
Manipulation of Focal Adhesion Signaling by Pathogenic Microbes


Abstract
:1. Introduction
2. Multiple Regulatory Mechanisms Control Focal Adhesion Dynamics
2.1. ECM Stiffness Sensing of FA Proteins
2.2. Tension Responsiveness of FA Proteins
2.3. Calpain and Caspase-Mediated Cleavage of FA Proteins
2.4. Autoinhibitory Mechanisms of FA Proteins
2.5. Nuclear Translocation of FA Proteins
2.6. Phosphorylation Events on Tyrosine and Serine/Threonine Residues of FA Proteins
3. Pathogenic Microbes Utilize “Outside–In” and “Inside–Out” Signaling during Host Remodeling
3.1. “Outside–In Signaling” upon Microbial Engagement with ECM or Integrin Receptors
3.1.1. TSA56
3.1.2. Opc
3.2. “Inside–Out Signaling” in Which Secreted Microbial Factors Signal from within the Cell
3.2.1. YopH
3.2.2. Certhrax
3.2.3. OspE
3.3. Effectors of Attaching and Effacing E. coli
3.3.1. EspO1
3.3.2. EspM
3.3.3. EspG
3.4. Vinculin-Mimetic Effectors
3.4.1. IpaA
3.4.2. Sca4
3.4.3. TarP
3.5. Manipulation of FA by Viral Proteins
3.5.1. KSHV TK
3.5.2. Tat
3.5.3. E7
4. Conclusions and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FA | Focal adhesion |
| ECM | Extracellular matrix |
| LD motif | Leucine–aspartic acid motif |
| VBD | Vinculin-binding domain |
| FAK | Focal adhesion kinase |
| ILK | Integrin-linked kinase |
| p130Cas | Crk-associated substrate |
| RIAM | Rap1-interacting adaptor molecule |
| FRET | Förster (Fluorescence) Resonance Energy Transfer |
| ABS | Actin-binding site |
| ROS | Reactive oxygen species |
| EPEC | Enteropathogenic Escherichia coli |
| EHEC | Enterohemorrhagic Escherichia coli |
| E4orf4 | E4 open reading frame 4 |
| CaM-LD | Calmodulin-like domain |
| iPalm | Interferometric photoactivation and localization microscopy |
| NES | Nuclear export sequence |
| NIS | Nuclear import sequence |
| PABP1 | Poly(A)-binding protein 1 |
| AR | Androgen receptor |
| ePABP | Polyadenylation binding protein |
| GR | Glucocorticoid receptor |
| NMP4 | Nuclear matrix protein 4 |
| HPV | Human papillomavirus |
| KSHV | Kaposi sarcoma herpesvirus |
| AGS | Adenocarcinoma gastric epithelial cells |
| RGD | Arg-Gly-Asp |
| PAI | Pathogenicity island |
| Vh | Vinculin head domain |
| Vt | Vinculin tail domain |
| EGFR | Epidermal growth factor receptor |
| ROCK | Rho-associated protein kinase |
| MLC | Myosin light chain |
| MEF | Mouse embryonic fibroblasts |
| HBMEC | Human brain microvascular endothelial cell |
| TK | Thymidine kinase |
References
- Press Release. World Health Organization, 29 July 2013. Available online: www.who.int/whr/1996/media_centre/press_release/en/ (accessed on 28 January 2021).
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking host cell highways: Manipulation of the host actin cytoskeleton by obligate intracellular bacterial pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caven, L.; Carabeo, R.A. Pathogenic puppetry: Manipulation of the host actin cytoskeleton by Chlamydia trachomatis. Int. J. Mol. Sci. 2020, 21, 90. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Garcia-del Portillo, F. Hijacking of eukaryotic functions by intracellular bacterial pathogens. Int. Microbiol. 2004, 7, 181–191. [Google Scholar]
- Flieger, A.; Frischknecht, F.; Häcker, G.; Hornef, M.W.; Pradel, G. Pathways of host cell exit by intracellular pathogens. Microb. Cell. 2018, 5, 525. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O.; Frame, M.C. Focal adhesion and actin dynamics: A place where kinases and proteases meet to promote invasion. Trends Cell Biol. 2004, 14, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. BBA-Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Schiller, H.B.; Fässler, R. Mechanosensitivity and compositional dynamics of cell–matrix adhesions. EMBO Rep. 2013, 14, 509–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doxey, A.C.; McConkey, B.J. Prediction of molecular mimicry candidates in human pathogenic bacteria. Virulence 2013, 4, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Gowthaman, U.; Eswarakumar, V.P. Molecular mimicry: Good artists copy, great artists steal. Virulence 2013, 4, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfenson, H.; Lavelin, I.; Geiger, B. Dynamic regulation of the structure and functions of integrin adhesions. Dev. Cell 2013, 24, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, K. Focal adhesions: A personal perspective on a half century of progress. FEBS J. 2017, 284, 3355–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C. Focal adhesion: A focal point in current cell biology and molecular medicine. Cell Adh. Migr. 2007, 1, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maziveyi, M.; Alahari, S.K. Cell matrix adhesions in cancer: The proteins that form the glue. Oncotarget 2017, 8, 48471. [Google Scholar] [CrossRef] [Green Version]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, G.; Leopizzi, M.; Urciuoli, E.; Peruzzi, B. Nuclear Functions of the Tyrosine Kinase Src. Int. J. Mol. Sci. 2020, 21, 2675. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.C.; Hu, H.F.; Hong, P.; Zhang, Q.H.; Xu, W.W.; He, Q.Y.; Li, B. Significance of integrin-linked kinase (ILK) in tumorigenesis and its potential implication as a biomarker and therapeutic target for human cancer. Am. J. Cancer Res. 2019, 9, 186. [Google Scholar]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef]
- Cooper, L.A.; Shen, T.L.; Guan, J.L. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell. Biol. 2003, 23, 8030–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.J.; Stacey, M.M.; Liu, B.A.; Pawson, T. Molecular mechanisms of SH2-and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a008987. [Google Scholar] [PubMed]
- Schlessinger, J. SH2/SH3 signaling proteins. Curr. Opin. Genet. Dev. 1994, 4, 25–30. [Google Scholar] [CrossRef]
- Hahn, S.; Kim, D. Transient protein-protein interaction of the SH3-peptide complex via closely located multiple binding sites. PLoS ONE 2012, 7, e32804. [Google Scholar] [CrossRef] [Green Version]
- Djinovic-Carugo, K.; Gautel, M.; Ylänne, J.; Young, P. The spectrin repeat: A structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002, 513, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Lenne, P.F.; Raae, A.J.; Altmann, S.M.; Saraste, M.; Hörber, J.K.H. States and transitions during forced unfolding of a single spectrin repeat. FEBS Lett. 2000, 476, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Villalobo, A.; González-Muñoz, M.; Berchtold, M.W. Proteins with calmodulin-like domains: Structures and functional roles. Cell. Mol. Life Sci. 2019, 76, 2299–2328. [Google Scholar] [CrossRef]
- Wang, K.K.; Villalobo, A.; Roufogalis, B.D. Calmodulin binding proteins as calpain substrates. Biochem. J. 1989, 262, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Shams, H.; Golji, J.; Garakani, K.; Mofrad, M.R. Dynamic regulation of α-actinin’s calponin homology domains on F-actin. Biophys. J. 2016, 110, 1444–1455. [Google Scholar]
- Kelly, D.F.; Taylor, D.W.; Bakolitsa, C.; Bobkov, A.A.; Bankston, L.; Liddington, R.C.; Taylor, K.A. Structure of the α-actinin-vinculin head domain complex determined by cryo-electron microscopy. J. Mol. Biol. 2006, 357, 562–573. [Google Scholar] [CrossRef]
- Bodeau, A.L.; Berrier, A.L.; Mastrangelo, A.M.; Martinez, R.; LaFlamme, S.E. A functional comparison of mutations in integrin β cytoplasmic domains: Effects on the regulation of tyrosine phosphorylation, cell spreading, cell attachment and β1 integrin conformation. J. Cell Sci. 2001, 114, 2795–2807. [Google Scholar] [PubMed]
- Baade, T.; Paone, C.; Baldrich, A.; Hauck, C.R. Clustering of integrin β cytoplasmic domains triggers nascent adhesion formation and reveals a protozoan origin of the integrin-talin interaction. Sci. Rep. 2019, 9, 5728. [Google Scholar] [CrossRef] [Green Version]
- Stout, T.J.; Foster, P.G.; Matthews, D.J. High-throughput structural biology in drug discovery: Protein kinases. Curr. Pharm. Des. 2004, 10, 1069–1082. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M.; Abrams, C.S. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002, 513, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Takala, H.; Ylänne, J. Binding properties and stability of the Ras-association domain of Rap1-GTP interacting adapter molecule (RIAM). PLoS ONE 2012, 7, e31955. [Google Scholar] [CrossRef]
- Velyvis, A.; Qin, J. LIM domain and its binding to target proteins. In Zinc Finger Proteins; Springer: Boston, MA, USA, 2005; pp. 99–105. [Google Scholar]
- Schiller, H.B.; Friedel, C.C.; Boulegue, C.; Fässler, R. Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 2011, 12, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Tumbarello, D.A.; Brown, M.C.; Turner, C.E. The paxillin LD motifs. FEBS Lett. 2002, 513, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Korenbaum, E.; Rivero, F. Calponin homology domains at a glance. J. Cell Sci. 2002, 115, 3543–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinweber, B.D.; Leavis, P.C.; Grabarek, Z.; Wang, C.L.A.; Morgan, K.G. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem. J. 1999, 344, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Weaver, J.D.; Sari, D.; Torres-Gomez, A.; Li, L.; Strauss, L.; Lafuente, E.M.; Boussiotis, V.A. The adaptor molecule RIAM integrates signaling events critical for integrin-mediated control of immune function and cancer progression. Sci. Signal. 2017, 10, 493. [Google Scholar] [CrossRef] [Green Version]
- Gough, R.E.; Goult, B.T. The tale of two talins–two isoforms to fine-tune integrin signalling. FEBS Lett. 2018, 592, 2108–2125. [Google Scholar] [CrossRef] [PubMed]
- Klapholz, B.; Brown, N.H. Talin–the master of integrin adhesions. J. Cell Sci. 2017, 130, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, J.L.; DeMali, K.A. Vinculin in cell–cell and cell–matrix adhesions. Cell. Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Miller, D.J.; Guibao, C.D.; Donato, D.M.; Hanks, S.K.; Zheng, J.J. Structural and functional insights into the interaction between the Cas family scaffolding protein p130Cas and the focal adhesion-associated protein paxillin. J. Biol. Chem. 2017, 292, 18281–18289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defilippi, P.; Di Stefano, P.; Cabodi, S. p130Cas: A versatile scaffold in signaling networks. Trends Cell Biol. 2006, 16, 257–263. [Google Scholar]
- Thomas, D.G.; Robinson, D.N. The fifth sense: Mechanosensory regulation of alpha-actinin-4 and its relevance for cancer metastasis. Semin. Cell Dev. Biol. 2017, 71, 68–74. [Google Scholar] [CrossRef]
