
Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-β1/SMAD Signaling Pathway


Abstract
1. Introduction
2. Results
2.1. Disruption of Npr1 Triggers the Enhanced mRNA Expression of Cardiac Fibrotic Markers and TGF-β1 Receptors
2.2. Ablation of Npr1 Induces the Expression of SMAD Group of Proteins and TAK-1 and ERK1/2 Proteins
2.3. Disruption of Npr1 Increases the Levels of MMPs, PCNA, and Fibrotic Markers Proteins
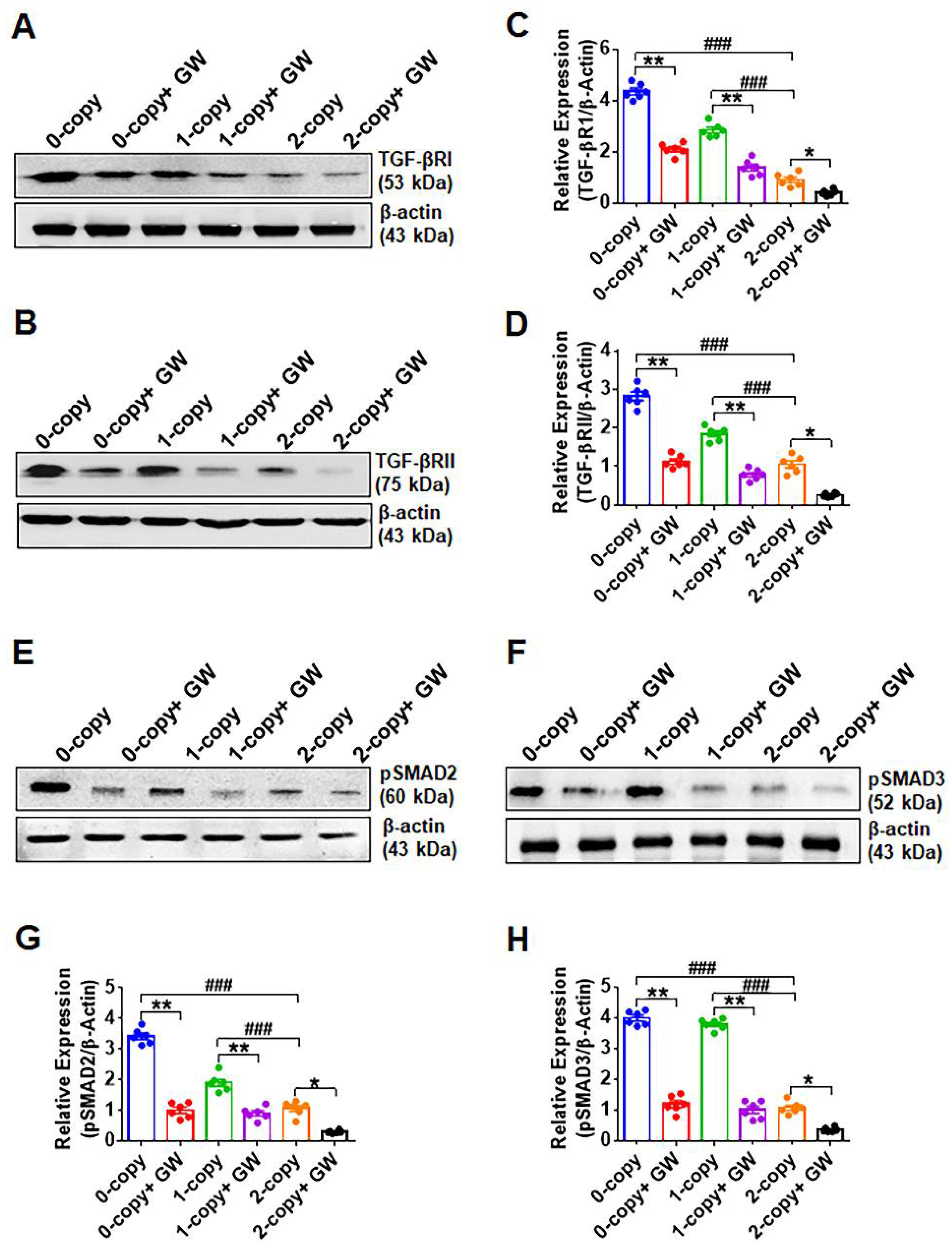
2.4. Disruption of Npr1 Increases the Levels of TGF-β1RI and TGF-β1RII
2.5. Disruption of Npr1 Enhances the Immunohistochemical Signals of TGF-β1RI, TGF-β1RII and pSMAD-2 in Heart Tissue
2.6. Treatment of Npr1−/− Mice with GW788388 Attenuates Systolic BP and Cardiac Dysfunction in a Sex-Dependent Manner
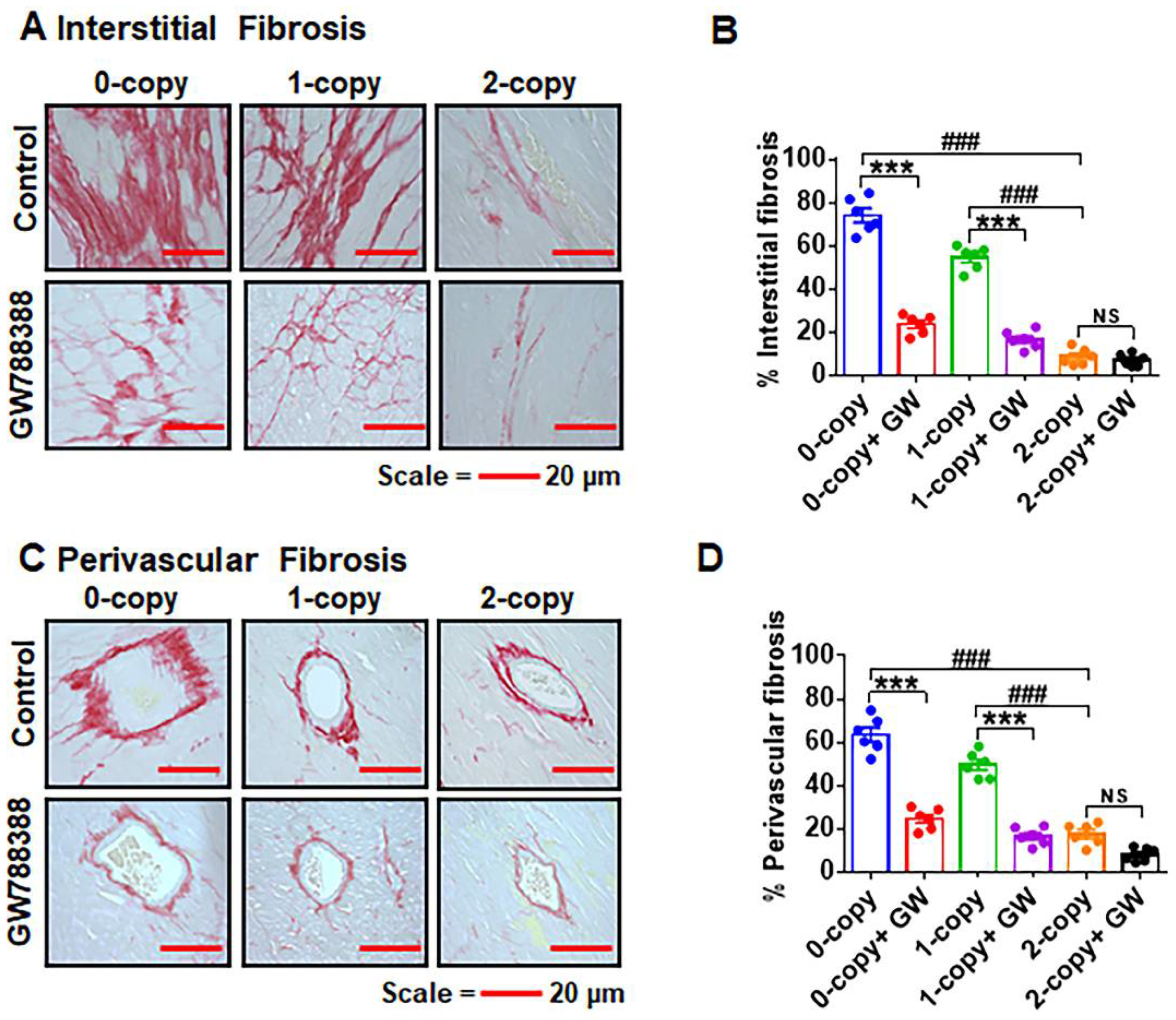
2.7. Ablation of Npr1 Triggers Interstitial and Perivascular Cardiac Tissue Fibrosis
2.8. Differential Hemodynamic Parameters and Echocardiographic Functional Outcome in Male and Female Mutant Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Generation and Genotyping of Mice
4.3. Experimental Animal Groups
4.4. Blood Pressure and Cardiac Function Analyses
4.5. Blood and Tissue Collection
4.6. Measurement of Cardiac Hypertrophy and Interstitial Fibrosis
4.7. Real-Time RT-PCR Analysis
4.8. Preparation of Cytosolic and Nuclear Extracts
4.9. Western Blot Analysis
4.10. Immunohistochemical Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Bold, A.J. Atrial natriuretic factor: A hormone produced by the heart. Science 1985, 230, 767–770. [Google Scholar] [CrossRef]
- Pandey, K.N. Biology of natriuretic peptides and their receptors. Peptides 2005, 26, 901–932. [Google Scholar] [CrossRef]
- Levin, E.R.; Gardner, D.G.; Samson, W.K. Natriuretic peptides. N. Engl. J. Med. 1998, 339, 321–328. [Google Scholar]
- Kishimoto, I.; Tokudome, T.; Nakao, K.; Kangawa, K. Natriuretic peptide system: An overview of studies using genetically engineered animal models. FEBS J. 2011, 278, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genom. 2018, 50, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.J.; Ellmers, L.J.; Lainchbury, J.G.; Maeda, N.; Smithies, O.; Richards, A.M.; Cameron, V.A. Influence of natriuretic peptide receptor-1 on survival and cardiac hypertrophy during development. Biochim. Biophys. Acta 2009, 1792, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Vellaichamy, E.; Khurana, M.L.; Fink, J.; Pandey, K.N. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J. Biol. Chem. 2005, 280, 19230–19242. [Google Scholar] [CrossRef]
- Pandey, K.N. Emerging roles of natriuretic peptides and their receptors in pathophysiology of hypertension and cardiovascular regulation. J. Am. Soc. Hypertens 2008, 2, 210–226. [Google Scholar] [CrossRef]
