1. Introduction
Endometrial cancer (EC) originates in the cellular layer of the endometrium and is one of the most common malignancies of the female reproductive tract [
1]. Although most patients are diagnosed after menopause, the incidence of EC has gradually increased in fertile women [
2,
3]. EC is generally classified as type I or type II based on its pathological and molecular features. Type I EC positively responds to estrogen receptors (ERs) and progesterone receptors (PRs) and accounts for 70–80% of EC. Type II EC has a negative or weakly positive response to ERs and PRs, and accounts for 20–30% of EC [
4,
5]. Standard surgical treatments, including hysterectomy and bilateral salpingo-oophorectomy, are not suitable for all patients with EC, especially for young patients with type I EC who wish to preserve their fertility. For these patients, progestin treatments are generally allowed after a rigorous evaluation. At present, the medication approved officially for EC therapy is medroxyprogesterone acetate (MPA) [
6]. However, some patients experience progestin resistance during the therapy, failing treatment for approximately 30% of patients [
7,
8,
9]. MPA has an initial response rate of 55–100% [
10,
11], but an overall response rate of only 35% during the therapy [
12]. Additionally, the complete response rate for the patients taking low-dose (200 mg/day) and high-dose (1000 mg/day) MPA has been shown to be 17% and 9%, respectively [
13,
14], demonstrating an opposite dose–response relationship. These pieces of evidence indicate the existence of progestin resistance. The down-regulation of PR and the abnormal activation of phosphatidylinositol-3-hydroxykinase (PI3K)/phosphorylated protein kinase B (Akt) are generally considered as the main factors inducing progestin resistance in EC [
15,
16]. However, the theory does not explain why some patients with ER- and PR-positive expression do not respond to progestins while others with ER- and PR-negative expression respond to hormonal treatment [
8]. Accordingly, it is plausible to speculate that unknown factors are associated with progestin resistance.
Nuclear thyroid hormone receptors (TRs) have two isoforms: thyroid hormone receptor α (TRα) and thyroid hormone receptor β (TRβ), encoded by THRA and THRB, respectively, mediating the effects of thyroid hormones (THs) [
17]. The state of transcriptional regulation for the target genes of TRs depends on triiodothyronine (T3), which is the ligand and agonist of TRs [
18]. Studies showed that hypothyroidism might be a risk factor for many tumors, including liver, breast, and thyroid [
19,
20,
21,
22], and the expression of TRα and TRβ could be one of the factors affecting cancer progression [
23,
24,
25,
26,
27]. Recent evidence shows that hypothyroidism may also correlate with the occurrence of EC because it is one of the comorbidities in patients with EC [
21]. Thyroid dysfunction occurs in about 8.2% of patients with EC. Its incidence ranks second to that of metabolic syndrome [
28] but the relationship between thyroid dysfunction and EC has not yet been elucidated. Earlier, it was reported that high doses of MPA could increase the uptake of T3 during the therapy for EC and renal carcinoma [
29]. This implies that MPA promotes the binding of more thyroxine-binding globulin to T4 or T3. Nevertheless, how THs and TRs influence the progression of EC and the therapeutic outcome of progestins are currently unknown.
The presented study focused on the role of THRB in the growth of EC cells and its impact on Akt/ mammalian target of rapamycin (mTOR) pathway in the absence and presence of progestins. Both MPA and nomegestrol acetate (NOMAc) were tested since NOMAc, one of the fourth-generation progestins, has demonstrated stronger chemical properties than MPA and exhibited a suppressive effect on mTOR and its downstream signaling pathway in previous experiments [
30].
3. Discussion
The present study was novel in demonstrating that THRB knockdown promoted the growth and motility of RL95-2 EC cells and attenuated the suppressive effect of the progestins via enhancing the activity of the mTOR-4EBP1/eIF4G pathway. Thus, progestins facilitated the growth of type I EC cells in the absence of THRB. We found lower serum levels of T3 in patients with EC compared with healthy women. Moreover, weaker expressions of TRβ were observed in the endometrium of patients with EC or EAH insensitive to progestin therapy compared with those sensitive to the therapy. Taken together, it was plausible to presume that the lower expression of THRB/TRβ was likely correlated with progestin resistance in EC therapy.