- Wang, Y.X.; Wang, D.Y.; Guo, Y.C.; Guo, J. Zyxin: A mechanotransductor to regulate gene expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 413–425. [Google Scholar]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439. [Google Scholar]
- Lele, T.P.; Thodeti, C.K.; Pendse, J.; Ingber, D.E. Investigating complexity of protein–protein interactions in focal adhesions. Biochem. Biophys. Res. Commun. 2008, 369, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Hervy, M.; Hoffman, L.; Beckerle, M.C. From the membrane to the nucleus and back again: Bifunctional focal adhesion proteins. Curr. Opin. Cell Biol. 2006, 18, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20–S23. [Google Scholar]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Prager-Khoutorsky, M.; Lichtenstein, A.; Krishnan, R.; Rajendran, K.; Mayo, A.; Kam, Z.; Geiger, B.; Bershadsky, A.D. Fibroblast polarization is a matrix-rigidity-dependent process controlled by focal adhesion mechano-sensing. Nat. Cell Biol. 2011, 13, 1457–1465. [Google Scholar] [CrossRef]
- Hetmanski, J.H.; de Belly, H.; Busnelli, I.; Waring, T.; Nair, R.V.; Sokleva, V.; Swift, J. Membrane tension orchestrates rear retraction in matrix-directed cell migration. Dev. Cell 2019, 51, 460–475. [Google Scholar]
- Bastounis, E.E.; Yeh, Y.T.; Theriot, J.A. Matrix stiffness modulates infection of endothelial cells by Listeria monocytogenes via expression of cell surface vimentin. Mol. Biol. Cell. 2018, 29, 1571–1589. [Google Scholar] [CrossRef]
- Yusko, E.C.; Asbury, C.L. Force is a signal that cells cannot ignore. Mol. Biol. Cell. 2014, 25, 3717–3725. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular mechanotransduction: From tension to function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Ballestrem, C.; Kam, Z.; Geiger, B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 2003, 116, 4605–4613. [Google Scholar] [CrossRef] [Green Version]
- Oakes, P.W.; Beckham, Y.; Stricker, J.; Gardel, M.L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 2012, 196, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching single talin rod molecules activates vinculin binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Anekal, P.; Lim, C.J.; Liu, C.-C.; Ginsberg, M.H. Two modes of integrin activation form a binary molecular switch in adhesion maturation. Mol. Biol. Cell. 2013, 24, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Vigouroux, C.; Henriot, V.; Le Clainche, C. Talin dissociates from RIAM and associates to vinculin sequentially in response to the actomyosin force. Nat. Commun. 2020, 11, 3116. [Google Scholar] [CrossRef]
- Kumar, A.; Ouyang, M.; Van den Dries, K.; McGhee, E.J.; Tanaka, K.; Anderson, M.D.; Schwartz, M.A. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol. 2016, 213, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Anderson, K.L.; Swift, M.F.; Hanein, D.; Volkmann, N.; Schwartz, M.A. Local tension on talin in focal adhesions correlates with F-actin alignment at the Nanometer Scale. Biophys. J. 2018, 115, 1569–1579. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [PubMed] [Green Version]
- Wang, H.B.; Dembo, M.; Hanks, S.K.; Wang, Y.L. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA 2001, 98, 11295–11300. [Google Scholar] [CrossRef] [Green Version]
- Michael, K.E.; Dumbauld, D.W.; Burns, K.L.; Hanks, S.K.; Garcia, A.J. Focal adhesion kinase modulates cell adhesion strengthening via integrin activation. Mol. Biol. Cell 2009, 20, 2508–2519. [Google Scholar] [CrossRef] [Green Version]
- Seong, J.; Tajik, A.; Sun, J.; Guan, J.L.; Humphries, M.J.; Craig, S.E.; Shekaran, A.; García, A.J.; Lu, S.; Lin, M.Z.; et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2009, 110, 19372–19377. [Google Scholar]
- Zhou, J.; Aponte-Santamaría, C.; Sturm, S.; Bullerjahn, J.T.; Bronowska, A.; Gräter, F. Mechanism of focal adhesion kinase mechanosensing. PLoS Comput. Biol. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.S.; Baumann, F.; Daday, C.; Redondo, P.; Durner, E.; Jobst, M.A.; Gräter, F. Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 6766–6774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidel-Bar, R.; Milo, R.; Kam, Z.; Geiger, B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 2007, 120, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, R.; Schmid, H.; Münzberg, C.; Maass, U.; Krndija, D.; Adler, G.; Seufferlein, T.; Liedert, A.; Ignatius, A.; Oswald, F.; et al. Phosphorylation and turnover of paxillin in focal contacts is controlled by force and defines the dynamic state of the adhesion site. Cytoskeleton 2015, 72, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Blankman, E.; Deakin, N.O.; Hoffman, L.M.; Jensen, C.C.; Turner, C.E.; Beckerle, M.C. LIM domains target actin regulators paxillin and zyxin to sites of stress fiber strain. PLoS ONE 2013, 8, e69378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Uemura, A.; Nguyen, T.N.; Steele, A.N.; Yamada, S. The LIM domain of zyxin is sufficient for force-induced accumulation of zyxin during cell migration. Biophys. J. 2011, 101, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.M.; Jensen, C.C.; Chaturvedi, A.; Yoshigi, M.; Beckerle, M.C. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol. Biol. Cell 2012, 23, 1846–1859. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; del Rio, A.; Puklin-Faucher, E.; Gauthier, N.C.; Biais, N.; Sheetz, M.P. Integrin-dependent force transmission to the extracellular matrix by -actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. USA 2013, 110, E1361–E1370. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Verma, D.; Meng, F.; Davidson, M.W.; Suffoletto, K.; Hua, S.Z. Direct observation of α-actinin tension and recruitment at focal adhesions during contact growth. Exp. Cell Res. 2014, 327, 57–67. [Google Scholar]
- Meacci, G.; Wolfenson, H.; Liu, S.; Stachowiak, M.R.; Iskratsch, T.; Mathur, A.; Ghassemi, S.; Gauthier, N.; Tabdanov, E.; Lohner, J.; et al. α-Actinin links extracellular matrix rigidity-sensing contractile units with periodic cell-edge retractions. Mol. Biol. Cell 2016, 27, 3471–3479. [Google Scholar] [CrossRef] [Green Version]
- Sawada, Y.; Tamada, M.; Dubin-Thaler, B.J.; Cherniavskaya, O.; Sakai, R.; Tanaka, S.; Sheetz, M.P. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell 2006, 127, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janoštiak, R.; Brábek, J.; Auernheimer, V.; Tatárová, Z.; Lautscham, L.A.; Dey, T.; Rösel, D. CAS directly interacts with vinculin to control mechanosensing and focal adhesion dynamics. Cell. Mol. Life Sci. 2014, 71, 727–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowd, G.C.; Mortuza, R.; Ireton, K. Molecular Mechanisms of Intercellular Dissemination of Bacterial Pathogens. Trends Microbiol. 2020, 2, 127–141. [Google Scholar] [CrossRef]
- Franco, S.J.; Huttenlocher, A. Regulating cell migration: Calpains make the cut. J. Cell Sci. 2005, 118, 3829–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, S.; Perrin, B.; Huttenlocher, A. Isoform specific function of calpain 2 in regulating membrane protrusion. Exp. Cell Res. 2004, 299, 179–187. [Google Scholar] [CrossRef]
- Bate, N.; Gingras, A.R.; Bachir, A.; Horwitz, R.; Ye, F.; Patel, B.; Goult, B.T.; Critchley, D.R. Talin contains a C-terminal calpain2 cleavage site important in focal adhesion dynamics. PLoS ONE 2012, 7, e34461. [Google Scholar] [CrossRef] [Green Version]
- Serrano, K.; Devine, D.V. Vinculin is proteolyzed by calpain during platelet aggregation: 95 kDa cleavage fragment associates with the platelet cytoskeleton. Cell Motil. Cytoskelet. 2004, 58, 242–252. [Google Scholar]
- Cortesio, C.L.; Boateng, L.R.; Piazza, T.M.; Bennin, D.A.; Huttenlocher, A. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. J. Biol. Chem. 2011, 286, 9998–10006. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.T.; Bennin, D.A.; Huttenlocher, A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 2010, 285, 11418–11426. [Google Scholar] [CrossRef] [Green Version]
- Selliah, N.; Brooks, W.H.; Roszman, T.L. Proteolytic cleavage of alpha-actinin by calpain in T cells stimulated with anti-CD3 monoclonal antibody. J. Immunol. 1996, 156, 3215–3221. [Google Scholar] [PubMed]
- Shim, S.R.; Kook, S.; Kim, J.I.; Song, W.K. Degradation of focal adhesion proteins paxillin and p130cas by caspases or calpains in apoptotic rat-1 and L929 cells. Biochem. Biophys. Res. Commun. 2001, 286, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.X.; Yu, K.; Dong, J.; Zhao, L.; Liu, Z.; Zhang, Q.; Li, S.; Du, Y.; Cheng, H. Precise prediction of calpain cleavage sites and their aberrance caused by mutations in cancer. Front. Genet. 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Calderwood, D.A.; Yaspan, B.; Ginsberg, M.H. Calpain Cleavage Promotes Talin Binding to the β3Integrin Cytoplasmic Domain. J. Biol. Chem. 2001, 276, 28164–28170. [Google Scholar] [PubMed] [Green Version]
- Cluzel, C.; Saltel, F.; Lussi, J.; Paulhe, F.; Imhof, B.A.; Wehrle-Haller, B. The mechanisms and dynamics of αvβ3 integrin clustering in living cells. J. Cell Biol. 2005, 171, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Rajfur, Z.; Yousefi, N.; Chen, Z.; Jacobson, K.; Ginsberg, M.H. Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat. Cell Biol. 2009, 11, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Franco, S.J.; Rodgers, M.A.; Perrin, B.J.; Han, J.; Bennin, D.A.; Critchley, D.R.; Huttenlocher, A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 2004, 6, 977–983. [Google Scholar]
- Zhang, F.; Saha, S.; Kashina, A. Arginylation dependent regulation of a proteolytic product of talin is essential for cell-cell adhesion. J. Cell Biol. 2012, 197, 819–836. [Google Scholar]
- Saxena, M.; Changede, R.; Hone, J.C.; Wolfenson, H.; Sheetz, M.P. Force-induced calpain cleavage of Talin is critical for growth, adhesion development, and rigidity sensing. Nano Lett. 2017, 17, 7242–7251. [Google Scholar] [CrossRef]
- Truttmann, M.C.; Misselwitz, B.; Huser, S.; Hardt, W.D.; Critchley, D.R.; Dehio, C. Bartonella henselae engages inside-out and outside-in signaling by integrin β1 and talin1 during invasome-mediated bacterial uptake. J. Cell Sci. 2011, 124, 3591–3602. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Rounds, S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc. Res. 2012, 83, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoug, N.K.; Keeble, J.A.; Lindsay, J.; Valentijn, A.J.; Zhang, L.; Mills, D.; Gilmore, A.P. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia–differential roles for paxillin and p130Cas. J. Cell Sci. 2009, 122, 357–367. [Google Scholar]