- Oliver, P.M.; Fox, J.E.; Kim, R.; Rockman, H.A.; Kim, H.S.; Reddick, R.L.; Pandey, K.N.; Milgram, S.L.; Smithies, O.; Maeda, N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc. Natl. Acad. Sci. USA 1997, 94, 14730–14735. [Google Scholar] [CrossRef]
- Vellaichamy, E.; Das, S.; Subramanian, U.; Maeda, N.; Pandey, K.N. Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner. Endocrinology 2014, 155, 1045–1056. [Google Scholar] [CrossRef]
- Pandey, K.N. Genetic Ablation and Guanylyl Cyclase/Natriuretic Peptide Receptor-A: Impact on the Pathophysiology of Cardiovascular Dysfunction. Int. J. Mol. Sci. 2019, 20, 3946. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Molecular Signaling Mechanisms and Function of Natriuretic Peptide Receptor-A in the Pathophysiology of Cardiovascular Homeostasis. Front. Physiol. 2021, 12, 693099. [Google Scholar] [CrossRef] [PubMed]
- Ellmers, L.J.; Scott, N.J.; Piuhola, J.; Maeda, N.; Smithies, O.; Frampton, C.M.; Richards, A.M.; Cameron, V.A. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J. Mol. Endocrinol. 2007, 38, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, U.; Kumar, P.; Mani, I.; Chen, D.; Kessler, I.; Periyasamy, R.; Raghavaraju, G.; Pandey, K.N. Retinoic acid and sodium butyrate suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene-disrupted haplotype mice. Physiol. Genom. 2016, 48, 477–490. [Google Scholar] [CrossRef]
- Li, Y.; Saito, Y.; Kuwahara, K.; Rong, X.; Kishimoto, I.; Harada, M.; Adachi, Y.; Nakanishi, M.; Kinoshita, H.; Horiuchi, M.; et al. Guanylyl cyclase-A inhibits angiotensin II type 2 receptor-mediated pro-hypertrophic signaling in the heart. Endocrinology 2009, 150, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Periyasamy, R.; Pandey, K.N. Activation of IKK/NF-kappaB provokes renal inflammatory responses in guanylyl cyclase/natriuretic peptide receptor-A gene-knockout mice. Physiol. Genom. 2012, 44, 430–442. [Google Scholar] [CrossRef][Green Version]
- Pandey, K.N. The functional genomics of guanylyl cyclase/natriuretic peptide receptor-A: Perspectives and paradigms. FEBS J. 2011, 278, 1792–1807. [Google Scholar] [CrossRef]
- Das, S.; Neelamegam, K.; Peters, W.N.; Periyasamy, R.; Pandey, K.N. Depletion of cyclic-GMP levels and inhibition of cGMP-dependent protein kinase activate p21(Cip1)/p27(Kip1) pathways and lead to renal fibrosis and dysfunction. FASEB J. 2020, 34, 11925–11943. [Google Scholar] [CrossRef]
- Periyasamy, R.; Das, S.; Pandey, K.N. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates renal (pro) renin receptor expression in Npr1 null mutant mice. Peptides 2019, 114, 17–28. [Google Scholar] [CrossRef]
- Vellaichamy, E.; Zhao, D.; Somanna, N.; Pandey, K.N. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates ACE and AT1 receptor gene expression and signaling: Role in cardiac hypertrophy. Physiol. Genom. 2007, 31, 193–202. [Google Scholar] [CrossRef]
