
Small Vessel Disease: Ancient Description, Novel Biomarkers


Abstract
:1. Introduction
2. Possible and Proved New Markers of Blood–Brain Barrier Leakage, Perivascular Enlargements, and Mitochondrial Alterations
3. Markers of Endothelial Dysfunction
4. Markers of Oxidative Damages in SVD
5. Inflammation and SVD
6. Potential Future Therapies Approach
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ab | Amyloid b-peptide |
| AD | Alzheimer′s Disease |
| Angpt2 | Angiopoietin 2 |
| ApoH | Apolipoprotein H |
| BBB | Blood–-brain barrier |
| CAA | Cerebral amyloid angiopathy |
| CBF | Cerebral blood flow |
| CCH | Chronic cerebral hypoperfusion |
| CNS | Central Nervous System |
| COX | Cyclooxygenase |
| CSF | Cerebrospinal fluid |
| cSVD | Cerebral small vessel disease |
| CVR | Cerebrovascular reactivity |
| EC | Endothelial cells |
| EDHF | Endothelium-derived hyperpolarizing factors |
| eNOS | Endothelial NO synthase |
| ENOV | Endothelium-derived nitric oxide-vasodilators |
| Fgfbp1 | Fibroblast growth factor binding protein |
| GABA | Gamma-aminobutyric acid |
| GPx | Glutathione Peroxidase |
| GSH | Glutathione |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL | Interleukin |
| ISF | Interstitial fluid |
| MBP | Myelin basic protein |
| MMPs | Matrix metalloproteinase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NfL | Neurofilament light chain |
| NO | Nitric oxide |
| ODMSMS | Oligodendrocyte-derived myelin sheath-like myelin lipid sulfatide |
| OPCs | Oligodendrocyte precursor cells |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PS | Permeability-surface area product |
| PVS | Perivascular spaces |
| ROCK | Rho-associated protein kinase |
| ROS | Reactive oxygen species |
| SA | Serum albumin |
| SVD | Small vessel disease |
| SOD | Super Oxide Dismutase |
| sTM | Soluble thrombomodulin |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TNF-α | Tumor necrosis factor-α |
| VCI | Vascular cognitive impairment |
| VE-cadherins | Vascular endothelium cadherins |
| VEGF Vascular | Vascular endothelial growth factor |
| vP | Plasma volume fraction |
| WMH | White matter hyperintensities |
References
- Hakim, A.M. Small Vessel Disease. Front. Neurol. 2019, 10, 1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uiterwijk, R.; Van Oostenbrugge, R.J.; Huijts, M.; De Leeuw, P.W.; Kroon, A.A.; Staals, J. Total Cerebral Small Vessel Disease MRI Score Is Associated with Cognitive Decline in Executive Function in Patients with Hypertension. Front. Aging Neurosci. 2016, 8, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.D.; Aizenstein, H.J.; Alexopoulos, G.S. The vascular depression hypothesis: Mechanisms linking vascular disease with depression. Mol. Psychiatry 2013, 18, 963–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinter, D.; Ritchie, S.J.; Doubal, F.; Gattringer, T.; Morris, Z.; Bastin, M.; Hernández, M.D.C.V.; Royle, N.A.; Corley, J.; Maniega, S.M.; et al. Impact of small vessel disease in the brain on gait and balance. Sci. Rep. 2017, 7, 41637. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Smith, E.F.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Haffner, C.; Malik, R.; Dichgans, M. Genetic factors in cerebral small vessel disease and their impact on stroke and dementia. J. Cereb. Blood Flow Metab. 2016, 36, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Anderson, R.L. Periventricular venous collagenosis: Association with leukoaraiosis. Radiology 1995, 194, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Vijayappa, M.; Lima, F.; Delgado, P.; Wendell, L.; Rosand, J.; Greenberg, S.M. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology 2008, 71, 1424–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Koizumi, K.; El Jamal, S.; Zhou, P.; Previti, M.L.; Van Nostrand, W.E.; Carlson, G.; Iadecola, C. Age-Dependent Neurovascular Dysfunction and Damage in a Mouse Model of Cerebral Amyloid Angiopathy. Stroke 2014, 45, 1815–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staals, J.; Booth, T.; Morris, Z.; Bastin, M.E.; Gow, A.J.; Corley, J.; Redmond, P.; Starr, J.M.; Deary, I.; Wardlaw, J.M. Total MRI load of cerebral small vessel disease and cognitive ability in older people. Neurobiol. Aging 2015, 36, 2806–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staals, J.; Makin, S.D.; Doubal, F.N.; Dennis, M.S.; Wardlaw, J.M. Stroke subtype, vascular risk factors, and total MRI brain small-vessel disease burden. Neurology 2014, 83, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Caruso, P. Small Vessel Disease-Related Dementia: An Invalid Neurovascular Coupling? Int. J. Mol. Sci. 2020, 21, 1095. [Google Scholar] [CrossRef] [Green Version]
- Tonet, E.; Pompei, G.; Faragasso, E.; Cossu, A.; Pavasini, R.; Passarini, G.; Tebaldi, M.; Campo, G. Coronary Microvascular Dysfunction: PET, CMR and CT Assessment. J. Clin. Med. 2021, 10, 1848. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Wallin, A.; Wardlaw, J.M.; Markus, H.S.; Montaner, J.; Wolfson, L.; Iadecola, C.; Zlokovic, B.V.; Joutel, A.; Dichgans, M.; et al. Consensus statement for diagnosis of subcortical small vessel disease. J. Cereb. Blood Flow Metab. 2016, 36, 6–25. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Markus, H.S. Magnetic Resonance Imaging in Cerebral Small Vessel Disease and its Use as a Surrogate Disease Marker. Int. J. Stroke 2011, 6, 47–59. [Google Scholar] [CrossRef]
- Erkinjuntti, T.; Inzitari, D.; Pantoni, L.; Wallin, A.; Scheltens, P.; Rockwood, K.; Roman, G.C.; Chui, H.; Desmond, D.W. Resaerch criteria for subcortical vascular dementia in clinical trials. J. Neur. Transm. Suppl. 2000, 59, 23–30. [Google Scholar]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]
- Potter, G.M.; Marlborough, F.J.; Wardlaw, J.M. Wide variation in definition, detection and description of lacunar lesions on imaging. Stroke 2011, 42, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wardlaw, J.M. Update on cerebral small vessel disease. A dynamic whole-brain disease. Stroke Vasc. Neurol. 2016, 1, e000035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardlaw, J.M.; Doubal, F.; Armitage, P.; Msc, F.C.; Carpenter, T.; Maniega, S.M.; Farrall, A.; Sudlow, C.; Dennis, M.; Dhillon, B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann. Neurol. 2009, 65, 194–202. [Google Scholar] [CrossRef]
- Englund, E. White matter pathology of vascular dementia. In Vascular Dementia; Chui, E., Ed.; M. Dunitz: London, UK, 2004; pp. 117–130. [Google Scholar]