- Gervais, F.G.; Thornberry, N.A.; Ruffolo, S.C.; Nicholson, D.W.; Roy, S. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J. Biol. Chem. 1998, 273, 17102–17108. [Google Scholar]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Garcia, F.; Serapio-Palacios, A.; Vidal, J.E.; Salazar, M.I.; Tapia-Pastrana, G. EspC promotes epithelial cell detachment by enteropathogenic Escherichia coli via sequential cleavages of a cytoskeletal protein and then focal adhesion proteins. Infect. Immun. 2014, 82, 2255–2265. [Google Scholar]
- Serapio-Palacios, A.; Navarro-Garcia, F. EspC, an autotransporter protein secreted by enteropathogenic Escherichia coli, causes apoptosis and necrosis through caspase and calpain activation, including direct procaspase-3 cleavage. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, J.N.; Champagne, C.; Gingras, M.C.; Robert, A. Adenovirus E4 open reading frame 4–induced apoptosis involves dysregulation of Src family kinases. J. Cell Biol. 2000, 150, 1037–1056. [Google Scholar] [CrossRef] [Green Version]
- Branton, P.E.; Roopchand, D.E. The role of adenovirus E4orf4 protein in viral replication and cell killing. Oncogene 2001, 20, 7855–7865. [Google Scholar]
- Robert, A.; Smadja-Lamère, N.; Landry, M.C.; Champagne, C.; Petrie, R.; Lamarche-Vane, N.; Lavoie, J.N. Adenovirus E4orf4 hijacks rho GTPase-dependent actin dynamics to kill cells: A role for endosome-associated actin assembly. Mol. Biol. Cell 2006, 17, 3329–3344. [Google Scholar] [CrossRef] [Green Version]
- Smadja-Lamère, N.; Boulanger, M.C.; Champagne, C.; Branton, P.E.; Lavoie, J.N. JNK-mediated phosphorylation of paxillin in adhesion assembly and tension-induced cell death by the adenovirus death factor E4orf4. J. Biol. Chem. 2008, 283, 34352–34364. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.B.; Goult, B.T. Adhesions Assemble!—Autoinhibition as a Major Regulatory Mechanism of Integrin-Mediated Adhesion. Front. Mol. Biosci. 2019, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yang, J.; Hirbawi, J.; Ye, S.; Perera, H.D.; Goksoy, E.; Dwivedi, P.; Plow, E.F.; Zhang, R.; Qin, J. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012, 22, 1533–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goult, B.T.; Xu, X.P.; Gingras, A.R.; Swift, M.; Patel, B.; Bate, N.; Kopp, P.M.; Barsukov, I.L.; Critchley, D.R.; Volkmann, N.; et al. Structural studies on full-length talin1 reveal a compact autoinhibited dimer: Implications for talin activation. J. Struct. Biol. 2013, 184, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedden, D.; Schumacher, S.; Kelley, C.F.; Zacharias, M.; Biertümpfel, C.; Fässler, R.; Mizuno, N. The Architecture of Talin1 reveals an autoinhibition mechanism. Cell 2019, 179, 120–131.e13. [Google Scholar] [CrossRef] [Green Version]
- Haage, A.; Goodwin, K.; Whitewood, A.; Camp, D.; Bogutz, A.; Turner, C.T.; Granville, D.J.; Lefebvre, L.; Plotnikov, S.; Goult, B.T.; et al. Talin autoinhibition regulates cell-ECM adhesion dynamics and wound healing in vivo. Cell Rep. 2018, 25, 2401–2416.e5. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.M.; Chen, H.; Johnson, R.P.; Choudhury, B.; Craig, S.W. Two distinct head-tail interfaces cooperate to suppress activation of vinculin by talin. J. Biol. Chem. 2005, 280, 17109–17117. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.M.; Kutscher, B.; Chen, H.; Murphy, D.B.; Craig, S.W. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J. Biol. Chem. 2006, 281, 16006–16015. [Google Scholar] [CrossRef] [Green Version]
- Carisey, A.; Tsang, R.; Greiner, A.M.; Nijenhuis, N.; Heath, N.; Nazgiewicz, A.; Kemkemer, R.; Derby, B.; Spatz, J.; Ballestrem, C. Vinculin regulates the recruitment and release of core focal adhesion proteins in a force-dependent manner. Curr. Biol. 2013, 23, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Chorev, D.S.; Volberg, T.; Livne, A.; Eisenstein, M.; Martins, B.; Kam, Z.; Jockusch, B.M.; Medalia, O.; Sharon, M.; Geiger, B. Conformational states during vinculin unlocking differentially regulate focal adhesion properties. Sci. Rep. 2018, 8, 2693. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.; Goult, B.T.; Chen, H.; Cong, P.; Sheetz, M.P.; Yan, J. Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep. 2014, 4, 4610. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Su, W.; Cho, E.A.; Zhang, H.; Huang, Q.; Philips, M.R.; Wu, J. Molecular basis for autoinhibition of RIAM regulated by FAK in integrin activation. Proc. Natl. Acad. Sci. USA 2019, 116, 3524–3529. [Google Scholar] [CrossRef] [Green Version]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.J.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Heim, J.B.; McDonald, C.A.; Wyles, S.P.; Sominidi-Damodaran, S.; Squirewell, E.J.; Li, M.; Meves, A. FAK auto-phosphorylation site tyrosine 397 is required for development but dispensable for normal skin homeostasis. PLoS ONE 2018, 13, e0200558. [Google Scholar] [CrossRef]
- Shenoy, S.; Chackalaparampil, I.; Bagrodia, S.; Lin, P.H.; Shalloway, D. Role of p34cdc2-mediated phosphorylations in two-step activation of pp60c-Src during mitosis. Proc. Natl. Acad. Sci. USA 1992, 89, 7237–7241. [Google Scholar] [CrossRef] [Green Version]
- Call, G.S.; Chung, J.Y.; Davis, J.A.; Price, B.D.; Primavera, T.S.; Thomson, N.C.; Wagner, M.V.; Hansen, M.D. Zyxin phosphorylation at serine 142 modulates the zyxin head–tail interaction to alter cell–cell adhesion. Biochem. Biophys. Res. Commun. 2011, 404, 780–784. [Google Scholar] [CrossRef]
- Young, P.; Gautel, M. The interaction of titin and α-actinin is controlled by a phospholipid-regulated intramolecular pseudoligand mechanism. EMBO J. 2000, 19, 6331–6340. [Google Scholar]
- Ribeiro, J.E.A.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjöblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle α-Actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Atherton, P.; Lausecker, F.; Carisey, A.; Gilmore, A.; Critchley, D.; Barsukov, I.; Ballestrem, C. Force-independent interactions of talin and vinculin govern integrin-mediated mechanotransduction. bioRxiv 2019. [Google Scholar] [CrossRef]
- Han, S.J.; Dean, K.M.; Whitewood, A.J.; Bachir, A.; Guttierrez, E.; Groisman, A.; Horwitz, A.R.; Goult, B.T.; Danuser, G. Formation of talin-vinculin pre-complexes dictates maturation of nascent adhesions by accelerated force transmission and vinculin recruitment. bioRxiv 2019, 735183. [Google Scholar] [CrossRef] [Green Version]
- Lemke, S.B.; Weidemann, T.; Cost, A.-L.L.; Grashoff, C.; Schnorrer, F. A small proportion of Talin molecules transmit forces at developing muscle attachments in vivo. PLoS Biol. 2019, 17, e3000057. [Google Scholar] [CrossRef] [Green Version]
- Case, L.B.; Baird, M.A.; Shtengel, G.; Campbell, S.L.; Hess, H.F.; Davidson, M.W.; Waterman, C.M. Molecular mechanism of vinculin activation and spatial organization in focal adhesions. Nat. Cell Biol. 2015, 17, 880–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yan, J.; Goult, B.T. Force-dependent binding constants. Biochemistry 2019, 58, 4696–4709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gilmore, T.D. Zyxin and paxillin proteins: Focal adhesion plaque LIM domain proteins go nuclear. BBA-Mol. Cell Res. 2003, 1593, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Hammes, S.R. Paxillin actions in the nucleus. Steroids 2018, 133, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [PubMed] [Green Version]
- Nix, D.A.; Fradelizi, J.; Bockholt, S.; Menichi, B.; Louvard, D.; Friederich, E.; Beckerle, M.C. Targeting of zyxin to sites of actin membrane interaction and to the nucleus. J. Biol. Chem. 2001, 276, 34759–34767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.J.; Roberts, M.S.; Choudhary, J.; Barry, S.T.; Mazaki, Y.; Sabe, H.; Norman, J.C. Paxillin associates with poly (A)-binding protein 1 at the dense endoplasmic reticulum and the leading edge of migrating cells. J. Biol. Chem. 2002, 277, 6428–6437. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.J.; Kantidakis, T.; Sabe, H.; Critchley, D.R.; Norman, J.C. Interaction of paxillin with poly (A)-binding protein 1 and its role in focal adhesion turnover and cell migration. Mol. Cell. Biol. 2005, 25, 3763–3773. [Google Scholar] [PubMed] [Green Version]
- Dong, J.M.; Lau, L.S.; Ng, Y.W.; Lim, L.; Manser, E. Paxillin nuclear-cytoplasmic localization is regulated by phosphorylation of the LD4 motif: Evidence that nuclear paxillin promotes cell proliferation. Biochem. J. 2009, 418, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.M.; Schwartz, M.A. Integrins regulate the association and phosphorylation of paxillin by c-Abl. J. Biol. Chem. 1998, 273, 14225–14230. [Google Scholar] [CrossRef] [Green Version]
- Sathe, A.R.; Shivashankar, G.V.; Sheetz, M.P. Nuclear transport of paxillin depends on focal adhesion dynamics and FAT domains. J. Cell Sci. 2016, 129, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Sabino, F.; Madzharova, E.; Auf dem Keller, U. Cell density-dependent proteolysis by HtrA1 induces translocation of zyxin to the nucleus and increased cell survival. Cell Death Dis. 2020, 11, 674. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Urciuoli, E.; Coletta, I.; Rizzuto, E.; De Vito, R.; Petrini, S.; D’Oria, V.; Peruzzi, B. Src nuclear localization and its prognostic relevance in human osteosarcoma. J. Cell. Physiol. 2018, 233, 1658–1670. [Google Scholar] [CrossRef]
- Le Roux, A.L.; Mohammad, I.L.; Mateos, B.; Arbesú, M.; Gairí, M.; Khan, F.A.; Pons, M. A myristoyl-binding site in the SH3 domain modulates c-Src membrane anchoring. Iscience 2019, 12, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Miedlich, S.U.; Taya, M.; Young, M.R.; Hammes, S.R. Paxillin and embryonic PolyAdenylation Binding Protein (ePABP) engage to regulate androgen-dependent Xenopus laevis oocyte maturation-A model of kinase-dependent regulation of protein expression. Mol. Cell. Endocrinol. 2017, 448, 87–97. [Google Scholar] [CrossRef]
- Fujimoto, N.; Yeh, S.; Kang, H.Y.; Inui, S.; Chang, H.C.; Mizokami, A.; Chang, C. Cloning and characterization of androgen receptor coactivator, ARA55, in human prostate. J. Biol. Chem. 1999, 274, 8316–8321. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Guerrero, J.; Hong, H.; DeFranco, D.B.; Stallcup, M.R. Interaction of the τ2 transcriptional activation domain of glucocorticoid receptor with a novel steroid receptor coactivator, Hic-5, which localizes to both focal adhesions and the nuclear matrix. Mol. Biol. Cell. 2000, 11, 2007–2018. [Google Scholar] [CrossRef]
- Marášek, P.; Dzijak, R.; Studenyak, I.; Fišerová, J.; Uličná, L.; Novák, P.; Hozák, P. Paxillin-dependent regulation of IGF2 and H19 gene cluster expression. J. Cell Sci. 2015, 128, 3106–3116. [Google Scholar] [CrossRef] [Green Version]