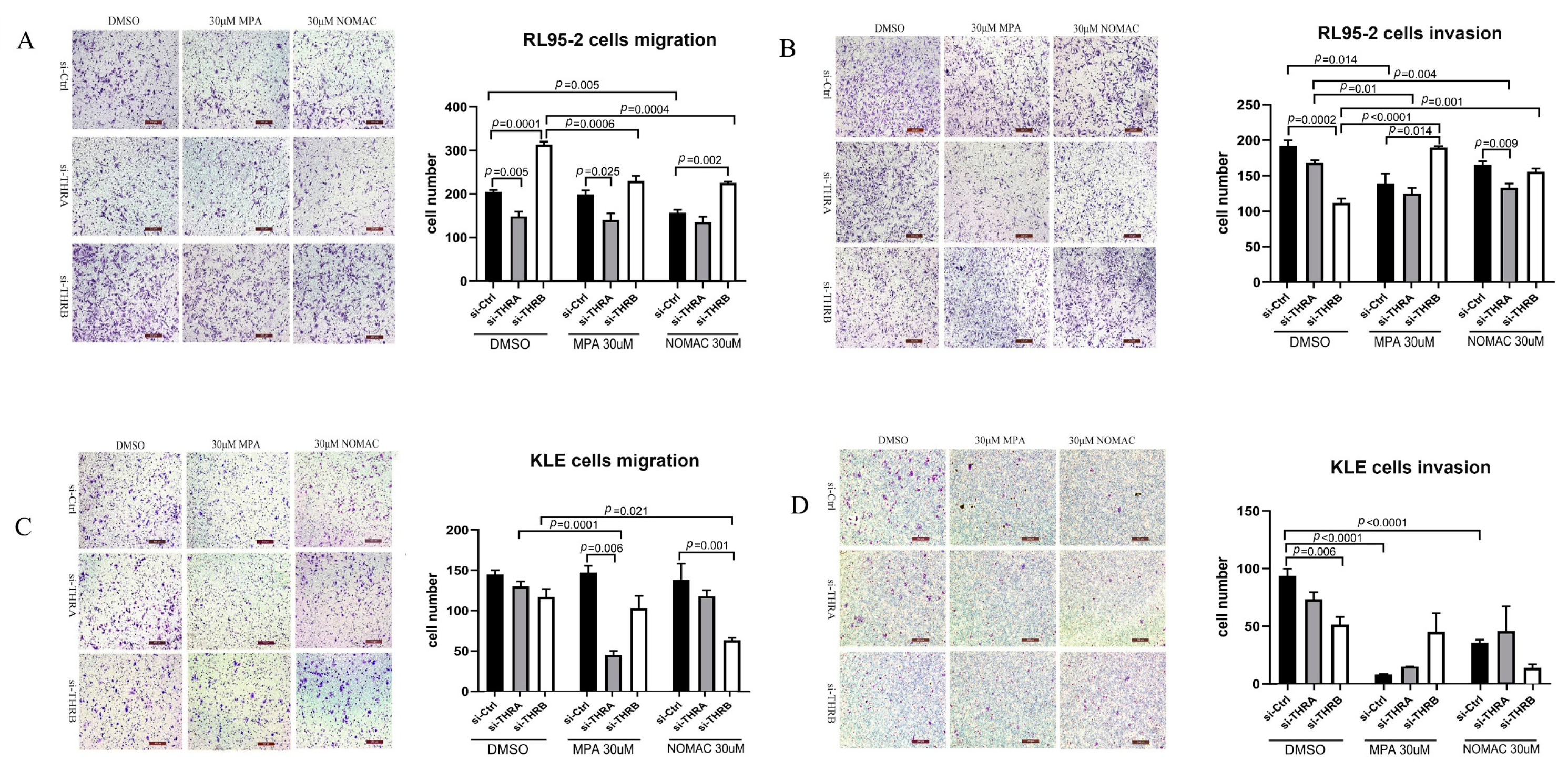
Human type I EC is generally described as hormone receptor-positive and sensitive to progestin therapy and type II EC is described as hormone receptor-negative and insensitive to progestin therapy. Accordingly, human-originated RL95-2 cells are defined as type I EC cells and KLE as type II EC cells, which are characterized by positive and negative expression of hormone receptors, respectively. In our previous study, the protein expression of ERα and PR was observed in RL95-2 cells but not in KLE cells, and the expression of p53 was detected in KLE cells but not in RL95-2 cells [
30]. In this study, therefore, we used both RL95-2 and KLE cells to investigate the effect of THRB on progestin treatment and found that RL95-2 cells were more sensitive to MPA and NOMAc than KLE cells, which was consistent with the previous findings [
30].
THRB has been reported to act as a transcription-suppressive factor in response to the changes in THs levels, lying at the crossroad of many cellular signaling pathways and playing a critical role in maintaining normal cell characteristics and tumor progression in thyroid cancer [
25]. THRB may act as a tumor suppressor in solid tumors, such as breast, hepatocyte, and thyroid, by inducing apoptosis and reducing the cell renewal capacity to restrain the growth of tumors [
25,
26]. The present study found that both TRα and TRβ were expressed in RL95-2 and KLE cells; however, it was THRB knockdown, rather than THRA, boosting the proliferation and motility in RL95-2 cells. It indicated that THRB exerted a suppressive effect on cell proliferation, when THRB was knocked down, the suppressive effect was attenuated.
The Akt-mTOR pathway is generally considered to play a role in the proliferation of tumor cells and is involved in progestin resistance [
16,
31]. Akt inhibitor has been used in treating ER/PR-positive breast cancer to disrupt the function of the PI3K/Akt/mTOR pathway and alleviate endocrine resistance [
16,
32]. The mTOR inhibitor has also been tested as an EC therapy in phase II clinical trials [
33,
34]. We previously found that activating the mTOR-4EBP1/eIF4G pathway could promote proliferation and inhibit apoptosis in RL95-2 and HEC-1A EC cells [
30]. The present study found that the growth and the motility of cells were enhanced, and mTOR and its downstream 4EBP1/eIF4G signaling pathway were significantly activated in THRB-silenced RL95-2 cells. In contrast, not the whole of the signaling pathway was activated in THRB-silenced KLE cells, which might result in little impact on the growth and motility of the cells. It is plausible to presume that
THRB likely plays a more critical role in regulating the growth of RL95-2 cells than KLE cells, and its action is correlated with the mTOR signaling pathway.