- Englund, E.A.; Person, B. Correlations between histopathologic white matter changes and proton MR relaxation times in dementia. Alzheimer Dis. Assoc. Disord. 1987, 1, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, A.; Oulhaj, A.; Joachim, C.; Christie, S.; Sloan, C.; Smith, A.D.; Esiri, M. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol. Appl. Neurobiol. 2012, 38, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Gold, G.; Kovari, E.; Herrmann, F.R.; Canuto, A.; Hof, P.R.; Michel, J.P.; Bouras, C.; Giannakopoulos, P. Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 2005, 36, 1184–1188. [Google Scholar] [CrossRef]
- Klassen, A.C.; Sung, J.H.; Stadlan, E.M. Histological changes in cerebral arteries with increasing age. J. Neuropathol. Exp. Neurol. 1968, 27, 607–623. [Google Scholar] [CrossRef]
- Cummings, J.L. Frontal-subcortical circuits and human behavior. Arch. Neurol. 1993, 50, 873–880. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L. Frontal-subcortical circuits and neuropsychiatric disorders. J. Neuropsychiatry Clin. Neurosci. 1994, 6, 358–370. [Google Scholar]
- Tak, S.; Yoon, S.J.; Jang, J.; Yoo, K.; Jeong, Y.; Ye, J.C. Quantitative analysis of hemodynamic and metabolic changes in subcortical vascular dementia using simultaneous near-infrared spectroscopy and FMRI measurements. Neuroimage 2011, 55, 176–184. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, J.H.; Reed, B.R.; Mungas, D.; Weiner, M.W.; Chui, H. Executive dysfunction in subcortical ischaemic vascular disease. J. Neurol. Neurosurg. Psychiatr. 2002, 72, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, E.; Ballard, C.; Stephens, S.; Kenny, R.A.; Kalaria, R.; Barber, R.; OʹBrien, J. Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement. Geriatr. Cogn. Disord. 2003, 16, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Tullberg, M.; Fletcher, E.; DeCarli, C.; Mungas, D.; Reed, B.R.; Harvey, D.J.; Weiner, M.W.; Chui, H.C.; Jagust, W.J. White matter lesions impair frontal lobe function regardless of their location. Neurology 2004, 63, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Tatemichi, T.K.; Erkinjuntti, T.; Cummings, J.L.; Masdeu, J.C.; Garcia, J.H.; Amaducci, L.; Orgogozo, J.M.; Brun, A.; Hofman, A. Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 1993, 43, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Chui, H.C.; Victoroff, J.I.; MArgolin, D.; Jagust, W.; Shankle, R.; Katzman, R. Criteria for the diagnosis of ischemic vascular dementia proposed by the state of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology 1992, 42, 473–480. [Google Scholar] [CrossRef]
- Kim, G.H.; Lee, J.H.; Seo, S.W.; Ye, B.S.; Cho, H.; Kim, H.J.; Noh, Y.; Yoon, C.W.; Chin, J.H.; Oh, S.J.; et al. Seoul criteria for PIB(-) subcortical vascular dementia based on clinical and MRI variables. Neurology 2014, 82, 1529–1535. [Google Scholar] [CrossRef]
- Fazekas, F.; Chawluk, J.B.; Alavi, A.; Hurtig, H.I.; Zimmermann, R.A. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Am. J. Roentgenol. 1987, 149, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Cleutjens, F.A.H.M.; Ponds, R.W.H.M.; Spruit, M.A.; Burgmans, S.; Jacobs, H.I.L.; Gronenchield, H.B.M.; Stalls, J.; Franssen, F.M.E.; Dijkstra, J.B.; Vanfleteren, L.E.G.M.; et al. The relationship between cerebral small vessel disease, hippocampal volume and cognitive functioning in patients with COPD: An MRI study. Front. Aging Neurosci. 2017, 9, 88. [Google Scholar] [CrossRef]
- Scheltens, P.; Barkhof, F.; Leys, D.; Pruvo, J.P.; Nauta, J.J.; Vermersch, P.; Steinling, M.; Valk, J. A semiquantative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J. Neurol. Sci. 1993, 114, 7–12. [Google Scholar] [CrossRef]
- Kim, K.W.; MacFall, J.R.; Payne, M.E. Classification of white matter lesions on magnetic resonance imaging in elderly persons. Biol. Psychiatry 2008, 64, 273–280. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.T.; Erkinjuntti, T.; Reisberg, B.; Roman, G.; Sawada, T.; Pantoni, L.; Bowler, J.V.; Ballard, C.; DeCarli, C.; Gorelick, P.B.; et al. Vascular cognitive impairment. Lancet Neurol. 2003, 2, 89–98. [Google Scholar] [CrossRef]
- Erkinjunnti, T.; Gauthier, S. Diagnosing vascular cognitive impairment and dementia. In Concepts and Controversies in Vascular Cognitive Impairment in Clinical Practice; Wahlund, L.O., Erkinjunnti, T., Gauthier, S., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 3–9. [Google Scholar]
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef]
- Skrobot, O.A.; O’Brien, J.; Black, S.; Chen, C.; DeCarli, C.; Erkinjuntti, T.; Ford, G.A.; Kalaria, R.N.; Pantoni, L.; Pasquier, F.; et al. The Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. 2017, 13, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Grinberg, L.T.; Attems, J. Vascular dementia: Different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp. Gerontol. 2012, 47, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Bowler, J.V. Vascular cognitive impairment. J. Neurol. Neurosurg. Psychiatr. 2005, 76, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259–281. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathomechanisms of Vascular Depression in Older Adults. Int. J. Mol. Sci. 2021, 23, 308. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, V.; Bramanti, A.; Lanza, G.; Cantone, M.; Vinciguerra, L.; Pennisi, M.; Bonanno, L.; Pennisi, G.; Bella, R. Impaired Cerebral Haemodynamics in Vascular Depression: Insights from Transcranial Doppler Ultrasonography. Front. Psychiatry 2018, 9, 316. [Google Scholar] [CrossRef] [Green Version]
- Vinciguerra, L.; Lanza, G.; Puglisi, V.; Pennisi, M.; Cantone, M.; Bramanti, A.; Pennisi, G.; Bella, R. Transcranial Doppler ultrasound in vascular cognitive impairment-no dementia. PLoS ONE 2019, 14, e0216162. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Cserr, H.F.; DePasquale, M.; Patlak, C.S. Regulation of brain water and electrolytes during acute hyperosmolality in rats. Am. J. Physiol. 1987, 253, F522–F529. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.A.; Dallas, A.D. Permeability of disrupted cerebral microvessels in the frog. J. Physiol. 1993, 461, 619–663. [Google Scholar] [CrossRef] [Green Version]