- Morinobu, M.; Nakamoto, T.; Hino, K.; Tsuji, K.; Shen, Z.J.; Nakashima, K.; Noda, M. The nucleocytoplasmic shuttling protein CIZ reduces adult bone mass by inhibiting bone morphogenetic protein–induced bone formation. J. Exp. Med. 2005, 201, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, H.; Marynen, P. Interaction partners for human ZNF384/CIZ/NMP4—zyxin as a mediator for p130CAS signaling? Exp. Cell Res. 2006, 312, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; McNally, B.T.; Igarashi, P. Zyxin regulates migration of renal epithelial cells through activation of hepatocyte nuclear factor-1β. Am. J. Phys. Renal Physiol. 2013, 305, F100–F110. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-T.; Mikolon, D.; Stupack, D.G.; Schlaepfer, D.D. FERM control of FAK function, Implications for cancer therapy. Cell Cycle 2008, 7, 2306–2314. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Nakayama, Y.; Togashi, Y.; Obata, Y.; Kuga, T.; Kasahara, K.; Yamaguchi, N. Nuclear localization of Lyn tyrosine kinase mediated by inhibition of its kinase activity. Exp. Cell Res. 2008, 314, 3392–3404. [Google Scholar] [CrossRef]
- Takahashi, A.; Obata, Y.; Fukumoto, Y.; Nakayama, Y.; Kasahara, K.; Kuga, T.; Yamaguchi, N. Nuclear localization of Src-family tyrosine kinases is required for growth factor-induced euchromatinization. Exp. Cell Res. 2009, 315, 1117–1141. [Google Scholar] [CrossRef]
- Degenhardt, Y.Y.; Silverstein, S. Interaction of zyxin, a focal adhesion protein, with the e6 protein from human papillomavirus type 6 results in its nuclear translocation. J. Virol. 2001, 75, 11791–11802. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Salgia, R.; Li, J.L.; Griffin, J.D.; Howley, P.M. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J. Biol. Chem. 1997, 272, 33373–33376. [Google Scholar] [CrossRef] [Green Version]
- Pol, S.B.V.; Brown, M.C.; Turner, C.E. Association of bovine papillomavirus type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene 1998, 16, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Howley, P.M. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 1997, 94, 4412–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, J.; Das, K.; Dasgupta, B.; Pol, S.B.V. Competitive binding to a charged leucine motif represses transformation by a papillomavirus E6 oncoprotein. Virology 2000, 271, 163–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, R.; Brimer, N.; Pol, S.V. Transformation by bovine papillomavirus type 1 E6 requires paxillin. J. Virol. 2008, 82, 5962–5966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, V.; Boyer, B.; Lentz, D.; Turner, C.E.; Thiery, J.P.; Vallés, A.M. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J. Cell Biol. 2000, 148, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, D.J.; Schroeder, M.J.; Brame, C.J.; Whitmore, L.; Shabanowitz, J.; Hunt, D.F.; Horwitz, A.R. Paxillin phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 2005, 118, 4925–4929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Zeid, N.; Vallés, A.M.; Boyer, B. Serine phosphorylation regulates paxillin turnover during cell migration. Cell Commun. Signal. 2006, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Yan, D.P.; Ge, B.X. JNK regulates cell migration through promotion of tyrosine phosphorylation of paxillin. Cell. Signal. 2008, 20, 2002–2012. [Google Scholar] [CrossRef]
- Ishibe, S.; Joly, D.; Zhu, X.; Cantley, L.G. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol. Cell 2003, 12, 1275–1285. [Google Scholar] [CrossRef]
- Ishibe, S.; Joly, D.; Liu, Z.X.; Cantley, L.G. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 2004, 16, 257–267. [Google Scholar] [CrossRef]
- Woodrow, M.A.; Woods, D.; Cherwinski, H.M.; Stokoe, D.; McMahon, M. Ras-induced serine phosphorylation of the focal adhesion protein paxillin is mediated by the Raf→ MEK→ ERK pathway. Exp. Cell Res. 2003, 287, 325–338. [Google Scholar] [CrossRef]
- Brown, M.C.; Perrotta, J.A.; Turner, C.E. Serine and threonine phosphorylation of the paxillin LIM domains regulates paxillin focal adhesion localization and cell adhesion to fibronectin. Mol. Biol. Cell. 1998, 9, 1803–1816. [Google Scholar]
- Brown, M.C.; Cary, L.A.; Jamieson, J.S.; Cooper, J.A.; Turner, C.E. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol. Biol. Cell. 2005, 16, 4316–4328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellis, S.L.; Perrotta, J.A.; Curtis, M.S.; Turner, C.E. Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem. J. 1997, 325, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratnikov, B.; Ptak, C.; Han, J.; Shabanowitz, J.; Hunt, D.F.; Ginsberg, M.H. Talin phosphorylation sites mapped by mass spectrometry. J. Cell Sci. 2005, 118, 4921–4923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goult, B.T.; Bouaouina, M.; Elliott, P.R.; Bate, N.; Patel, B.; Gingras, A.R.; Grossmann, J.G.; Roberts, G.C.K.; Calderwood, D.A.; Critchley, D.R.; et al. Structure of a double ubiquitin-like domain in the talin head: A role in integrin activation. EMBO J. 2010, 29, 1069–1080. [Google Scholar] [CrossRef]
- Elliott, P.R.; Goult, B.T.; Kopp, P.M.; Bate, N.; Grossmann, J.G.; Roberts, G.C.; Critchley, D.R.; Barsukov, I.L. The structure of the talin head reveals a novel extended conformation of the FERM domain. Structure 2010, 18, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Luo, X.; Sun, Y.; Cui, Z.; Liu, Y.; Liu, R.; Guo, X. High stoichiometry phosphorylation of talin at T144/T150 or S446 produces contrasting effects on calpain-mediated talin cleavage and cell migration. J. Cancer 2016, 7, 1645. [Google Scholar] [CrossRef] [Green Version]
- Katzemich, A.; Long, J.Y.; Panneton, V.; Fisher, L.A.; Hipfner, D.; Schöck, F. Slik phosphorylation of Talin T152 is crucial for proper Talin recruitment and maintenance of muscle attachment in Drosophila. Development 2019, 146, dev176339. [Google Scholar] [CrossRef] [Green Version]
- Möhl, C.; Kirchgeßner, N.; Schäfer, C.; Küpper, K.; Born, S.; Diez, G.; Goldmann, W.H.; Merkel, R.; Hoffmann, B. Becoming stable and strong: The interplay between vinculin exchange dynamics and adhesion strength during adhesion site maturation. Cell Motil. Cytoskelet. 2009, 66, 350–364. [Google Scholar] [CrossRef]
- Golji, J.; Wendorff, T.; Mofrad, M.R. Phosphorylation primes vinculin for activation. Biophys. J. 2012, 102, 2022–2030. [Google Scholar] [CrossRef] [Green Version]
- Auernheimer, V.; Lautscham, L.A.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Fabry, B.; Goldmann, W.H. Vinculin phosphorylation at residues Y100 and Y1065 is required for cellular force transmission. J. Cell Sci. 2015, 128, 3435–3443. [Google Scholar]
- Modzelewska, K.; Newman, L.P.; Desai, R.; Keely, P.J. Ack1 mediates Cdc42-dependent cell migration and signaling to p130Cas. J. Biol. Chem. 2006, 281, 37527–37535. [Google Scholar] [CrossRef] [Green Version]
- Palanisamy, A.P.; Suryakumar, G.; Panneerselvam, K.; Willey, C.D.; Kuppuswamy, D. A Kinase-Independent Function of c-Src Mediates p130Cas Phosphorylation at the Serine-639 Site in Pressure Overloaded Myocardium. J. Cell. Biochem. 2015, 116, 2793–2803. [Google Scholar] [CrossRef] [Green Version]
- Eide, B.L.; Turck, C.W.; Escobedo, J.A. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol. Cell. Biol. 1995, 15, 2819–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calalb, M.B.; Zhang, X.; Polte, T.R.; Hanks, S.K. Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem. Biophys. Res. Commun. 1996, 228, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Mikolon, D.; Molina, J.E.; Hsia, D.A.; Hanson, D.A.; Chi, A.; Lim, S.T.; Bernard-Trifilo, J.A.; Ilic, D.; Stupack, D.G.; et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene 2006, 25, 5969–5984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.H.; Chan, P.C.; Chen, C.L.; Chen, H.C. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene 2011, 30, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 2005, 331, 1–14. [Google Scholar] [CrossRef]
- Stover, D.R.; Furet, P.; Lydon, N.B. Modulation of the SH2 binding specificity and kinase activity of Src by tyrosine phosphorylation within its SH2 domain. J. Biol. Chem. 1997, 271, 12481–12487. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.M.; Wang, Y.; Fallen, C.J. Cell transformation and activation of pp60 c-src by overexpression of a protein tyrosine phosphatase. Nature 1992, 359, 336–339. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Obara, Y.; Labudda, K.; Dillon, T.J.; Stork, P.J. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. J. Cell Sci. 2004, 117, 6085–6094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, T.; Shao, H.; Joughin, B.A.; Lauffenburger, D.A.; Wells, A.; Camacho, C.J. Tandem phosphorylation within an intrinsically disordered region regulates ACTN4 function. Sci. Signal. 2015, 8, ra51. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Wu, C.; Wells, A. Phosphorylation of α-actinin 4 upon epidermal growth factor exposure regulates its interaction with actin. J. Biol. Chem. 2010, 285, 2591–2600. [Google Scholar] [CrossRef] [Green Version]
- Sari, D.; Tsopoulidis, N.; Asara, J.M.; Patsoukis, N.; Boussiotis, V.A. Phosphorylation of Tyrosine 340 in the Plekstrin Homology Domain of RIAM Is Required for Translocation of RIAM to the Plasma Membrane, Phosphorylation of RIAM-Associated PLC-g1 and LFA-1 Activation. Blood 2014, 124, 2743. [Google Scholar] [CrossRef]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK–Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef]
