
The Arabidopsis thaliana–Streptomyces Interaction Is Controlled by the Metabolic Status of the Holobiont
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
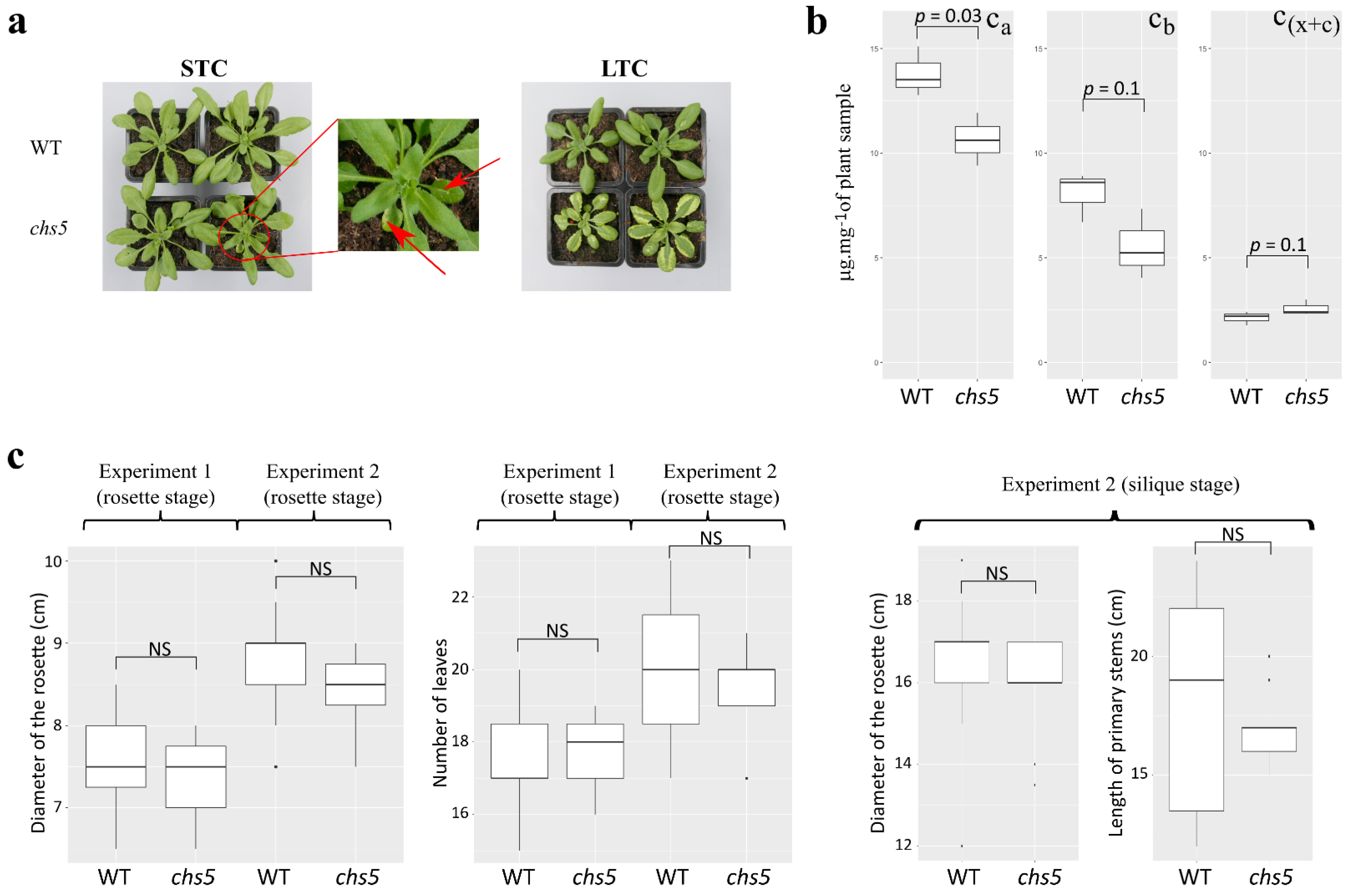
2.1. The chs5 Mutant Exhibits Changes in Carotenoids, Xanthophylls, Phenylpropanoids, and Lipid Profiles
2.2. Plant–Bacteria Interactions Are Changed in the chs5 Mutant
2.2.1. The chs5 Mutant Is More Sensitive Than the Wild Type to the Phytopathogen DC3000 Infection
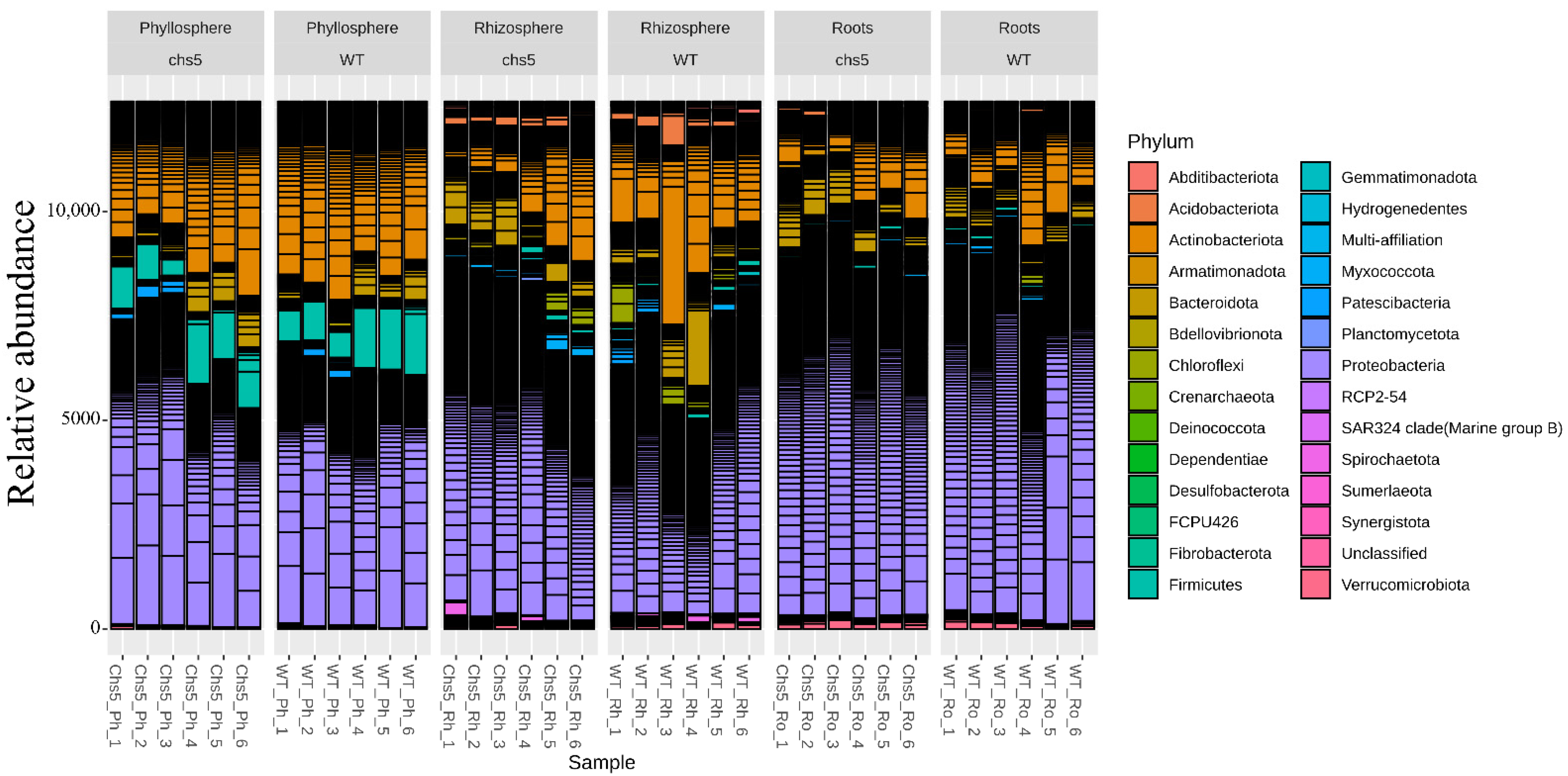
2.2.2. The Global Structures of the Wild-Type and chs5 Microbiota Are Similar When Grown in STC or in LTC
2.2.3. The Colonization of Specific Bacteria Is Reduced in the chs5 Mutant as Compared to the Wild-Type Plant, in STC at Both Stages, and in LTC
2.2.4. The Variation in Abundance According to the Stage of Development Is Different for Some Specific Taxa, in the Wild-Type and the Mutant Plants
2.2.5. Streptomyces Species Are Impaired in Their Colonization or Growth When Associated with the chs5 Mutant
3. Discussion
4. Materials and Methods
4.1. Horticultural Growth Conditions
4.2. Infection with Pseudomonas syringae pv. tomato DC3000 (Pto DC3000)
4.3. Metabolomic Analysis
4.3.1. Extraction and Quantification of Chlorophylls, Carotenoids, and Phytosterols
4.3.2. Non-Targeted Metabolomic Analysis
4.4. Microbiota Profiling
4.5. Gene Capture
4.6. Phylogenetic Analysis
4.7. DNA Sequence Comparisons
4.8. Statistical Methods
4.8.1. Non-Targeted Metabolomics
4.8.2. Infection Experiments, Analysis of Stigmasterol, and Chlorophyll Accumulation
4.8.3. Microbiota Profiling
4.8.4. Growth of the Wild Type or the chs5 Mutant in Holoxenic Conditions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial Interactions within the Plant Holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Uroz, S.; Courty, P.E.; Oger, P. Plant Symbionts Are Engineers of the Plant-Associated Microbiome. Trends Plant Sci. 2019, 24, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the Plant Microbiome: Looking Back and Future Perspectives. Front. Microbiol. 2014, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeis, S.L. Greater than the Sum of Their Parts: Characterizing Plant Microbiomes at the Community-Level. Curr. Opin. Plant Biol. 2015, 24, 82–86. [Google Scholar] [CrossRef]
- Simon, J.-C.; Marchesi, J.R.; Mougel, C.; Selosse, M.-A. Host-Microbiota Interactions: From Holobiont Theory to Analysis. Microbiome 2019, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The Importance of the Microbiome of the Plant Holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional Overlap of the Arabidopsis Leaf and Root Microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Guttman, D.S.; McHardy, A.C.; Schulze-Lefert, P. Microbial Genome-Enabled Insights into Plant-Microorganism Interactions. Nat. Rev. Genet. 