Abstract
6-Hydroxyquinoline and 3-hydroxyisoquinoline as N-containing naphthol analogues were tested in modified Mannich reactions (mMr’s). In the case of 6-hydroxyquinoline, the outcomes of the attempted Mannich reactions were strongly influenced by the amine components. Aminoalkylation of this substrate with reagents 1-naphthaldehyde and N-benzylmethylamine led to the isolation of a diol regarded as a stabilised water adduct of an ortho-quinone methide (o-QM), of which formation can be ascribed to the presence of a hydroxide ion in a relatively higher concentration generated by the bulky and basic amine component with decreased nucleophilicity. The classical Mannich base was isolated as a single product when the amine component was replaced for morpholine, featuring nucleophilicity rather than basic character under the applied reaction conditions. Starting from the isomer substrate 3-hydroxyisoquinoline, independently on the nucleophile (methanol or morpholine) besides the formation of the classical Mannich base, the nucleophilic attack at position one of the heterocyclic substrate was also observed. The DFT analysis of the acceptor molecular orbitals of the potential electrophilic components and the thermodynamics of the assumed-possible transformations demonstrated that this regioselective addition is a feasible process on the investigated heterocyclic skeleton. DFT modelling studies also suggest that besides the steric bulk, the orbital-controlled electronic properties of the aryl group, originating from the aldehyde components, have a strong influence on the ratios and the NMR-monitored interconversions of the C-1-substituted 3-hydroxyisoquinolines and the classical Mannich bases formed in multistep reaction sequences. On the basis of the DFT analysis of the thermodynamics of alternative pathways, a reaction mechanism was proposed for the rationalization of these characteristic substrate-controlled interconversions.
1. Introduction
The Mannich reaction is a widely used multicomponent reaction, providing a carbon–carbon bond formation under relatively mild reaction conditions [1,2]. According to the original reaction, a C–H acid, formaldehyde and a secondary amine were applied. In the modified Mannich reaction (mMr), the C–H acid is replaced with an electron-rich aromatic compound, such as 1- or 2-naphthol [3,4], quinolinol or isoquinolinol [3,5], and, in a similar manner, formaldehyde with benzaldehyde. The formation of the Mannich product may be approached in two ways. The first is the nucleophilic addition of an electron-rich aromatic compound to the Schiff base [4], while the other assumes, first, the formation of an ortho-quinone methide (o-QM) that will be stabilised by reacting it with an amine component as a nucleophilic agent [6].
Quinolinols and isoquinolinols have a wide range of biological activity. 3-Hydroxyquinolines are cytotoxic [7], and 3-hydroxyisoquinolines are matrix metalloproteinase inhibitors [8]. Meanwhile, 6- and 7-hydroxyquinolines are anti-hepatitis B virus agents [9,10]. Moreover, 6-hydroxyisoquinolines are Na+, K+-ATPase inhibitors [11], and 7-hydroxyisoquinolines are selective kappa-opioid receptor antagonists [12].
In our previous study [13], 2-naphthol, 2-naphthaldehyde and N-benzylmethylamine were supposed to follow the mechanism of the mMr’s. In contrast, the unexpected formation of 1-(hydroxy(naphthalen-1-yl)methyl)naphthalen-2-ol (a formal hydrate of an o-QM) was observed, instead of the classical Mannich base (Figure 1). Our aim in this work was to investigate the scope and limitations of the latter reaction (the stabilised benzylidene derivative vs. the Mannich base) starting from 6-hydroxyquinoline and 3-hydroxyisoquinoline as N-containing 2-naphthol substrates.
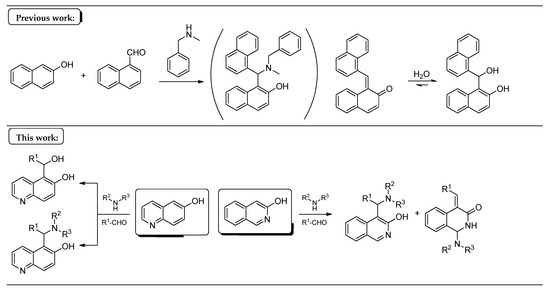
Figure 1.
The summary of the present work with the previous findings.
2. Results and Discussion
In our first experiments, 6-hydroxyquinoline (1) was reacted with 1-naphthaldehyde (2) and N-benzylmethylamine (3) under neat conditions (Scheme 1). Of the tested temperatures (60 °C, 80 °C and 100 °C), 60 °C was found to be optimal, which was provided by classical heating (an oil bath). After 8 h, in the course of the workup of the reaction mixture, the crude product was subjected to column chromatography. Subsequent crystallisation from methanol gave a mixture with hydrate 5 as the main component, which is the stabilised form of ortho-quinone methide 4 as disclosed by combined NMR methods. An analogous product was isolated in our previous work when 2-naphthol was attempted to be aminoalkylated with N-benzylmethylamine in the presence of 1-naphthaldehyde [12]. In order to evaluate the role of the amines, morpholine (6) as a secondary cyclic amine was reacted with 6-hydroxyquinoline and 1-naphthaldehyde under conditions that were previously optimised (Scheme 1). Thin-layer chromatography (TLC) confirmed the formation of a single product, which was then isolated by crystallisation. The NMR spectra provided evidence that the use of morpholine as an amine component led to the formation of the classical Mannich product 7. The differences between the reaction pathways can be explained in terms of the different steric demands and the basicity of the applied amines (pKb = 4.25 and 5.64 for 3 and 6, respectively). Since 3 is bulkier and at least tenfold more basic than 6, it can be more prone to activate water molecules rather than to attack as a nucleophile on the potential electrophilic species present in the reaction mixture.