- Zhao, D.; Das, S.; Pandey, K.N. Interactive roles of Npr1 gene-dosage and salt diets on cardiac angiotensin II, aldosterone and pro-inflammatory cytokines levels in mutant mice. J. Hypertens. 2013, 31, 134–144. [Google Scholar] [CrossRef]
- Zhao, D.; Vellaichamy, E.; Somanna, N.K.; Pandey, K.N. Guanylyl cyclase/natriuretic peptide receptor-A gene disruption causes increased adrenal angiotensin II and aldosterone levels. Am. J. Physiol. Ren. Physiol. 2007, 293, F121–F127. [Google Scholar] [CrossRef] [PubMed]
- Gogulamudi, V.R.; Mani, I.; Subramanian, U.; Pandey, K.N. Genetic disruption of Npr1 depletes regulatory T cells and provokes high levels of proinflammatory cytokines and fibrosis in the kidneys of female mutant mice. Am. J. Physiol. Ren. Physiol. 2019, 316, F1254–F1272. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Gogulamudi, V.R.; Periasamy, R.; Raghavaraju, G.; Subramanian, U.; Pandey, K.N. Inhibition of HDAC enhances STAT acetylation, blocks NF-kappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am. J. Physiol. Ren. Physiol. 2017, 313, F781–F795. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Au, E.; Krazit, S.T.; Pandey, K.N. Targeted disruption of guanylyl cyclase-A/natriuretic peptide receptor-A gene provokes renal fibrosis and remodeling in null mutant mice: Role of proinflammatory cytokines. Endocrinology 2010, 151, 5841–5850. [Google Scholar] [CrossRef]
- Pandey, K.N.; Vellaichamy, E. Regulation of cardiac angiotensin-converting enzyme and angiotensin AT1 receptor gene expression in Npr1 gene-disrupted mice. Clin. Exp. Pharm. Physiol. 2010, 37, e70–e77. [Google Scholar] [CrossRef]
- Adwent, I.; Grabarek, B.O.; Kojs-Mrożkiewicz, M.; Brus, R.; Staszkiewicz, R.; Plewka, A.; Stasiowski, M.; Lyssek-Boroń, A. The Influence of Adalimumab and Cyclosporine A on the Expression Profile of the Genes Related to TGFβ Signaling Pathways in Keratinocyte Cells Treated with Lipopolysaccharide A. Mediat. Inflamm. 2020, 2020, 3821279. [Google Scholar] [CrossRef]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Ganesan, R.; Sivalingam, N. Transforming Growth Factor Beta 2 Inhibits Growth and Proliferation Potential of Smad4 and p53 Mutated Human Colon Adenocarcinoma Cells. Pathol. Oncol. Res. 2019, 25, 819–821. [Google Scholar] [CrossRef] [PubMed]
- Grafe, I.; Alexander, S.; Peterson, J.R.; Snider, T.N.; Levi, B.; Lee, B.; Mishina, Y. TGF-beta Family Signaling in Mesenchymal Differentiation. Cold Spring Harb. Perspect. Biol. 2018, 10, a022202. [Google Scholar] [CrossRef]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Differential expression and clinical significance of transforming growth factor-beta isoforms in gbm tumors. Int. J. Mol. Sci. 2018, 19, 1113. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Ueno, H. Regulation of human helper T cell subset differentiation by cytokines. Curr. Opin. Immunol. 2015, 34, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Manabe, I. Cellular Interplay between Cardiomyocytes and Nonmyocytes in Cardiac Remodeling. Int. J. Inflam. 