2014, 15, 797–813. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the Core Arabidopsis thaliana Root Microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Schlaeppi, K.; Dombrowski, N.; Oter, R.G.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Quantitative Divergence of the Bacterial Root Microbiota in Arabidopsis thaliana Relatives. Proc. Natl. Acad. Sci. USA 2014, 111, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaiero, J.R.; McCall, C.A.; Thompson, K.A.; Day, N.J.; Best, A.S.; Dunfield, K.E. Inside the Root Microbiome: Bacterial Root Endophytes and Plant Growth Promotion. Am. J. Bot. 2013, 100, 1738–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The Hidden World within Plants: Ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorholt, J.A. Microbial Life in the Phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Viaene, T.; Langendries, S.; Beirinckx, S.; Maes, M.; Goormachtig, S. Streptomyces as a Plant’s Best Friend? FEMS Microbiol. Ecol. 2016, 92, fiw119. [Google Scholar] [CrossRef] [Green Version]
- van der Meij, A.; Willemse, J.; Schneijderberg, M.A.; Geurts, R.; Raaijmakers, J.M.; van Wezel, G.P. Inter- and Intracellular Colonization of Arabidopsis Roots by Endophytic Actinobacteria and the Impact of Plant Hormones on Their Antimicrobial Activity. Antonie Leeuwenhoek 2018, 111, 679–690. [Google Scholar] [CrossRef] [Green Version]
- van der Meij, A.; Worsley, S.F.; Hutchings, M.I.; van Wezel, G.P. Chemical Ecology of Antibiotic Production by Actinomycetes. FEMS Microbiol. Rev. 2017, 41, 392–416. [Google Scholar] [CrossRef] [Green Version]
- Worsley, S.F.; Newitt, J.; Rassbach, J.; Batey, S.F.D.; Holmes, N.A.; Murrell, J.C.; Wilkinson, B.; Hutchings, M.I. Streptomyces Endophytes Promote Host Health and Enhance Growth across Plant Species. Appl. Environ. Microbiol. 2020, 86, e01053-20. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Q.; He, F.; Li, Y.; Xue, Q.; Lai, H. Biocontrol of Root Diseases and Growth Promotion of the Tuberous Plant Aconitum Carmichaelii Induced by Actinomycetes Are Related to Shifts in the Rhizosphere Microbiota. Microb. Ecol. 2020, 79, 134–147. [Google Scholar] [CrossRef]
- Vergnes, S.; Gayrard, D.; Veyssière, M.; Toulotte, J.; Martinez, Y.; Dumont, V.; Bouchez, O.; Rey, T.; Dumas, B. Phyllosphere Colonization by a Soil Streptomyces sp. Promotes Plant Defense Responses Against Fungal Infection. Mol. Plant Microbe Interact. 2020, 33, 223–234. [Google Scholar] [CrossRef]
- Ayswaria, R.; Vasu, V.; Krishna, R. Diverse Endophytic Streptomyces Species with Dynamic Metabolites and Their Meritorious Applications: A Critical Review. Crit. Rev. Microbiol. 2020, 46, 750–758. [Google Scholar] [CrossRef]