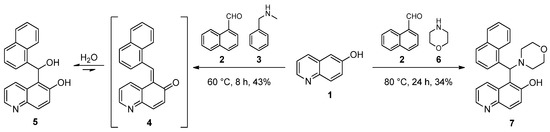
Scheme 1.
Formation of ortho-quinone methide (4) and its stabilisation to hydrate (5) and aminoalkylation of 6-hydroxyquinoline (1).
In order to explore further structure–reactivity relationships, 3-hydoxyisoquinoline (8), a N-containing electron-rich aromatic substrate, was selected for the subsequent studies. Accordingly, 3-hydroxyisoquinoline (8), 1-naphthaldehyde (2) and N-benzylmethylamine (3) were reacted under neat conditions. After 30 minutes of reaction time at 60 °C, the prepared TLC was multi-spotted, and no well-defined component could be isolated from the reaction mixture. Thereafter, N-benzylmethylamine was switched to morpholine. A prolonged reaction time (6 h) at 80 °C was necessary for the appearance of a few new spots on the TLC. By means of column chromatography, using EtOAc:MeOH (20:1) as an eluent, we isolated methoxy-substituted lactam 10 as a distinct compound. In the 1H NMR spectrum of 10, the typical singlet of 3H intensity at 3.35 ppm and the amide-type NH doublet at 9.25 ppm along with 1H-13C-HMBC connectivity between signal pairs OCH3/C-1, H-1/OCH3, H-1/C-3, NH/C-3 and NH/C-4 unambiguously proved its structure. The formation of this product can be explained via the formation of the ortho-quinone methide (9), which reacted at the most electrophilic N-acylimine site with methanol present in the solvent mixture used in the chromatographic purification (Scheme 2).
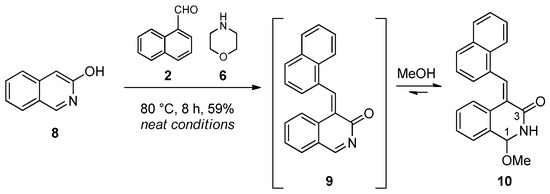
Scheme 2.
Selective substitution of 3-hydroxyisoquinoline (8).
To investigate the influence of the arylidene moiety on the apparently unexpected nucleophilic attack of MeOH at position 1, 3-hydroxyisoquinoline (8) and methanol were stirred at 80 °C. Since, even after a prolonged reaction time (20 h), TLC indicated only the presence of the starting compounds, the reaction was repeated at 100 °C and 120 °C. Because mixing 8 with MeOH at a higher temperature resulted again in the presence of the unchanged starting compounds, we focused our attention on the evaluation of the effects of stronger nucleophiles, such as N-benzylmethylamine and morpholine, on the outcome of the reactions. First, N-benzylmethylamine (3) and 3-hydroxyisoquinoline (8) were reacted at 80 °C under neat conditions. Surprisingly, after 12 h of reaction time, TLC showed the formation of a multicomponent mixture.
After purification by column chromatography, two components with characteristic compositions (11a and 11b: Scheme 3) could be isolated and identified by combined NMR methods, including the use of highly diagnostic 1H-13C-HMBC. The apparently intriguing transformations can be rationalised by the equilibrium formation of tautomer 12, the key intermediate of the subsequent transformations. On one hand, the nucleophilic attack of tautomer 8 on the highly-electrophilic C-1 centre of this activated N-acyl-imine results in the formation of the racemic mixture of the chiral dimer 11a. On the other hand, upon nucleophilic attack of 3, tautomer 12 is converted into adduct 13. The latter undergoes a synchronous imine- and dihydrogen-forming fragmentation, leading to Schiff base 14 with the simultaneous regeneration of 12. Hydrolysis of 14 gives benzaldehyde 15 and methylamine. Upon condensation with 15, saturated aminal adduct 13 is transformed into 16. This enone-containing aminal intermediate then undergoes sequential addition and elimination of 3, eventually affording 11b, the other isolated aromatic product. Finally, either of the two equilibrating tautomers of the heterocyclic precursor (8 or 12) might also react with 15 to construct enone 17. The Aza-Michael addition of the latter with 3 also leads to 11b.
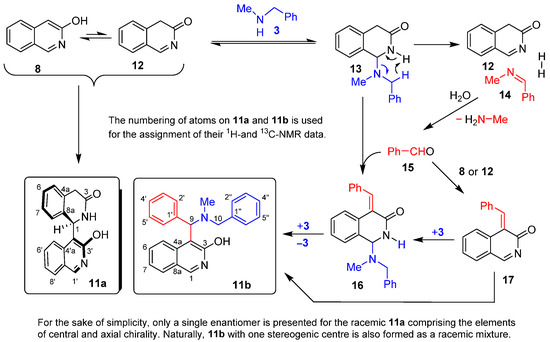
Scheme 3.
Unexpected transformations of 3-hydroxyisoquinoline (8) in the presence of N-benzylmethylamine (3).
Since N-benzylmethylamine was expected to undergo uncontrolled decomposition, leading to a wide range of side products, morpholine was applied in our subsequent experiments. This stable secondary cyclic amine was stirred with 3-hydroxyisoquinoline at 80 °C under neat conditions. After 2 h, the spot indicating 3-hydroxyisoquinoline disappeared on a TLC; therefore, the mixture was worked up by neutral column chromatography to produce aminal 18 as a single isolated product (Scheme 4). In its 1H-NMR spectrum, the separated AB signals (J = 20 Hz) of the diastereotopic H-4 protons discernible at 3.37 ppm and 3.64 ppm indicated its lactame-type skeletal structure with a stereogenic centre at position 1. This structure was also supported by the chemical shift of C-1 (171.4 ppm) and by HMBC cross peaks revealing correlations between 1H/13C coupled pairs H-4A/C-1, H-4B/C-1, H-1/C-1 and NH/C-1 (Scheme 4).