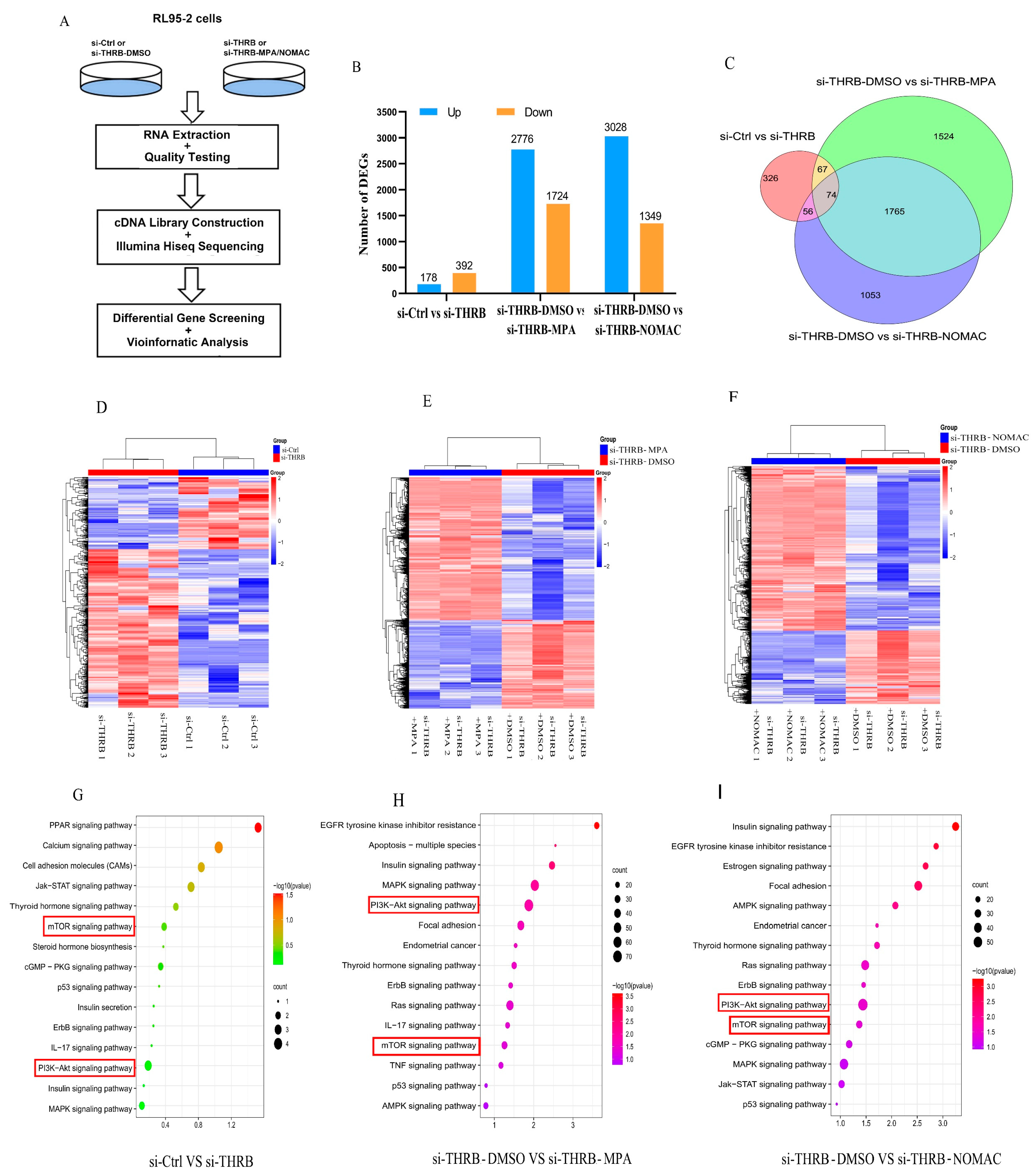
Progestin resistance caused by EC cells insensitive to the treatment of progestins has been a major obstacle for patients who wish to preserve fertility, and the mechanism involves the activation of the PI3K/Akt pathway and abnormal proliferation of tumor cells [
31,
35,
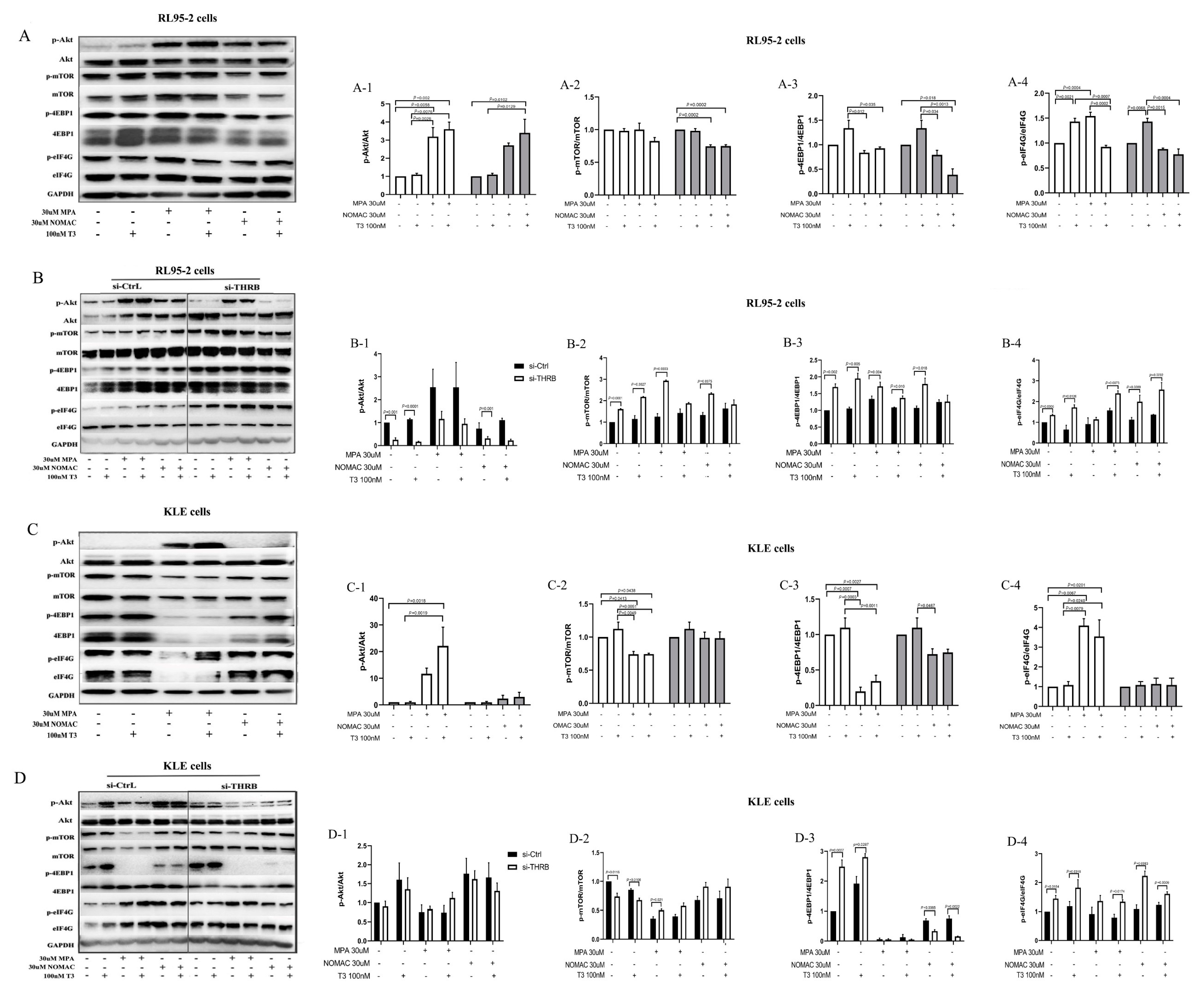
36]. Previously, we found that NOMAc effectively restrained the growth of RL95-2 and HEC-1A cells by suppressing the activity of mTOR-4EBP1/eIF4G. In this study, we further found that several proliferation-related signaling pathways, including PI3K/Akt, mTOR, Ras, MAPK, and TP53, were markedly enriched in the progestin treatment of THRB-silenced RL95-2 cells using the transcriptomic assay. Accordingly, we focused on Akt and its downstream mTOR signaling pathway to explore how progestins induced cell growth. We found that both MPA and NOMAc suppressed the activity of Akt but significantly facilitated the activity of mTOR and its substrates 4EBP1 and eIF4G in THRB-silenced RL95-2 cells. Additionally, the supplementation of T3 partially abrogated the pronounced boosting effect of the progestins on mTOR and 4EBP1. In THRB-silenced KLE cells, the progestins did not affect the activity of Akt but increased the activity of mTOR or eIF4G. It suggests that MPA and NOMAc activate mTOR/4EBP1/eIF4G rather than Akt in the THRB-silenced EC cells. In view of the remarkable promoting effects of both progestins on cell growth and motility in the THRB-silenced RL95-2 cells, it was plausible to presume that the down-regulation of THRB might be one of the critical factors associated with progestin resistance in type I EC cells. The underlying mechanism was probably via activating mTOR and its downstream 4EBP1/eIF4G signaling pathway rather than upstream Akt, but more related studies are warranted in the future.
Activating Akt, the upstream signaling of mTOR was partially ascribed to MPA-induced resistance in EC and breast cancer [
16,