- Bridges, L.R.; Andoh, J.; Lawrence, A.; Khoong, C.H.; Poon, W.W.; Esiri, M.M.; Markus, H.S.; Hainsworth, A.H. Blood-Brain Barrier Dysfunction and Cerebral Small Vessel Disease (Arteriolosclerosis) in Brains of Older People. J. Neuropathol. Exp. Neurol. 2014, 73, 1026–1033. [Google Scholar] [CrossRef]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.-D.D.; Maki, T.; Ayata, C.; Kim, K.-W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Wardlaw, J.M.; Sandercock, P.A.G.; Dennis, M.S.; Starr, J. Ithe s breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke 2003, 34, 806–812. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Doubal, F.N.; Valdes-Hernandez, M.; Wang, X.; Chappell, F.M.; Shuler, K.; Armitage, P.A.; Carpenter, T.C.; Dennis, M.S. Blood–Brain Barrier Permeability and Long-Term Clinical and Imaging Outcomes in Cerebral Small Vessel Disease. Stroke 2013, 44, 525–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdo, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef] [Green Version]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Aging and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Zhang, X.; Shi, Q.; Yang, S.; Fan, H.; Qin, W.; Yang, L.; Yuan, J.; Jiang, T.; et al. Higher blood-brain barrier permeability is associated with ha igher white matter hyperintensities burden. J. Neurol. 2017, 264, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, M.; Zuo, L.; Shi, Q.; Qin, W.; Yang, L.; Jiang, T.; Hu, W. Compromised blood-brain barrier integrity is associated with the total magnetic resonance imaging burden of cerebral small vessel disease. Front. Neurol. 2018, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.S.; Heye, A.K.; Armitage, P.A.; Chappell, F.; Hernández, M.D.C.V.; Makin, S.D.J.; Sakka, E.; Thrippleton, M.J.; Wardlaw, J.M. Tracer kinetic assessment of blood-brain barrier leakage and blood volume in cerebral small vessel disease: Associations with disease burden and vascular risk factors. NeuroImage 2021, 32, 102883. [Google Scholar] [CrossRef]
- Thrippleton, M.J.; Backes, W.H.; Sourbron, S.; Ingrisch, M.; Osch, M.J.P.; Dichgans, M.; Fazekas, F.; Ropele, S.; Frayne, R.; Oostenbrugge, R.J.; et al. Quantifying blood-brain barrier leakage in small vessel disease: Review and consensus recommendations. Alzheimer’s Dement. 2019, 15, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Manning, C.; Stringer, M.; Dickie, B.; Clancy, U.; Hernandez, M.C.V.; Wiseman, S.J.; Garcia, D.J.; Sakka, E.; Backes, W.H.; Ingrisch, M.; et al. Sources of systematic error in DCE-MRI estimation of low-level blood-brain barrier leakage. Magn. Reason. Med. 2021, 86, 1888–1903. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stringer, M.S.; Shi, Y.; Thrippleton, M.J.; Wardlaw, J.M. Associations Between White Matter Hyperintensity Burden, Cerebral Blood Flow and Transit Time in Small Vessel Disease: An Updated Meta-Analysis. Front. Neurol. 2021, 12, 647848. [Google Scholar] [CrossRef] [PubMed]
- Heye, A.K.; Thrippleton, M.J.; Armitage, P.A.; Hernandez, M.D.C.V.; Makin, S.D.; Glatz, A.; Sakka, E.; Wardlaw, J.M. Tracer kinetic modelling for DCE-MRI quantification of subtle blood-brain barrier permeability. Neuroimage 2016, 125, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Mäe, M.A.; He, L.; Nordling, S.; Vazquez-Liebanas, E.; Nahar, K.; Jung, B.; Li, X.; Tan, B.C.; Chin Foo, J.; Cazenave-Gassiot, A.; et al. Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circ. Res. 2021, 128, e46–e62. [Google Scholar] [CrossRef]
- Zhang, E.T.; Inman, C.B.; Weller, R.O. Interrelationships of the pia mater and the perivascular (Wirchov- Robin) spaces in the human cerebrum. J. Anat. 1990, 170, 111–123. [Google Scholar]
- Iadecola, C. The neurovascular Unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Hendrikx, D.; Smits, A.; Lavanga, M.; De Wel, O.; Thewissen, L.; Jansen, K.; Caicedo, A.; Van Huffe, S.; Naulaers, G. Measurement of Neurovascular Coupling in Neonates. Front. Physiol. 2019, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef] [Green Version]
- Huijts, M.; Duits, A.; Staals, J.; Kroon, A.A.; De Leeuw, P.W.; Van Oostenbrugge, R.J. Basal ganglia enlarged perivascular spaces are linked to cognitive function in patients with cerebral small vessel disease. Curr. Neurovasc. Res. 2014, 11, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, T.; Alarcon-Martinez, L. Cerebral micro-vascular signaling in health and disease. Brain Res. 2015, 1623, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Benveniste, H.; Nedergaard, M.; Zlokovic, B.V.; Mestre, H.; Lee, H.; Doubal, F.N.; Brown, R.; Ramirez, J.; MacIntosh, B.J.; et al. Perivascular spaces in the brain: Anatomy, physiology and pathology. Nat. Rev. Neurol. 2020, 16, 137–153. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, Y.; Wang, J.; Gong, X.; Chen, Z.; Zhang, X.; Cai, J.; Chen, S.; Fang, L.; Sun, J.; et al. Glymphatic clearance function in patients with cerebral small vessel disease. Neuroimage 2021, 238, 118257. [Google Scholar] [CrossRef]
- Benveniste, H.; Nedergaard, M. Cerebral small vessel disease: A glymphopathy? Curr. Opin. Neurobiol. 2022, 72, 15–21. [Google Scholar] [CrossRef]
- Jiménez-Balado, J.; Riba-Llena, I.; Garde, E.; Valor, M.; Gutiérrez, B.; Pujadas, F.; Delgado, P. Prevalence of hippocampal enlarged perivascular spaces in a sample of patients with hypertension and their relation with vascular risk factors and cognitive function. J. Neurol. Neurosurg. Psychiatry 2018, 89, 651–656. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Gold, G.; Kowaru, E.; von Gunten, A.; Imhof, A.; Bouras, C. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience. Acta Neuropathol. 2007, 113, 1–12. [Google Scholar] [CrossRef]
- Van der Veen, P.H.; Muller, M.; Vinken, K.L.; Hendrikse, J.; Mali, W.P.; van der Graaf, Y.; Geerlings, M.I.; SMART Study Group. Longitudinal relationship between cerebral small vessel disease and cerebral blood flow. The second manifestations of arterial disease-magnetic resonance study. Stroke 2015, 46, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado-Godia, E.; Dwivedi, P.; Sharma, S.; Santiago, A.O.; Roquer Gonzalez, J.; Balcells, M.; Laird, J.; Turk, M.; Suri, H.S.; Nicolaides, A.; et al. Cerebral Small Vessel Disease: A Review Focusing on Pathophysiology, Biomarkers, and Machine Learning Strategies. J. Stroke 2018, 20, 302–320. [Google Scholar] [CrossRef]
