E.; Severin, A.J.; May, G.D.; Cianzio, S.R.; Lin, S.F.; Lauter, N.C.; Shoemaker, R.C. Identification of candidate genes underlying an iron efficiency quantitative trait locus in soybean. Plant Physiol. 2012, 158, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.A.; Graham, M.A.; Vodkin, L.; Gonzalez, D.O.; Cianzio, S.R.; Shoemaker, R.C. Recovering from iron deficiency chlorosis in near-isogenic soybeans: A microarray study. Plant Physiol. Biochem. 2007, 45, 287–292. [Google Scholar] [CrossRef]
- O’Rourke, J.A.; Charlson, D.V.; Gonzalez, D.O.; Vodkin, L.O.; Graham, M.A.; Cianzio, S.R.; Grusak, M.A.; Shoemaker, R.C. Microarray analysis of iron deficiency chlorosis in near-isogenic soybean lines. BMC Genom. 2007, 8, 476. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.A.; Nelson, R.T.; Grant, D.; Schmutz, J.; Grimwood, J.; Cannon, S.; Vance, C.P.; Graham, M.A.; Shoemaker, R.C. Integrating microarray analysis and the soybean genome to understand the soybeans iron deficiency response. BMC Genom. 2009, 10, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran Lauter, A.N.; Peiffer, G.A.; Yin, T.; Whitham, S.A.; Cook, D.; Shoemaker, R.C.; Graham, M.A. Identification of candidate genes involved in early iron deficiency chlorosis signaling in soybean (Glycine max) roots and leaves. BMC Genom. 2014, 15, 702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran Lauter, A.N.; Rutter, L.; Cook, D.; O’Rourke, J.A.; Graham, M.A. Examining short-term responses to a long-term problem: RNA-seq analyses of iron deficiency chlorosis tolerant soybean. Int. J. Mol. Sci. 2020, 21, 3591. [Google Scholar] [CrossRef] [PubMed]
- Atencio, L.; Salazar, J.; Moran Lauter, A.N.; Gonzales, M.D.; O’Rourke, J.A.; Graham, M.A. Characterizing short and long term iron stress responses in iron deficiency tolerant and susceptible soybean (Glycine max L. Merr.). Plant Stress 2021, 2, 100012. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control [version 2; referees: 4 approved]. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.W.; Verma, R.; Kim, M.; Lee, J.Y.; Kim, Y.K.; Bang, J.W.; Reiter, W.D.; Pai, H.S. Depletion of UDP-D-apiose/UDP-D-xylose synthases results in rhamnogalacturonan-II deficiency, cell wall thickening, and cell death in higher plants. J. Biol. Chem. 2006, 281, 13708–13716. [Google Scholar] [CrossRef] [Green Version]
- López-Millán, A.F.; Morales, F.; Gogorcena, Y.; Abadía, A.; Abadía, J. Metabolic responses in iron deficient tomato plants. J. Plant Physiol. 2009, 166, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wang, L.; Wang, Y.; Yang, G.; Gao, C.; Yu, Z.; Li, T.; Zhang, X.; Ma, L.; Xu, X.; et al. Overexpression of Malus xiaojinensis CS1 gene in tobacco affects plant development and increases iron stress tolerance. Sci. Hortic. 2013, 150, 65–72. [Google Scholar] [CrossRef]
- Schröder, F.; Lisso, J.; Lange, P.; Müssig, C. The extracellular EXO protein mediates cell expansion in Arabidopsis leaves. BMC Plant Biol. 2009, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Lisso, J.; Schröder, F.; Müssig, C. EXO modifies sucrose and trehalose responses and connects the extracellular carbon status to growth. Front. Plant Sci. 2013, 4, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Katsir, L.; Schilmiller, A.L.; Staswick, P.E.; Sheng, Y.H.; Howe, G.A. COI1 is a critical component of a receptor for jasmonate and the bacterial virulence factor coronatine. Proc. Natl. Acad. Sci. USA 2008, 105, 7100–7105. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.U.; Song, W.Y.; Hong, D.; Ko, D.; Yamaoka, Y.; Jang, S.; Yim, S.; Lee, E.; Khare, D.; Kim, K.; et al. Plant ABC transporters enable many unique aspects of a terrestrial plant’s lifestyle. Mol. Plant 2016, 9, 338–355. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Vishal, B.; Ho, W.J.; Lok, F.C.J.; Lee, F.S.M.; Kumar, P.P. Regulation of a cytochrome P450 gene CYP94B1 by WRKY33 transcription factor controls apoplastic barrier formation in roots to confer salt tolerance. Plant Physiol. 2020, 184, 2199–2215. [Google Scholar] [CrossRef] [PubMed]