31]. Consistently, we also observed that MPA enhanced the activity of Akt and eIF4G in both RL95-2 and KLE cells. In contrast, NOMAc facilitated the activity of Akt in RL95-2 cells but not in KLE cells. The results could partially expound the reason why NOMAc exhibited stronger inhibition than MPA on the growth of KLE cells. In THRB-silenced EC cells, both MPA and NOMAc demonstrated similar boosting effects on p-mTOR and p-eIF4G, except that NOMAc notably decreased the activity of 4EBP1 in THRB-silenced KLE cells. It suggests that MPA and NOMAc display subtly different effects on the proliferation-related signaling pathways in both wild-type and THRB-silenced EC cells.
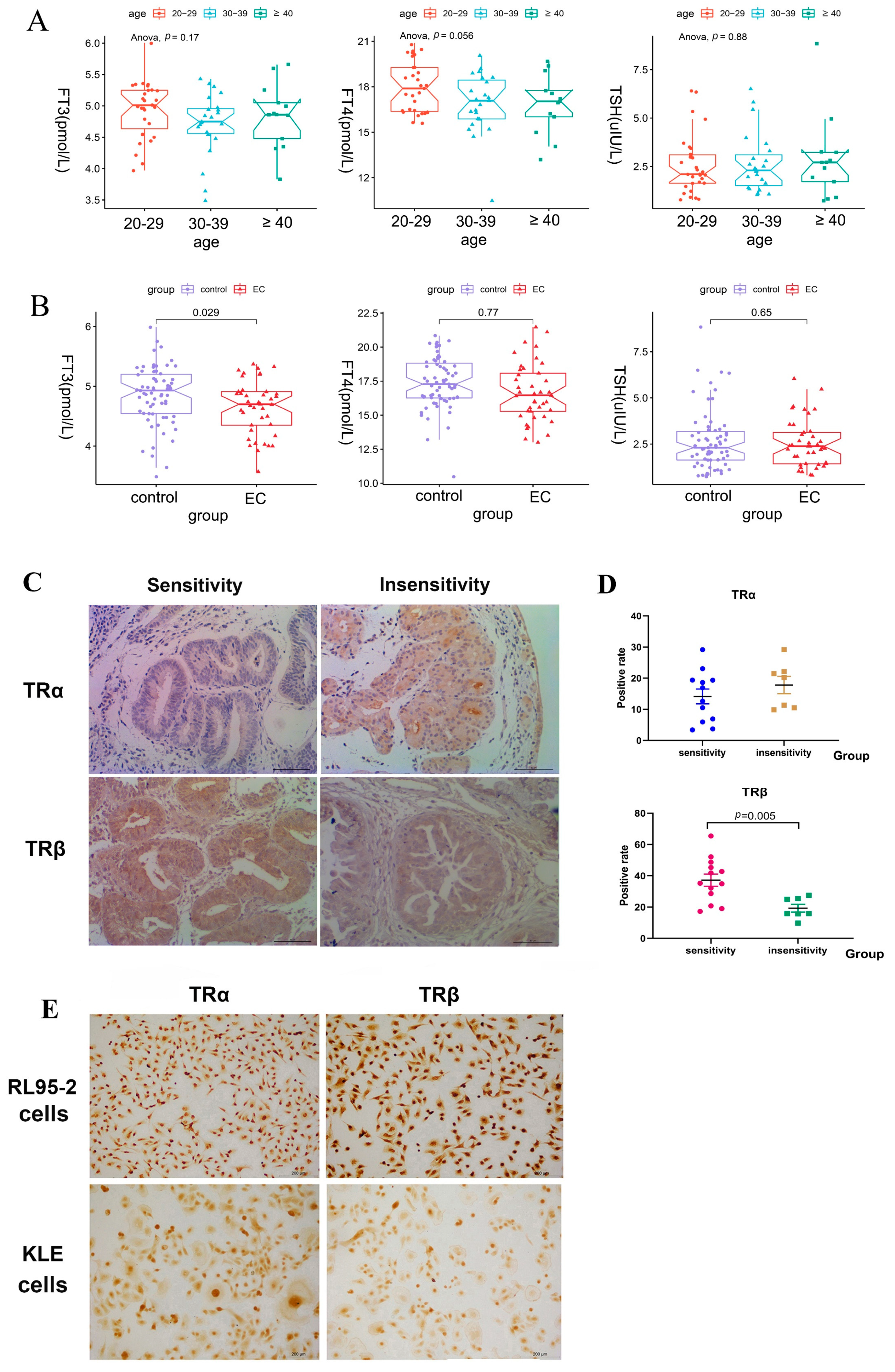
Consistent with the findings of the cell experiments, we found a disparity in the protein expression of TRα and TRβ in patients sensitive or insensitive to progestin therapy. Both endometrium tissues were detected in the study because the number of patients with EAH was more than that of patients with EC. Notably, the expression of TRβ in progestin-sensitive EAH and EC tissues was significantly stronger than that in progestin-insensitive tissues. In contrast, no significant difference was found in the expression of TRα between progestin-sensitive and progestin-insensitive tissues. It suggested that the expression of TRβ, rather than TRα, might influence the effect of progestins in treating of EAH/EC, and a stronger expression of TRβ was more likely correlated with the effective therapy using progestins.
Generally, hypothyroidism is diagnosed based on changes in the levels of TSH, total thyroxine (TT4), and free thyroxine (FT4) [
21]. However, we found that the serum levels of FT3, rather than FT4 and TSH, significantly declined in patients with EC compared with healthy women in this study. Although low-T3 syndrome has been observed in many patients with chronic disease or cancer, including type II diabetes mellitus [
37] and chronic lymphocytic leukemia [
38], whether the low levels of T3 are correlated with the occurrence of EC is unclear. Nevertheless, the result provides evidence that a relationship that exists between EC and abnormal thyroid systemic function. More clinical investigations are warranted in the future.