- Zhang, C.E.; Wong, S.M.; van de Haar, H.J.; Staals, J.; Jansen, J.F.; Jeukens, C.R.; Hofman, P.A.; van Oostenbrugge, R.J.; Backes, W.H. Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology 2017, 88, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Huisa, B.N.; Caprihan, A.; Thompson, J.; Prestopnik, J.; Qualls, C.R.; Rosenberg, G.A. Long-term blood-brain barrier permeability changes in Binswanger disease. Stroke 2015, 46, 2413–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardlaw, J.M.; Makin, S.J.; Hernández, M.C.V.; Armitage, P.A.; Heye, A.K.; Chappell, F.M.; Muñoz-Maniega, S.; Sakka, E.; Shuler, K.; Dennis, M.S.; et al. Blood-brain barrier failure as a core mechanism in cerebral small vessel disease and dementia: Evidence from a cohort study. Alzheimer’s Dement. 2017, 13, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Ihara, M.; Yamamoto, Y. Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 2016, 47, 554–560. [Google Scholar] [CrossRef] [Green Version]
- Rajani, R.M.; Williams, A. Endothelial cell-oligodendrocyte interactions in small vessel disease and aging. Clin. Sci. 2017, 131, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Sameshima, H.; Yang, L.; Hariskuma, M.; Ikenoue, T. Regional differences of microglial accumulation within 72 hours of hypoxia-ischemia and the effect of acetylcholine receptor agonist on brain damage and microglial activation in newborn rats. Brain Res. 2014, 1562, 52–58. [Google Scholar] [CrossRef]
- Petito, C.K. Transformation of postisichemic perineuronal glial cells. J. Cereb. Blood Flow Metabol. 1986, 6, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Petito, C.K.; Olarte, J.P.; Roberts, B.; Nowak, T.S.; Pulsinelli, W.A. Selective glial vulnerability following transient global ischemia in rat brain. J. Neuropathol. Exp. Neurol. 1998, 57, 231–238. [Google Scholar] [CrossRef]
- Masuda, T.; Croom, D.; Hida, H.; Kirov, S.A. Capillary blood flow around microglial somata determines dynamics of microglial processes in ischemic conditions. Glia 2011, 59, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Ju, F.; Ran, Y.; Zhu, L.; Cheng, X.; Gao, H.; Xi, X.; Yang, Z.; Zhang, S. Increased BBB Permeability Enhances Activation of Microglia and Exacerbates Loss of Dendritic Spines after Transient Global Cerebral Ischemia. Front. Cell Neurosci. 2018, 12, 236. [Google Scholar] [CrossRef]
- Zhang, S. Microglial activation after ischaemic stroke. Stroke Vasc. Neurol. 2019, 4, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filous, A.S.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.M.E.; Zhang, Y.; Reiners, J.; Ander, M.; Niedermayer, A.; Fang, L.; Neugebauer, H.; Kassubek, J.; Katona, I.; Weis, J.; et al. Endothelial damage, vascular bagging and remodeling of the microvascular bed in human microangiopathy with deep white matter lesions. Acta Neuropathol. Commun. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.-C.; Ma, L.-S.; Chu, Z.-H.; Xu, H.; Wu, W.-Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; Donka, G.; de Vous, R.A.I.; Mihaly, A.; Bari, F.; Luiten, P.G.M. Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 2004, 108, 57–64. [Google Scholar] [CrossRef]
- Paolini Paoletti, F.; Simoni, S.; Parnetti, L.; Gaetani, L. The Contribution of Small Vessel Disease to Neurodegeneration: Focus on Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 4958. [Google Scholar] [CrossRef]
- Fredman, P.; Wallin, A.; Blennow, K.; Davidsson, P.; Gottfries, C.; Svennerholm, L. Sulfatide as a biochemical marker in cerebrospinal fluid of patients with vascular dementia. Acta Neurol. Scand. 1992, 85, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Tullberg, M.; Månsson, J.E.; Fredman, P.; Lekman, A.; Blennow, K.; Ekman, R.; Rosengren, L.E.; Tisell, M.; Wikkelso, C. CSF sulfatide distinguishes between normal pressure hydrocephalus and subcortical arteriosclerotic encephalopathy. J. Neurol. Neurosurg. Psychiatry 2000, 69, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibert, K.A.; Raymond, G.V.; Nascene, D.R.; Miller, W.P.; Tolar, J.; Orchard, P.J.; Lund, T.C. Cerebrospinal fluid matrix metalloproteinases are elevated in cerebral adrenoleukodystrophy and correlate with MRI severity and neurologic dysfunction. PLoS ONE 2012, 7, e50430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, M.; Zetterberg, H.; Van Straaten, E.; Lind, K.; Syversen, S.; Edman, Å.; Blennow, K.; Rosengren, L.; Pantoni, L.; Inzitari, D.; et al. Cerebrospinal fluid biomarkers of white matter lesions—Cross-sectional results from the LADIS study. Eur. J. Neurol. 2010, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, M.; Andreasson, U.; Rolstad, S.; Nordlund, A.; Lind, K.; Zetterberg, H.; Edman, Å.; Blennow, K.; Wallin, A. Subcortical vascular dementia biomarker pattern in mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 348–356. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Andreasson, U.; Simon, A.; Zetterberg, H.; Edman, Å.; Potter, W.; Holder, D.; Devanarayan, V.; Seeburger, J.; Smith, A.D.; et al. Screening for New Biomarkers for Subcortical Vascular Dementia and Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2011, 1, 31–42. [Google Scholar] [CrossRef]
- Peters, A.; Sethares, C. Age-related changes in the morphology of cerebral capillaries do not correlate with cognitive decline. J. Comp. Neurol. 2012, 520, 1339–1347. [Google Scholar] [CrossRef]
- Iejima, D.; Itabashi, T.; Kawamura, Y.; Noda, T.; Yuasa, S.; Fukuda, K.; Oka, C.; Iwata, T. HTRA1 (high temperature requirement A serine peptidase 1) gene is transcriptionally regulated by insertion/deletion nucleotides located at the 3′ end of the ARMS2 (age related maculopathy susceptibility 2) gene in patients with age-related macular degeneration. J. Biol. Chem. 2015, 290, 2784–2797. [Google Scholar]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–728. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Prisby, R.D.; Ramsey, M.W.; Behnke, B.J.; Dominguez, J.M.; Donato, A.J.; Allen, M.R.; Delp, M.D. Aging reduces skeletal blood flow endothelium dependent vasodilation, and NO bioavailability in Rats. J. Bone Miner. Res. 2007, 22, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.T.; Vaa, B.; Hesse, C.; Eisenach, J.H.; Joyner, M.J. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension 2009, 53, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, E.J.; Prins, N.D.; Vermeer, S.E.; Vrooman, H.A.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M.B. C-reactive protein and cerebral small-vessel disease: The Rotterdam Scan Study. Circulation 2005, 112, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Long, D.A.; Newaz, M.A.; Prabahakar, S.S.; Price, K.L.; Truong, L.; Feng, L.; Mu Oyekan, A.O.; Johnson, R.J. