Abstract
Nonalcoholic fatty liver disease (NAFLD) is a progressive condition that encompasses a spectrum of liver disorders, beginning with the simple steatosis, progressing to nonalcoholic steatohepatitis (NASH), and possibly leading to more severe diseases, including liver cirrhosis and hepatocellular carcinoma (HCC). In recent years, the prevalence of NAFLD has increased due to a shift towards energy-dense dietary patterns and a sedentary lifestyle. NAFLD is also strongly associated with metabolic disorders such as obesity and hyperlipidemia. The progression of NAFLD could be influenced by a variety of factors, such as diet, genetic factors, and even epigenetic factors. In contrast to genetic factors, epigenetic factors, including histone modifications, exhibit dynamic and reversible features. Therefore, the epigenetic regulation of the initiation and progression of NAFLD is one of the directions under intensive investigation in terms of pathogenic mechanisms and possible therapeutic interventions. This review aims to discuss the possible mechanisms and the crucial role of histone modifications in the framework of epigenetic regulation in NAFLD, which may provide potential therapeutic targets and a scientific basis for the treatment of NAFLD.
1. Introduction
1.1. Pathogenesis of Nonalcoholic Fatty Liver Disease
Nonalcoholic fatty liver disease (NAFLD) is an increasingly common condition [1], which is strongly associated with metabolic diseases such as obesity, type 2 diabetes mellitus (T2DM), hyperlipidemia, and atherosclerosis [2,3]. NAFLD patients with metabolic syndrome or T2DM typically have an increased risk of death [2]. The prevalence of NAFLD ranges from 13.5% in Africa to 31.8% in the Middle East, and NAFLD is now the leading cause of chronic liver disease worldwide. In Western countries, the prevalence of NAFLD is 20–40% in adults, and 10–30% of the NAFLD patients eventually develop nonalcoholic steatohepatitis (NASH) [4]. Researchers have indicated that NASH patients who progress to the stage of cirrhosis have an increased risk of hepatocellular carcinoma (HCC) [5]. However, there is no approved pharmacotherapy for the treatment of NASH or liver cirrhosis. Therefore, NAFLD is a prevalent disease with a high social burden and unmet medical need.
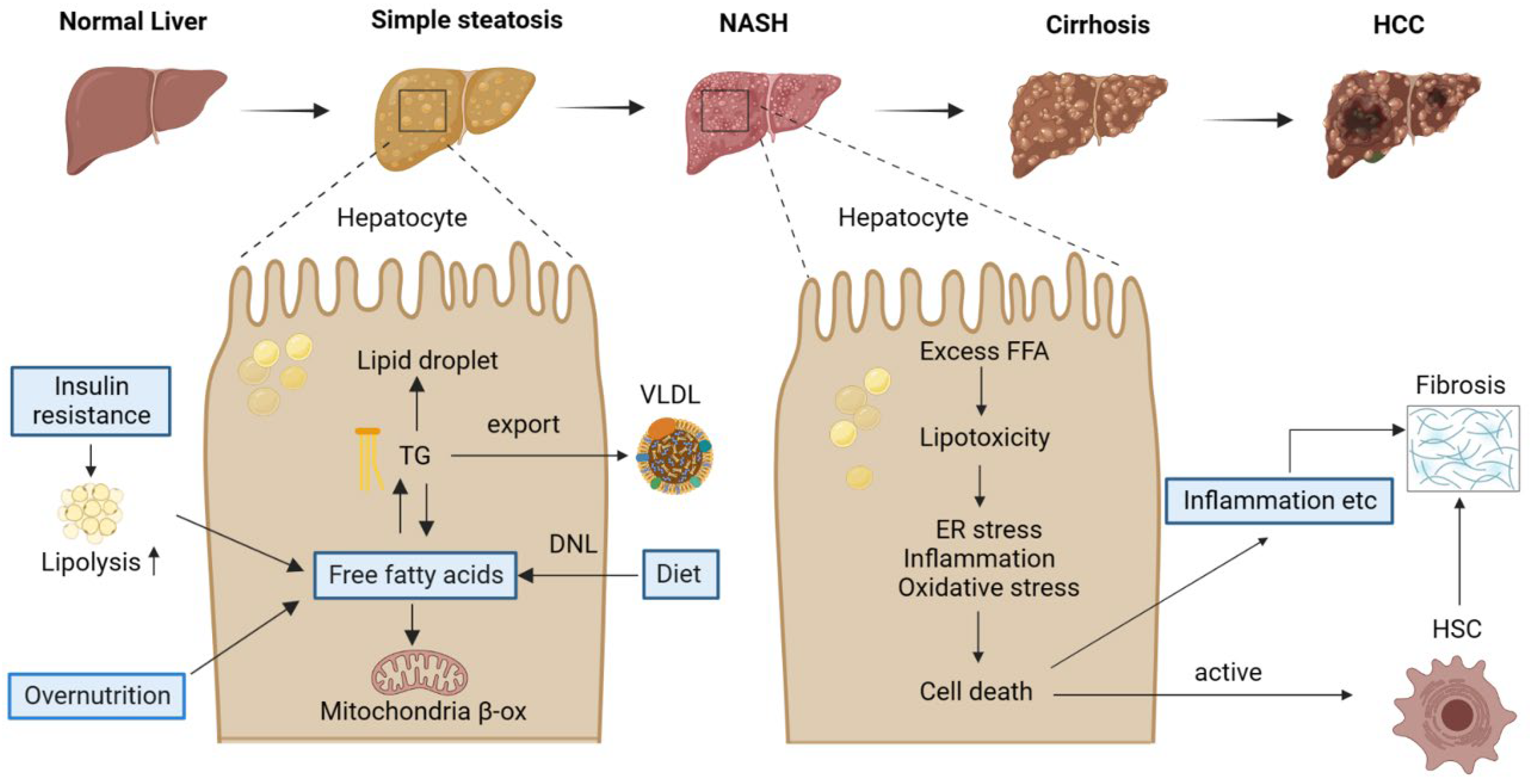
From simple steatosis to NASH, cirrhosis, and hepatocellular cancer, NAFLD is a progressive disease [6] (Figure 1). Simple steatosis, or nonalcoholic fatty liver (NAFL), is characterized by the accumulation of triglycerides (TG) in hepatocytes. Hepatic steatosis can occur due to various factors. Excess dietary fat is certainly one. In addition, dietary carbohydrates raise blood glucose and insulin levels, leading to the activation of carbohydrate responsive element-binding protein (ChREBP) and sterol regulatory element-binding protein-1c (SREBP-1c) in the liver. These are two major transcription factors upregulating genes in fatty acid synthesis, thereby promoting de novo lipogenesis (DNL) [7,8,9], which produces free fatty acids (FFAs) from acetyl-coenzyme A (CoA) and leads to the accumulation of lipid droplets in hepatocytes upon activation. Furthermore, insulin resistance (IR), which is a common condition in patients with obesity or T2DM, could decrease the capacity of adipose tissue to store lipids, thereby increasing the amount of FFAs in the blood and, eventually, in the liver. In addition to producing energy through oxidation and storage in hepatocytes as TG, hepatic FFAs can be coupled to apolipoproteins and secreted in very-low-density lipoproteins (VLDL). Hepatic steatosis occurs when the amount of hepatocyte triglyceride production increases more than the amount of VLDL triglyceride release [10]. Therefore, an imbalance in lipid homeostasis causes the accumulation of lipids in the liver.
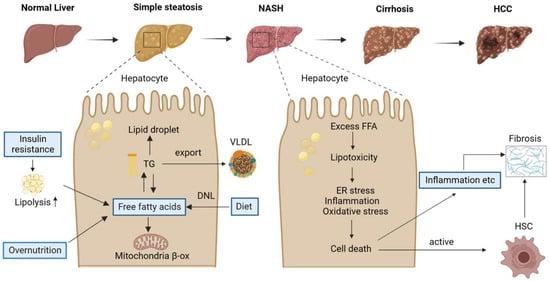
Figure 1.