- ChunJuan, D.; JinYuan, L. The Arabidopsis EAR-motif-containing protein RAP2.1 functions as an active transcriptional repressor to keep stress responses under tight control. BMC Plant Biol. 2010, 10, 47. [Google Scholar]
- Shi, H.; Liu, W.; Yao, Y.; Wei, Y.; Chan, Z. Alcohol dehydrogenase 1 (ADH1) confers both abiotic and biotic stress resistance in Arabidopsis. Plant Sci. 2017, 262, 24–31. [Google Scholar] [CrossRef]
- Lucena, C.; Porras, R.; García, M.J.; Alcántara, E.; Pérez-Vicente, R.; Zamarreño, Á.M.; Bacaicoa, E.; García-Mina, J.M.; Smith, A.P.; Romera, F.J. Ethylene and phloem signals are involved in the regulation of responses to Fe and P deficiencies in roots of strategy I plants. Front. Plant Sci. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front. Plant Sci. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taran, N.; Okanenko, A.; Musienko, N. Sulpholipid reflects plant resistance to stress-factor action. Biochem. Soc. Trans. 2000, 28, 922–924. [Google Scholar] [CrossRef]
- Klecker, M.; Gasch, P.; Peisker, H.; Dörmann, P.; Schlicke, H.; Grimm, B.; Mustroph, A. A shoot-specific hypoxic response of Arabidopsis sheds light on the role of the phosphate-responsive transcription factor PHOSPHATE STARVATION RESPONSE1. Plant Physiol. 2014, 165, 774–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimm, O.; Essigmann, B.; Kloska, S.; Altmann, T.; Buckhout, T.J. Response of Arabidopsis to iron deficiency stress as revealed by microarray analysis. Plant Physiol. 2001, 127, 1030–1043. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Garcia, N.; Da-Silva, C.J.; Cocco, K.L.T.; Pomagualli, D.; de Oliveira, F.K.; da Silva, J.V.L.; de Oliveira, A.C.B.; do Amarante, L. Waterlogging tolerance of five soybean genotypes through different physiological and biochemical mechanisms. Environ. Exp. Bot. 2020, 172, 103975. [Google Scholar] [CrossRef]
- Charron, J.-B.F.; Ouellet, F.; Houde, M.; Sarhan, F. The plant Apolipoprotein D ortholog protects Arabidopsis against oxidative stress. BMC Plant Biol. 2008, 8, 86. [Google Scholar] [CrossRef] [Green Version]
- García, M.J.; Suárez, V.; Romera, F.J.; Alcántara, E.; Pérez-Vicente, R. A new model involving ethylene, nitric oxide and Fe to explain the regulation of Fe-acquisition genes in Strategy I plants. Plant Physiol. Biochem. 2011, 49, 537–544. [Google Scholar] [CrossRef]
- Santamaria, M.; Thomson, C.J.; Read, N.D.; Loake, G.J. The promoter of a basic PR1-like gene, AtPRB1, from Arabidopsis establishes an organ-specific expression pattern and responsiveness to ethylene and methyl jasmonate. Plant Mol. Biol. 2001, 47, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Ebisu, Y.; Kinoshita, T.; Doi, M.; Okuma, E.; Murata, Y.; Shimazaki, K.I. bHLH transcription factors that facilitate K+ uptake during stomatal opening are repressed by abscisic acid through phosphorylation. Sci. Signal. 2013, 6, ra48. [Google Scholar] [CrossRef]