2011, 2011, 535241. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Kumar, P.; Garg, R.; Lindsey, S.H.; Katakam, P.V.; Bloodworth, M.; Pandey, K.N. Transforming growth factor beta1 antagonizes the transcription, expression and vascular signaling of guanylyl cyclase/natriuretic peptide receptor A—Role of deltaEF1. FEBS J. 2016, 283, 1767–1781. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, Y.Y. Understanding the role of transforming growth factor-beta signalling in the heart: Overview of studies using genetic mouse models. Clin. Exp. Pharm. Physiol. 2008, 35, 335–341. [Google Scholar] [CrossRef]
- Shi, S.J.; Vellaichamy, E.; Chin, S.Y.; Smithies, O.; Navar, L.G.; Pandey, K.N. Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion. Am. J. Physiol. Ren. Physiol. 2003, 285, F694–F702. [Google Scholar] [CrossRef]
- Rodriguez, F.H.; Book, W.M. Management of the adult Fontan patient. Heart 2020, 106, 105–110. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Guo, J.; Hua, Y.; Huang, K.; Magaye, R.; Cornell, J.; Kelly, D.J.; Reid, C.; Liew, D.; Zhou, Y.; et al. Cardiac fibrosis in the ageing heart: Contributors and mechanisms. Clin. Exp. Pharm. Physiol. 2017, 44 (Suppl. S1), 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Meng, Q.; James, J.; Osinska, H.; Gulick, J.; Valiente-Alandi, I.; Sargent, M.A.; Bhuiyan, M.S.; Blaxall, B.C.; Molkentin, J.D.; et al. Cardiac Fibrosis in Proteotoxic Cardiac Disease is Dependent Upon Myofibroblast TGF -beta Signaling. J. Am. Heart Assoc. 2018, 7, e010013. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar]
- Esebanmen, G.E.; Langridge, W.H.R. The role of TGF-beta signaling in dendritic cell tolerance. Immunol. Res. 2017, 65, 987–994. [Google Scholar] [CrossRef]
- Tian, M.; Neil, J.R.; Schiemann, W.P. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011, 23, 951–962. [Google Scholar] [CrossRef]
- Cho, N.; Razipour, S.E.; McCain, M.L. Featured Article: TGF-beta1 dominates extracellular matrix rigidity for inducing differentiation of human cardiac fibroblasts to myofibroblasts. Exp. Biol. Med. 2018, 243, 601–612. [Google Scholar] [CrossRef]
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J. Mol. Cell Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef]
- Zhang, J.; Chang, L.; Chen, C.; Zhang, M.; Luo, Y.; Hamblin, M.; Villacorta, L.; Xiong, J.W.; Chen, Y.E.; Zhang, J.; et al. Rad GTPase inhibits cardiac fibrosis through connective tissue growth factor. Cardiovasc. Res. 2011, 91, 90–98. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, W.; Ao, X.; Dai, H.; Yuan, L.; Huang, X.; Zhou, Q. TAK-242, a Toll-Like receptor 4 antagonist, protects against aldosterone-induced cardiac and renal injury. PLoS ONE 2015, 10, e0142456. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, Q.; Cui, J.; Zou, J.; Xia, X.; Wang, M.; Tong, Y.; Hui, W.; Liu, D.; Su, B.; et al. TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 2012, 36, 43–54. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Jimenez, S.A. Abrogation of transforming growth factor-beta-induced tissue fibrosis in TBRIcaCol1a2Cre transgenic mice by the second generation tyrosine kinase inhibitor SKI-606 (Bosutinib). PLoS ONE 2018, 13, e0196559. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, N.; Danner, T.; Zaiman, A.L.; Pinto, Y.M.; Rowell, J.; Mankowski, J.; Zhang, D.; Nakamura, T.; Takimoto, E.; Kass, D.A. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Investig. 2011, 121, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Buxton, I.L.; Duan, D. Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor-beta-induced nuclear Smad translocation: A key antifibrogenic mechanism of atrial natriuretic peptide. Circ. Res. 2008, 102, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.F. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef]
- Chen, J.; Wang, D.; Wang, F.; Shi, S.; Chen, Y.; Yang, B.; Tang, Y.; Huang, C. Exendin-4 inhibits structural remodeling and improves Ca(2+) homeostasis in rats with heart failure via the GLP-1 receptor through the eNOS/cGMP/PKG pathway. Peptides 2017, 90, 69–77. [Google Scholar] [CrossRef]
- Gong, K.; Xing, D.; Li, P.; Hilgers, R.H.; Hage, F.G.; Oparil, S.; Chen, Y.F. cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Mol. Endocrinol. 2011, 25, 1794–1803. [Google Scholar] [CrossRef]
- Shen, H.; Wang, J.; Min, J.; Xi, W.; Gao, Y.; Yin, L.; Yu, Y.; Liu, K.; Xiao, J.; Zhang, Y.F.; et al. Activation of TGF-beta1/alpha-SMA/Col I Profibrotic Pathway in Fibroblasts by Galectin-3 Contributes to Atrial Fibrosis in Experimental Models and Patients. Cell. Physiol. Biochem. 2018, 47, 851–863. [Google Scholar] [CrossRef]
- Tan, W.Q.; Fang, Q.Q.; Shen, X.Z.; Giani, J.F.; Zhao, T.V.; Shi, P.; Zhang, L.Y.; Khan, Z.; Li, Y.; Li, L.; et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-beta-activated kinase 1 (TAK1) pathways in mice. Br. J. Pharm. 2018, 175, 4239–4252. [Google Scholar] [CrossRef] [PubMed]
- Becerra, A.; Rojas, M.; Vallejos, A.; Villegas, V.; Perez, L.; Cabello-Verrugio, C.; Simon, F. Endothelial fibrosis induced by suppressed STAT3 expression mediated by signaling involving the TGF-beta1/ALK5/Smad pathway. Lab. Investig. 2017, 97, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Bjornstad, J.L.; Skrbic, B.; Marstein, H.S.; Hasic, A.; Sjaastad, I.; Louch, W.E.; Florholmen, G.; Christensen, G.; Tonnessen, T. Inhibition of SMAD2 phosphorylation preserves cardiac function during pressure overload. Cardiovasc. Res. 2012, 93, 100–110. [Google Scholar] [CrossRef]