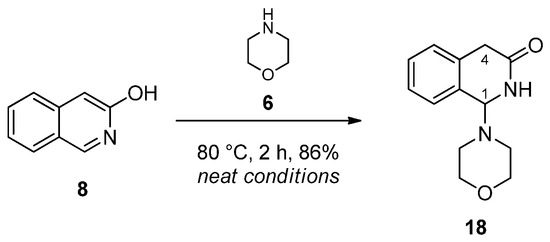
Scheme 4.
Reaction of 3-hydroxyisoquinoline (8) and morpholine (6).
In order to unambiguously clarify the role of the amine and the aldehyde in this reaction and to avoid the rather unpredictable reactivity and decomposition of N-benzylmethylamine, 3-hydroxyisoquinoline (8) was reacted with morpholine (6) in the presence of different aromatic aldehydes under neat conditions. Since the incorporation of the methoxy group in the case of 10 was also undesirable, the composition of the crude reaction mixtures was analysed using 1H NMR without any further purification. In accord with our previous findings with morpholine as the amine nucleophile, in addition to 18, we detected two types of products (23A and 23B) in a time-dependent manner. Thus, the crude products were analysed at five different reaction times (1 h, 2 h, 4 h, 8 h and 16 h). In general, the first-appearing NMR signals can unambiguously be assigned to the protons of 18. In the progress of the experiments, the ratio of 18 was gradually decreased, while the ratios of the other products were increased. As a representative example, the transformation of 23d is demonstrated in Figure 2.
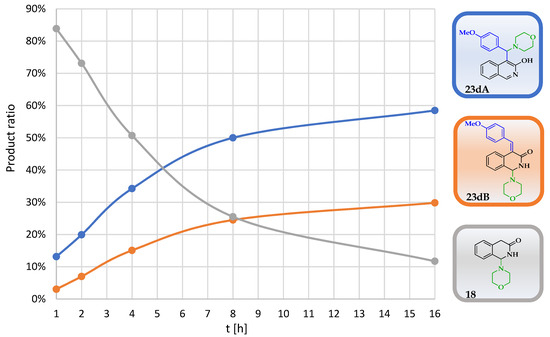
Figure 2.
The time-dependent ratios of the morpholino-hydroxyquinoline adduct 18, the Mannich product 23dA and the 4-methoxybenzylidene derivative 23dB as determined by 1H NMR.
The ratio of the regioisomers was obtained as the relative intensity of the diagnostic proton signals of particular components. Namely, a singlet in the range of 5.3–5.5 ppm is characteristic for Mannich adducts type 23A, and a doublet in the range of 5.0–5.1 ppm is diagnostic for enone aminals of type 23B (Table 1). The relative amount of 18 could also be calculated by the intensity of the characteristic proton signals at 5.03 ppm and 8.43 ppm.

Table 1.
The characteristic proton signals discernible in the 1H-NMR spectra of the reaction mixtures.
The 1H NMR analyses revealed the parallel formation of the classical Mannich product (23b–dA) and the corresponding arylidene derivative (23b–dB) when benzaldehyde (15), 2-naphthaldehyde (19) and 4-methoxybenzaldehyde (20) were applied as the carbonyl component (Figure 3). However, independently of the reaction time, when 1-naphthaldehyde (2) was used as a coupling partner, the reactions led to the formation of the arylidene derivative 23aB, which could be isolated as a single product.
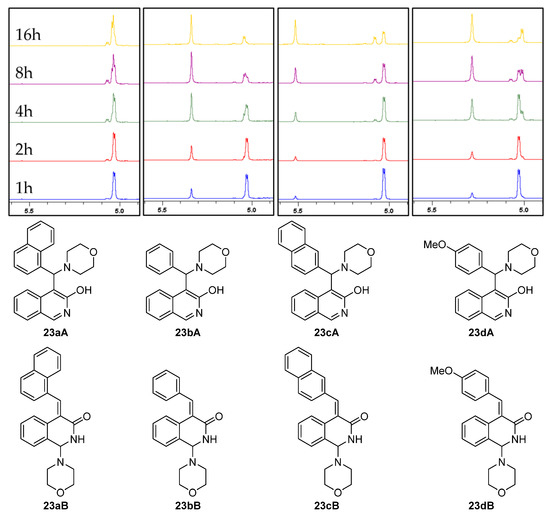
Figure 3.
Structures and diagnostic 1H NMR signals of Mannich products (type 23A) and arylidene derivatives (type 23B).
It must be pointed out that enones 23b–dB were slowly transformed into the appropriate Mannich-type product 23b–dA as highlighted by the 1H NMR spectra of the reaction mixtures that were registered at different times (Figure 3).
In order to rationalise all the aforementioned aryl-group-dependent experimental findings as well as the exceptional reactivity of 4-nitrobenzaldehyde 26 manifested in its analogue transformation to attempt to obtain the Mannich-type product (discussed later), we undertook comparative DFT modelling studies on the aryl-group-dependent progress of the formation and possible interconversion of 18 and compound types 23A and 23B. We hypothesised that these transformations might take place along the pathways outlined on Scheme 5. The feasibility of this assumption was assessed by the relative energetics of particular elementary steps and by the analysis of the acceptor orbitals (LUMO and LUMO + 1) of the possible electrophilic species involved in these Mannich-type and related conversions (Figure 4).