- Li, J.; Brader, G.; Palva, E.T. Kunitz trypsin inhibitor: An antagonist of cell death triggered by phytopathogens and fumonisin B1 in Arabidopsis. Mol. Plant 2008, 1, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.Y.; Thor, K.; Gubler, A.; Meier, S.; Dietrich, D.; Weichert, A.; Grotemeyer, M.S.; Tegeder, M.; Rentsch, D. AtPTR1 and AtPTR5 transport dipeptides in planta. Plant Physiol. 2008, 148, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Rubin, G.; Tohge, T.; Matsuda, F.; Saito, K.; Scheible, W.R. Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 2009, 21, 3567–3584. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Wang, S.; Dao, Y.; Wang, J.; Wang, K. Arabidopsis thaliana trehalose-6-phosphate phosphatase gene TPPI enhances drought tolerance by regulating stomatal apertures. J. Exp. Bot. 2020, 71, 4285–4297. [Google Scholar] [CrossRef] [Green Version]
- Hindt, M.N.; Guerinot, M. Lou Getting a sense for signals: Regulation of the plant iron deficiency response. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Yang, Y.; Liu, K.; Zhang, L.; Guo, H.; Sun, T.; Wang, H. Involvement of endogenous salicylic acid in iron-deficiency responses in Arabidopsis. J. Exp. Bot. 2016, 67, 4179–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.E.; O’Rourke, J.A.; Peiffer, G.A.; Yin, T.; Majumder, M.; Zhang, C.; Cianzio, S.R.; Hill, J.H.; Cook, D.; Whitham, S.A.; et al. Replication protein A subunit 3 and the iron efficiency response in soybean. Plant Cell Environ. 2014, 37, 213–234. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.Y.; Ye, Y.Q.; Fan, S.K.; Jin, C.W.; Zheng, S.J. Increased sucrose accumulation regulates iron-deficiency responses by promoting auxin signaling in Arabidopsis plants. Plant Physiol. 2016, 170, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Butenhoff, K.J. QTL Mapping and GWAS Identify Sources of Iron Deficiency Chlorosis and Canopy Wilt Tolerance in the Fiskeby III X Mandarin (Ottawa) Soybean Population. Ph.D. Thesis, University of Minnesota Digital Conservancy, Minneapolis, MN, USA, 2015. [Google Scholar]
- Shanmugam, S.; Zhao, S.; Nandy, S.; Srivastava, V.; Khodakovskaya, M. Modification of soybean growth and abiotic stress tolerance by expression of truncated ERECTA protein from Arabidopsis thaliana. PLoS ONE 2020, 15, e0233383. [Google Scholar] [CrossRef]
- Chuang, H.W.; Feng, J.H.; Feng, Y.L.; Wei, M.J. An Arabidopsis WDR protein coordinates cellular networks involved in light, stress response and hormone signals. Plant Sci. 2015, 241, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.W.; Wu, X.L.; Liu, Z.H.; Luo, H.B.; Huang, R.Z. Abiotic stresses and phytohormones regulate expression of FAD2 gene in Arabidopsis thaliana. J. Integr. Agric. 2012, 11, 62–72. [Google Scholar] [CrossRef]
- Chen, X.; Han, H.; Jiang, P.; Nie, L.; Bao, H.; Fan, P.; Lv, S.; Feng, J.; Li, Y. Transformation of β-lycopene cyclase genes from Salicornia europaea and Arabidopsis conferred salt tolerance in Arabidopsis and tobacco. Plant Cell Physiol. 2011, 52, 909–921. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.A.; McCabe, C.E.; Graham, M.A. Dynamic gene expression changes in response to micronutrient, macronutrient, and multiple stress exposures in soybean. Funct. Integr. Genom. 2019, 20, 321–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissen, R.; Winge, P.; Tran, D.H.T.; Jørstad, T.S.; Størseth, T.R.; Christensen, T.; Bones, A.M. Transcriptional profiling of an Fd-GOGAT1/GLU1 mutant in Arabidopsis thaliana reveals a multiple stress response and extensive reprogramming of the transcriptome. BMC Genom. 2010, 11, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Gu, M.; Lu, Y.; Zhang, Y.; Chen, C.; Ling, H.Q.; Wu, H. Glutamate synthase 1 is involved in iron-deficiency response and long-distance transportation in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 1925–1941. [Google Scholar] [CrossRef] [PubMed]