- Moese, S.; Selbach, M.; Brinkmann, V.; Karlas, A.; Haimovich, B.; Backert, S.; Meyer, T.F. The Helicobacter pylori CagA protein disrupts matrix adhesion of gastric epithelial cells by dephosphorylation of vinculin. Cell. Microbiol. 2007, 9, 1148–1161. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Takahashi, A.; Azuma, T.; Higashi, H.; Hatakeyama, M. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol. Cell. Biol. 2006, 26, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Lucciarini, R.; Amantini, C.; Jacobelli, J.; Spreghini, E.; Ballarini, P.; Piccoli, M.; Gismondi, A. Candida albicans expresses a focal adhesion kinase-like protein that undergoes increased tyrosine phosphorylation upon yeast cell adhesion to vitronectin and the EA.hy 926 human endothelial cell line. Infect. Immun. 2002, 70, 3804–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogan, T.V.; Jadoun, J.; Mittelman, L.; Hirschberg, K.; Osherov, N. Involvement of secreted Aspergillus fumigatus proteases in disruption of the actin fiber cytoskeleton and loss of focal adhesion sites in infected A549 lung pneumocytes. J. Infect. Dis. 2004, 189, 1965–1973. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, M. Enforcing host cell polarity: An apicomplexan parasite strategy towards dissemination. Curr. Opin. Microbiol. 2011, 14, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Konkel, M.E.; Talukdar, P.K.; Negretti, N.M.; Klappenbach, C.M. Taking Control: Campylobacter jejuni Binding to Fibronectin Sets the Stage for Cellular Adherence and Invasion. Front. Microbiol. 2020, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.L.; Samuelson, D.R.; Eucker, T.P.; O’Loughlin, J.L.; Konkel, M.E. The fibronectin-binding motif within FlpA facilitates Campylobacter jejuni adherence to host cell and activation of host cell signaling. Emerg. Microbes Infect 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Monteville, M.R.; Yoon, J.E.; Konkel, M.E. Maximal adherence and invasion of INT 407 cells by Campylobacter jejuni requires the CadF outer-membrane protein and microfilament reorganization. Microbiology 2003, 149, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slanina, H.; König, A.; Hebling, S.; Hauck, C.R.; Frosch, M.; Schubert-Unkmeir, A. Entry of Neisseria meningitidis into mammalian cells requires the Src family protein tyrosine kinases. Infect. Immun. 2010, 78, 1905–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cue, D.; Southern, S.O.; Southern, P.J.; Prabhakar, J.; Lorelli, W.; Smallheer, J.M.; Mousa, S.A.; Cleary, P.P. A nonpeptide integrin antagonist can inhibit epithelial cell ingestion of Streptococcus pyogenes by blocking formation of integrin alpha5beta1–fibronectin– M1 protein complexes. Proc. Natl. Acad. Sci. USA 2000, 97, 2858–2863. [Google Scholar]
- Ozeri, V.; Rosenshine, I.; Ben-Ze’Ev, A.; Bokoch, G.M.; Jou, T.S.; Hanski, E. De novo formation of focal complex-like structures in host cells by invading Streptococci. Mol. Microbiol. 2001, 41, 561–573. [Google Scholar] [CrossRef]
- Sinha, B.; Francois, P.P.; Nusse, O.; Foti, M.; Hartford, O.M.; Vaudaux, P.; Foster, T.J.; Lew, D.P.; Herrmann, M.; Krause, K.H. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell. Microbiol. 1999, 1, 101–117. [Google Scholar] [CrossRef]
- Agerer, F.; Lux, S.; Michel, A.; Rohde, M.; Ohlsen, K.; Hauck, C.R. Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalization. J. Cell Sci. 2005, 118, 2189–2200. [Google Scholar] [CrossRef] [Green Version]
- El Tahir, Y.; Skurnik, M. YadA, the multifaceted Yersinia adhesin. Int. J. Med. Microbiol. 2001, 291, 209–218. [Google Scholar] [CrossRef]
- Eitel, J.; Heise, T.; Thiesen, U.; Dersch, P. Cell invasion and IL-8 production pathways initiated by YadA of Yersinia pseudotuberculosis require common signalling molecules (FAK, c-Src, Ras) and distinct cell factors. Cell. Microbiol. 2005, 7, 63–77. [Google Scholar] [CrossRef]
- Nägele, V.; Heesemann, J.; Schielke, S.; Jiménez-Soto, L.F.; Kurzai, O.; Ackermann, N. Neisseria meningitidis Adhesin NadA Targets β1 Integrins functional similarity to Yersinia Invasin. J. Biol. Chem. 2011, 286, 20536–20546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamburger, Z.A.; Brown, M.S.; Isberg, R.R.; Bjorkman, P.J. Crystal structure of invasin: A bacterial integrin-binding protein. Science 1999, 286, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Alrutz, M.A.; Isberg, R.R. Involvement of focal adhesion kinase in invasin-mediated uptake. Proc. Natl. Acad. Sci. USA 1998, 95, 13658–13663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemann, T.; Hofbaur, S.; Tegtmeyer, N.; Huber, S.; Sewald, N.; Wessler, S.; Backert, S.; Rieder, G. Helicobacter pylori CagL dependent induction of gastrin expression via a novel αvβ5-integrin–integrin linked kinase signalling complex. Gut 2012, 61, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Buß, M.; Tegtmeyer, N.; Schnieder, J.; Dong, X.; Li, J.; Springer, T.A.; Niemann, H.H. Specific high affinity interaction of Helicobacter pylori CagL with integrin αVβ6 promotes type IV secretion of CagA into human cells. FEBS J. 2019, 286, 3980–3997. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Hartig, R.; Delahay, R.M.; Rohde, M.; Brandt, S.; Conradi, J.; Backert, S. A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem. 2010, 285, 23515–23526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.A.; Cho, N.H.; Seong, S.Y.; Choi, M.S.; Kim, I.S. Intracellular invasion by Orientia tsutsugamushi is mediated by integrin signaling and actin cytoskeleton rearrangements. Infect. Immun. 2010, 78, 1915–1923. [Google Scholar] [PubMed] [Green Version]
- Urbinati, C.; Bugatti, A.; Giacca, M.; Schlaepfer, D.; Presta, M.; Rusnati, M. αvβ3-integrin-dependent activation of focal adhesion kinase mediates NF-κB activation and motogenic activity by HIV-1 Tat in endothelial cells. J. Cell Sci. 2005, 118, 3949–3958. [Google Scholar] [CrossRef] [Green Version]
- Black, D.S.; Bliska, J.B. Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO J. 1997, 16, 2730–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, C.; Carballeira, N.; Wolf-Watz, H.; Fällman, M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997, 16, 2307–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, D.S.; Montagna, L.G.; Zitsmann, S.; Bliska, J.B. Identification of an amino-terminal substrate-binding domain in the Yersinia tyrosine phosphatase that is required for efficient recognition of focal adhesion targets. Mol. Microbiol. 1998, 29, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Hamid, N.; Gustavsson, A.; Andersson, K.; McGee, K.; Persson, C.; Rudd, C.E.; Fällman, M. YopH dephosphorylates Cas and Fyn-binding protein in macrophages. Microb Pathog. 1999, 27, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Black, D.S.; Marie-Cardine, A.; Schraven, B.; Bliska, J.B. The Yersinia tyrosine phosphatase YopH targets a novel adhesion-regulated signalling complex in macrophages. Cell Microbiol. 2000, 2, 401–414. [Google Scholar] [CrossRef]
- Deleuil, F.; Mogemark, L.; Francis, M.S.; Wolf-Watz, H.; Fällman, M. Interaction between the Yersinia protein tyrosine phosphatase YopH and eukaryotic Cas/Fyb is an important virulence mechanism. Cell. Microbiol. 2003, 5, 53–64. [Google Scholar] [CrossRef]
- Bourdet-Sicard, R.; Rüdiger, M.; Jockusch, B.M.; Gounon, P.; Sansonetti, P.J.; Van Nhieu, G.T. Binding of the Shigella protein IpaA to vinculin induces F-actin depolymerization. EMBO J. 1999, 18, 5853–5862. [Google Scholar] [CrossRef]
- Tran Van Nhieu, G.; Sansonetti, P.J. Mechanism of Shigella entry into epithelial cells. Curr. Opin. Microbiol. 1999, 2, 51–55. [Google Scholar] [CrossRef]
- Izard, T.; Tran Van Nhieu, G.; Bois, P.R.J. Shigella applies molecular mimicry to subvert vinculin and invade host cells. J. Cell Biol. 2006, 175, 465–475. [Google Scholar] [CrossRef]
- Park, H.J.; Valencia-Gallardo, C.; Sharff, A.; Van Nhieu, G.T.; Izard, T. Novel vinculin binding site of the IpaA invasin of Shigella. J. Biol. Chem. 2011, 286, 23214–23221. [Google Scholar] [CrossRef] [Green Version]
- DeMali, K.A.; Jue, A.L.; Burridge, K. IpaA targets β1 integrins and rho to promote actin cytoskeleton rearrangements necessary for Shigella entry. J. Biol. Chem. 2006, 281, 39534–39541. [Google Scholar] [CrossRef] [Green Version]
- Thwaites, T.R.; Pedrosa, A.T.; Peacock, T.P.; Carabeo, R.A. Vinculin Interacts with the Chlamydia Effector TarP via a Tripartite Vinculin Binding Domain to Mediate Actin Recruitment and Assembly at the Plasma Membrane. Front. Cell. Infect. Microbiol. 2015, 5, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrosa, A.T.; Murphy, K.N.; Nogueira, A.T.; Brinkworth, A.J.; Thwaites, T.R.; Aaron, J.; Carabeo, R.A. A post-invasion role for Chlamydia type III effector TarP in modulating the dynamics and organization of host cell focal adhesions. J. Biol. Chem. 2020, 295, 14763–14779. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, J.H.; Gouin, E.; Cossart, P.; Izard, T. The Rickettsia surface cell antigen 4 applies mimicry to bind to and activate vinculin. J. Biol. Chem. 2011, 286, 35096–35103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamason, R.L.; Bastounis, E.; Kafai, N.M.; Serrano, R.; Theriot, J.A.; Welch, M.D. Rickettsia Sca4 Reduces Vinculin-Mediated Intercellular Tension to Promote Spread. Cell 2016, 167, 670–683.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visschedyk, D.; Rochon, A.; Tempel, W.; Dimov, S.; Park, H.-W.; Merrill, A.R. Certhrax toxin, an anthrax-related ADP-ribosyltransferase from Bacillus cereus. J. Biol. Chem. 2012, 287, 41089–44110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.C.; Vergis, J.M.; Ebrahimi, A.V.; Ventura, C.L.; O’Brien, A.D.; Barbieri, J.T. Host cell cytotoxicity and cytoskeleton disruption by CerADPr, an ADP-ribosyltransferase of Bacillus cereus G9241. Biochemistry 2013, 52, 2309–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.C.; Barbieri, J.T. Bacillus cereus Certhrax ADP-ribosylates vinculin to disrupt focal adhesion complexes and cell adhesion. J. Biol. Chem. 2014, 289, 10650–10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avraham, H.K.; Jiang, S.; Lee, T.-H.; Prakash, O.; Avraham, S. HIV-1 Tat-Mediated Effects on Focal Adhesion Assembly and Permeability in Brain Microvascular Endothelial Cells. J. Immunol. 2004, 173, 6228–6233. [Google Scholar] [CrossRef] [Green Version]
- Thwaites, T.; Nogueira, A.T.; Campeotto, I.; Silva, A.P.; Grieshaber, S.S.; Carabeo, R.A. The Chlamydia effector TarP mimics the mammalian leucine-aspartic acid motif of paxillin to subvert the focal adhesion kinase during invasion. J. Biol. Chem. 2014, 289, 30426–30442. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.B.; Turner, R.; Stevenson, P.G.; Way, M. KSHV—TK is a tyrosine kinase that disrupts focal adhesions and induces Rho-mediated cell contraction. EMBO J. 2015, 34, 448–465. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Terajima, J.; Izumiya, H.; Mitobe, J.; Komano, T.; Watanabe, H. OspE2 of Shigella sonnei is required for the maintenance of cell architecture of bacterium-infected cells. Infect. Immun. 2006, 74, 2587–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Ogawa, M.; Fujita, Y.; Yoshikawa, Y.; Nagai, T.; Koyama, T.; Nagai, S.; Lange, A.; Fässler, R.; Sasakawa, C. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 2009, 459, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Morita-Ishihara, T.; Miura, M.; Iyoda, S.; Izumiya, H.; Watanabe, H.; Ohnishi, M.; Terajima, J. EspO1-2 regulates EspM2-mediated RhoA activity to stabilize formation of focal adhesions in enterohemorrhagic Escherichia coli-infected host cells. PLoS ONE 2013, 8, e55960. [Google Scholar] [CrossRef] [PubMed]