- Dias, M.P.; Bastos, M.S.; Xavier, V.B.; Cassel, E.; Astarita, L.V.; Santarém, E.R. Plant Growth and Resistance Promoted by Streptomyces spp. in Tomato. Plant Physiol. Biochem. 2017, 118, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Newitt, J.T.; Prudence, S.M.M.; Hutchings, M.I.; Worsley, S.F. Biocontrol of Cereal Crop Diseases Using Streptomycetes. Pathogens 2019, 8, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanrewaju, O.S.; Babalola, O.O. Streptomyces: Implications and Interactions in Plant Growth Promotion. Appl. Microbiol. Biotechnol. 2019, 103, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Moreno, Z.R.; Vinchira-Villarraga, D.M.; Vergara-Morales, D.I.; Castellanos, L.; Ramos, F.A.; Guarnaccia, C.; Degrassi, G.; Venturi, V.; Moreno-Sarmiento, N. Plant-Growth Promotion and Biocontrol Properties of Three Streptomyces spp. Isolates to Control Bacterial Rice Pathogens. Front. Microbiol. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Cordovez, V.; Carrion, V.J.; Etalo, D.W.; Mumm, R.; Zhu, H.; van Wezel, G.P.; Raaijmakers, J.M. Diversity and Functions of Volatile Organic Compounds Produced by Streptomyces from a Disease-Suppressive Soil. Front. Microbiol. 2015, 6, 1081. [Google Scholar] [CrossRef] [Green Version]
- Conn, V.M.; Walker, A.R.; Franco, C.M.M. Endophytic Actinobacteria Induce Defense Pathways in Arabidopsis thaliana. Mol. Plant Microbe Interact. 2008, 21, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Kaur, T.; Rani, R.; Manhas, R.K. Biocontrol and Plant Growth Promoting Potential of Phylogenetically New Streptomyces sp. MR14 of Rhizospheric Origin. AMB Express 2019, 9, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome Definition Re-Visited: Old Concepts and New Challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Thoms, D.; Liang, Y.; Haney, C.H. Maintaining Symbiotic Homeostasis: How Do Plants Engage with Beneficial Microorganisms While at the Same Time Restricting Pathogens? Mol. Plant Microbe Interact. 2021, 34, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Cotton, T.E.A.; Pétriacq, P.; Cameron, D.D.; Meselmani, M.A.; Schwarzenbacher, R.; Rolfe, S.A.; Ton, J. Metabolic Regulation of the Maize Rhizobiome by Benzoxazinoids. ISME J. 2019, 13, 1647–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; van der Heijden, M.G.A.; et al. Root Exudate Metabolites Drive Plant-Soil Feedbacks on Growth and Defense by Shaping the Rhizosphere Microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Garcia, B.J.; Furches, A.; Tuskan, G.A.; Jacobson, D. Plant Host-Associated Mechanisms for Microbial Selection. Front. Plant Sci. 2019, 10, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; del Rio, T.G.; Jones, C.D.; Tringe, S.G.; et al. Salicylic Acid Modulates Colonization of the Root Microbiome by Specific Bacterial Taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voges, M.J.E.E.E.