- Farrow, S.C.; Facchini, P.J. Functional diversity of 2-oxoglutarate/Fe(II)-dependent dioxygenases in plant metabolism. Front. Plant Sci. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigani, G.; Morandini, P.; Murgia, I. Searching iron sensors in plants by exploring the link among 2′-OG-dependent dioxygenases, the iron deficiency response and metabolic adjustments occurring under iron deficiency. Front. Plant Sci. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohani, M.M.; Schenk, P.M.; Schultz, C.J.; Schmidt, O. Phylogenetic and transcriptional analysis of a strictosidine synthase-like gene family in Arabidopsis thaliana reveals involvement in plant defence responses. Plant Biol. 2009, 11, 105–117. [Google Scholar] [CrossRef]
- Zhang, J.; Naik, H.S.; Assefa, T.; Sarkar, S.; Reddy, R.V.C.; Singh, A.; Ganapathysubramanian, B.; Singh, A.K. Computer vision and machine learning for robust phenotyping in genome-wide studies. Sci. Rep. 2017, 7, 44048. [Google Scholar] [CrossRef] [PubMed]
- Kollist, H.; Zandalinas, S.I.; Sengupta, S.; Nuhkat, M.; Kangasjärvi, J.; Mittler, R. Rapid responses to abiotic stress: Priming the landscape for the signal transduction network. Trends Plant Sci. 2019, 24, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckhout, T.J.; Yang, T.J.W.; Schmidt, W. Early iron-deficiency-induced transcriptional changes in Arabidopsis roots as revealed by microarray analyses. BMC Genomics 2009, 10, 147. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Castro-Guerrero, N.A.; McInturf, S.A.; Nguyen, N.T.; Dame, A.N.; Wang, J.; Bindbeutel, R.K.; Joshi, T.; Jurisson, S.S.; Nusinow, D.A.; et al. Changes in iron availability in Arabidopsis are rapidly sensed in the leaf vasculature and impaired sensing leads to opposite transcriptional programs in leaves and roots. Plant Cell Environ. 2018, 41, 2263–2276. [Google Scholar] [CrossRef]
- Lin, F.; Zhao, M.; Baumann, D.D.; Ping, J.; Sun, L.; Liu, Y.; Zhang, B.; Tang, Z.; Hughes, E.; Doerge, R.W.; et al. Molecular response to the pathogen Phytophthora sojae among ten soybean near isogenic lines revealed by comparative transcriptomics. BMC Genom. 2014, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, D.; Jiménez-Gómez, J.M.; Kimura, S.; Fulop, D.; Chitwood, D.H.; Headland, L.R.; Kumar, R.; Covington, M.F.; Devisetty, U.K.; Tat, A.V.; et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. USA 2013, 110, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Xu, Q.; Ma, D.; Zhang, W.; Xu, Z.; Zhao, M.; Guo, Z. Transcriptomics profiling in response to cold stress in cultivated rice and weedy rice. Gene 2019, 685, 96–105. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Li, W.; Gu, Y.; Lai, Y.; Bi, Y.; He, C. Transcriptomic comparison reveals genetic variation potentially underlying seed developmental evolution of soybeans. J. Exp. Bot. 2018, 69, 5089–5104. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.J.; Waters, B.M. Use of natural variation reveals core genes in the transcriptome of iron-deficient Arabidopsis thaliana roots. J. Exp. Bot. 2012, 63, 1039–1055. [Google Scholar] [CrossRef] [Green Version]