The pathological spectrum and pathogenesis of NAFLD. NAFLD is a progressive disease, including a broad spectrum of liver conditions, from simple steatosis to NASH, with the potential to progress to more severe stages such as cirrhosis and HCC. High-fat or -fructose diets could increase the free fatty acids in the liver. Free fatty acids have a key role in the development of NAFLD, and this proceeds three ways in the liver. (1) FFAs enter the mitochondria and are oxidized to produce energy and ketone bodies. (2) Esterified to TG and stored in lipid droplets. (3) Secreted and excreted as VLDL. NASH is characterized by steatosis, inflammation, and fibrosis. Cellular stresses due to lipotoxicity cause cell death and liver injury, which ultimately leads to hepatic fibrosis after the activation of HSCs. NASH, nonalcoholic steatohepatitis; HCC, hepatocellular carcinoma; TG, triglyceride; VLDL, very-low-density lipoprotein; β-ox, β-oxidization; DNL, de novo lipogenesis; FFA, free fatty acid; HSC, hepatic stellate cell.
Unlike simple steatosis, NASH is a state in which inflammation and fibrosis occur in addition to the lipid accumulation in the liver. Therefore, NASH is characterized by the accumulation of excessive FFAs, increased oxidative/endoplasmic reticulum (ER)stress, fibrosis, and inflammation in the liver [11,12]. It is defined clinically by the presence of steatosis, hepatocellular ballooning, and lobular inflammation, accompanied by variable degrees of fibrosis upon a liver biopsy [13]. When there is an increased uptake/synthesis or hampered removal of fatty acids, they can be used as substrates for the production of lipotoxic species that induce cellular stress, hepatocyte apoptosis, and liver injury [14]. Indeed, the occurrence of NASH is associated with a number of cellular stresses, such as endoplasmic reticulum (ER) stress, mitochondrial damage, and oxidative stress, which is related to the