- Daly, A.C.; Randall, R.A.; Hill, C.S. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol. Cell. Biol. 2008, 28, 6889–6902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, Y.; Zhang, P.; Chen, Y.; Li, C.; Chen, J.; Wang, Y.; Li, Y. The crucial role of activin A/ALK4 pathway in the pathogenesis of Ang-II-induced atrial fibrosis and vulnerability to atrial fibrillation. Basic Res. Cardiol. 2017, 112, 47. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Zhang, Y.; Connelly, K.A.; Gilbert, R.E.; Kelly, D.J. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1415–H1425. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; Van Waes, C. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Goldstein, J.A.; Bogdanovich, S.; Beiriger, A.; Wren, L.M.; Rossi, A.E.; Gao, Q.Q.; Gardner, B.B.; Earley, J.U.; Molkentin, J.D.; McNally, E.M. Excess SMAD signaling contributes to heart and muscle dysfunction in muscular dystrophy. Hum. Mol. Genet. 2014, 23, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef] [PubMed]
- Kotlarz, D.; Marquardt, B.; Baroy, T.; Lee, W.S.; Konnikova, L.; Hollizeck, S.; Magg, T.; Lehle, A.S.; Walz, C.; Borggraefe, I.; et al. Human TGF-beta1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat. Genet. 2018, 50, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Umbarkar, P.; Singh, A.P.; Gupte, M.; Verma, V.K.; Galindo, C.L.; Guo, Y.; Zhang, Q.; McNamara, J.W.; Force, T.; Lal, H. Cardiomyocyte SMAD4-Dependent TGF-beta Signaling is Essential to Maintain Adult Heart Homeostasis. JACC Basic Transl. Sci. 2019, 4, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; Maier, S.K.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res. Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef] [PubMed]
- John, S.W.; Krege, J.H.; Oliver, P.M.; Hagaman, J.R.; Hodgin, J.B.; Pang, S.C.; Flynn, T.G.; Smithies, O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 1995, 267, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Newton-Cheh, C.; Larson, M.G.; Vasan, R.S.; Levy, D.; Bloch, K.D.; Surti, A.; Guiducci, C.; Kathiresan, S.; Benjamin, E.J.; Struck, J.; et al. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat. Genet. 2009, 41, 348–353. [Google Scholar] [CrossRef]
- Rubattu, S.; Bigatti, G.; Evangelista, A.; Lanzani, C.; Stanzione, R.; Zagato, L.; Manunta, P.; Marchitti, S.; Venturelli, V.; Bianchi, G.; et al. Association of atrial natriuretic peptide and type a natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J. Am. Coll. Cardiol. 2006, 48, 499–505. [Google Scholar] [CrossRef]
- Vandenwijngaert, S.; Ledsky, C.D.; Lahrouchi, N.; Khan, M.A.F.; Wunderer, F.; Ames, L.; Honda, T.; Diener, J.L.; Bezzina, C.R.; Buys, E.S.; et al. Blood Pressure-Associated Genetic Variants in the Natriuretic Peptide Receptor 1 Gene Modulate Guanylate Cyclase Activity. Circ. Genom. Precis. Med. 2019, 12, e002472. [Google Scholar] [CrossRef]
- Nakayama, T.; Soma, M.; Takahashi, Y.; Rehemudula, D.; Kanmatsuse, K.; Furuya, K. Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ. Res. 2000, 86, 841–845. [Google Scholar] [CrossRef]