- Waters, B.M.; McInturf, S.A.; Stein, R.J. Rosette iron deficiency transcript and microRNA profiling reveals links between copper and iron homeostasis in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 5903–5918. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.; Messina, C.D. Can we harness “enviromics” to accelerate crop improvement by integrating breeding and agronomy? Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Merry, R.; Butenhoff, K.; Campbell, B.W.; Michno, J.-M.; Wang, D.; Orf, J.H.; Lorenz, A.J.; Stupar, R.M. Identification and fine-mapping of a soybean quantitative trait locus on chromosome 5 conferring tolerance to iron deficiency chlorosis. Plant Genome 2019, 12, 190007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Hellmann, H.A.; Liu, Y.; Wang, W. Editorial: Protein quality controlling systems in plant responses to environmental stresses. Front. Plant Sci. 2018, 9, 1–3. [Google Scholar] [CrossRef]
- Singh, H.; Kaur, K.; Singh, M.; Kaur, G.; Singh, P. Plant cyclophilins: Multifaceted proteins with versatile roles. Front. Plant Sci. 2020, 11, 1–30. [Google Scholar] [CrossRef]
- Yuen, C.Y.L.; Matsumoto, K.O.; Christopher, D.A. Variation in the subcellular localization and protein folding activity among Arabidopsis thaliana homologs of protein disulfide isomerase. Biomolecules 2013, 3, 848–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, V.B.; D’Silva, P. Arabidopsis thaliana J-class heat shock proteins: Cellular stress sensors. Funct. Integr. Genom. 2009, 9, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, J.; Muroya, M.; Shirakami, N.; Takahashi, S.; Higuchi, C.; Muranaka, A.; Takami, T.; Watanabe, S. Overexpression of BUNDLE SHEATH DEFECTIVE 2 improves the efficiency of photosynthesis and growth in Arabidopsis. Plant J. 2020, 102, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wen, R.; Wang, J.; Xiang, D.; Wang, Q.; Zang, Y.; Wang, Z.; Huang, S.; Li, X.; Datla, R.; et al. Arabidopsis UBC13 differentially regulates two programmed cell death pathways in responses to pathogen and low-temperature stress. New Phytol. 2019, 221, 919–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skelly, M.J.; Malik, S.I.; Le Bihan, T.; Bo, Y.; Jiang, J.; Spoel, S.H.; Loake, G.J. A role for S-nitrosylation of the SUMO-conjugating enzyme SCE1 in plant immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 17090–17095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawkar, G.M.; Lee, E.S.; Shelake, R.M.; Park, J.H.; Ryu, S.W.; Kang, C.H.; Lee, S.Y. Activation of the transducers of unfolded protein response in plants. Front. Plant Sci. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.H. Evolution of the unfolded protein response in plants. Plant Cell Environ. 2021, 44, 2625–2635. [Google Scholar] [CrossRef]
- Deepika, D.; Singh, A. Plant phospholipase D: Novel structure, regulatory mechanism, and multifaceted functions with biotechnological application. Crit. Rev. Biotechnol. 2021, 1–19. [Google Scholar] [CrossRef]
- Urrea Castellanos, R.; Friedrich, T.; Petrovic, N.; Altmann, S.; Brzezinka, K.; Gorka, M.; Graf, A.; Bäurle, I. FORGETTER2 protein phosphatase and phospholipase D modulate heat stress memory in Arabidopsis. Plant J. 2020, 104, 7–17. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hazama, K.; Ishida, K.; Komori, M.; Nishimura, K.; Matsuo, S. Inhibition of PLD1 activity causes ER stress via regulation of COPII vesicle formation. Biochem. Biophys. Res. Commun. 2017, 490, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Uzilday, B.; Ozgur, R.; Sekmen, A.H.; Turkan, I. Endoplasmic reticulum stress regulates glutathione metabolism and activities of glutathione related enzymes in Arabidopsis. Funct. Plant Biol. 2018, 45, 284–296. [Google Scholar] [CrossRef]