production of reactive oxygen species (ROS) (Figure 1). It is noteworthy that NASH is progressive, and more severe forms may occur, such as liver cirrhosis and HCC [11]. HCC is the fifth-most prevalent form of cancer and the third leading cause of cancer-related death [15], highlighting the importance of preventing the progression of hepatic steatosis to HCC.
1.2. Factors Influencing NAFLD
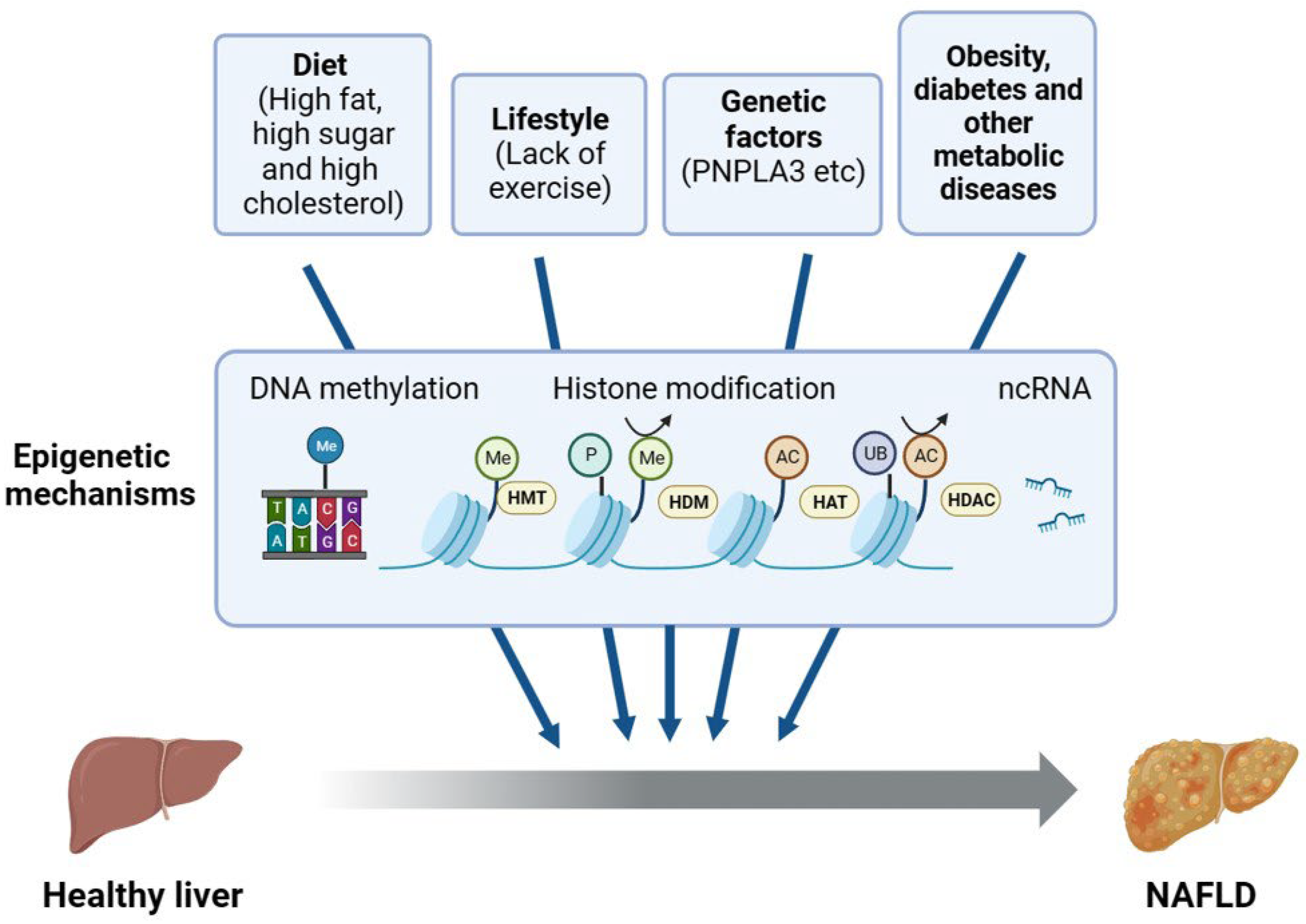
NAFLD is a complex disease resulting from multiple aspects of lifestyle, diet, genetic variants, and epigenetic factors (Figure 2). The lack of physical exercise has been associated with increased body weight, insulin resistance, and an increased risk of metabolic syndrome and NAFLD [16]. Unhealthy diets may lead to overnutrition characterized by the excessive intake of fats or fructose, which are metabolized in the liver through DNL and converted to TGs. Consequently, this metabolic pathway contributes to the accumulation of fat in the liver. Insulin resistance has long been acknowledged as a crucial factor in the development of NAFLD [17]. There is a growing body of evidence supporting that the microbiome also plays a critical role in NAFLD [18,19,20]. The gut–liver axis has been connected to a number of disorders associated with obesity, including NAFLD, and these two organs are interdependent at many levels [21].
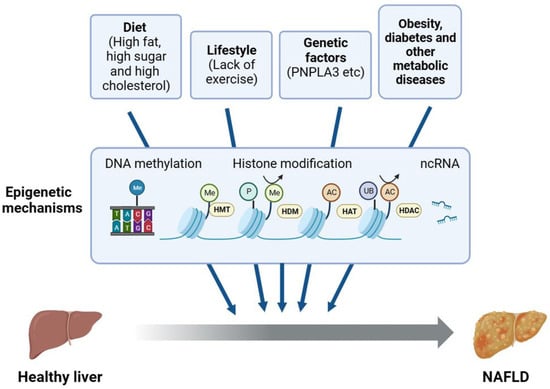
Figure 2.
Epigenetic regulation of NAFLD. The factors influencing NAFLD include diet; lifestyle; and basal conditions such as metabolic syndrome, genetic variants, and epigenetic factors. In addition, non-epigenetic factors may influence NAFLD through epigenetic mechanisms, such as DNA methylation, histone modifications, and ncRNA. PNPLA3, patatin-like phospholipase domain-containing 3; HMT, histone methyltransferase; HDM, histone demethylase; HAT, histone acetyltransferase; HDAC, histone deacetylase; ncRNA, non-coding RNA.