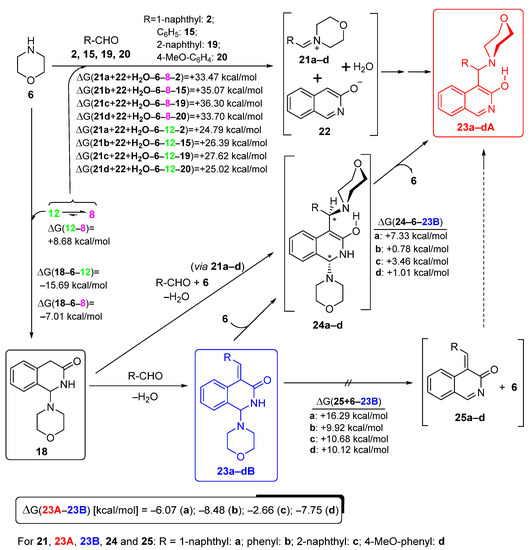
Scheme 5.
Mechanism of the formation of Mannich and benzylidene products (types 23A and 23B, respectively) proposed on the basis of the aryl group-dependent relative thermodynamics of the assumed transformations obtained using DFT modelling.
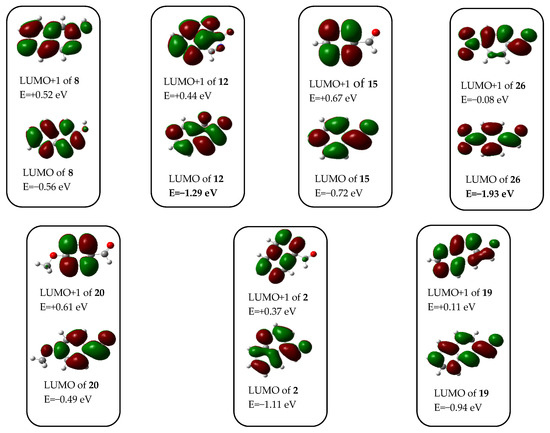
Figure 4.
Acceptor orbitals (LUMO and LUMO + 1) of the electrophilic components involved in the Mannich-type reactions obtained using MO analysis of the optimized structures.
First, the classical Mannich products were expected to be formed through an ion pair incorporating iminium cations 21a–d and a heterocyclic anion 22 generated by the nucleophilic attack of 6 on the aldehyde component followed by the 3-hydroxyisoquinoline-promoted elimination of water. However, on the basis of the assumed relatively small amount of the ion pair 21b–d/22 [cf., the changes in Gibbs free energy (ΔG) accompanying their formation by implication of either tautomers 8 or 12, as presented on Scheme 6] and the dominance of 18 in the reaction mixtures obtained after 1 h, the share of these pathways was negligible in the construction of compounds type 21A. On the other hand, the rapid and dominant primary formation of 18 could be ascribed to the pronounced electrophilicity of 12, mainly ascribed to its low-energy LUMO, with a significant share on the C-1 atom. Accordingly, except for 4-nitrobenzaldehyde 26, the other arylaldehyde components had markedly higher LUMO energy relative to that of 12. This oxo-tautomer could be regarded as the main precursor of 18, even though the calculated ΔG value suggested that its tautomer 8 must be present in the reaction mixture in a substantially higher concentration. In the subsequent steps, 18 could have undergone condensation with the appropriate aldehyde to construct arylidenes 23B, or, with the involvement of 6 generating iminium ions 21a–d, it could have been converted into bis-morpholine intermediates type 24. Morpholine elimination from the aminal residue of 24 gave the corresponding Mannich product (23A). It is of note that due to the thermodynamically unfavoured formation of 21a–d under slightly acidic conditions (cf., data presented in Scheme 6), this condensation–elimination sequence can be considered as a less feasible pathway. Since the relative energetic data calculated for all investigated isomer pairs 23A/23B [(ΔG(23B-23A), see Scheme 5] suggested that 23a–dA are formed under thermodynamic control, the isomerisation of 23B into 23A can also be taken into account as a realistic process taking place under the applied conditions. In this regard, the calculated energetic data [(ΔG(25+6-23B)] practically rule out the elimination–addition sequence 23B→25→23A as a realistic pathway because the competitive addition–elimination sequence proceeding via bis-morpholine intermediates 24 seems to be more feasible as indicated again by the relative energetics calculated for the critical addition step [(ΔG(24-6-23B)].
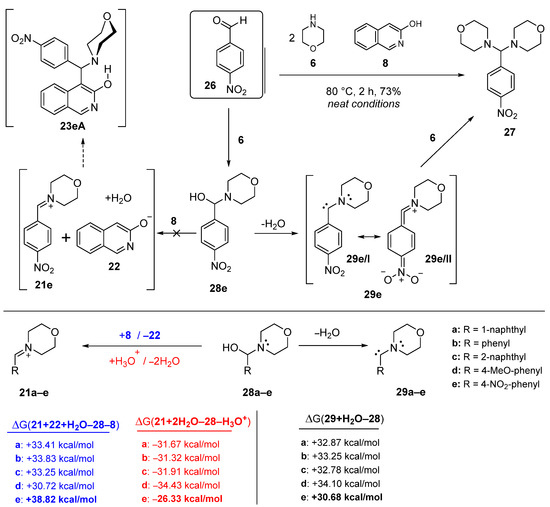
Scheme 6.
DFT-supported rationale of the unique reactivity of 4-nitrobenzaldehyde (26) manifested in its attempted Mannich condensation with morpholine (6) and 3-hydroxyisoquinoline (8) conducted under neat conditions.
In this context, the relatively increased reluctance of 23aB to undergo isomerisation into 23aA seems to be the consequence of the pronounced endoergic addition step 23aB + 6→24a, which might be associated not only with its enhanced electron-donating character, but also with the steric bulk of the 1-naphthyl group.