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-Derived Coumarins Shape the Composition of an Arabidopsis Synthetic Root Microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Concepción, M.; Boronat, A. Breaking New Ground in the Regulation of the Early Steps of Plant Isoprenoid Biosynthesis. Curr. Opin. Plant Biol. 2015, 25, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Sharma, R.A. Plant Terpenes: Defense Responses, Phylogenetic Analysis, Regulation and Clinical Applications. 3 Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef] [Green Version]
- Burdon, R.C.F.; Junker, R.R.; Scofield, D.G.; Parachnowitsch, A.L. Bacteria Colonising Penstemon digitalis Show Volatile and Tissue-Specific Responses to a Natural Concentration Range of the Floral Volatile Linalool. Chemoecology 2018, 28, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Senthil-Kumar, M.; Ryu, C.-M.; Kang, L.; Mysore, K.S. Phytosterols Play a Key Role in Plant Innate Immunity against Bacterial Pathogens by Regulating Nutrient Efflux into the Apoplast. Plant Physiol. 2012, 158, 1789–1802. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Jiang, T.; Liu, Y.-X.; Bai, Y.-C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.-W.; Bai, Y.; Osbourn, A. A Specialized Metabolic Network Selectively Modulates Arabidopsis Root Microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, M. The Discovery of a Mevalonate-Independent Pathway for Isoprenoid Biosynthesis in Bacteria, Algae and Higher Plants. Nat. Prod. Rep. 1999, 16, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.C.; Suzanne, H.; Somerville, C.R. Chilling-Sensitive Mutants of Arabidopsis. Plant Mol. Biol. Rep. 1995, 13, 11–17. [Google Scholar] [CrossRef]
- Araki, N.; Kusumi, K.; Masamoto, K.; Niwa, Y.; Iba, K. Temperature-Sensitive Arabidopsis Mutant Defective in 1-Deoxy-d-Xylulose 5-Phosphate Synthase within the Plastid Non-Mevalonate Pathway of Isoprenoid Biosynthesis. Physiol. Plant. 2000, 108, 19–24. [Google Scholar] [CrossRef]
- Wright, L.P.; Rohwer, J.M.; Ghirardo, A.; Hammerbacher, A.; Ortiz-Alcaide, M.; Raguschke, B.; Schnitzler, J.-P.; Gershenzon, J.; Phillips, M.A. Deoxyxylulose 5-Phosphate Synthase Controls Flux through the Methylerythritol 4-Phosphate Pathway in Arabidopsis. Plant Physiol. 2014, 165, 1488–1504. [Google Scholar] [CrossRef]
- Baltrus, D.A.; McCann, H.C.; Guttman, D.S. Evolution, Genomics and Epidemiology of Pseudomonas syringae: Challenges in Bacterial Molecular Plant Pathology. Mol. Plant Pathol. 