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in ageing. Kidney Int. 2005, 68, 2154–2163. [Google Scholar] [CrossRef] [Green Version]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.B.; Achschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callendere, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T. Enhanced peroxynitrite formation is associated with vascular ageing. J. Exp. Med. 2000, 18, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Puca, A.A.; Carrizzo, A.; Ferrario, A.; Villa, F.; Vecchione, C. Endothelial nitric oxide synthase, vascular integrity and human exceptional longevity. Immun. Ageing 2012, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small GTPases and Their Role in Vascular Disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Szulcek, R.; Beckers, C.M.; Hodzic, J.; de Wit, J.; Chen, Z.; Grob, T.; Musters, R.J.; Minshall, R.D.; van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc. Res. 2013, 99, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuw Amerongen, G.P.; Beckers, C.M.; Achekar, I.D.; Zeeman, S.; Musters, R.J.; van Hinsbergh, V.W. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2332–2339. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, H.; Chen, B.; Li, Q.; Huang, X.; Wang, L.; Guo, X.; Huang, Q. RhoA/ROCK-dependent moesin phosphorylation regulates AGE-induced endothelial cellular response. Cardiovasc. Diabetol. 2012, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Breslin, J.W.; Zhu, J.; Yuan, S.Y.; Wu, M.H. Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation 2006, 13, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Fernando, M.S.; Clark, L.; Ince, P.G.; Matthews, F.; Forster, G.; O’Brien, J.T.; Barber, R.; Kalaria, R.N.; Brayne, C.; et al. White matter lesions in an unselected cohort of the elderly: Astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol. Appl. Neurobiol. 2007, 33, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Pantoni, L.; Inzitari, D.; Pracucci, G.; Lolli, F.; Giordano, G.; Bracco, L.; Amaducci, L. Cerebrospinal fluid proteins in patients with leucoaraiosis: Possible abnormalities in blood-brain barrier function. J. Neurol. Sci. 1993, 115, 125–131. [Google Scholar] [CrossRef]
- Musaeus, C.S.; Gleerup, H.S.; Høgh, P.; Waldemar, G.; Hasselbalch, S.G.; Simonsen, A.H. Cerebrospinal Fluid/Plasma Albumin Ratio as a Biomarker for Blood-Brain Barrier Impairment Across Neurodegenerative Dementias. J. Alzheimers Dis. 2020, 75, 429–436. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Chatzopoulou, D.; Tsivgoulis, G.; Petridou, E.T. Albuminuria and cerebral small vessel disease: A systematic review and meta-analysis. J. Am. Geriatr. Soc. 2018, 66, 509–517. [Google Scholar] [CrossRef]
- Wada, M.; Takahashi, Y.; Iseki, C.; Kawanami, T.; Daimon, M.; Kato, T. Plasma fibrinogen, global cognitive function, and cerebral small vessel disease: Results of a cross-sectional study in community-dwelling Japanese elderly. Intern. Med. 2011, 50, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Kulikauskas, M.R.; Shaka, X.; Bautch, V.L. The versatility and paradox of BMP signaling in endothelial cell behaviors and blood vessel function. Cell. Mol. Life Sci. 2022, 79, 77. [Google Scholar] [CrossRef]
- Knottnerus, I.L.; Govers-Riemslag, J.W.; Hamulyak, K.; Rouhl, R.P.; Staals, J.; Spronk, H.M. Endothelial activation in lacunar stroke subtypes. Stroke 2010, 41, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, I.L.; Cate, H.; Lodder, J.; Kessels, F.; van Oostenbrugge, R.J. Endothelial dysfunction in lacunar stroke: A systematic review. Cerebrovasc. Dis. 2009, 27, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, S.F.; Doubal, F.N.; Shuler, K.; Wardlaw, J.M. A systematic review of dynamic cerebral andperipheral endothelial function in lacunar stroke versus controls. Stroke 2010, 41, e434–e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markus, H.S.; Hunt, B.; Palmer, K.; Enzinger, C.; Schmidt, H.; Schmidt, R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: Longitudinal results of the Austrian Stroke Prevention Study. Stroke 2005, 36, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162, Erratum in Redox Biol. 2018, 14, 694–696. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, B.; Nie, K.; Jia, Y.; Yu, J. Effects of acupuncture on declined cerebral blood flow, impaired mitochondrial respiratory function and oxidative stress in multi-infarct dementia rats. Neurochem. Int. 2014, 65, 23–29. [Google Scholar] [CrossRef]
- Huang, J.L.; Fu, S.T.; Jiang, Y.Y.; Cao, Y.B.; Guo, M.L.; Wang, Y.; Xu, Z. Protective effects of Nicotiflorin on reducing memory dysfunction, energy metabolism failure, and oxidative stress in multi-infarct dementia model rats. Pharmacol. Biochem. Behav. 2007, 86, 741–748. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Brandes, R.P. The Nox family of NADPH oxidases: Friend or foe of the vascular system? Curr. Hypertens. Rep. 2012, 14, 70–78. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; DeSilva, T.M.; Mast, A.E.; Hickey, H.; Williams, J.P.; Broughton, B.R.; Sobey, C.G. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: Role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H220–H225. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J. Cerebral hypoperfusion and cognitive impairment: The pathogenic role of vascular oxidative stress. Int. J. Neurosci. 2012, 122, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3kinase, NADPH-oxidase, and nitric oxide synthase. J. Cereb. Blood Flow Metab. 2006, 26, 836–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Silva, T.M.; Brait, V.H.; Drummond, G.R.; Sobey, C.G.; Miller, A.A. Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS ONE 2011, 6, e28393. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Raz, N. The aging brain: Structural changes and their implications for cognitive aging. In New Frontiers in Cognitive Aging; Dixon, R., Bäckman, L., Nilsson, L., Eds.; Oxford University Press: Telangana, India, 2004; pp. 115–134. [Google Scholar]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Valiente-Pallejà, A.; Tortajada, J.; Bulduk, B.K.; Vilella, E.; Garrabou, G.; Muntané, G.; Martorell, L. Comprehensive summary of mitochondrial DNA alterations in the postmortem human brain: A systematic review. EBioMedicine 2022, 76, 103815. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef]