The reaction of 8 and 6 with p-nitrobenzaldehyde (26, [14,15,16]) was an exceptional case, because neither the expected Mannich product 23eA nor the appropriate benzylidene product 23eB could be isolated. Instead, 4,4′-((4-nitrophenyl)methylene)dimorpholine (27) was formed as the main product (Scheme 6). This unique conversion of 26, proceeding without the implication of equilibrating heterocyclic tautomers (8 and 12), can be interpreted by its LUMO energy being markedly lower than that of 12 (Figure 4). Furthermore, the relatively low-energy LUMO+1, also available to nucleophilic attack with significant share on the carbonyl group, further attenuates the electrophilicity of this reactive aldehyde. On the other hand, the tendency in the calculated energetic data—in accord with the general qualitative expectations—clearly shows that 3-hydroxyisoquinoline-promoted generation of the ion pair 21e/22 is a less favoured process, than the formation of the ion pairs 21a–d/22 containing a positively charged nitrogen atom without a strong electron-withdrawing substituent on the aryl group. It must be pointed out here that the readiness of iminium generation is strongly dependent on the proton source as shown by the energetic data obtained by replacing 8 with a hydroxonium ion in the calculations. The extreme difference between the two series of energetic data, otherwise both demonstrating the same tendency in the function of the electronic character of the aryl groups, can be considered as a strong indication of the dramatic dependence of the thermochemistry of ion-pair formation on a number of factors that might influence the stability of the charged particles, including, e.g., coulombic interaction, solvation and other intermolecular contacts. Avoiding unrealistically demanding computations, except for the estimated polarity of the reaction mixture (cf., description of the computational model in the “Materials and Methods” section), the aforementioned factors were neglected in the course of our DFT modelling studies.
Finally, we proposed a mechanism for the formation of 27, accounting for taking place without the implication of 8. Thus, according to our assumption, reductive elimination of the elements of the water molecule from the selectively generated adduct 28e gives carbene 29e stabilised by the 4-nitrophenyl group as represented by resonance hybrids 29e/I and 29e/II, which then reacts with 6 in an oxidative addition step affording 27 as the single isolable product (Scheme 6). In this sequence, carbene formation is the critical step with indispensable assistance of the nitrophenyl group. This view was supported by the comparison of the relative energetics of water elimination from adducts 28a–e (Scheme 6). The calculated data [(ΔG(29+H2O-28)] unambiguously indicated that the process 28e→29e+H2O is significantly less endoergic, and, consequently, it is more feasible than the analogous reaction steps 28a–d→29a–d+H2O. It must be emphasised again, that it is the tendency of the relative energetics that must be regarded to have diagnostic value in the assessment of the substituent effect, as solvation of the involved species along with other stabilizing and destabilizing interactions—which were not modelled in the calculations—are also expected to determine the real thermochemistry of the studied conversions. It is also of note that carbene 29e reacts with 6 rather than with 8. This substrate selectivity might be associated with the enhanced nucleophilicity of 6 compared to the neutral 8 of which the deprotonation-generating ion pair 21e/22 with the highly reactive anion 22, a real competitor of 6, is suppressed by the nitrophenyl group as discussed above.
3. Materials and Methods
The 1H and 13C NMR spectra were recorded in DMSO and D2O solutions in 5 mm tubes, at room temperature, with a Bruker spectrometer at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as the internal standard.
Melting points were determined on a Hinotek X-4 melting point apparatus. Elemental analyses were performed with a Perkin-Elmer 2400 CHNS elemental analyser. Merck Kieselgel 60F254 plates were used for TLC.
In the course of DFT calculations, the optimisation and frequency calculations on molecular structures were carried out with M06-2X global functional [17] using 6-31 G(d,p) basis set [18] supplemented by IEFPCM solvent model [19]. For these computations, dielectric constant (ε) was set to 10 (an arbitrarily modified value of 7.3 and an accessible parameter for morpholine), approximately representing the polarity of the modelled reaction mixtures composed of 3:1:1 molar ratio of morpholine, 3-hydroxyisoquinoline and the corresponding arylaldehyde. The energetic profile of the transformations was given by the changes in Gibbs free energy (ΔG). The free energy values of the optimised structures were obtained by correcting the computed total energy with zero-point vibrational energy (ZPE) and thermal corrections calculated at the same level of theory.
All calculations were carried out using the Gaussian 09 software (Gaussian Incorporation, Pittsburgh, PA, USA) package [20]. The optimised structures are available from the authors.
The NMR spectra of 11a and 11b are described in more detailed because of its unexpected and complex structure.
3.1. Synthesis of 5-(hydroxy(naphthalen-1-yl)methyl)quinolin-6-ol (5)
6-Hydroxyquinoline (50 mg, 0.35 mmol), 1-naphtaldehyde (54 mg, 0.35 mmol) and N-benzylmethylamine (42 mg, 0.35 mmol) were placed in a 10 mL reaction vial and heated in an oil bath at 60 °C for 8 h under neat conditions. The crude mixture was purified by column chromatography (EtOAc: MeOH, 20:1) and crystallised from MeOH (5 mL). Yellowish solid; yield: 43% (45 mg); m.p.: 84–87 °C. 1H NMR (DMSO-d6): δ = 6.50 (1H, s); 7.26 (1H, dd, J = 8.1 Hz and 4.5 Hz); 7.37 (1H, br ~t, J ~ 8 Hz); 7.43–7.47 (2H, m); 7.60 (1H, t, J = 7.8 Hz); 7.84–7.89 (4H, m); 7.97 (1H, d, J = 7.8 Hz); 8.18 (1H, dd, J = 8.1 Hz and 1.8 Hz); 8.56 (1H, dd, J = 4.5 Hz and 1.9 Hz). 13C NMR (DMSO-d6): δ = 63.1; 117.1; 117.3; 121.7; 125.9; 126.5; 127.4; 127.9; 129.1; 130.2; 129.2; 130.8; 132.5; 134.1; 136.2; 144.0; 147.1; 156.3. (Figures S1–S3).