2017, 18, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, F.; Thilmony, R.; He, S.Y. The Arabidopsis thaliana-Pseudomonas syringae Interaction. Arab. Book 2002, 1, e0039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, M.W. Microbial Communities, Structure, and Function. In Encyclopedia of Geobiology; Reitner, J., Thiel, V., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 592–595. ISBN 978-1-4020-9212-1. [Google Scholar]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Barupal, D.K.; Fiehn, O. Chemical Similarity Enrichment Analysis (ChemRICH) as Alternative to Biochemical Pathway Mapping for Metabolomic Datasets. Sci. Rep. 2017, 7, 14567. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.A.; León, P.; Boronat, A.; Rodríguez-Concepción, M. The Plastidial MEP Pathway: Unified Nomenclature and Resources. Trends Plant Sci. 2008, 13, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Schenke, D.; Utami, H.P.; Zhou, Z.; Gallegos, M.-T.; Cai, D. Suppression of UV-B Stress Induced Flavonoids by Biotic Stress: Is There Reciprocal Crosstalk? Plant Physiol. Biochem. 2019, 134, 53–63. [Google Scholar] [CrossRef]
- Huang, X.-X.; Wang, Y.; Lin, J.-S.; Chen, L.; Li, Y.-J.; Liu, Q.; Wang, G.-F.; Xu, F.; Liu, L.; Hou, B.-K. The Novel Pathogen-Responsive Glycosyltransferase UGT73C7 Mediates the Redirection of Phenylpropanoid Metabolism and Promotes SNC1-Dependent Arabidopsis Immunity. Plant J. 2021, 107, 149–165. [Google Scholar] [CrossRef]
- Stassen, M.J.J.; Hsu, S.-H.; Pieterse, C.M.J.; Stringlis, I.A. Coumarin Communication Along the Microbiome–Root–Shoot Axis. Trends Plant Sci. 2021, 26, 169–183. [Google Scholar] [CrossRef]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Görlach, J. Growth Stage-Based Phenotypic Analysis of Arabidopsis: A Model for High Throughput Functional Genomics in Plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Gasc, C.; Peyret, P. Hybridization Capture Reveals Microbial Diversity Missed Using Current Profiling Methods. Microbiome 2018, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Nisar, N.; Li, L.; Lu, S.; Khin, N.C.; Pogson, B.J. Carotenoid Metabolism in Plants. Mol. Plant 2015, 8, 68–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandon, K.; Nazaret, F.; Farajzadeh, D.; Alloing, G.; Frendo, P. Redox Regulation in Diazotrophic Bacteria in Interaction with Plants. Antioxidants 2021, 10, 880. [Google Scholar] [CrossRef]
- Stec, N.; Banasiak, J.; Jasiński, M. Abscisic Acid—An Overlooked Player in Plant-Microbe Symbioses Formation? Acta Biochim. Pol. 2016, 63, 53–58. [Google Scholar] [CrossRef]
- Eichmann, R.; Richards, L.; Schäfer, P. Hormones as Go-Betweens in Plant Microbiome Assembly. Plant J. 2021, 105, 518–541. [Google Scholar] [CrossRef]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.A.H.M.; Feussner, I.; Pieterse, C.M.J. MYB72-Dependent Coumarin Exudation Shapes Root Microbiome Assembly to Promote Plant Health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, A.; Fischer, D.; Kinzel, L.; Chowdhury, S.P.; Hofmann, A.; Baldani, J.I.; Rothballer, M. Assessment