- Taylor, S.D.; Ericson, N.G.; Burton, J.N.; Prolla, T.A.; Silber, J.R.; Shendure, J.; Bielas, J.H. Targeted enrichment and high-resolution digital profiling of mitochondrial DNA deletions in human brain. Aging Cell 2014, 13, 29–38. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, 1003794. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Bayona-Bafaluy, M.P.; Rana, M.; Mora, M.; Hao, H.; Moraes, C.T. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002, 30, 4626–4633. [Google Scholar] [CrossRef]
- Guo, L.; Tian, J.; Du, H. Mitochondrial dysfunction and synaptic transmission failure in Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1071–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballif, B.C.; Theisen, A.; Coppinger, J.; Gowans, G.C.; Hersh, J.H.; Madan-Khetarpal, S.; Schmidt, K.R.; Tervo, R.; Escobar, L.F.; Friedrich, C.A.; et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal microduplication. Mol. Cytogenet. 2008, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, G.; Cantone, M.; Musso, S.; Borgione, E.; Scuderi, C.; Ferri, R. Early-onset subcortical ischemic vascular dementia in an adult with mtDNA mutation 3316G>A. J. Neurol. 2018, 265, 968–969. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Chinnery, P.F. Mitochondrial disorders overview. In GeneReviews [Internet]; Margaret, P.A., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Karen, S.A.A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 2020; p. 1993. [Google Scholar]
- Basel, D. Mitochondrial DNA Depletion Syndromes. Clin. Perinatol. 2020, 47, 123–141. [Google Scholar] [CrossRef]
- Coskun, P.E.; Wyrembak, J.; Derbereva, O.; Melkonian, G.; Doran, E.; Lott, I.T.; Head, E.; Cotman, C.W.; Wallace, D.C. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J. Alzheimers Dis. 2010, 20, 293–310. [Google Scholar] [CrossRef] [Green Version]
- Roca-Bayerri, C.; Robertson, F.; Pyle, A.; Hudson, G.; Payne, B.A.I. Mitochondrial DNA damage and brain aging in human immunodeficiency virus. Clin. Infect. Dis. 2021, 73, e466–e473. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Mayhan, W.G.; Arrick, D.M.; Sharpe, G.M.; Sun, H. Age-related alterations in reactivity of cerebral arterioles: Role of oxidative stress. Microcirculation 2008, 15, 225–236. [Google Scholar] [CrossRef]
- Dong, Y.F.; Kataoka, K.; Toyama, K.; Sueta, D.; Koibuchi, N.; Yamamoto, E.; Yata, K.; Tomimoto, H.; Ogawa, H.; Kim-Mitsuyama, S. Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension 2011, 58, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Santhanam, A.V.; d’Uscio, L.V.; Katusic, Z.S. Erythropoietin increases bioavailability of tetrahydrobiopterin and protects cerebral microvasculature against oxidative stress induced by eNOS uncoupling. J. Neurochem. 2014, 131, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Ray, P.E.; Short, B.L. NF-kappa B activation plays a role in superoxide-mediated cerebral dysfunction after hypoxia/reoxygenation. Stroke 2005, 36, 1047–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS ONE 2009, 4, e8045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraco, G.; Moraga, A.; Moore, J.; Anrather, J.; Pickel, V.M.; Iadecola, C. Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension 2013, 62, 759–766. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Philippova, M.; Oskolkova, O.; Kadl, A.; Furnkranz, A.; Karabeg, E.; Afonyushkin, T.; Gruber, F.; Breuss, J.; Minchenko, A.; et al. Oxidized phospholipids stimulate angiogenesis via autocrine mechanisms, implicating a novel role for lipid oxidation in the evolution of atherosclerotic lesions. Circ. Res. 2006, 99, 900–908. [Google Scholar] [CrossRef]
- Tai, L.M.; Thomas, R.; Marottoli, F.M.; Koster, K.P.; Kanekiyo, T.; Morris, A.W.; Bu, G. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016, 131, 709–723. [Google Scholar] [CrossRef] [Green Version]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and micro-hemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [Green Version]
- Grochowski, C.; Litak, J.; Kamieniak, P.; Maciejewski, R. Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radic. Res. 2018, 52, 1–13. [Google Scholar] [CrossRef]
- Han, S.; Wu, H.; Li, W.; Gao, P. Protective effects of genistein in homocysteine-induced endothelial cell inflammatory injury. Mol. Cell. Biochem. 2015, 403, 43–49. [Google Scholar] [CrossRef]
- Fleszar, M.G.; Wiśniewski, J.; Zboch, M.; Diakowska, D.; Gamian, A.; Krzystek-Korpacka, M. Targeted metabolomic analysis of nitric oxide/L-arginine pathway metabolites in dementia: Association with pathology, severity, and structural brain changes. Sci. Rep. 2019, 9, 1376. [Google Scholar] [CrossRef]
- Gao, Q.; Fan, Y.; Mu, L.-Y.; Ma, L.; Song, Z.-Q.; Zhang, Y.-N. S100B and ADMA in cerebral small vessel disease and cognitive dysfunction. J. Neurol. Sci. 2015, 354, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, L.; Lanza, G.; Puglisi, V.; Fisicaro, F.; Pennisi, M.; Bella, R.; Cantone, M. Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. Int. J. Mol. Sci. 2020, 21, 2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-J.; Li, Q.; Du, H.-P. Homocysteine Triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Giuffré, M.; Caruso, P.; Gazzin, S.; Tiribelli, C. Homocysteine in Neurology: A Possible Contributing Factor to Small Vessel Disease. Int. J. Mol. Sci. 2021, 22, 2051. [Google Scholar] [CrossRef]
- Ahmad, S.; Siddiqi, M.I. Insights from molecular modeling into the selective inhibition of cathepsin S by its inhibitor. J. Mol. Model. 2017, 23, 92. [Google Scholar] [CrossRef]
- Leng, Y.P.; Ma, Y.S.; Li, X.G.; Chen, R.F.; Zeng, P.Y.; Li, X.H.; Qiu, C.F.; Li, Y.P.; Zhang, Z.; Chen, A.F. l-Homocysteine-induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. Br. J. Pharmacol. 2018, 175, 1157–1172. [Google Scholar] [CrossRef]
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homcysteine in neurology: From endothelium to neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175. [Google Scholar] [CrossRef]
- Deng, J.; Lu, S.; Li, H. Homocysteine activates B cells via regulating PKM-2 dependent metabolic reprogramming. J. Immunol. 2017, 198, 170–183. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Dayal, S.; Wilson, K.M.; Leo, L.; Arning, E.; Bottiglieri, T.; Lentz, S.R. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood 2006, 108, 2237–2243. [Google Scholar] [CrossRef] [Green Version]