3.2. Synthesis of 5-(morpholino(naphthalen-1-yl)methyl)quinoline-6-ol (7)
6-Hydroxyisoquinoline (250 mg, 1.72 mmol), 1-naphtaldehyde (268 mg, 1.72 mmol) and morpholine (180 mg, 2.06 mmol) were placed in a 50 mL round-bottom flask and stirred at 80 °C for 24 h. The crude mixture was crystallised from EtOAc (7 mL) and recrystallised from iPr2O (8 mL). Beige solid; yield: 34% (194 mg); m.p.: 189–192 °C. 1H NMR (DMSO-d6): δ = 3.59–3.68 (4H, m); 6.37 (1H, s); 7.21–7.26 (1H, m); 7.39–7.46 (2H, m); 7.60 (1H, t, J = 7.8 Hz); 7.70–7.78 (2H, m); 7.82–7.88 (2H, m); 7.97 (1H, d, J = 8.4 Hz); 8.14 (1H, brs); 8.56 (1H, s); 8.92 (1H, s). 13C NMR (DMSO-d6): δ = 63.7; 66.8; 116.6; 121.7; 123.6; 123.8; 126.3; 126.4; 127.3; 127.5; 127.9; 129.2; 129.6; 130.8; 132.4; 133.9; 143.9; 147.1. (Figures S4 and S5).
3.3. Synthesis of (E)-1-methoxy-4-(naphthalen-1-ylmethylene)-1,2-dihydroisoquinolin-3(4H)-one (10)
3-Hydroxyisoquinoline (50 mg, 0.34 mmol), 1-naphtaldehyde (53 mg, 0.34 mmol) and N-benzylmethylamine (41 mg, 0.34 mmol) were heated under solvent-free conditions in an oil bath at 80 °C for 8 h. The mixture was purified by column chromatography (EtOAc:MeOH, 20:1) and crystallised from MeOH (10 mL). Pale brown solid; yield: 59% (64 mg); m.p.: 187–190 °C. 1H NMR (DMSO-d6): δ = 3.36 (3H, s); 5.42 (1H, d, J = 4.5 Hz); 6.75 (1H, d, J = 8.1 Hz); 6.91 (1H, t, J = 7.3 Hz); 7.20 (1H, t, J = 7.7 Hz); 7.40 (1H, d, J = 7.3 Hz); 7.43–7.59 (4H, m); 7.90–8.01 (3H, m); 8.13 (1H, s); 9.28 (1H, d, J = 4.5 Hz). 13C NMR (DMSO-d6): δ = 54.1; 83.0; 124.7; 126.2; 126.6; 126.9; 127.2; 127.5; 128.2; 128.2; 129.1; 129.2; 130.9; 131.1; 131.3; 133.7; 133.8; 134.0; 167.6. (Figures S6 and S7).
3.4. Reaction of 3-Hydroxyisoquinoline with N-Benzylmethylamine
The mixture of N-benzylmethylamine (168 mg, 1.39 mmol) and 3-hydroxyisoquinoline (50 mg, 0.35 mmol) was placed in a 10 mL reaction vial and heated in an oil bath at 80 °C for 12 h. Dichloromethane (50 mL) and water (50 mL) were added to the mixture that was then extracted with 2% hydrochloric acid. After extraction, the organic fractions were collected, dried with anhydrous Na2SO4 and filtered, and the solvent was removed under reduced pressure. The residue was crystallised with n-hexane (15 mL) and recrystallised from iPr2O (10 mL) to afford 11a. The aqueous phase was alkalised with Na2CO3 and extracted with dichloromethane (2 × 50 mL). The organic fractions were collected, dried on Na2SO4, filtered and evaporated. The residue was purified by column chromatography (eluent EtOAc) to afford 11b.
3.4.1. 3′-Hydroxy-1,2-dihydro-[1,4′-biisoquinolin]-3(4H)-one (11a)
Yield: 13% (13 mg); m.p.: 181–184 °C. 1H NMR (DMSO-d6): δ = 3.67 (1H, d, J = 21.6 Hz, H-4A); 3,71 (1H, d, J = 21.6 Hz, H-4B); 6.53 (1H, br s, H-1); 6.65 (1H, br d, J = 7.8 Hz, H-8); 7.03 (1H, t, J = 7.8 Hz, H-7); 7.20 (1H, t, J = 7.8 Hz, H-6); 7.25–7.28 (2H, two overlapping m’s, H-5 and H-5′); 7.35 (1H, m, H-7′); 7.47 (1H, br m, H-6′); 7.96 (1H, d, J = 8.0 Hz, H-8′); 8.15 (1H, br s, H-2); 8.88 (1H, s, H-1′). 13C NMR (DMSO-d6): δ = 36.6 (C-4); 51.2 (C-1); 111.3 (C-4′); 123.0 (C-5′); 123.2 (C-7′); 123.7 (C-8′a); 125.4 (C-8); 127.1 (C-7); 127.3 (C-6); 128.0 (C-5); 129.3 (C-8′); 131.2 (C-6′); 132.8 (C-4a); 134.9 (C-8a); 137.6 C-4′a); 149.4 (C-1′); 159.3 (C-3′); 169.9 (C-3). (Figures S8–S10).