- Cantarelli, V.V.; Takahashi, A.; Yanagihara, I.; Akeda, Y.; Imura, K.; Kodama, T.; Honda, T. Talin, a host cell protein, interacts directly with the translocated intimin receptor, Tir, of enteropathogenic Escherichia coli, and is essential for pedestal formation. Cell. Microbiol. 2001, 3, 745–751. [Google Scholar] [CrossRef]
- Valencia-Gallardo, C.; Bou-Nader, C.; Aguilar-Salvador, D.I.; Carayol, N.; Quenech’Du, N.; Pecqueur, L.; Park, H.; Fontecave, M.; Izard, T.; Van Nhieu, G.T. Shigella IpaA binding to talin stimulates filopodial capture and cell adhesion. Cell Rep. 2019, 26, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Arbeloa, A.; Bulgin, R.R.; MacKenzie, G.; Shaw, R.K.; Pallen, M.J.; Crepin, V.F.; Berger, C.N.; Frankel, G. Subversion of actin dynamics by EspM effectors of attaching and effacing bacterial pathogens. Cell Microbiol. 2008, 10, 1429–1441. [Google Scholar] [CrossRef] [Green Version]
- Simovitch, M.; Sason, H.; Cohen, S.; Zahavi, E.E.; Melamed-Book, N.; Weiss, A.; Aroeti, B.; Rosenshine, I. EspM inhibits pedestal formation by enterohaemorrhagic Escherichia coli and enteropathogenic E. coli and disrupts the architecture of a polarized epithelial monolayer. Cell Microbiol. 2010, 12, 489–505. [Google Scholar] [CrossRef]
- Todorovic, B.; Nichols, A.C.; Chitilian, J.M.; Myers, M.P.; Shepherd, T.G.; Parsons, S.J.; Barrett, J.W.; Banks, L.; Mymryk, J.S. The Human Papillomavirus E7 Proteins Associate with p190RhoGAP and Alter Its Function. J. Virol. 2014, 88, 3653–3663. [Google Scholar] [CrossRef] [Green Version]
- Stones, D.H.; Krachler, A.M. Fatal attraction: How bacterial adhesins affect host signaling and what we can learn from them. Int. J. Mol. Sci. 2015, 16, 2626–2640. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, C.; Ohlsen, K.; Hauck, C.R. Integrin-mediated uptake of fibronectin-binding bacteria. Eur. J. Cell Biol. 2011, 90, 891–896. [Google Scholar] [CrossRef]
- Hamzaoui, N.; Kerneis, S.; Caliot, E.; Pringault, E. Expression and distribution of b1 integrins in vitro-induced M cells: Implications for Yersinia adhesion to Peyer’s patch epithelium. Cell. Microbiol. 2004, 6, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Scibelli, A.; Matteoli, G.; Roperto, S.; Alimenti, E.; Di Pineto, L.; Pavone, L.M.; Della Morte, R.; Menna, L.F.; Fioretti, A.; Staiano, N. Flavoridin inhibits Yersinia enterocolitica uptake into fibronectinadherent HeLa cells. FEMS Microbiol. Lett. 2005, 247, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuschle, E.; Keller, B.; Siegfried, A.; Manncke, B.; Spaeth, T.; Köberle, M.; Autenrieth, I.B. Role of β1 integrins and bacterial adhesins for Yop injection into leukocytes in Yersinia enterocolitica systemic mouse infection. Int. J. Med. Microbiol. 2016, 306, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytönen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, K.; Chen, M. Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases 2013, 4, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huveneers, S.; Danen, E.H. Adhesion signaling–crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eucker, T.P.; Konkel, M.E. The cooperative action of bacterial fibronectin-binding proteins and secreted proteins promote maximal Campylobacter jejuni invasion of host cells by stimulating membrane ruffling. Cell Microbiol. 2012, 14, 226–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.A.; Wass, C.A.; Kim, K.S.; Schlaepfer, D.D.; Prasadarao, N.V. Involvement of Focal Adhesion Kinase in Escherichia coli Invasion of Human Brain Microvascular Endothelial Cells. Infect. Immun. 2000, 68, 6423–6430. [Google Scholar] [CrossRef]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. OipA plays a role in Helicobacter pylori-induced focal adhesion kinase activation and cytoskeletal re-organization. Cell. Microbiol. 2008, 10, 1008–1020. [Google Scholar] [CrossRef] [Green Version]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Paxillin is a novel cellular target for converging Helicobacter pylori-induced cellular signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G601–G611. [Google Scholar] [CrossRef] [Green Version]
- Kee, S.H.; Cho, K.A.; Kim, M.K.; Lim, B.U.; Chang, W.H.; Kang, J.S. Disassembly of focal adhesions during apoptosis of endothelial cell line ECV304 infected with Orientia tsutsugamushi. Microb. Pathog. 1999, 27, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Unkmeir, A.; Latsch, K.; Dietrich, G.; Wintermeyer, E.; Schinke, B.; Schwender, S.; Frosch, M. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 2002, 46, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Slanina, H.; Hebling, S.; Hauck, C.R.; Schubert-Unkmeir, A. Cell invasion by Neisseria meningitidis requires a functional interplay between the focal adhesion kinase, Src and cortactin. PLoS ONE 2012, 7, e39613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambotin, M.; Hoffmann, I.; Laran-Chich, M.P.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Invasion of endothelial cells by Neisseria meningitidis requires cortactin recruitment by a phosphoinositide-3-kinase/Rac1 signalling pathway triggered by the lipo-oligosaccharide. J. Cell Sci. 2005, 118, 3805–3816. [Google Scholar] [CrossRef] [Green Version]
- Guan, K.L.; Dixon, J.E. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science 1990, 249, 553–556. [Google Scholar] [CrossRef]
- Bliska, J.B.; Guan, K.L.; Dixon, J.E.; Falkow, S. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc. Natl. Acad. Sci. USA 1991, 88, 1187–1191. [Google Scholar] [CrossRef] [Green Version]
- Bliska, J.B.; Clemens, J.C.; Dixon, J.E.; Falkow, S. The Yersinia tyrosine phosphatase: Specificity of a bacterial virulence determinant for phosphoproteins in the J774A. 1 macrophage. J. Exp. Med. 1992, 176, 1625–1630. [Google Scholar] [CrossRef]
- Andersson, K.; Carballeira, N.; Magnusson, K.E.; Persson, C.; Stendahl, O.; Wolf-Watz, H.; Fällman, M. YopH of Yersinia pseudotuberculosis interrupts early phosphotyrosine signalling associated with phagocytosis. Mol. Microbiol. 1996, 20, 1057–1069. [Google Scholar] [CrossRef]
- Persson, C.; Nordfelth, R.; Andersson, K.; Forsberg, Å.; Wolf-Watz, H.; Fällman, M. Localization of the Yersinia PTPase to focal complexes is an important virulence mechanism. Mol. Microbiol. 1999, 33, 828–838. [Google Scholar] [CrossRef]
- Mogemark, L.; McGee, K.; Yuan, M.; Deleuil, F.; Fällman, M. Disruption of target cell adhesion structures by the Yersinia effector YopH requires interaction with the substrate domain of p130Cas. Eur. J. Cell Biol. 2005, 84, 477–489. [Google Scholar] [CrossRef]
- Yi, C.R.; Allen, J.E.; Russo, B.; Lee, S.Y.; Heindl, J.E.; Baxt, L.A.; Herrera, B.B.; Kahoud, E.; MacBeath, G.; Goldberg, M.B. Systematic analysis of bacterial effector-postsynaptic density 95/disc large/zonula occludens-1 (PDZ) domain interactions demonstrates Shigella OspE protein promotes protein kinase C activation via PDLIM proteins. J. Biol. Chem. 2014, 289, 30101–30113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shifrin, Y.; Kirschner, J.; Geiger, B.; Rosenshine, I. Enteropathogenic Escherichia coli induces modification of the focal adhesions of infected host cells. Cell Microbiol. 2002, 4, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.P.; Frankel, G.M. The Locus of Enterocyte Effacement and Associated Virulence Factors of Enterohemorrhagic Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Whelan, R.; McVicker, G.; Leo, J.C. Staying out or going in? The interplay between type 3 and type 5 secretion systems in adhesion and invasion of enterobacterial pathogens. Int. J. Mol. Sci. 2020, 21, 4102. [Google Scholar] [CrossRef]
- Arbeloa, A.; Garnett, J.; Lillington, J.; Bulgin, R.R.; Berger, C.N.; Lea, S.M.; Matthews, S.; Frankel, G. EspM2 is a RhoA guanine nucleotide exchange factor. Cell Microbiol. 2010, 12, 654–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, T.; Kuwae, A.; Yoshida, S.; Sasakawa, C.; Abe, A. Enteropathogenic Escherichia coli activates the RhoA signaling pathway via the stimulation of GEF-H1. EMBO J. 2004, 23, 3570–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran Van Nhieu, G.; Ben-Ze’ev, A.; Sansonetti, P.J. Modulation of bacterial entry into epithelial cells by association between vinculin and the Shigella IpaA invasin. EMBO J. 1997, 16, 2717–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, C.A.; Bryan, B.A. Rho kinase proteins-pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011, 31, 3645–3657. [Google Scholar] [PubMed]
- Hamiaux, C.; van Eerde, A.; Parsot, C.; Broos, J.; Dijkstra, B.W. Structural mimicry for vinculin activation by IpaA, a virulence factor of Shigella flexneri. EMBO Rep. 2006, 7, 794–799. [Google Scholar] [CrossRef] [Green Version]
- Tran Van Nhieu, G.; Izard, T. Vinculin binding in its closed conformation by a helix addition mechanism. EMBO J. 2007, 26, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Ramarao, N.; Le Clainche, C.; Izard, T.; Bourdet-Sicard, R.; Ageron, E.; Sansonetti, P.J.; Carlier, M.F.; Tran Van Nhieu, G. Capping of actin filaments by vinculin activated by the Shigella IpaA carboxyl-terminal domain. FEBS Lett. 2007, 581, 853–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewett, T.J.; Dooley, C.A.; Mead, D.J.; Hackstadt, T. Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem. Biophys. Res. Commun. 2008, 371, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Lane, B.J.; Mutchler, C.; Al Khodor, S.; Grieshaber, S.S.; Carabeo, R.A. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 2008, 4, e1000014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewett, T.J.; Miller, N.J.; Dooley, C.A.; Hackstadt, T. The conserved Tarp actin binding domain is important for chlamydial invasion. PLoS Pathog. 2010, 6, e1000997. [Google Scholar] [CrossRef] [Green Version]
- Jiwani, S.; Alvarado, S.; Ohr, R.J.; Romero, A.; Nguyen, B.; Jewett, T.J. Chlamydia trachomatis Tarp Harbors Distinct G and F Actin Binding Domains That Bundle Actin Filaments. J. Bacteriol. 2013, 195, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Braun, C.; Alcázar-Román, A.R.; Laska, A.; Mölleken, K.; Fleig, U.; Hegemann, J.H. CPn0572, the C. pneumoniae ortholog of TarP, reorganizes the actin cytoskeleton via a newly identified F-actin binding domain and recruitment of vinculin. PLoS ONE 2019, 14, e0210403. [Google Scholar] [CrossRef] [PubMed]
- Whitewood, A.J.; Singh, A.K.; Brown, D.G.; Goult, B.T. Chlamydial virulence factor TarP mimics talin to disrupt the talin-vinculin complex. FEBS Lett. 2018, 592, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluger, C.; Braun, L.; Sedlak, S.M.; Pippig, D.A.; Bauer, M.S.; Miller, K.; Milles, L.F.; Gaub, H.E.; Vogel, V. Different Vinculin Binding Sites Use the Same Mechanism to Regulate Directional Force Transduction. Biophys. J. 2020, 118, 1344–1356. [Google Scholar] [CrossRef] [Green Version]