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Y.; Li, J.; Yang, X.; Zhang, H.; Qin, X.; Hu, Y.; Mo, Z. Serum Homocysteine Concentration Is Significantly Associated with Inflammatory/Immune Factors. PLoS ONE 2015, 10, e0138099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, V.S.; Trinath, J.; Reddy, G.B. Implication of homocysteine in protein quality control processes. Biochimie 2019, 165, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Lucero, J.; Abumiya, T.; Koziol, J.A.; Copeland, B.R.; del Zoppo, G.J. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhu, W.; Yun, W.; Wang, Q.; Cheng, M.; Zhang, Z.; Liu, X.; Zhou, X.; Xu, G. Correlation of matrix metalloproteinase-2 single nucleotide polymorphisms with the risk of small vessel disease (SVD). J. Neurol. Sci. 2015, 356, 61–64. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Stamenkovic, I. Extracellular matrix remodeling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef]
- English, W.R.; Suarez-Puente, X.S.; Freije, J.M.; Knauper, V.; Amour, A.; Merryweather, A.; López-Otín, C.; Murphy, G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J. Biol. Chem. 2000, 275, 14046–14055. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Choi, D.H.; Block, M.L.; Lorenzl, S.; Yang, L.; Kim, Y.J.; Sugama, S.; Cho, B.P.; Hwang, O.; Browne, S.E.; et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. 2007, 21, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Woo, M.S.; Park, J.S.; Choi, I.Y.; Kim, W.K.; Kim, H.S. Inhibition of MMP-3 or -9 suppresses lipopolysaccharide-induced expression of proinflammatory cytokines and iNOS in microglia. J. Neurochem. 2008, 106, 770–780. [Google Scholar] [CrossRef]
- Powell, W.C.; Fingleton, B.; Wilson, C.L.; Boothby, M.; Matrisian, L.M. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr. Biol. 1999, 9, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zheng, G.; Xu, M.; Li, Y.; Chen, X.; Zhu, W.; Tong, Y.; Chung, S.K.; Liu, K.J.; Shen, J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood–brain barrier permeability in focal cerebral ischemia and reperfusion injury. J. Neurochem. 2012, 120, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Chandler, S.; Miller, K.M.; Clements, J.M.; Lury, J.; Corkill, D.; Anthony, D.C.C.; Adams, S.E.; Gearing, A.J.H. Matrix metalloproteinases, tumor necrosis factor and multiple sclerosis: An overview. J. Neuroimmunol. 1997, 72, 155–161. [Google Scholar] [CrossRef]
- Inzitari, D.; Giusti, B.; Nencini, P.; Gori, A.M.; Nesi, M.; Palumbo, V.; Piccardi, B.; Armillis, A.; Pracucci, G.; Bono, G.; et al. MMP9 Variation After Thrombolysis Is Associated with Hemorrhagic Transformation of Lesion and Death. Stroke 2013, 44, 2901–2903. [Google Scholar] [CrossRef] [PubMed]
- Montaner, J.; Molina, C.A.; Monasterio, J.; Abilleira, S.; Arenillas, J.F.; Ribo, M.; Quintana, M.; Alvarez-Sabin, J. Matrix Metalloproteinase-9 Pretreatment Level Predicts Intracranial Hemorrhagic Complications After Thrombolysis in Human Stroke. Circulation 2003, 107, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Arba, F.; Piccardi, B.; Palumbo, V.; Giusti, B.; Nencini, P.; Gori, A.M.; Sereni, A.; Nesi, M.; Pracucci, G.; Bono, G.; et al. Small Vessel Disease Is Associated with Tissue Inhibitor of Matrix Metalloproteinase-4 After Ischaemic Stroke. Transl. Stroke Res. 2018, 10, 44–51. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Thompson, J.; Taheri, S.; Grossetete, M.; Adair, J.C.; Edmonds, E.; Prestopnik, J.; Wills, J.; Rosenberg, G.A. Matrix Metalloproteinases Are Associated with Increased Blood–Brain Barrier Opening in Vascular Cognitive Impairment. Stroke 2011, 42, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A.; Sullivan, N.; Esiri, M.M. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke 2001, 32, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A. Inflammation and white matter damage in vascular cognitive impairment. Stroke 2009, 40, S20–S23. [Google Scholar] [CrossRef] [Green Version]
- Ketsawatsomkron, P.; Keen, H.L.; Davis, D.R.; Lu, K.T.; Stump, M.; De Silva, T.M.; Hilzendeger, A.M.; Grobe, J.L.; Faraci, F.M.; Sigmund, C.D. Protective role for tissue inhibitor of Metalloproteinase-4, a novel peroxisome proliferator-activated receptor-γ target gene, in smooth muscle in Deoxycorticosterone acetate-salt hypertension. Hypertension 2016, 67, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Radomski, A.; Jurasz, P.; Sanders, E.J.; Overall, C.M.; Bigg, H.F.; Edwards, D.R.; Radomski, M.W. Identification, regulation and role of tissue inhibitor of metalloproteinases-4 (TIMP-4) in human platelets. Br. J. Pharmacol. 2002, 137, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Osaki, A.; Hayashi, M.; Yamamoto, Y. Coagulation activation in patients with Binswanger disease. Arch. Neurol. 1999, 56, 1104–1108. [Google Scholar] [CrossRef]
- Iwamoto, T.; Kubo, H.; Takasaki, M. Platelet activation in the cerebral circulation in different subtypes of ischaemic stroke and Binswanger’s disease. Stroke 1995, 26, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, M.; Zetterberg, H.; Edman, A.; Blennow, K.; Wallin, A.; Andreasson, U. Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, N.; Yang, C. Effects of rosuvastatin in combination with nimodipine in patients with mild cognitive impairment caused by cerebral small vessel disease. Panminerva Med. 2019, 61, 439–443. [Google Scholar] [CrossRef]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cazzato, G.; Pizzolato, G. Different responses to rivastigmine in subcortical vascular dementia and multi-infarct dementia. Am. J. Alzheimers Dis. Other Demenentias 2008, 23, 167–176. [Google Scholar] [CrossRef]
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730. [Google Scholar] [CrossRef]
- De Silva, T.M.; Miller, A.A. Cerebral Small Vessel Disease: Targeting Oxidative Stress as a Novel Therapeutic Strategy? Front. Pharmacol. 2016, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Pretnar-Oblak, J.; Sebestjen, M.; Sabovic, M. Statin treatment improves cerebral more than systemic endothelial dysfunction in patients with arterial hypertension. Am. J. Hypertens. 2008, 21, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Amarenco, P.; Benavente, O.; Goldstein, L.B.; Callahan, A.; Sillesen, H.; Hennerici, M.G.; Gilbert, S.; Rudolph, A.E.; Simunovic, L.; Zivin, J.A.; et al. Stroke Prevention by Aggressive Reduction in CholesterolLevels Investigators. Results of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) trial by stroke subtypes. Stroke 2009, 40, 1405–1409. [Google Scholar] [CrossRef] [Green Version]