We also investigated the effect of T3 and found that both 10 and 100 nM T3 demonstrated similar effects on the EC cells and did not influence the growth of the two types of EC cells but antagonized the suppressive effects of progestins in RL95-2 cells rather than in KLE cells. Therefore, 100 nM T3 was used as an agonist of THRB for subsequent investigation to distinguish the effects induced by exogenous T3, rather than endogenous T3, in FBS because normal FBS (possibly containing THs), rather than hormone-depleted serum, was used in the present study to avoid more severe injury to the cells in the presence of transfection reagents. In THRB-silenced RL95-2 cells, we found that T3 did not change the viability of the cells but abolished cell proliferation induced by progestins, which might be ascribed to the inconsistent or even inversed regulation of T3 on the activity of Akt and mTOR/4EBP1/eIF4G. T3 treatment significantly enhanced the activity of mTOR-4EBP1/eIF4G but not of Akt; while a combination with the progestins increased the activity of either p-4EBP1 or p-eIF4G and markedly decreased the activity Akt. As a result, the proliferative effects of progestins were mildly abolished. These pieces of evidence indicate that T3 supplement likely restored the suppressive effects of the progestins in THRB-silenced RL95-2 cells but not in THRB-expressing cells. Moreover, despite the fact that T3 and progestins demonstrated facilitative or suppressive effects on the signaling of Akt/mTOR-4EBP1/eIF4G in THRB-silenced EC cells, no significant difference was observed between DMSO and T3 or progestins treatment groups. It suggests that the alteration of the signaling pathway arose from the knockdown of THRB rather than induced by T3 or progestins themselves. Taken together, lower expression of THRB is likely one of the crucial factors causing progestin resistance.
5. Materials and Methods
5.1. Compounds
Medroxyprogesterone acetate (MPA) and nomegestrol acetate (NOMAc) were provided by Xianju Pharmaceutical Co., Ltd. (Taizhou, China). Triiodothyronine (T3) was purchased from Sigma-Aldrich Co., LLC. (St. Louis, MD, USA).