- Beauclair, G.; Naimo, E.; Dubich, T.; Rückert, J.; Koch, S.; Dhingra, A.; Wirth, D.; Schulz, T.F. Targeting Kaposi’s Sarcoma-Associated Herpesvirus ORF21 Tyrosine Kinase and Viral Lytic Reactivation by Tyrosine Kinase Inhibitors Approved for Clinical Use. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Ghosh-Choudhury, N.; Moyer, M.L.; Mott, G.E.; Thomas, C.A.; Foster, B.A.; Kreisberg, J.I. Role of RhoA activation in the growth and morphology of a murine prostate tumor cell line. Oncogene 1999, 18, 4120–4130. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F.; Ashworth, A.; Hall, A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 1995, 269, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Bagrodia, S.; Chackalaparampil, I.; Kmiecik, T.E.; Shalloway, D. Altered tyrosine 527 phosphorylation and mitotic activation of p60 c-src. Nature 1991, 349, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Malik, R.K.; Hildebrand, J.D.; Parsons, J.T. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: A role for paxillin tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6906–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanini, L.; Ye, F.; Snider, A.K.; Sarabakhsh, K.; Piatt, R.; Paul, D.S.; Petrich, B.G. A talin mutant that impairs talin-integrin binding in platelets decelerates αIIbβ3 activation without pathological bleeding. Blood 2014, 123, 2722–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingras, A.R.; Bate, N.; Goult, B.T.; Hazelwood, L.; Canestrelli, I.; Grossmann, J.G.; Hanein, D. The structure of the C-terminal actin-binding domain of talin. EMBO J. 2008, 27, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.X.; Egile, C.; Li, R.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef]
- Schafer, D.A.; Weed, S.A.; Binns, D.; Karginov, A.V.; Parsons, J.T.; Cooper, J.A. Dynamin2 and cortactin regulate actin assembly and filament organization. Curr. Biol. 2002, 12, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Ishizaki, T.; Uehata, M.; Tamechika, I.; Keel, J.; Nonomura, K.; Maekawa, M.; Narumiya, S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 2000, 57, 976–983. [Google Scholar]
- Kovács, M.; Tóth, J.; Hetényi, C.; Málnási-Csizmadia, A.; Sellers, J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004, 279, 35557–35563. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Han, R. Genistein suppresses adhesion-induced protein tyrosine phosphorylation and invasion of B16-BL6 melanoma cells. Cancer Lett. 1998, 129, 117–124. [Google Scholar] [CrossRef]
- Hanke, J.H.; Gardner, J.P.; Changelian, P.S.; Brissette, W.H.; Weringer, E.J.; Pollock, D.A.; Connelly, P.A. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 1996, 271, 695–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blake, R.A.; Broome, M.A.; Liu, X.; Wu, J.; Gishizky, M.; Sun, L.; Courtneidge, S.A. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 2000, 20, 9018–9027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, A.F.; Godman, G.C.; Deitch, A.D.; Tanenbaum, S.W. Action of cytochalasin D on cells of established lines: I. Early events. J. Cell Biol. 1974, 61, 481–500. [Google Scholar] [CrossRef] [Green Version]
- Spector, I.; Shochet, N.R.; Blasberger, D.; Kashman, Y. Latrunculins-novel marine macrolides that disrupt microfilament organization and affect cell growth: I. comparison with cytochalasin D. Cell Motil. Cytoskelet. 1989, 13, 127–144. [Google Scholar] [CrossRef]
- Bubb, M.R.; Senderowicz, A.M.; Sausville, E.A.; Duncan, K.L.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef]
- Peterson, J.R.; Bickford, L.C.; Morgan, D.; Kim, A.S.; Ouerfelli, O.; Kirschner, M.W.; Rosen, M.K. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Biol. 2004, 11, 747–755. [Google Scholar] [CrossRef]
- Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Aizawa, S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 1995, 377, 539–544. [Google Scholar]
- Thievessen, I.; Fakhri, N.; Steinwachs, J.; Kraus, V.; McIsaac, R.S.; Gao, L.; Oldenbourg, R. Vinculin is required for cell polarization, migration, and extracellular matrix remodeling in 3D collagen. FASEB J. 2015, 29, 4555–4567. [Google Scholar] [CrossRef] [Green Version]
- Priddle, H.; Hemmings, L.; Monkley, S.; Woods, A.; Patel, B.; Sutton, D.; Critchley, D.R. Disruption of the talin gene compromises focal adhesion assembly in undifferentiated but not differentiated embryonic stem cells. J. Cell Biol. 1998, 142, 1121–1133. [Google Scholar]
- Kumar, A.; Jaggi, A.S.; Singh, N. Pharmacology of Src family kinases and therapeutic implications of their modulators. Fund. Clin. Pharm. 2015, 29, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Li, S.; Docheva, D.; Grashoff, C.; Sakai, K.; Kostka, G.; Fässler, R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003, 17, 926–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagel, M.; George, E.L.; Kim, A.; Tamimi, R.; Opitz, S.L.; Turner, C.E.; Thomas, S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 2002, 22, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, L.M.; Jensen, C.C.; Kloeker, S.; Wang, C.L.A.; Yoshigi, M.; Beckerle, M.C. Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J. Cell Biol. 2006, 172, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapproth, S.; Sperandio, M.; Pinheiro, E.M.; Prünster, M.; Soehnlein, O.; Gertler, F.B.; Moser, M. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired β2 integrin function in mice. Blood J. Am. Soc. Hematol. 2015, 126, 2704–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennerberg, K.; Lohikangas, L.; Gullberg, D.; Pfaff, M.; Johansson, S.; Fässler, R. β1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 1996, 132, 227–238. [Google Scholar] [CrossRef]





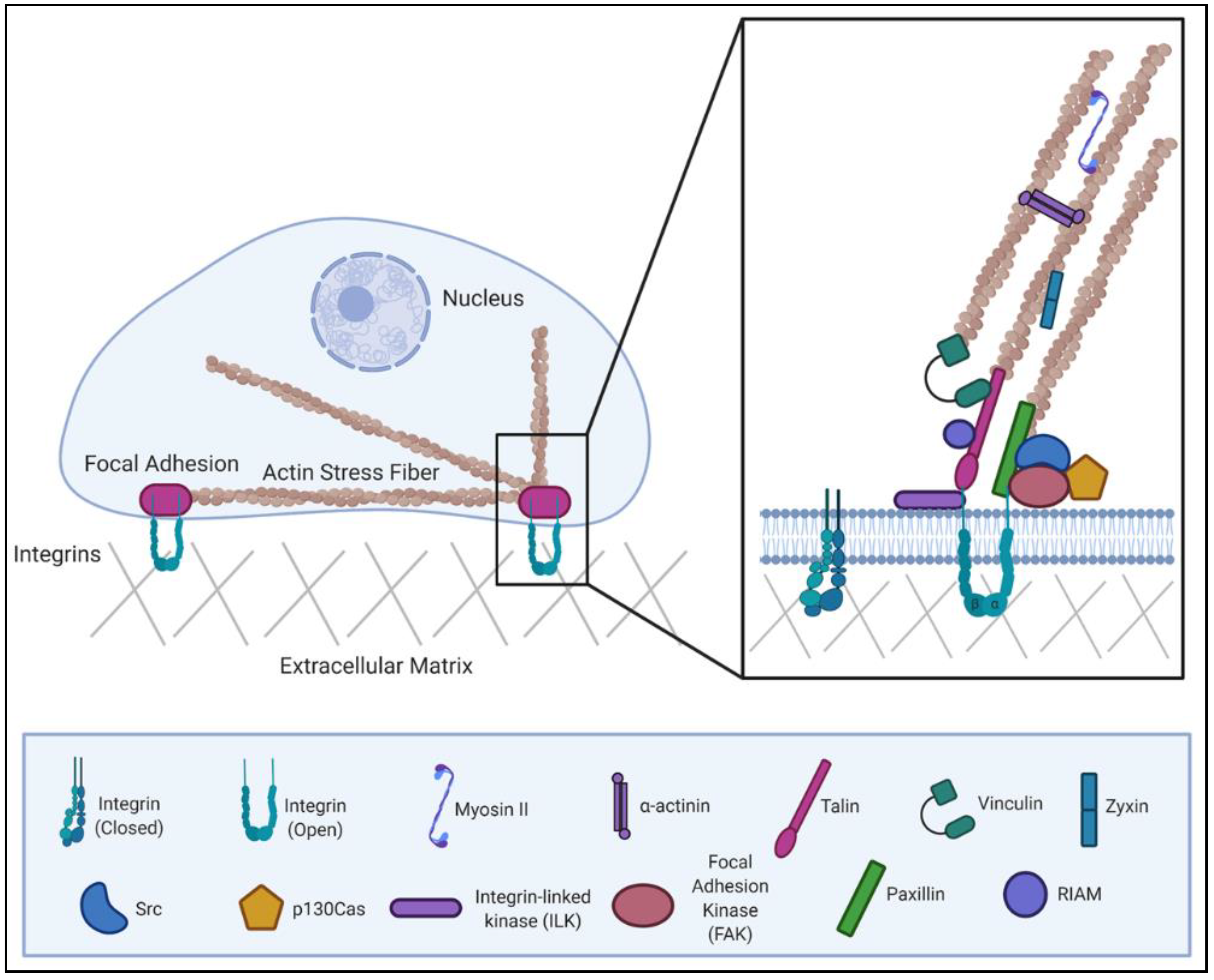{kind=link}
{kind=link}
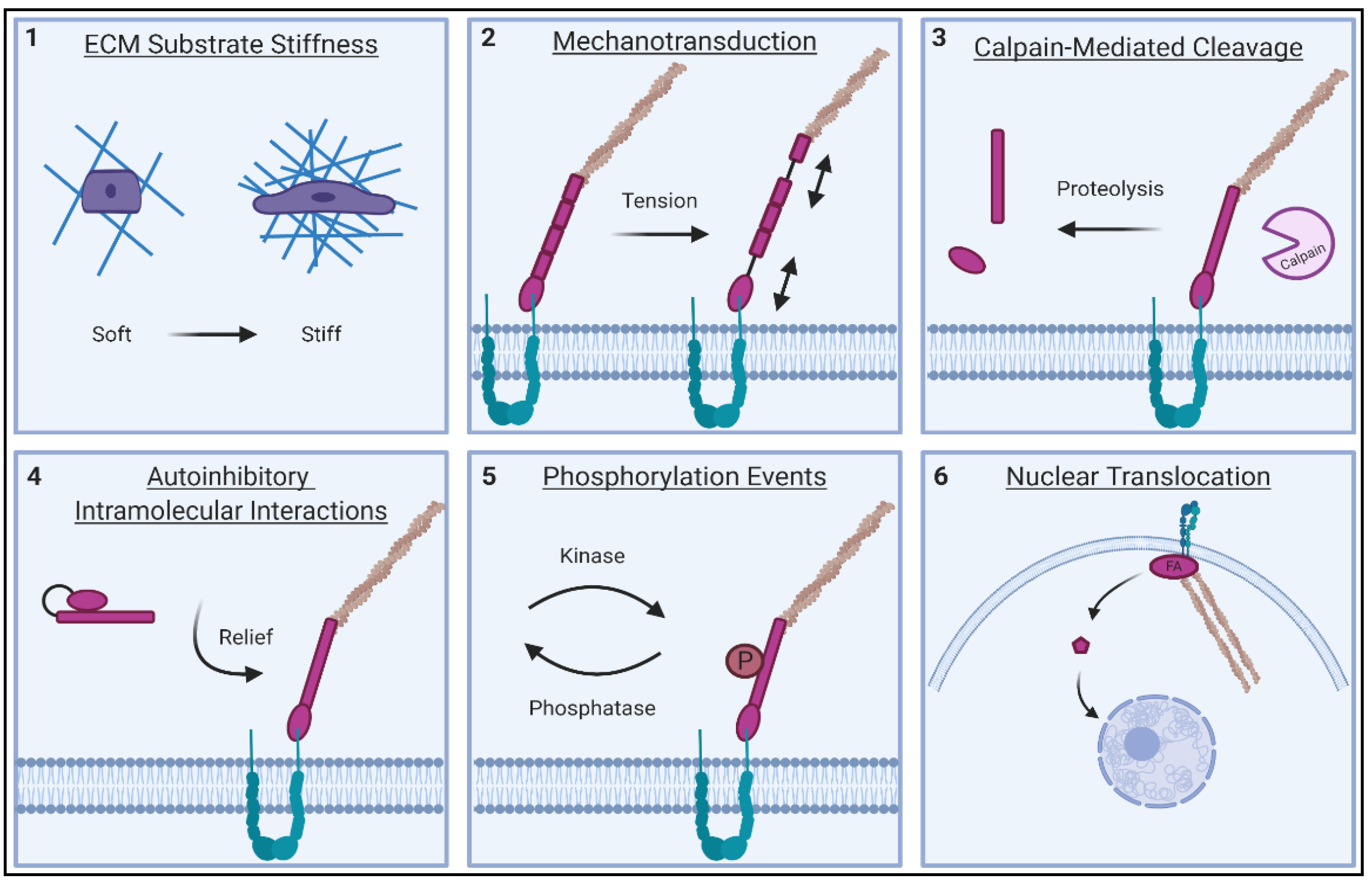{kind=link}
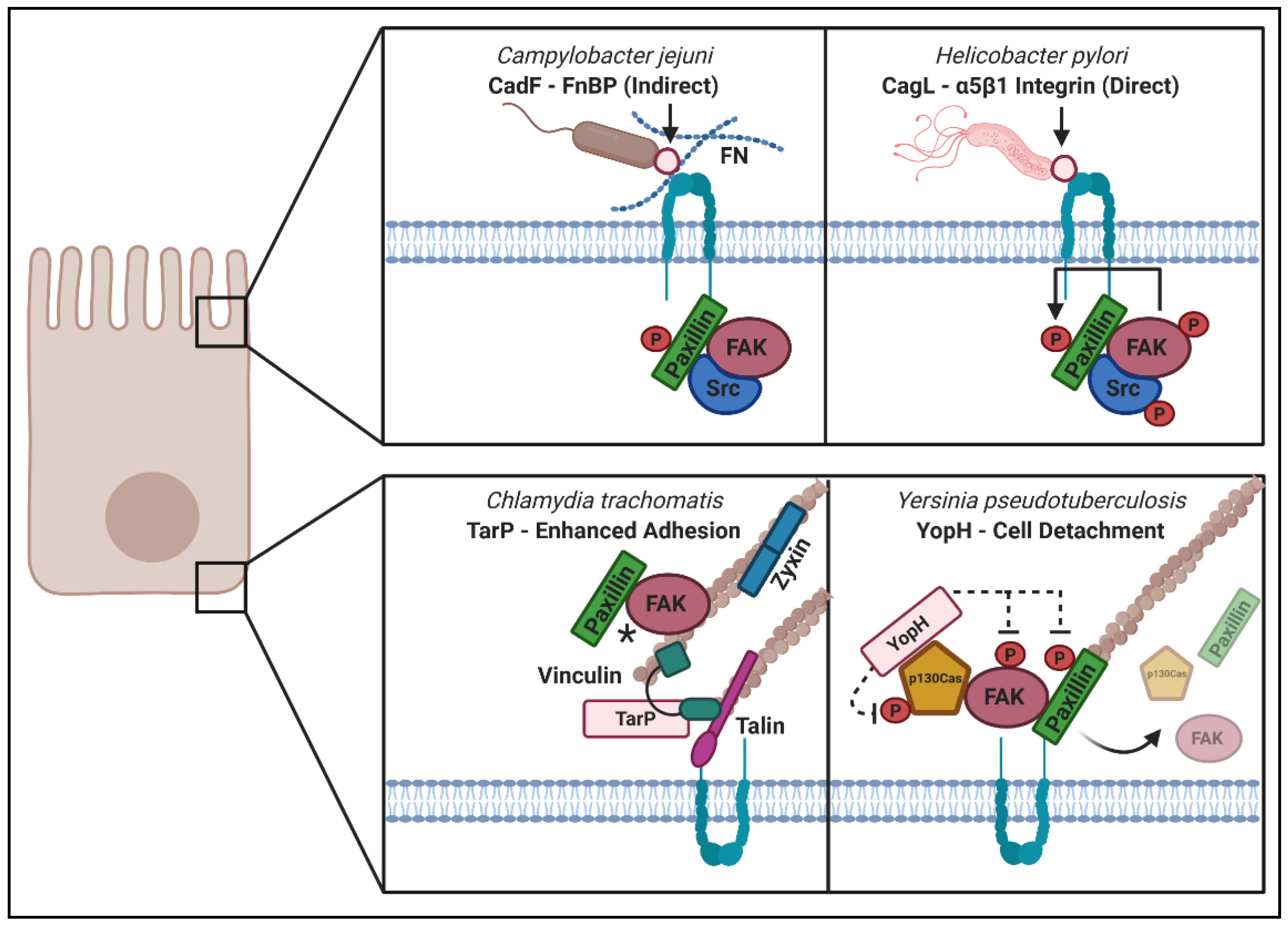{kind=link}
| FA Protein Phosphorylation Events with an Assigned Biological Function | |
|---|---|
| Phosphorylation Site | Function |
| Paxillin—As summarized in [46] | |
| Tyr 31; Tyr 118; Ser 178 | Regulation of Cell Migration and FA Turnover [165,166,167,168] |
| Ser 106; Ser 231; Ser 290 | Regulation of Pax disassembly from FAs [169,170] |
| Ser 126; Ser 130 | Translocation of Pax from FAs to the cytosol [171] |
| Ser 398; Ser 403; Ser 457; Ser 481 | Adhesion regulation; Pax localization at FAs [172,173] |
| Ser 272 | Regulation of Ras activity; adhesion/protrusion; inhibition of nuclear export [142] |
| Ser 188; Ser 190 | Integrin Activation [174] |
| Talin—As mapped in [175] | |
| Thr 114; Thr 150 | Negative regulation of integrin activation; Regulation of calpain-mediated cleavage of talin and FA turnover [176,177,178] |
| Thr 152 | Talin recruitment to integrin adhesion sites and maintaining muscle attachment in Drosophila [179] |
| Ser 425 | Inhibits binding to Smurf1—thereby preventing talin head ubiquitylation and degradation; Favors adhesion assembly, talin activity and integrin activation [98] |
| Ser 446 | Regulation of calpain-mediated cleavage of talin and FA turnover [178] |
| Vinculin | |
| Tyr 100; Tyr 1065 | Favors activation by promoting talin and actin binding; Regulates binding to the Arp2/3 complex; Focal adhesion development and maturation [180,181,182] |
| Ser 1033; Ser 1045 | Favors activation by promoting talin and actin binding; Focal adhesion development and maturation [181] |
| p130Cas | |
| Tyr 249; Tyr 410 | Binding sites for Crk SH2 domain [183] |
| Ser 139; Ser 437; Ser 639 | Intracellular localization [184] |
| Focal Adhesion Kinase (FAK) | |
| Tyr 397 | Major site of FAK autophosphorylation; Binding site for the SH2 domain of Src [185] |
| Tyr 861 | Major site of phosphorylation by Src [186] |
| Tyr 576; Tyr 577 | Promotes open FAK conformation; Facilitates scaffold and kinase functions [124] |
| Tyr 925 | Promotes Grb2 SH2-mediated binding [187] |
| Tyr 194 | Activation through relief of autoinhibition [188] |
| Src—As summarized in [189] | |
| Tyr 213 | Activation [190] |
| Tyr 527 | Autoinhibitory phosphorylation site—promotes an inactive conformation [191] |
| Tyr 416 | Activation from autophosphorylation [192] |
| Ser 17 | Facilitates activation of the small G protein Rap1 [126] |
| Thr 34; Thr 46; Ser 72 | Activation of pTyrosine527 Src [193] |
| α-actinin-4 | |
| Tyr 4; Tyr 31 | Reduces actin binding behavior [194] |
| Tyr 265 | Enhanced actin binding behavior; Susceptibility to calpain-mediated cleavage [195] |
| Zyxin | |
| Ser 142 | Regulates release from autoinhibitory head: tail interaction; cell-cell adhesion regulation [127] |
| RIAM | |
| Tyr 340 | Translocation to plasma membrane; β2 integrin designated lymphocyte functional antigen 1 (LFA-1) activation [196] |
| Tyr 45 | Release of autoinhibitory configuration [123] |
| Microbial Adhesins That Modulate “Outside-In” Signaling | ||||
|---|---|---|---|---|
| Fibronectin | ||||
| Adhesin/Microbe | Interactions Detected | Effect on Focal Signaling/Complex/Adhesion | Microbial Advantage | References |
| FlpA/Campylobacter jejuni | Fibronectin: FlpA | Required for ERK 1/2 phosphorylation | Invasion | [204,205] |
| CadF/Campylobacter jejuni | Fibronectin: CadF | Increased paxillin phosphorylation and actin polymerization | Invasion | [204,206] |
| Opc/Neisseria meningitidis | OpC: serum factors | Increased phosporylation of Src and FA-associated proteins | Invasion | [207] |
| SfbI/Streptococcus pyrogenes | Fibronectin: SfbI | Induces integrin clustering which results in the recruitment of paxillin, FAK, and other FA proteins to site of entry; intiates FAK autophosphorylation | Invasion | [208,209] |
| FnBPA-B/Staphylococcus aureus | Fibronectin: FnBPA-B | Activation of integrin signalling, including FAK-Src mediated phosphorylation of cortactin which resulted in actin rearrangement and induced uptake | Invasion | [210,211] |
| YadA/Enteropathogenic Yersinia | Fibronectin: YadA | Triggers FAK-Src complex formation and subsequent Ras activation | Invasion | [212,213] |
| Beta-integrins | ||||
| NadA/Neisseria meningitidis | NadA: α5β1 integrin | Uncharacterized | Invasion | [214] |
| Invasin/Enteropathogenic Yersinia | Invasin: β1 integrins | FAK recruitment of a Src family kinase and phosphorylation of paxillin | Invasion | [215,216] |
| CagL/Helicobacter pylori | CagL: α5β1/αVβ6 | Activates FAK and Src and triggers focal adhesion formation | Invasion | [217,218,219] |