- Caruso, G.; Godos, J.; Privitera, A.; Lanza, G.; Castellano, S.; Chillemi, A.; Bruni, O.; Ferri, R.; Caraci, F.; Grosso, G. Phenolic Acids and Prevention of Cognitive Decline: Polyphenols with a Neuroprotective Role in Cognitive Disorders and Alzheimer’s Disease. Nutrients 2022, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, A.E.; Centner, A.M.; Deitado, R.; Salazar, J.F.A. The Impact of Dietary Supplementation of Whole Foods and Polyphenols on Atherosclerosis. Nutrients 2020, 12, 2069. [Google Scholar] [CrossRef] [PubMed]
- Kesse-Guyot, E.; Fezeu, L.; Andreeva, V.A.; Touvier, M.; Scalbert, A.; Hercberg, S.; Galan, P. Total and specific polyphenol intakes in midlife are associated with cognitive function measured 13 years later. J. Nutr. 2012, 142, 76–83. [Google Scholar] [CrossRef]
- Goni, L.; Fernández-Matarrubia, M.; Romanos-Nanclares, A.; Razquin, C.; Ruiz-Canela, M.; Martínez-González, M.Á.; Toledo, E. Polyphenol intake and cognitive decline in the Seguimiento Universidad de Navarra (SUN) Project. Br. J. Nutr. 2021, 126, 43–52. [Google Scholar] [CrossRef]
- Shakoor, H.; Feehan, J.; Apostolopoulos, V.; Platat, C.; Al Dhaheri, A.S.; Ali, H.I.; Ismail, L.C.; Bosevski, M.; Stojanovska, L. Immunomodulatory Effects of Dietary Polyphenols. Nutrients 2021, 13, 728. [Google Scholar] [CrossRef]
- Godos, J.; Caraci, F.; Micek, A.; Castellano, S.; D’Amico, E.; Paladino, N.; Ferri, R.; Galvano, F.; Grosso, G. Dietary Phenolic Acids and Their Major Food Sources Are Associated with Cognitive Status in Older Italian Adults. Antioxidants 2021, 10, 700. [Google Scholar] [CrossRef]
- Ran, L.S.; Liu, W.H.; Fang, Y.Y.; Xu, S.B.; Li, J.; Luo, X.; Pan, D.J.; Wang, M.H.; Wang, W. Alcohol, coffee and tea intake and the risk of cognitive deficits: A dose-response meta-analysis. Epidemiol. Psychiatr. Sci. 2021, 30, e13. [Google Scholar] [CrossRef]
- Mallik, S.B.; Mudgal, J.; Nampoothiri, M.; Hall, S.; Dukie, S.A.; Grant, G.; Rao, C.M.; Arora, D. Caffeic acid attenuates lipopolysaccharide-induced sickness behaviour and neuroinflammation in mice. Neurosci. Lett. 2016, 632, 218–223. [Google Scholar] [CrossRef]
- Lee, A.Y.; Wu, T.T.; Hwang, B.R.; Lee, J.; Lee, M.-H.; Lee, S.; Cho, E.J. The Neuro-Protective Effect of the Methanolic Extract of Perilla frutescens var. japonicaand Rosmarinic Acid against H2O2-Induced Oxidative Stress in C6 Glial Cells. Biomol. Ther. 2016, 24, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mello Andrade, J.M.; Dos Santos Passos, C.; Kieling Rubio, M.A.; Mendonça, J.N.; Lopes, N.P.; Henriques, A.T. Combining in vitro and in silico approaches to evaluate the multifunctional profile of rosmarinic acid from Blechnum brasiliense on targets related to neurodegeneration. Chem. Biol. Interact. 2016, 254, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Rahbardar, M.G.; Amin, B.; Mehri, S.; Mirnajafi-Zadeh, S.J.; Hosseinzadeh, H. Anti-inflammatory effects of ethanolic extract of Rosmarinus officinalis L. and rosmarinic acid in a rat model of neuropathic pain. Biomed. Pharmacother. 2017, 86, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Dragomanova, S.; Pavlov, S.; Marinova, D.; Hodzev, Y.; Petralia, M.C.; Fagone, P.; Nicoletti, F.; Lazarova, M.; Tzvetanova, E.; Alexandrova, A.; et al. Neuroprotective Effects of Myrtenal in an Experimental Model of Dementia Induced in Rats. Antioxidants 2022, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.; Bramanti, P.; Cantone, M.; Pennisi, M.; Pennisi, G.; Bella, R. Vascular Cognitive Impairment through the Looking Glass of Transcranial Magnetic Stimulation. Behav. Neurol. 2017, 2017, 1421326. [Google Scholar] [CrossRef]
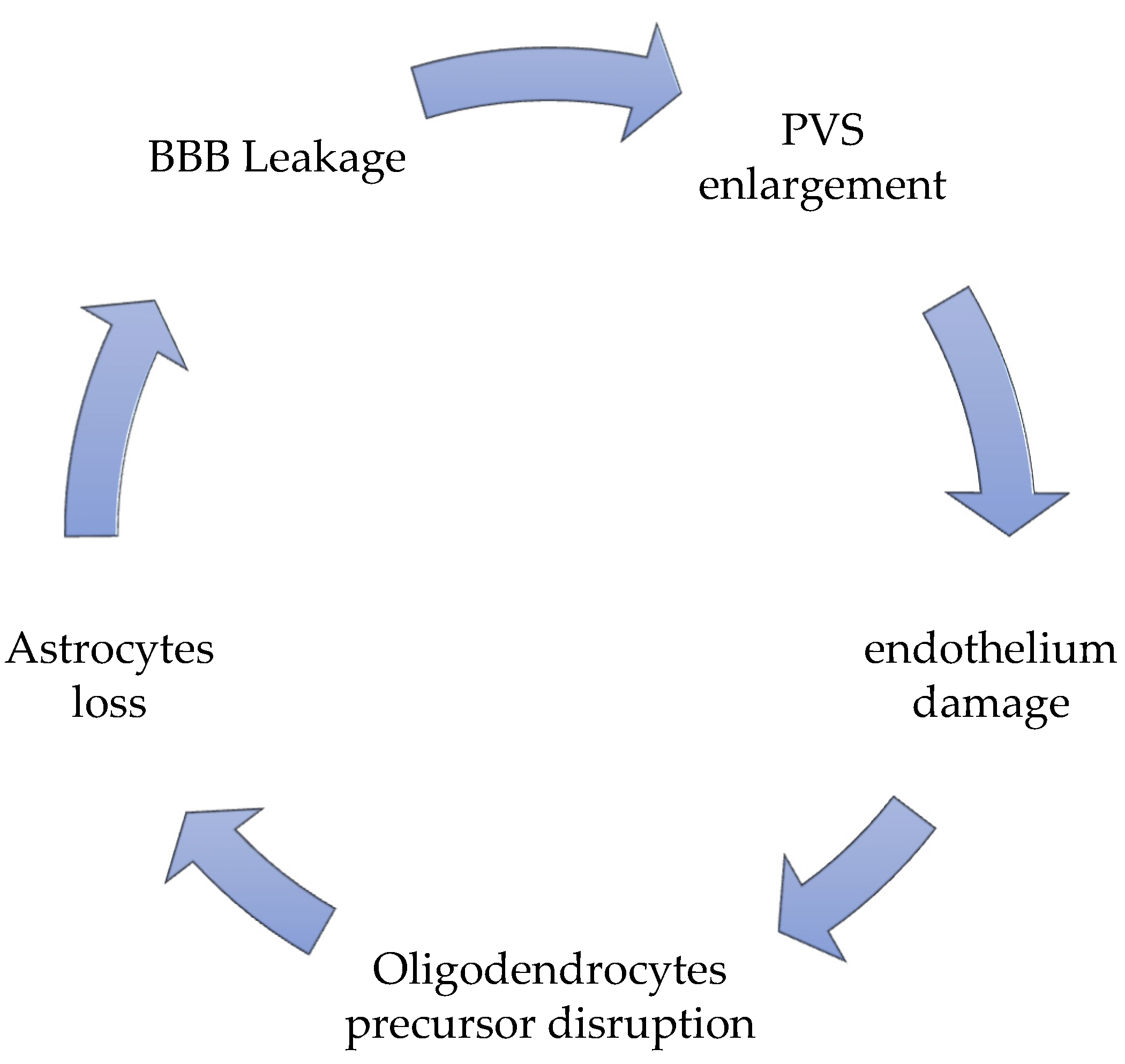
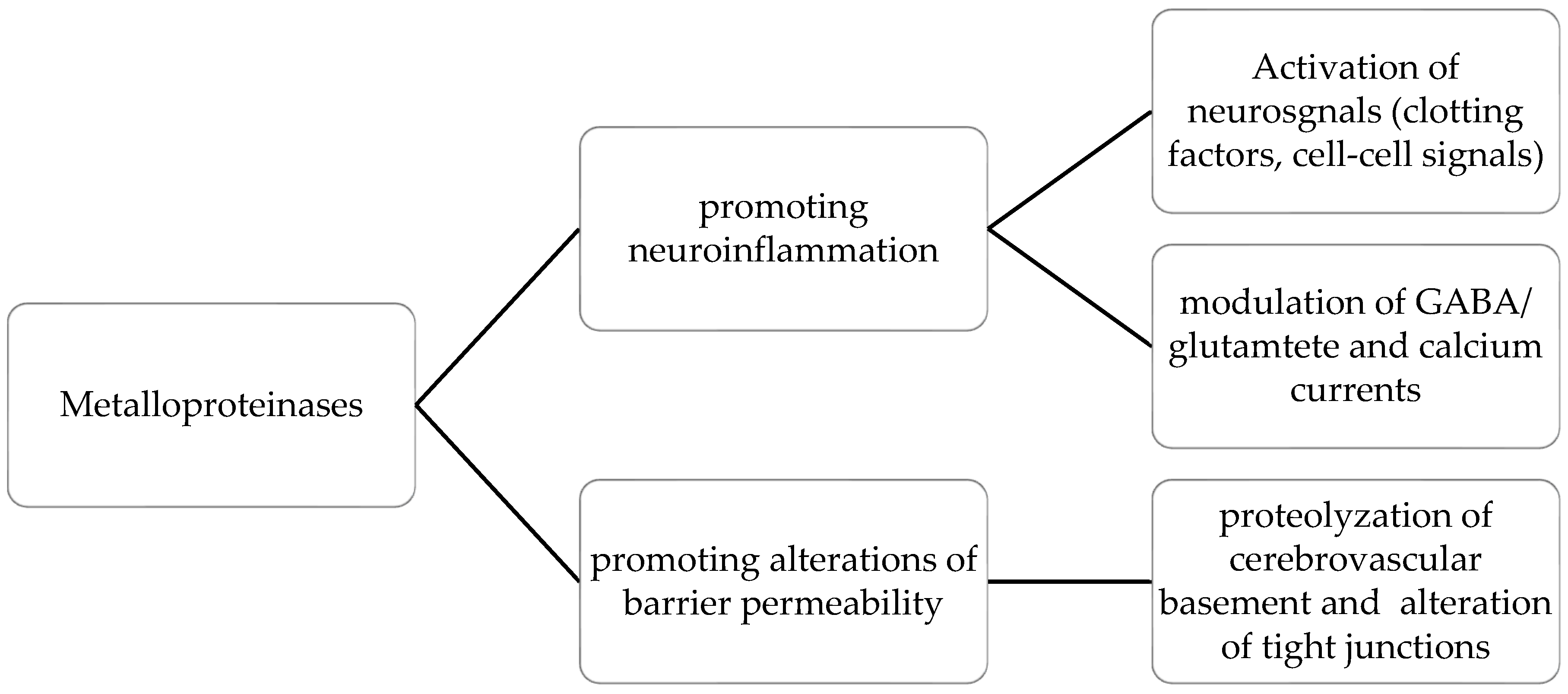

{kind=link}
{kind=link}
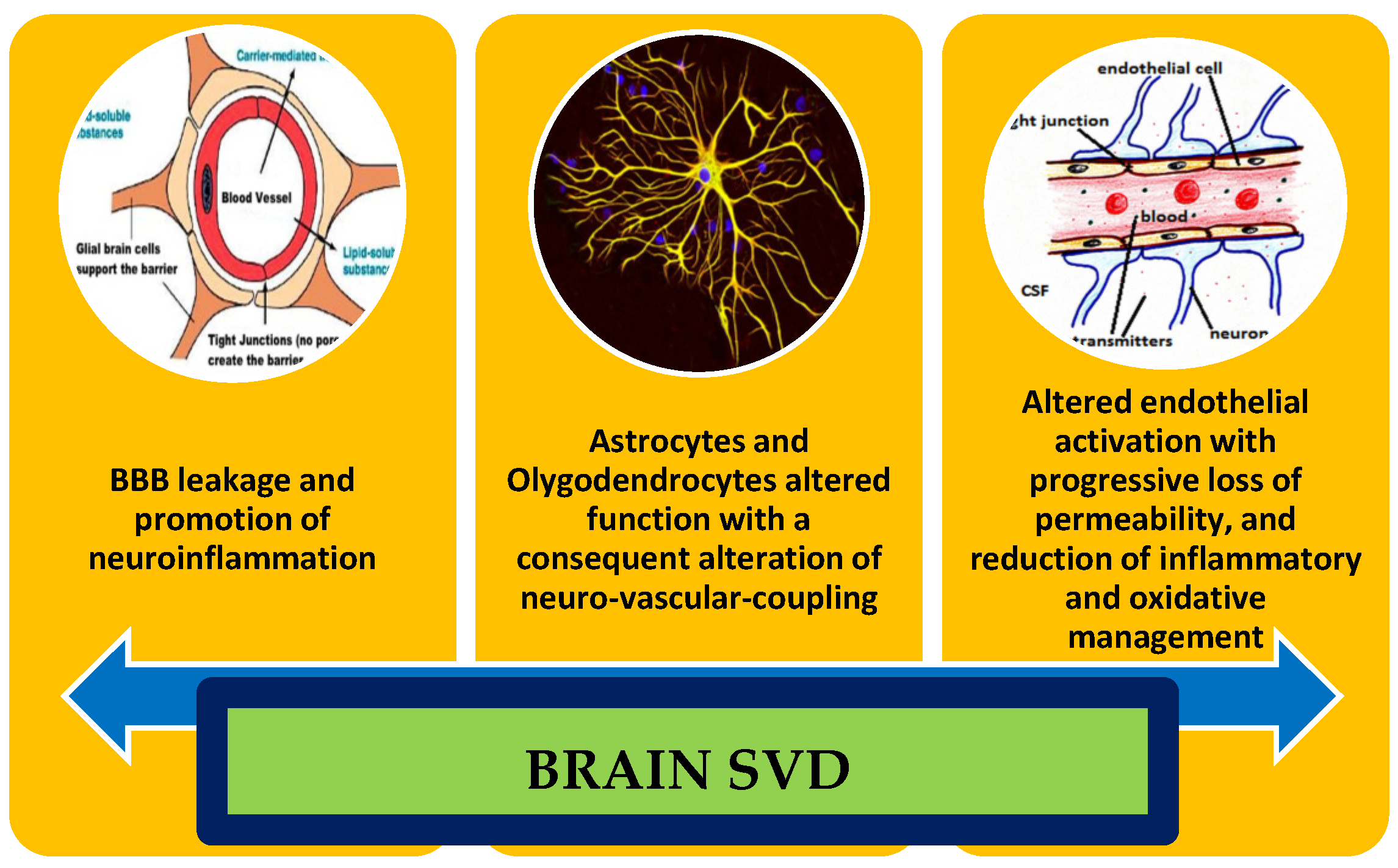
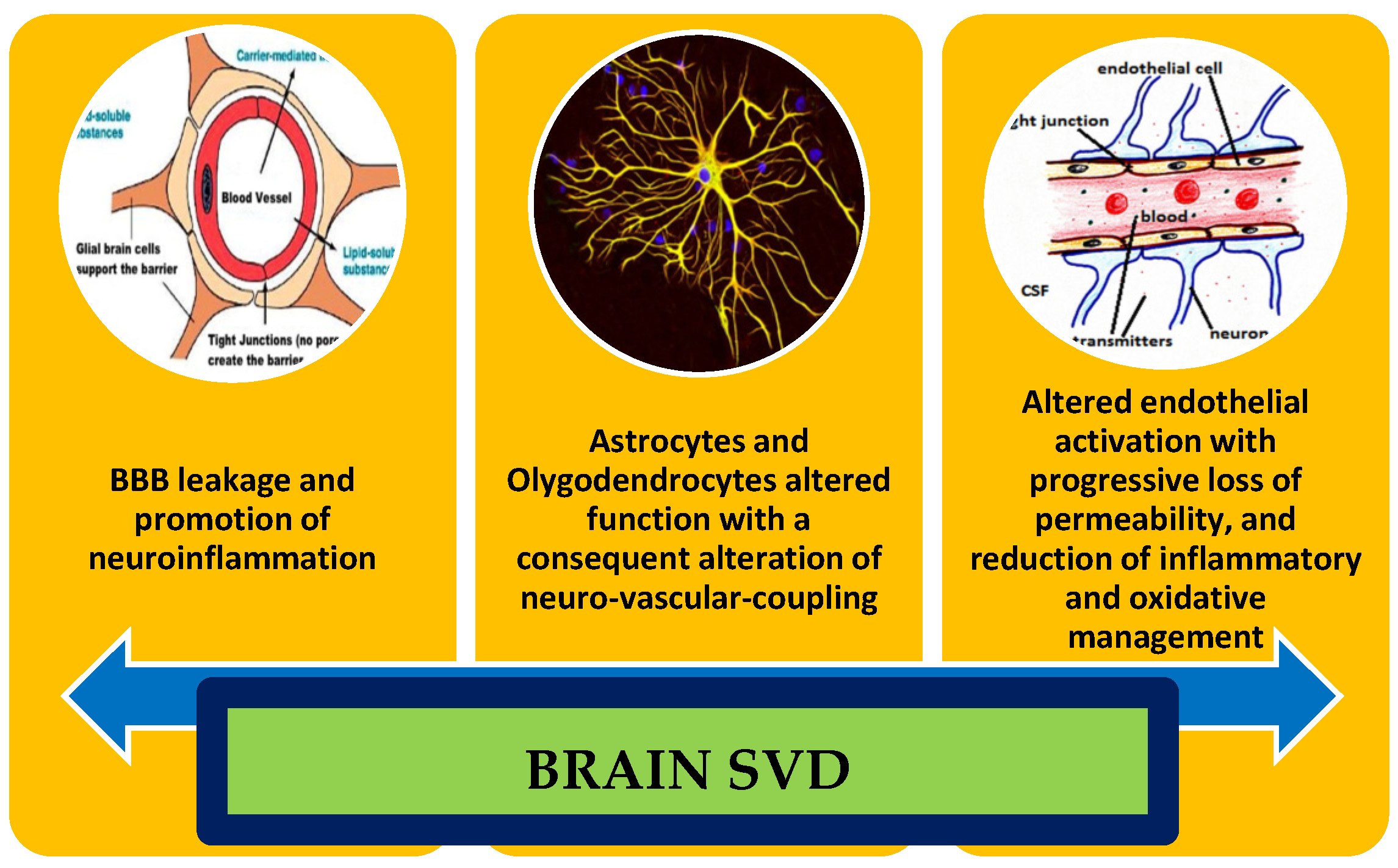{kind=link}
| Functional Domain | Markers | Effectiveness on SVD |
|---|---|---|
| BBB Leakage | DCE-MRI technique: Increase in permeability surface area Increase in white matter alterations Lower blood plasma volume in white matter altered regions | Demonstrate diagnostic confirmation Demonstrate the amount and the progression of SVD Determine the PS increasing together with a lowering of blood vP |
| Loss of pericytes: upregulation of FGFBP1 and ANGPT2 | Altered angiogenesis and demonstration of a venous-shifted molecular pattern of BBB, due to the altered arterial regulatory properties | |
| Enlargement of PVS | Alteration of the glymphatic system | |
| M1 activation: increase in TNF-alpha, Il-23, IL-1 beta, and IL-12 | Strong and chronic neuroinflammatory condition, shifted to a M1 vs. M2 activation | |
| General increment in caspase-3 RNA; of MMP-2 | Promoting and overwhelming the active neuroinflammation condition | |
| Endothelial dysfunction | Decrease in ENOV, prostacyclins, NO, eNOS, and VE-cadherins | Altered production of NO, due to decrease in its production and increment in its consumption, due to increment in ROS |
| Increase in C-protein, EDHF, VEGF, ICAM-1, sTM, Il-6, PA-1, von Willebrand Factors, HIF-1 alpha; VEGFR, and Neuroglobin | Expression of endothelial altered activation, with important flawless permeability and activation of thrombotic pattern | |
| Increase in homocysteine | Endothelial toxicity, promotion of oxidative and inflammatory damages | |
| Increase in CSF/plasma albumin ratio | Proof of endothelial altered permeability | |
| albuminuria | Indirect proof of endothelial altered permeability | |
| Oxidative damage | Increase in SOD, prostacyclin, and Hydrogen peroxide | Altered response to oxidative stress, with damages to mitochondria, altered oxygen delivery, and endothelial degeneration promotion |
| Decrease in NOX2 NADPH oxidase | Further reduction of proper response to ROS accumulation; their decrease is proportional to endothelial inflammation and alteration | |
| APOE4 | Promotion of endothelial reduced resistance to ROS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretti, R.; Caruso, P. Small Vessel Disease: Ancient Description, Novel Biomarkers. Int. J. Mol. Sci. 2022, 23, 3508. https://doi.org/10.3390/ijms23073508
Moretti R, Caruso P. Small Vessel Disease: Ancient Description, Novel Biomarkers. International Journal of Molecular Sciences. 2022; 23(7):3508. https://doi.org/10.3390/ijms23073508
Chicago/Turabian StyleMoretti, Rita, and Paola Caruso. 2022. "Small Vessel Disease: Ancient Description, Novel Biomarkers" International Journal of Molecular Sciences 23, no. 7: 3508. https://doi.org/10.3390/ijms23073508
APA StyleMoretti, R., & Caruso, P. (2022). Small Vessel Disease: Ancient Description, Novel Biomarkers. International Journal of Molecular Sciences, 23(7), 3508. https://doi.org/10.3390/ijms23073508