- Webber, M.A.; Marder, S.R. Better pharmacotherapy for schizophrenia: What does the future hold? Curr. Psychiatry Rep. 2008, 10, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Wang, S.; Wang, H.; Sun, K.; Song, X.; Zhang, W.; Fu, C.; Han, Y.; Hui, R. Atrial natriuretic peptide gene promoter polymorphism is associated with left ventricular hypertrophy in hypertension. Clin. Sci. 2008, 114, 131–137. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pitzalis, M.V.; Sarzani, R.; Dessi-Fulgheri, P.; Iacoviello, M.; Forleo, C.; Lucarelli, K.; Pietrucci, F.; Salvi, F.; Sorrentino, S.; Romito, R.; et al. Allelic variants of natriuretic peptide receptor genes are associated with family history of hypertension and cardiovascular phenotype. J. Hypertens. 2003, 21, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Usami, S.; Kishimoto, I.; Saito, Y.; Harada, M.; Kuwahara, K.; Nakagawa, Y.; Nakanishi, M.; Yasuno, S.; Kangawa, K.; Nakao, K. Association of CT dinucleotide repeat polymorphism in the 5′-flanking region of the guanylyl cyclase (GC)-A gene with essential hypertension in the Japanese. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2008, 31, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N.; Oliver, P.M.; Maeda, N.; Smithies, O. Hypertension associated with decreased testosterone levels in natriuretic peptide receptor-A gene-knockout and gene-duplicated mutant mouse models. Endocrinology 1999, 140, 5112–5119. [Google Scholar] [CrossRef]
- de Oliveira, F.L.; Araujo-Jorge, T.C.; de Souza, E.M.; de Oliveira, G.M.; Degrave, W.M.; Feige, J.J.; Bailly, S.; Waghabi, M.C. Oral administration of GW788388, an inhibitor of transforming growth factor beta signaling, prevents heart fibrosis in Chagas disease. PLoS Negl. Trop. Dis. 2012, 6, e1696. [Google Scholar] [CrossRef]
- Levolger, S.; Wiemer, E.A.C.; van Vugt, J.L.A.; Huisman, S.A.; van Vledder, M.G.; van Damme-van Engel, S.; Ambagtsheer, G.; JNM, I.J.; de Bruin, R.W.F. Inhibition of activin-like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Sci. Rep. 2019, 9, 9826. [Google Scholar] [CrossRef]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar]
- Oakes, J.M.; Xu, J.; Morris, T.M.; Fried, N.D.; Pearson, C.S.; Lobell, T.D.; Gilpin, N.W.; Lazartigues, E.; Gardner, J.D.; Yue, X. Effects of Chronic Nicotine Inhalation on Systemic and Pulmonary Blood Pressure and Right Ventricular Remodeling in Mice. Hypertension 2020, 75, 1305–1314. [Google Scholar]
- Shi, S.J.; Nguyen, H.T.; Sharma, G.D.; Navar, L.G.; Pandey, K.N. Genetic disruption of atrial natriuretic peptide receptor-A alters renin and angiotensin II levels. Am. J. Physiol. Ren. Physiol. 2001, 281, F665–F673. [Google Scholar] [CrossRef]
- Pandey, K.N.; Kumar, R.; Li, M.; Nguyen, H. Functional domains and expression of truncated atrial natriuretic peptide receptor-A: The carboxyl-terminal regions direct the receptor internalization and sequestration in COS-7 cells. Mol. Pharm. 2000, 57, 259–267. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Application | Catalog No. | Dilutions | Company |