5.2. Collection of Human Sera Data and Endometrial Tissues
This study was approved by the Ethics Committee of the Shanghai Institute of Planned Parenthood Research (SIPPR) and the approval No. is PJ2019-10. The data of serum THs were collected from the medical history forms of the patients hospitalized at the Obstetrics and Gynecology Hospital of Fudan University from June 2016 to December 2017, including 41 patients with EC. Adult women diagnosed with EC by primary examination of B-ultrasound and subsequent curettage, hysteroscopy, or surgical pathology were included in the EC group. All 41 patients with EC included were diagnosed for the first time, except for 2 women who were re-diagnosed and 2 women who were found to have EC after surgery for multiple cancers. The serum samples were taken by venipuncture after at least 12 h fasting when they accepted routine hematological examination. None of them had undergone medication treatment. The control group consisted of 67 healthy women from routine physical examinations during the same time. All women had been excluded from malignant tumor-related diseases and thyroid diseases. The thyroid function tests were assessed by extracting peripheral venous blood at fasting status, and serum TSH, free triiodothyronine (FT3), and free thyroxine (FT4) levels were measured using the electrochemiluminescence method.
Moreover, we collected endometrial tissues from the patients hospitalized at the Obstetrics and Gynecology Hospital of Fudan University, including 8 patients with EAH and 12 patients with EC (aged 21–39 years), who underwent hysteroscopy between December 2018 and December 2019. Among them, EAH had 3 progestin-insensitive and 5 progestin-sensitive tissues and EC had 4 progestin-insensitive and 8 progestin-sensitive tissues. All the data were obtained after obtaining informed oral consent.
5.3. Cell Cultures
Human RL95-2 EC cell line was purchased from Baili Biotechnology Co., Ltd. (Shanghai, China). The RL95-2 cell line was derived from EC tissues of a 65-year-old white woman. Human KLE EC cell line was derived from the China Type Culture Collection (Wuhan, China). KLE cell line was derived from EC tissue of a 64-year-old woman. Both RL95-2 and KLE cells were cultured in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 1:1 (DMEM/F12; Gibco, Carlsbad, CA, USA) media supplemented with 10% FBS (Gibco, New Zealand). All cells were cultured in a humidified incubator at 37 °C with 5% CO2 in the air. The culture media were replaced every 2 or 3 days until the cells reached approximately 70–80% confluence, and then the cells were subcultured.
5.4. Cell Viability Assays
RL95-2 and KLE cells were seeded in 96-well plates (8000 cells/well), and cells were treated with MPA and NOMAc at concentrations of 1, 3, 10, 30, and 100 µM for 48 h for measurement of half-maximal inhibitory concentration (IC50). In other assays, the cells were treated with T3 (10 or 100 nM), MPA or NOMAc (30 µM), and MPA/NOMAc (30 µM) plus T3 (10 or 100 nM) for 48 h. Control cells were treated with dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MD, USA) and the final concentration in the culture media was 1% (v/v). According to the manufacturer’s protocol, 10μL CCK-8 solution (Dojindo Laboratories, Kumamoto Prefecture, Kyushu Island, Japan) was added to each well and incubating in a 37 °C incubator for 2 h, and then measured OD value at 450 nm on a microplate reader (BioTek ELX-800). Cell viability was calculated by the formula: cell viability (%) = OD of treatment cells/OD of control cells ×100%. Final results were presented as IC50 with 95% confidence intervals (95% CI), which were calculated from a nonlinear regression model based on log(inhibitor) vs. normalized response/variable slope dose–response curves using GraphPad Prism 8.0 (GraphPad Sofware Inc., La Jolla, CA, USA).
5.5. Immunohistochemistry
The biopsied uterine tissues taken from the patients with AC and EAH sensitive or insensitive to the therapy of progestins, including MPA, Mirena
®, or megestrol (MA), were fixed in formalin and embedded in paraffin. Progestin insensitivity was defined as disease progression at any time during treatment, stable disease after 7 months of treatment, or no complete response (CR) after 10 months of treatment. Other patients who achieved CR within 10 months of treatment were regarded as progestin sensitive. A SABC immunohistochemistry kit (Boster Bio, Wuhan, China) was used to detect the expression of TRα and TRβ in the endometrium following the manufacturer’s instruction. Briefly, the sections were deparaffinized and immersed in antigen retrieval solution (containing Tris 12.1 g, urea 50 g, and ultrapure water 1 L, pH = 9.5) for 10 min at 95 °C. Then, diluted antibodies (TRα 1:100 or TRβ 1:100; Sigma-Aldrich, St. Louis, MD, USA) were dropped onto the tissues and incubated overnight in a humidified chamber at 4 °C. Finally, the sections were stained with DAB working solution (Boster bio, Wuhan, China) for 5–8 min. The double-blind readings were performed by two experienced technicians. The expression of TRα or TRβ in each section was evaluated using histoscores and ratio of positive area. Histoscores were calculated using the following formula: scores of the positive cells multiplied by the grade of staining intensity. The scores of ≤3 points indicated negative expression, and scores of >3 points indicated positive expression. Moreover, the scores of >3 and ≤6 points denoted weakly positive, and scores of >6 denoted strong positive [
39]. The number of positive cells in each section was determined by the number of stained cells in 100 cells of 5 random fields under a microscope at 200× magnification. The grade of positive staining intensity was defined as follows: 1 point stood for weak immunostaining and was demonstrated in light yellow color, 2 points for moderate immunostaining and demonstrated in brown color, and 3 points for strong immunostaining and demonstrated in tan color. The criteria for the scores of positive cells were described as follows: no positively stained cells were scored as 0 points, 10–25 stained cells as 1 point, 26–50 stained cells as 2 points, and more than 50 stained cells as 3 points. In addition, the ratio of the positive area in each section was analyzed using Image J 1.48 (Rawak Software, Stuttgart, Germany) and its plug-in IHC Tools. The expression of TRα and TRβ in the sections of progestin-sensitive and progestin-insensitive tissues were then statistically analyzed.