| TSA56/Orientia tsutsugamushi | TSA56: α5β1 integrin | Fibronectin-binding domain of TSA56 blocked bacterial internalization | Invasion | [220] |
| Tat/HIV-1 | α5β3-integrin | Increased FAK phosphorylation, RhoA and Src activation | Spread | [221] |
| Secreted Effectors that modulate “inside-out” signaling | ||||
| p130Cas | ||||
| Effector/Microbe | Interactions detected | Effect on focal signaling/complex/adhesion | Microbial Advantage | References |
| YopH/Y. pseudotuberculosis | YopH: p130Cas | Cas disassembles focal complexes in presence of YopH | Cell detachment/spread | [222] |
| YopH PTPase activity | Dephosphorylation of p130Cas, paxillin and FAK | Cell detachment/spread | [223,224] | |
| Relocalization of FAK and Cas to the cytosol | Cell detachment/spread | [223] | ||
| Dephosphorylation of of FYB, SKAP-HOM, p55 | Cell detachment/spread | [225,226] | ||
| YopH(C403A) | Fyn and p130Cas localize to FAs in presence of kinase-dead YopH | Cell detachment/spread | [225] | |
| YopH(Q11) | Does not bind p130Cas and is readily phagocytized | Colonization | [227] | |
| YopH (N-term deleted) | Decreased substrate binding and decreased virulence in IP mouse model | Colonization | [224,227] | |
| Paxillin | ||||
| BE6/Bovine Papilloma Virus-1 | BE6: Paxillin | Depolymerizes actin stress fibers | Cell transformation | [160] |
| BE6: Paxillin (LD1) | Blocks paxillin interaction with vinculin and FAK | Cell transformation | [162] | |
| E6/HPV-16 | E6: Paxillin | LD1 domain of E6 interacts with Paxillin and decreases FA formation | Cell transformation | [161,162] |
| EspC/Enteropathogenic E. coli | EspC: Paxillin | Serine protease activity of EspC cleaves Paxillin | Cell detachment/spread | [107] |
| Vinculin | ||||
| IpaA/Shigella dysentirae | IpaA (N-term): Vinculin | Blocks vinculin: talin interaction, vinculin binds F-actin and induces depolymerization of stress fibers | Invasion | [228] |
| IpaA/Shigella flexneri | IpaA: Vinculin | Focal complexes form at invasion site | Invasion | [229] |
| IpaA (VBS1–2): Vinculin | Recruitment of Vinculin to invasion site forms actin-vinculin cup around Shigellae | Invasion | [230] | |
| IpaA (VBS3): Vinculin | Inhibition of head and tail domains and blocks vinculin-actin association | Invasion | [231] | |
| IpaA: Vinculin | Vinculin-binding excludes Talin, preventing Talin: B1-integrin interaction | Cell rounding | [232] | |
| TarP/Chlamydia caviae | TarP (VBD1–3): Vinculin | TarP binds vinculin, inducing actin polymerization at the invasion site | Invasion | [233] |
| TarP/Chlamydia trachomatis | TarP (LDVBD): Vinculin | TarP C-term recruitment to FAs requires vinculin, and increases FA size and stability. | Adhesion | [234] |
| Sca4/Rickettsia rickettsii | Sca4: Vinculin | Two VBD sites in Sca4 enable it to outcompetes alpha-catenin for vinculin binding, causing vinculin to relocalize from the cell periphery to internal sites. | Spread | [235] |
| Sca4/Rickettsia pakerii | Sca4: Vinculin | Sca4 interaction with vinculin blocks its ability to increase tension and barrier function at cell-cell junctions. | Spread | [236] |
| Certhrax/Bacillus cereus | Certhrax: Vinculin | Certhrax interacts with and ADP-ribosylates vinculin at R433 | Spread | [237,238,239] |
| FAK | ||||
| Tat/HIV-1 | Tat | Tat exposure increases phosphorylation of FAK-Y20 and increased FA numbers | Spread | [240] |
| TarP/Chlamydia caviae | TarP (LD): FAK | TarP orchestrates signaling through FAK-cdc42-Arp2/3 to induce actin polymerization | Invasion | [241] |
| EspC/EPEC | EspC: FAK | Serine protease activity of EspC cleaves FAK | Cell rounding | [107] |
| TK (ORF21)/KSHV | TK: FAK | Kinase activity of TK was required for relocalization of pFAK from peripheral FAs to internal sites, increasing cell detachment | Cell rounding | [242] |
| ILK | ||||
| OspE/Shigella flexneri | OspE: ILK | OspE-ILK localizes to FA, which results in increased FA size and stability, and decreased motility. OspE decreased phosphorylation of ILK targets, FAK and paxillin. OspE is required for efficient Shigella colonization of guinea pig colons. | Adhesion | [243,244] |
| EspO1/EHEC | EspO1: ILK | EspO1–1 localizes with ILK and prevents FA disassembly | Adhesion | [245] |
| Talin | ||||
| Tir/EPEC | Tir: Talin | Actin polymerization at EPEC adherence site | Invasion | [246] |
| IpaA/Shigella flexneri | IpaA: Talin | IpaA recruits talin to coat Shigella at invasion sites, and leads to the formation of extra long filopodia. | Invasion | [247] |
| Zyxin | ||||
| E6/HPV-6 | E6: Zyxin | E6 interaction with LIM3 domain of zyxin increases its nuclear translocation. | Unknown | [159] |
| Src | ||||
| E4orf4/Adenovirus 2 | E4:c-Src | Modulation of Src kinase activity increases phosphorylation of coractin and leads to decreased phosphorylation of FAK and paxillin. | Cell death | [109] |
| E4orf4 interaction with c-Src increases phosphorylation of MLC. E4orf4-Src localize to juxtanuclear actin complex that involves Rho GTPases. | Cell death | [111] | ||
| CagA/Helicobacter pylori | CagA: Src | Src phosphorylates CagA, then phosphorylated CagA inhibits the catalytic activity of Src. Vinculin does not get phosphorylated and lamellipodia formation is reduced. | Spread | [199] |
| RhoA | ||||
| EspM2/EHEC | EspM2: RhoA | GEF activity of EspM2 activates RhoA-ROCK-LIMK signaling, increasing stress fiber and FA formation. | Spread | [248] |
| EspM2: RhoA | EspM2 increases extrusion of cells and causes a redistribution of B1-integrin and ZO-1/tight junctions in a RhoA-dependent manner | Spread | [249] | |
| EspO1-2/EHEC | EspO1-2: EspM2 | Suppresses EspM2 activity, preventing RhoA activation and cell contraction | Adhesion | [245] |
| EspG and Orf3/ EPEC | EspG: tubulin | Causes microtubule destabilization which releases the H1-GEF and in turn activates RhoA and increases stress fiber formation. | Adhesion | [245] |
| E7/HPV-16 | E7(CR3): p190RhoGAP | Interaction wtith p190RhoGAP, a RhoA inhibiting hydrolase decreased F-actin levels and cell area in transfected cells. | Adhesion | [250] |
| Genetic Alterations | |||
|---|---|---|---|
| FA-Associated Protein | Mutation/Deletion | Phenotype | References |
| RhoA | T19N | Dominant negative | [292] |
| RhoA | V14 | Constitutively active | [293] |
| Src | K297M | Kinase-inactive | [207] |
| Src | K297M, Y529F | Dominant negative | [294] |
| Src | Y527F | Constitutively active | [294] |
| FAK | Y397F | Constitutively active; Cannot be auto-phosphorylated | [125] |
| FAK | D562A | Kinase-inactive | [216] |
| FAK | K464R | Kinase-inactive | [216] |
| FRNK | C-terminal domain | Dominant negative—blocks kinase activity and autophosphorylation of Y397 | [295] |
| Talin1 | L325R (FERM domain) | Compromises ability of talin1 to activate integrins without affecting binding to the NPxY motif | [296] |
| Talin1 | R2526G | Blocks talin1 dimerization and reduces activity of C-terminal actin-binding site | [297] |
| Talin1 | L432G | Resistant to calpain2 cleavage | [89] |
| Talin1 | E1770A | Constitutively active; F3-R9 interaction disrupted | [117] |
| RIAM | E60A/D63A | Constiutively active; Disrupts inhibitory region (IN) and RA domain interaction | [123] |
| Vinculin | “T12” and “T12K” | Constitutively active; Destabilize vinculin head-tail interaction | [120,121] |
| Cortactin | W22A (NTA domain) | Cannot bind Arp2/3 complex | [298] |
| Cortactin | W525K (SH3 domain) | Impaired dynamin binding | [299] |
| Zyxin | S142D | Constitutively active; Disrupts interaction between ActA and LIM regions | [127] |
| α-actinin | “NEECK” | Constitutively active | [128] |
| Chemical inhibitors for investigating FA signaling and dynamics | |||
| Inhibitor | Target | Function inhibited | Reference |
| Y27632 | ROCK | Competes with ATP for binding to ROCK’s catalytic site | [300] |
| Blebbistatin | Myosin II | Inhibits myosin ATPase activity | [301] |
| Genistein | Protein Tyrosine Kinases | Inhibits signaling molecules within the Receptor-MAPK or Receptor-PI3K/AKT cascades | [302] |
| PP2 | Src | Selectively inhibits Src-family kinases | [303] |
| SU6656 | Src | Broadly inhibits Src-family kinases | [304] |
| PF 573882 | FAK | Targets FAK catalytic activity by interaction with ATP-binding pocket; blocks Y397 phosphorylation | [305] |
| Cytochalaisin D | Actin | Inhibits actin polymerization by preventing actin-cofilin interaction | [306] |
| Latrunculin B | Actin | Inhibits actin polymerization by sequestering G-actin | [307] |
| Jasplakinolide | Actin | Induces actin polymerization and stabilizes F-actin | [308] |
| Wikostatin | N-WASP | Stabilizes N-WASP in its autoinhibited state | [309] |
| Leptomycin B | CRM1-mediated translocation | Inhibits the export of proteins from the nucleus to the cytoplasm | [138] |
| Established cell lines | |||
| Name of cell line | Deletion | Phenotype | Reference |
| MEF | FAK | Larger and more numerous focal adhesions; Reduced cell motility | [310] |
| MEF/ASML | Vinculin | Reduction in directionally persistent migration and traction force generation | [311] |
| MEF/Mouse ES | Talin1 | Unable to assemble vinculin or paxillin containing focal adhesions | [312] |
| MEF | Src, Yes, Fyn | Reduction in the tyrosine phosphorylation levels of FAK, p130Cas and paxillin | [313] |
| Mouse ES Cells | ILK | Impaired cell spreading and delayed formation of adhesions | [314] |
| MEF | Paxillin | Decreased cell spreading and migration; Abnormal FAs; Inefficient localization and phosphorylation of FAK | [315] |
| MEF | Zyxin | Reduced Ena/VASP proteins at adhesion sites; Increased motility; Deficits in actin stress fiber remodeling | [316] |
| MEF | p130Cas | Slow disassembly of focal adhesions at the cell front | [48] |
| PMNs | RIAM | Defects in adherence and cell spreading; Impaired activation of β2 integrins | [317] |
| GD25 (mouse) | β1-integrin | Low level of attachment to ECM substrates | [318] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, K.N.; Brinkworth, A.J. Manipulation of Focal Adhesion Signaling by Pathogenic Microbes. Int. J. Mol. Sci. 2021, 22, 1358. https://doi.org/10.3390/ijms22031358
Murphy KN, Brinkworth AJ. Manipulation of Focal Adhesion Signaling by Pathogenic Microbes. International Journal of Molecular Sciences. 2021; 22(3):1358. https://doi.org/10.3390/ijms22031358
Chicago/Turabian StyleMurphy, Korinn N., and Amanda J. Brinkworth. 2021. "Manipulation of Focal Adhesion Signaling by Pathogenic Microbes" International Journal of Molecular Sciences 22, no. 3: 1358. https://doi.org/10.3390/ijms22031358
APA StyleMurphy, K. N., & Brinkworth, A. J. (2021). Manipulation of Focal Adhesion Signaling by Pathogenic Microbes. International Journal of Molecular Sciences, 22(3), 1358. https://doi.org/10.3390/ijms22031358