of the Structural and Functional Diversities of Plant Microbiota: Achievements and Challenges—A Review. J. Adv. Res. 2019, 19, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhao, J.; Wen, T.; Zhao, M.; Li, R.; Goossens, P.; Huang, Q.; Bai, Y.; Vivanco, J.M.; Kowalchuk, G.A.; et al. Root Exudates Drive the Soil-Borne Legacy of Aboveground Pathogen Infection. Microbiome 2018, 6, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahlbrock, K.; Bednarek, P.; Ciolkowski, I.; Hamberger, B.; Heise, A.; Liedgens, H.; Logemann, E.; Nürnberger, T.; Schmelzer, E.; Somssich, I.E.; et al. Non-Self Recognition, Transcriptional Reprogramming, and Secondary Metabolite Accumulation during Plant/Pathogen Interactions. Proc. Natl. Acad. Sci. USA 2003, 100 (Suppl. 2), 14569–14576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarek, P.; Osbourn, A. Plant-Microbe Interactions: Chemical Diversity in Plant Defense. Science 2009, 324, 746–748. [Google Scholar] [CrossRef]
- Boivin, C.; Malpica, C.; Rosenberg, C.; Dénarié, J.; Goldmann, A.; Fleury, V.; Maille, M.; Message, B.; Tepfer, D. Metabolic Signals in the Rhizosphere: Catabolism of Calystegins and Trigonelline by Rhizobium Meliloti. In Signal Molecules in Plants and Plant-Microbe Interactions; Lugtenberg, B.J.J., Ed.; Springer: Berlin/Heidelberg, Germany, 1989; pp. 401–407. [Google Scholar]
- Feng, H.; Fu, R.; Hou, X.; Lv, Y.; Zhang, N.; Liu, Y.; Xu, Z.; Miao, Y.; Krell, T.; Shen, Q.; et al. Chemotaxis of Beneficial Rhizobacteria to Root Exudates: The First Step towards Root–Microbe Rhizosphere Interactions. Int. J. Mol. Sci. 2021, 22, 6655. [Google Scholar] [CrossRef]
- Vannier, N.; Agler, M.; Hacquard, S. Microbiota-Mediated Disease Resistance in Plants. PLoS Pathog. 2019, 15, e1007740. [Google Scholar] [CrossRef] [Green Version]
- Thomma, B.P.; Penninckx, I.A.; Broekaert, W.F.; Cammue, B.P. The Complexity of Disease Signaling in Arabidopsis. Curr. Opin. Immunol. 2001, 13, 63–68. [Google Scholar] [CrossRef]
- Zaynab, M.; Fatima, M.; Abbas, S.; Sharif, Y.; Umair, M.; Zafar, M.H.; Bahadar, K. Role of Secondary Metabolites in Plant Defense against Pathogens. Microb. Pathog. 2018, 124, 198–202. [Google Scholar] [CrossRef]
- Zaynab, M.; Fatima, M.; Sharif, Y.; Zafar, M.H.; Ali, H.; Khan, K.A. Role of Primary Metabolites in Plant Defense against Pathogens. Microb. Pathog. 2019, 137, 103728. [Google Scholar] [CrossRef]