3.4.2. 4-((Benzyl(methyl)amino)(phenyl)methyl)isoquinolin-3-ol (11b)
Yield: 15% (18 mg); m.p.: 153–156 °C. 1H NMR (DMSO-d6): δ = 2.09 (3H, s, NCH3); 3.58 (2H, br s, H-10); 5.47 (1H, s, H-9); 7.21 (1H, t, J = 7.8 Hz, H-7); 7.27–7.40 (9H, overlapping m’s, H-5, H-3′,5′, H-4′, and H-2″-6″); 7.65 (1H, t, J = 7.8 Hz, H-6), 7.71 (2H, d, J = 7.7 Hz, H-2′,6′); 7.94 (1H, d, J = 7.8 Hz, H-8); 8.88 (1H, s, H-1). 13C NMR (DMSO-d6): δ = 40.0 (NCH3); 60.0 (C-10); 68.6 (C-9); 110.7 (C-4); 124.0 (C-5); 128.1 (C-3′,5′); 128.3 (C-7); 128.4 (C-8a); 128.5 (C-4′); 128.6 (C-2′,6′); 129.0 (C-4″); 129.4 (C-3″,5″); 129.6 (C-2″,6″); 129.9 (C-8); 131.3 (C-6); 136.7 (C-4a); 138.1 (C-1″); 141.5 (C-1′); 151.0 (C-1); 159.5 (C-3). (Figures S11–S13).
3.5. Synthesis of 1-morpholino-1,2-dihydroisoquinolin-3(4H)-one (18)
3-Hydroxyisoquinoline (50 mg, 0.34 mmol) and morpholine (120 mg, 1.38 mmol) were placed in a 25 mL round-bottom flask and were stirred at 80 °C for 2 h under neat conditions in an oil bath. The crude mixture was crystallised with EtOAc (12 mL). Beige solid; yield: 86% (69 mg); m.p.: 166–170 °C. 1H NMR (DMSO-d6): δ = 2.40 (4H, t, J = 4.5 Hz); 3.37 (1H, d, J = 20.0 Hz); 3.46–3.55 (4H, m); 3.63 (1H, d, J = 20.0 Hz); 5.03 (1H, d, J = 3.5 Hz); 7.18 (1H, d, J = 7.1 Hz); 7.25–7.31 (2H, m); 7.36 (1H, d, J = 7.2 Hz); 8.44 (1H, d, J = 3.1 Hz). 13C NMR (DMSO-d6): δ = 36.3; 47.7; 66.6; 74.2; 126.7; 127.7; 128.3; 128.3; 132.0; 133.8; 171.4. (Figures S14 and S15).
3.6. Synthesis of Compounds 23
The mixture of 0.34 mmol aldehyde [1-naphthaldehyde (53 mg), 2-naphthaldehyde (53 mg), benzaldehyde (36 mg), 4-methoxybenzaldehyde (47 mg) or 4-nitrobenzaldehyde (52 mg)], morpholine (90 mg, 1.03 mmol) and 3-hydroxyisoquinoline (50 mg, 0.34 mmol) were placed in a 10 mL reaction vial and heated in an oil bath at 80 °C under neat conditions.
3.6.1. (E)-1-Morpholino-4-(naphthalen-1-ylmethylene)-1,2-dihydroisoquinolin-3(4H)-one (23aB)
The crude mixture was purified by column chromatography (EtOAc:Aceton, 3:1) and crystallised from Et2O/EtOAc (10 mL). Beige solid; yield: 24% (31 mg); m.p.: 127–129 °C. 1H NMR (DMSO-d6): δ = 2.55–2.65 (4H, m); 3.56–3.66 (4H, m); 5.04 (1H, s); 6.72 (1H, d, J = 7.7 Hz); 6.87 (1H, t, J = 7.6 Hz); 7.19 (1H, t, J = 7.2 Hz); 7.38 (2H, t, J = 7.3 Hz); 7.47 (1H, t, J = 7.9 Hz); 7.53 (1H, t, J = 7.5 Hz); 7.58 (1H, t, J = 7.8 Hz); 7.94 (2H, d, J = 8.1 Hz); 8.00 (1H, d, J = 7.9 Hz); 8.12 (1H, s); 8.86 (1H, s). 13C NMR (DMSO-d6): δ = 48.0; 66.7; 73.2; 124.7; 126.1; 126.3; 127.0; 127.2; 127.6; 128.1; 129.0; 129.1; 129.1; 131.1; 131.3; 132.0; 133.0; 133.2; 133.8; 134.0; 167.5. (Figures S16 and S17).
3.6.2. 4-(Morpholino(phenyl)methyl)isoquinolin-3-ol (23bA)
The crude mixture was purified by column chromatography (EtOAc:Aceton, 4:1, and DCM:Aceton, 19:1) and crystallised from Et2O (9 mL). Beige solid; yield: 17% (15 mg); m.p.: 165–168 °C. 1H NMR (DMSO-d6): δ = 2.38–2.48 (4H, m); 3,67 (4H, s); 5.34 (1H, s); 7.20 (1H, t, J = 7.4 Hz); 7.27–7.35 (3H, m); 7.58–7.65 (3H, m); 7.92 (1H, d, J = 8.3 Hz); 8.33 (1H, brs); 8.86 (1H,s). 13C NMR (DMSO-d6): δ = 29.5; 52.5; 66.6; 68.5; 123.8; 124.7; 128.2; 128.7; 129.1; 129.2; 131.2; 136.8; 159.3. (Figures S18 and S19).
4. Conclusions
In conclusion, 6-hydroxyquinoline and 3-hydroxyisoquinoline as N-containing naphthol analogues were tested in the mMr. By the reaction of 6-hydroxyquinoline and 1-naphthaldehyde in the presence of N-benzylmethylamine, diol 5 was isolated. By replacing the amine compounds, the classical Mannich base formed as a single product. The difference between the products was interpreted on the basis of the different basicity of the starting amines. When 3-hydroxyisoquinoline was aminoalkylated in the presence of aromatic aldehydes, in addition to the classical Mannich base, the parallel formation of the C-1-substituted 3-hydroxyisoquinolines was observed. On the basis of comparative DFT modelling studies, it was concluded that, besides the steric bulk, the orbital-controlled electronic properties of the potential electrophilic components including aldehydes and, in particular, the carbonyl tautomer of 3-hydroxyisoquinoline (12) have a strong influence on the ratios and interconversions of the C-1-substituted 3-hydroxyisoquinolines and the classical Mannich bases. The unique reactivity of 4-nitrobenzaldehyde manifested under the same conditions was also rationalised by a distinct mechanism proposed on the basis of DFT modelling studies. The characteristic structure–reactivity relationships disclosed in this contribution along with the mechanisms we proposed for the intriguing transformations might be taken into account in the design of related multicomponent reactions targeted to construct valuable products with complex molecular architectures including, e.g., compounds of biological relevance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232012329/s1.