The RL95-2 and KLE cells were seeded onto poly-lysine-coated coverslips (Boster Bio, Wuhan, China) inserted in a 24-well plate at 100,000 cells/well, and cultured in incubator at 37 °C with 5% CO2 for 24 h. The coverslips were incubated in 4% paraformaldehyde for 10 min and then immersed in antigen retrieval solution at 95 ℃ for 10 min. The cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, St. Louis, MD, USA), incubated with goat serum blocking solution for 30 min, and then incubated with diluted antibodies targeting TRα (1:40; R&D system, Emeryville, CA, USA) or TRβ (1:200; Abcam, Burlingame, CA, USA) overnight at 4 °C in a humidified chamber; finally, the cells were stained with DAB working solution for 5–8 min. The coverslips were dehydrated and photographed under a microscope (Leica DMi8, Hesse, Wetzlar, Germany)
5.6. Small Interfering RNA (siRNA) Transfection
RL95-2 and KLE cells were seeded into six-well culture plates at a density of five million cells/well for 24 h. Transfection experiments were performed when the cell confluence reached 70–90%. Then, 200 pmol of siRNA negative control or siRNA against THRA or THRB (Sangon Biotech, Shanghai, China) was added to Opti MEM (Thermo Scientific, Waltham, MA, USA), followed by Lipo3000 (Invitrogen, Carlsbad, CA, USA) for dilution. The solutions were mixed gently to prepare the Lipo3000 siRNA Transfection Reagent–siRNA complex. Subsequently, the complex was evenly dropped onto RL95-2 and KLE cells and then transfected for 48 h. THRA and THRB siRNA sequences in RL95-2 cells were as follows: THRA: 5′-CAAACACAACAUUCCGCAUUTT-3′; THRB: 5′-GCCUGUGUUGAGAGAAUAGAATT-3′. THRA and THRB siRNA sequences in KLE cells were as follows: THRA: 5′-GCGUAAGCUGAUUGAGCAGAATT-3′; THRB: 5′-GCCUGUGUUGAGAGAAUAGAATT-3′.
5.7. RNA Isolation and Quantitative Reverse Transcriptase–Polymerase Chain Reaction Analysis
Trizol (Invitrogen, Carlsbad, CA, USA) was added into RL95-2 and KLE cells or silenced THRA- or THRB-RL95-2 and KLE cells. The cells were collected and incubated in Trizol for 5 min. Chloroform (Sangon Biotech, Shanghai, China) was added to the lysed cells, vigorously shaken and mixed, and then centrifuged at 12,000×
g for 15 min at 4 °C. The supernatant was discarded, and isopropanol (Sangon Biotech, Shanghai, China) was added to the sediment, shaken, and centrifuged at 7500×
g for 10 min at 4 °C. After drying, added 15 μL of RNase-free water (Sangon Biotech, Shanghai, China) was added to dissolve, and the concentration on an RNA concentration meter (Thermo Scientific, Waltham, MA, USA) was measured. RNA was reverse transcribed into cDNA on a PCR machine (ABI Veriti) following the instructions of the TAKARA (TAKARA, Tokyo, Japan) reverse transcription kit. The DNA template, primers (THRA, THRB, and ACTB genes) (Sangon Biotech, Shanghai, China), and reagents such as TB Green Premix Ex Taq were mixed, and qPCR was performed following the manufacturer’s instructions (TAKARA, Tokyo, Japan). The primers designed in the experiment are shown in
Table 1 (Sangon Biotech, Shanghai, China). The melt curves and Ct values were analyzed using Roche LC480 software (Roche, Basel, Switzerland). The fold change of gene silencing efficiency was calculated using the formula 2
−△△Ct. The primer sequences used were as follows: ACTB (sense, 5′-CCTGGCACCCAGCACAAT-3′; antisense, 5′-GGGCCGGACTCGTCATAC -3′); THRA (sense, 5′-GATGACACGGAAGTGGCTCTGC-3′; antisense, 5′-AATGTTGTGTTTGCGGTGGTTGAC-3′); and THRB (sense, 5′-CAACTTTTTGGCAAAATCCACC-3′; antisense, 5′-GATGACACGGAAGT GGCTCTGC-3′).
5.8. Cell Migration Assay
After silencing THRA or THRB for 48 h, the silenced RL95-2 or KLE cells and negative siRNA control cells were digested and washed with HBSS (Thermo Scientific, Waltham, MA, USA) once or twice, and then resuspended in serum-free media. The densities of the cells were adjusted to 50,000 cells/well. The cell suspension was transferred to a Transwell chamber (Corning, New York, NY, USA) and 600 μL of media containing 10% FBS was added to the lower chamber. Then, 30 μM MPA or NOMAc was added to the cells and incubated at 37 °C in a 5% CO2 incubator for 12 h. The control cells were treated with the same volume of DMSO. After 12 h, the residual media in the chamber were discarded, and the chambers were washed twice with PBS (Corning, New York, NY, USA). The remnant cells in the upper chambers were wiped off with a cotton swab and fixed with 4% paraformaldehyde (dissolved in PBS) (Tansoole, Shanghai, China) for 15 min, and then rinsed slowly twice with water for 2 min each time. The cells were infiltrated with 0.1% crystal violet solution (Sangon Biotech, Shanghai, China) for 10 min and then washed with water twice. The staining cells were observed under a microscope at 20× magnification, and five fields of view were randomly selected for photographing and counting. The cells were counted using Image J 1.48 software (Rawak Software, Stuttgart, Germany).