- Huang, W.; Wang, Y.; Li, X.; Zhang, Y. Biosynthesis and Regulation of Salicylic Acid and N-Hydroxypipecolic Acid in Plant Immunity. Mol. Plant 2020, 13, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Fujioka, S.; Kwon, M.; Jeon, J.; Choe, S. Arabidopsis Brassinosteroid-Overproducing Gulliver3-D/Dwarf4-D Mutants Exhibit Altered Responses to Jasmonic Acid and Pathogen. Plant Cell Rep. 2013, 32, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Jaber, R.; Laggoun, F.; Lehner, A.; Remy-Jouet, I.; Pamlard, O.; Beaupierre, S.; Leprince, J.; Follet-Gueye, M.-L.; Vicré-Gibouin, M.; et al. Holaphyllamine, a Steroid, Is Able to Induce Defense Responses in Arabidopsis thaliana and Increases Resistance against Bacterial Infection. Planta 2017, 246, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The Plant Immune System. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalev, O.; Ashkenazy, H.; Neumann, M.; Weigel, D. Commensal Pseudomonas Protect Arabidopsis thaliana from a Coexisting Pathogen via Multiple Lineage-Dependent Mechanisms. ISME J. 2021, 16, 1235–1244. [Google Scholar] [CrossRef]
- Shalev, O.; Karasov, T.L.; Lundberg, D.S.; Ashkenazy, H.; Pramoj Na Ayutthaya, P.; Weigel, D. Commensal Pseudomonas Strains Facilitate Protective Response against Pathogens in the Host Plant. Nat. Ecol. Evol. 2022, 6, 383–396. [Google Scholar] [CrossRef]
- Vogel, C.M.; Potthoff, D.B.; Schäfer, M.; Barandun, N.; Vorholt, J.A. Protective Role of the Arabidopsis Leaf Microbiota against a Bacterial Pathogen. Nat. Microbiol. 2021, 6, 1537–1548. [Google Scholar] [CrossRef]
- Innerebner, G.; Knief, C.; Vorholt, J.A. Protection of Arabidopsis thaliana against Leaf-Pathogenic Pseudomonas Syringae by Sphingomonas Strains in a Controlled Model System. Appl. Environ. Microbiol. 2011, 77, 3202–3210. [Google Scholar] [CrossRef] [Green Version]
- Lichtenthaler, H.K.; Buschmann, C. Chlorophylls and Carotenoids: Measurement and Characterization by UV-VIS Spectroscopy. Curr. Protoc. Food Anal. Chem. 2001, 1, F4.3.1–F4.3.8. [Google Scholar] [CrossRef]
- Forestier, E.; Romero-Segura, C.; Pateraki, I.; Centeno, E.; Compagnon, V.; Preiss, M.; Berna, A.; Boronat, A.; Bach, T.J.; Darnet, S.; et al. Distinct Triterpene Synthases in the Laticifers of Euphorbia lathyris. Sci. Rep. 2019, 9, 4840. [Google Scholar] [CrossRef]
- Villette, C.; Zumsteg, J.; Schaller, H.; Heintz, D. Non-Targeted Metabolic Profiling of BW312 Hordeum vulgare Semi Dwarf Mutant Using UHPLC Coupled to QTOF High Resolution Mass Spectrometry. Sci. Rep. 2018, 8, 13178. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An Improved Dual-Indexing Approach for Multiplexed 16S rRNA Gene Sequencing on the Illumina MiSeq Platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [Green Version]
- Parisot, N.; Denonfoux, J.; Dugat-Bony, E.; Peyret, P.; Peyretaillade, E. KASpOD--a Web Service for Highly Specific and Explorative Oligonucleotide Design. Bioinformatics 2012, 28, 3161–3162. [Google Scholar] [CrossRef]
- Jaziri, F.; Parisot, N.; Abid, A.; Denonfoux, J.; Ribière, C.; Gasc, C.; Boucher, D.; Brugère, J.-F.; Mahul, A.; Hill, D.R.C.; et al. PhylOPDb: A 16S rRNA Oligonucleotide Probe Database for Prokaryotic Identification. Database 2014, 2014, bau036. [Google Scholar] [CrossRef] [Green Version]