|---|---|---|---|---|
| α-SMA | WB | sc-32251 | 1:250 | Santa Cruz Biotechnology |
| CTGF | WB | sc-365970 | 1:300 | Santa Cruz Biotechnology |
| ERK 1 | WB | sc-271269 | 1:200 | Santa Cruz Biotechnology |
| ERK 2 | WB | sc-1647 | 1:250 | Santa Cruz Biotechnology |
| ERK1/2 | WB | sc-514302 | 1:300 | Santa Cruz Biotechnology |
| MMP-2 | WB | sc-13595 | 1:300 | Santa Cruz Biotechnology |
| MMP-9 | WB | sc-393859 | 1:300 | Santa Cruz Biotechnology |
| PCNA | WB | sc-25280 | 1:250 | Santa Cruz Biotechnology |
| pERK1/2 | WB | sc-81492 | 1:250 | Santa Cruz Biotechnology |
| pSMAD2 | WB | #3108 | 1:250 | Cell Signaling Technology |
| pSMAD3 | WB | #9520 | 1:300 | Cell Signaling Technology |
| SMAD-1 | WB | #9743 | 1:400 | Cell Signaling Technology |
| SMAD-2 | WB | #5339 | 1:300 | Cell Signaling Technology |
| SMAD-3 | WB | #9513 | 1:250 | Cell Signaling Technology |
| SMAD-4 | WB | #46535 | 1:250 | Cell Signaling Technology |
| SMAD-6 | WB | #9519 | 1:250 | Cell Signaling Technology |
| TGF-β1 | WB | sc-130348 | 1:350 | Santa Cruz Biotechnology |
| TGF-β1R1 | WB | sc-101574 | 1:250 | Santa Cruz Biotechnology |
| TGF-β1RII | WB | sc-17792 | 1:250 | Santa Cruz Biotechnology |
| TAK1 | WB | #4505 | 1:250 | Cell Signaling Technology |
| pTAK1 | WB | #4536 | 1:250 | Cell Signaling Technology |
| Goat anti-mouse IgG2a-HRP | WB | sc-2061 | 1:5000 | Santa Cruz Biotechnology |
| Goat anti-rabbit | ||||
| IgG-HRP | WB | sc-2004 | 1:5000 | Santa Cruz Biotechnology |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramanian, U.; Ramasamy, C.; Ramachandran, S.; Oakes, J.M.; Gardner, J.D.; Pandey, K.N. Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-β1/SMAD Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 11487. https://doi.org/10.3390/ijms231911487
Subramanian U, Ramasamy C, Ramachandran S, Oakes JM, Gardner JD, Pandey KN. Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-β1/SMAD Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(19):11487. https://doi.org/10.3390/ijms231911487
Chicago/Turabian StyleSubramanian, Umadevi, Chandramohan Ramasamy, Samivel Ramachandran, Joshua M. Oakes, Jason D. Gardner, and Kailash N. Pandey. 2022. "Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-β1/SMAD Signaling Pathway" International Journal of Molecular Sciences 23, no. 19: 11487. https://doi.org/10.3390/ijms231911487
APA StyleSubramanian, U., Ramasamy, C., Ramachandran, S., Oakes, J. M., Gardner, J. D., & Pandey, K. N. (2022). Genetic Disruption of Guanylyl Cyclase/Natriuretic Peptide Receptor-A Triggers Differential Cardiac Fibrosis and Disorders in Male and Female Mutant Mice: Role of TGF-β1/SMAD Signaling Pathway. International Journal of Molecular Sciences, 23(19), 11487. https://doi.org/10.3390/ijms231911487