5.9. Cell Invasion Assay
Matrigels (Corning, New York, NY, USA) were pre-cooled to 0 °C in advance and then diluted to the concentration of 200–300 μg/mL using serum-free media. Then, 100 μL of Matrigel was added to each transwell chamber. The gel was placed in a 37 °C incubator for 1 h, and the upper liquid media was discarded and used to culture RL95-2 and KLE cells until the logarithmic growth phase. After silencing of THRA or THRB for 48 h, the silenced RL95-2 or KLE cells and negative siRNA control cells were digested. The media were discarded by centrifugation and washed once or twice with HBSS, and then resuspended in serum-free media. The densities of the cells were adjusted to 100,000 cells/well. The procedures for cell inoculation, drug treatment, staining, and cell counting were the same as for the cell migration assay.
5.10. Transcriptomic Analysis
RL95-2 cells were transfected with si-THRB or solvent for 48 h and then treated with DMSO, or 30 μM MPA or NOMAc for another 48 h. The cells were harvested and RNA was extracted and checked for quality. After quality inspection, cDNA libraries were constructed. The cells treated with si-Ctrl, si-THRB or si-THRB-DMSO, si-THRB-MPA, or si-THRB-NOMAc, were subjected to high-throughput sequencing after the silencing of THRB using the paired-end sequencing method of the Illumina Hiseq sequencing platform (Juran Biotech, Shanghai, China). For all samples, the raw sequence numbers of known genes were calculated using StringTie software and the expression of known genes was calculated using fragments per kilobase of transcript per million fragments mapped (FPKM). FPKM = total fragments/(mapped reads(millions) × exon length(KB)). The DESeq2 package was used to screen DEGs between different sample groups. The DEGs were screened out by calculating
p values with Fisher’s exact test. The calculated
p values were used to determine whether the KEGG functional set in the target genes were significantly enriched or not, and the
p values were corrected by Benjamini & Hochberg’s multiple tests to obtain a false discovery rate (FDR). The data satisfying |log2FC| ≥ 1 and
p value ≤ 0.05 were used to screen the DEGs between the two groups. The DEG screened out between si-Ctrl versus si-THRB, si-THRB-DMSO versus si-THRB-MPA, and si-THRB-DMSO versus si-THRB-NOMAc groups were further analyzed using the KEGG signaling pathway. KEGG functional analysis was performed via functional annotation and classification for the pathways in which these genes were involved. The enrichment results were visualized using an online tool (
http://www.bioinformatics.com, accessed on 26 April 2022 and 8 October 2022). Final data were from three independent experiments.
5.11. Western Blot
The cells silenced with si-THRB for 48 h or the cells were treated with T3 (100 nM), MPA or NOMAc (30 µM), and MPA/NOMAc (30 µM) plus T3 (100 nM) for 48 h, respectively. Control cells were treated with the DMSO. The cells were harvested and suspended in rapid cell-tissue lysis buffer (RIPA) (Invitrogen, Carlsbad, CA, USA) containing 1% protease and phosphatase inhibitors (Thermo Scientific, Waltham, MA, USA). The extracted proteins were boiled at 100 °C for 5 min and then stored at −20 °C. Protein concentration was determined using a BCA protein detection kit (Sangon Biotech, Shanghai, China), and 15 μg of total proteins were electrophoresed and loaded on to 10% SDS-PAGE gels (Sangon Biotech, Shanghai, China) and then transferred to PVDF membranes (Millipore) for 1.5 h. Membranes were blocked with 5% nonfat milk powder (TBST) for 1 h and incubated overnight at 4 °C. Primary antibodies against phospho-Akt (Ser 473) (#9271, 60 kDa), Akt (Pan) (#4691, 60 kDa), phospho-mTOR (Ser2448) (# 5536, 289 kDa), mTOR (# 2983, 289 kDa), phospho-4EBP1 (Ser65) (#9451, 15–20 kDa), 4EBP1 (#9644, 15–20 kDa), phospho-eIF4G (Ser1108) (#2441, 220 kDa), eIF4G (#2469, 220 kDa) and β-actin (#4970, 45 kDa) were diluted at 1:1000 (CST, Danvers, Massachusetts, USA). TRα (R&D Systems, Emeryville, CA, USA, #PP-H2804-00) and TRβ (sigma, St. Louis, MD, USA, #SAB4502969) were diluted at 1:1000; GAPDH (#ab181602, 36 kDa) were diluted at 1:10,000. The PVDF membranes (Millipore, Bedford, MA, USA) were then washed three times with TBST solution and incubated in peroxidase-conjugated goat anti-rabbit IgG (immunoglobulin G) (#7074) with dilution of 1:3000 at room temperature for 1 h. After washing with TBST three times, protein bands were visualized using the ECL SuperSignal West Femto Detection Kit (Thermo Scientific, Waltham, Massachusetts, USA). All antibodies were purchased from Cell Signaling Technology (Beverly). We performed grayscale analysis of the bands using Image Lab 4.0 (Sydney, Australia), and data were analyzed using the method of (p-protein/GAPDH)/(protein/GAPDH).
5.12. Statistical Analysis
Data were presented as the mean ± standard error of mean (SEM) of triplicate or three independent experiments, and all statistical analyses were performed with PRISM 8.0 (GraphPad Software, Inc., La Jolla, CA, USA). Multiple comparisons among groups were analyzed using one-way ANOVA (and nonparametric) followed by post-test of Tukey’s or Dunnett’s multiple comparison test. Two-tailed unpaired t-test was used to compare the serum levels of THs between the patients and healthy women, as well as the viability of the cells and the changes of protein expression and transcriptomics prior to and after progestins treatment. Data were considered as statistically significant at p-values less than 0.05.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}