- Ribière, C.; Beugnot, R.; Parisot, N.; Gasc, C.; Defois, C.; Denonfoux, J.; Boucher, D.; Peyretaillade, E.; Peyret, P. Targeted Gene Capture by Hybridization to Illuminate Ecosystem Functioning. Methods Mol. Biol. 2016, 1399, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Günther, B.; Marre, S.; Defois, C.; Merzi, T.; Blanc, P.; Peyret, P.; Arnaud-Haond, S. Capture by Hybridization for Full-Length Barcode-Based Eukaryotic and Prokaryotic Biodiversity Inventories of Deep Sea Ecosystems. Mol. Ecol. Resour. 2021, 22, 623–637. [Google Scholar] [CrossRef]
- Militon, C.; Rimour, S.; Missaoui, M.; Biderre, C.; Barra, V.; Hill, D.; Moné, A.; Gagne, G.; Meier, H.; Peyretaillade, E.; et al. PhylArray: Phylogenetic Probe Design Algorithm for Microarray. Bioinformatics 2007, 23, 2550–2557. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.S.; Handley, K.M.; Wrighton, K.C.; Frischkorn, K.R.; Thomas, B.C.; Banfield, J.F. Short-Read Assembly of Full-Length 16S Amplicons Reveals Bacterial Diversity in Subsurface Sediments. PLoS ONE 2013, 8, e56018. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2′s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [Green Version]
- Gascuel, O. BIONJ: An Improved Version of the NJ Algorithm Based on a Simple Model of Sequence Data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.Fr: Robust Phylogenetic Analysis for the Non-Specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Dereeper, A.; Audic, S.; Claverie, J.-M.; Blanc, G. BLAST-EXPLORER Helps You Building Datasets for Phylogenetic Analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graindorge, S.; Villette, C.; Koechler, S.; Groh, C.; Comtet-Marre, S.; Mercier, P.; Magerand, R.; Peyret, P.; Heintz, D.; Schaller, H.; et al. The Arabidopsis thaliana–Streptomyces Interaction Is Controlled by the Metabolic Status of the Holobiont. Int. J. Mol. Sci. 2022, 23, 12952. https://doi.org/10.3390/ijms232112952
Graindorge S, Villette C, Koechler S, Groh C, Comtet-Marre S, Mercier P, Magerand R, Peyret P, Heintz D, Schaller H, et al. The Arabidopsis thaliana–Streptomyces Interaction Is Controlled by the Metabolic Status of the Holobiont. International Journal of Molecular Sciences. 2022; 23(21):12952. https://doi.org/10.3390/ijms232112952
Chicago/Turabian StyleGraindorge, Stéfanie, Claire Villette, Sandrine Koechler, Chloé Groh, Sophie Comtet-Marre, Pierre Mercier, Romaric Magerand, Pierre Peyret, Dimitri Heintz, Hubert Schaller, and et al. 2022. "The Arabidopsis thaliana–Streptomyces Interaction Is Controlled by the Metabolic Status of the Holobiont" International Journal of Molecular Sciences 23, no. 21: 12952. https://doi.org/10.3390/ijms232112952