- Hollien, J. Evolution of the unfolded protein response. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 2458–2463. [Google Scholar] [CrossRef] [Green Version]
- Depaepe, T.; Hendrix, S.; Janse van Rensburg, H.C.; Van den Ende, W.; Cuypers, A.; Van Der Straeten, D. At the crossroads of survival and death: The reactive oxygen species–ethylene–sugar triad and the unfolded protein response. Trends Plant Sci. 2021, 26, 338–351. [Google Scholar] [CrossRef]
- RS Team. RStudio: Integrated Development Environment for R; RStudio, Inc.: Boston, Mass, USA, 2021. [Google Scholar]
- RC Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Chaney, R.L.; Coulombe, B.A.; Bell, P.F.; Angle, J.S.; Coulombe, B.A.; Bell, P.F.; Angle, J.S. Detailed method to screen dicot cultivars for resistance to Fe-chlorosis using FeDTPA and bicarbonate in nutrient solutions. J. Plant Nutr. 1992, 15, 2063–2083. [Google Scholar] [CrossRef]
- Lin, S.F.; Baumer, J.; Ivers, D.; Cianzio, S.; Shoemaker, R. Nutrient solution screening of fe chlorosis resistance in soybean evaluated by molecular characterization. J. Plant Nutr. 2000, 23, 1915–1928. [Google Scholar] [CrossRef]
- Buffalo, V. Scythe—A Bayesian Adapter Trimmer. Available online: https://github.com/ucdavis-bioinformatics/scythe (accessed on 24 October 2021).
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). Available online: https://github.com/ucdavis-bioinformatics/sickle (accessed on 24 October 2021).
- Dobin, A.; Gingeras, T.R. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, 1–4. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Grant, D.; Nelson, R.T.; Cannon, S.B.; Shoemaker, R.C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2009, 38, 843–846. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PS I-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Huala, E.; Dickerman, A.W.; Garcia-Hernandez, M.; Weems, D.; Reiser, L.; LaFond, F.; Hanley, D.; Kiphart, D.; Zhuang, M.; Huang, W.; et al. The Arabidopsis Information Resource (TAIR): A comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 2001, 29, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Libault, M.; Joshi, T.; Valliyodan, B.; Nguyen, H.T.; Xu, D.; Stacey, G.; Cheng, J. SoyDB: A knowledge database of soybean transcription factors. BMC Plant Biol. 2010, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. The Design of Experiment, 8th ed.; Oliver and Boyd: Edinburgh, UK; London, UK, 1966. [Google Scholar]
- Bonferroni, C.E. Il calcolo delle assicurazioni su gruppi di teste. Studi Onore Del Profr. Salvatore Ortu Carboni 1935, 13–60. [Google Scholar]
- Graham, M.A.; Silverstein, K.A.T.; Cannon, S.B.; VandenBosch, K.A. Computational identification and characterization of novel genes from legumes. Plant Physiol. 2004, 135, 1179–1197. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohlhase, D.R.; McCabe, C.E.; Singh, A.K.; O’Rourke, J.A.; Graham, M.A. Comparing Early Transcriptomic Responses of 18 Soybean (Glycine max) Genotypes to Iron Stress. Int. J. Mol. Sci. 2021, 22, 11643. https://doi.org/10.3390/ijms222111643
Kohlhase DR, McCabe CE, Singh AK, O’Rourke JA, Graham MA. Comparing Early Transcriptomic Responses of 18 Soybean (Glycine max) Genotypes to Iron Stress. International Journal of Molecular Sciences. 2021; 22(21):11643. https://doi.org/10.3390/ijms222111643
Chicago/Turabian StyleKohlhase, Daniel R., Chantal E. McCabe, Asheesh K. Singh, Jamie A. O’Rourke, and Michelle A. Graham. 2021. "Comparing Early Transcriptomic Responses of 18 Soybean (Glycine max) Genotypes to Iron Stress" International Journal of Molecular Sciences 22, no. 21: 11643. https://doi.org/10.3390/ijms222111643