Author Contributions
Conceptualisation, A.C. and I.S.; investigation, O.C., P.B. and I.S.; writing—original draft preparation, O.C., P.B. and I.S.; writing—review and editing, A.C. and I.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated and analyzed during our research are not available in any public database or repository but will be shared by the corresponding author upon reasonable request.
Acknowledgments
The authors’ thanks are due to the Hungarian Scientific Research Foundation (OTKA No. K-138871 and OTKA No. K-129037), the Ministry of Human Capacities, Hungary grant, TKP-2021-EGA-32 and the Gedeon Richter Plc. Centenarial Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Biersack, B.; Ahmed, K.; Padhye, S.; Schobert, R. Recent Developments Concerning the Application of the Mannich Reaction for Drug Design. Expert Opin. Drug Discov. 2018, 13, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Filho, J.F.A.; Lemos, B.C.; de Souza, A.S.; Pinheiro, S.; Greco, S.J. Multicomponent Mannich Reactions: General Aspects, Methodologies and Applications. Tetrahedron 2017, 73, 6977–7004. [Google Scholar] [CrossRef]
- Szatmári, I.A.; Fülöp, F. Syntheses, Transformations and Applications of Aminonaphthol Derivatives Prepared via Modified Mannich Reactions. Tetrahedron 2013, 69, 1255–1278. [Google Scholar] [CrossRef]
- Cimarelli, C.; Fratoni, D.; Mazzanti, A.; Palmieri, G. Enantiopure α-Imino Glyoxylate: A Versatile Substrate for the Spontaneous Asymmetric Synthesis of Unnatural Hydroxyaryl Glycinates. Tetrahedron Asymmetry 2011, 22, 591–596. [Google Scholar] [CrossRef]
- Szatmári, I.; Fülöp, F. Solvent-Free Synthesis of 1-(Hydroxyquinolyl)-and 1-(Hydroxyisoquinolyl)-1,2,3,4-Tetrahydroisoquinolines by Modified Mannich Reaction. Synthesis 2011, 5, 745–748. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, M.K.; Kumar, M. Non-Ionic Surfactant Catalyzed Synthesis of Betti Base in Water. Tetrahedron Lett. 2010, 51, 1582–1584. [Google Scholar] [CrossRef]
- Shaaban, K.A.; Shepherd, M.D.; Ahmed, T.A.; Nybo, S.E.; Leggas, M.; Rohr, J. Pyramidamycins A-D and 3-Hydroxyquinoline-2-Carboxamide; Cytotoxic Benzamides from Streptomyces Sp. DGC1. J. Antibiot. 2012, 65, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Bunker, A.M.; Sliskovic, D.R. 3-Isoquinolinone Derivatives as Matrix Metalloproteinase Inhibitors. U.S. Patent 6,974,822, 13 December 2005. [Google Scholar]
- Liu, Y.; Zhao, Y.; Zhai, X.; Feng, X.; Wang, J.; Gong, P. Synthesis and Anti-Hepatitis B Virus Evaluation of Novel Ethyl 6-Hydroxyquinoline-3-Carboxylates in Vitro. Bioorg. Med. Chem. 2008, 16, 6522–6527. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, Y.; Zhai, X.; Liu, X.; Sun, L.; Ren, Y.; Gong, P. Synthesis and Anti-HBV Activities Evaluation of New Ethyl 8-Imidazolylmethyl-7-Hydroxyquinoline-3-Carboxylate Derivatives in Vitro. Arch. Pharm. Chem. Life Sci. 2008, 341, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Cerri, A.; Mauri, P.; Mauro, M.; Melloni, P. Synthesis of 1-(4-Substituted)Benzyl-6-Hydroxyisoquinolines with Potential Activity on Na+, K+-ATPase. J. Heterocycl. Chem. 1993, 30, 1581–1591. [Google Scholar] [CrossRef]
- Schmitt, S.; Colloc’h, N.; Perrio, C. Novel Fluoroalkyl Derivatives of Selective Kappa Opioid Receptor Antagonist JDTic: Design, Synthesis, Pharmacology and Molecular Modeling Studies. Eur. J. Med. Chem. 2015, 90, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Szatmári, I.; Barta, P.; Csámpai, A.; Fülöp, F. Synthesis and Detailed Conformational Analysis of New Naphthoxazino[2,3-a]Benz[c]Azepine and Naphthoxazino[2,3-a]Thieno[3,2-c]Pyridine Derivatives. Tetrahedron 2017, 73, 4790–4804. [Google Scholar] [CrossRef]
- Henry, R.A.; Dehn, W.M. New Compounds. Some Derivatives of Morpholine. J. Am. Chem. Soc. 1949, 71, 2271–2272. [Google Scholar] [CrossRef]
- Sakoda, R.; Matsumoto, H.; Seto, K. Novel Synthesis of α-Acetylstyrylphosphonates. Synthesis 1993, 1993, 705–713. [Google Scholar] [CrossRef]
- Pereira, J.G.; António, J.P.M.; Mendonça, R.; Gomes, R.F.A.; Afonso, C.A.M. Rediscovering Aminal Chemistry: Copper(II) Catalysed Formation under Mild Conditions. Green Chem. 2020, 22, 7484–7490. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Hehre, J.W.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, G.A.; Petersson, H.; Nakatsuji, X. Gaussian; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).