In addition to external or environmental cues, genetic variants significantly influence the progression of NAFLD [22]. One solid example is the genetic variant in the PNPLA3 gene (patatin-like phospholipase domain-containing 3, rs738409), a substitution of cytosine with guanine resulting in a change of codon 148 from isoleucine to methionine. PNPLA3 is involved in the lipolysis of lipid droplets in hepatocytes. The I148M variant escapes proteasome degradation, accumulates on lipid droplets, and blocks the function of adipose triglyceride lipase and lipolysis [23]. PNPLA3-I148M has exhibited a robust correlation with increased hepatic TG accumulation and hepatic inflammation in human genome-wide association studies [24]. PNPLA3-I148M expression in liver resulted in an increase in the liver triglyceride content in a transgenic mouse model [25]. Thus, it is an important genetic risk factor and a valid therapeutic target for NAFLD [26]. Similarly, a few other genetic risk factors have been identified, such as a splice variant (rs72613567 T>A) in HSD17B13 and an E to K substitution (rs58542926 C>T) in TM6SF2, which have been well summarized recently [27,28].
Due to the fact that epigenetic alterations can be dynamic and reversible, epigenetic mechanisms have been widely implicated in the initiation and progression of NAFLD, even in collaboration with other environmental cues (Figure 2). Epigenetic inheritance studies the changes in heritable gene expression or cellular expression caused by specific mechanisms without altering the DNA sequence [29]. Alterations in DNA methylation, histone variants and modifications, chromatin remodeling, and non-coding RNA-based mechanisms may all result in epigenetic changes. Several epigenetic mechanisms are of significant importance in the NAFLD spectrum of diseases [30]. For example, the cytosine methylation (5mC) of mitochondrial DNA (mtDNA) had been found in the liver biopsies of patients with NAFLD, and patients with NASH had higher levels of DNA methyltransferase 1 [31]. In another study, AAV-miR-20b administration induced hepatic steatosis and reduced FA oxidation in HFD-fed mice, possibly by decreasing the level of PPARα [32]. More broadly, histone modification changes may lead to the dysregulation of multiple biological processes associated with NAFLD, such as hepatic lipid accumulation, ER stress, oxidative stress, mitochondrial damage, and inflammation, which may be used as a single mechanism or work in synergy with the environmental factors on the development of NAFLD [22]. Due to length limitations, in this review, we focus on the regulatory mechanism of histone modifications, their pathological implications, and the potential therapeutic applications in the treatment of NAFLD.
2. Regulation of NAFLD by Histone Modifications
The histone core is an octamer, comprising two H2A, H2B, H3, and H4 molecules. The histone core and the DNA coiled on it constitute the nucleosome, serving as the basic structural unit of chromatin. The N-terminal tails of histone H2A, H2B, H3, and H4 may extensively modified post-translationally and serve as a hub receiving diverse regulatory signals from diet, lifestyle, and other environmental cues. With the technical improvements and scientific advancements, the list of modification types has expanded, encompassing not only classical acetylation, methylation, ubiquitination, and phosphorylation but also newly discovered ones, including lactylation and dopaminylation [33,34]. Among the classical modifications, acetylation, methylation, and ubiquitination primarily happen on lysine or arginine, while phosphorylation happens on serine or threonine. Distinct modification types may alter the chromatin structure and gene expression differentially. For example, histone acetylation generally correlates with transcriptional activation, whereas deacetylation tends to exert a transcriptionally repressive role. From the perspectives of their relevance in NAFLD, the classical histone modifications have been extensively investigated, and therefore, more details are summarized below.
2.1. Histone Methylation in NAFLD and its Therapeutic Implications
Histone methylation is catalyzed by histone methyltransferases (HMTs), and the histone demethylases (HDMs) remove the methylation marks on lysine or arginine residues. Notably, distinct lysine or arginine residuals require specific HMT and HDM enzymes, which also provide a strong specificity toward individual methyltransferase or demethylase, thereby reenforcing their distinct functional impacts.
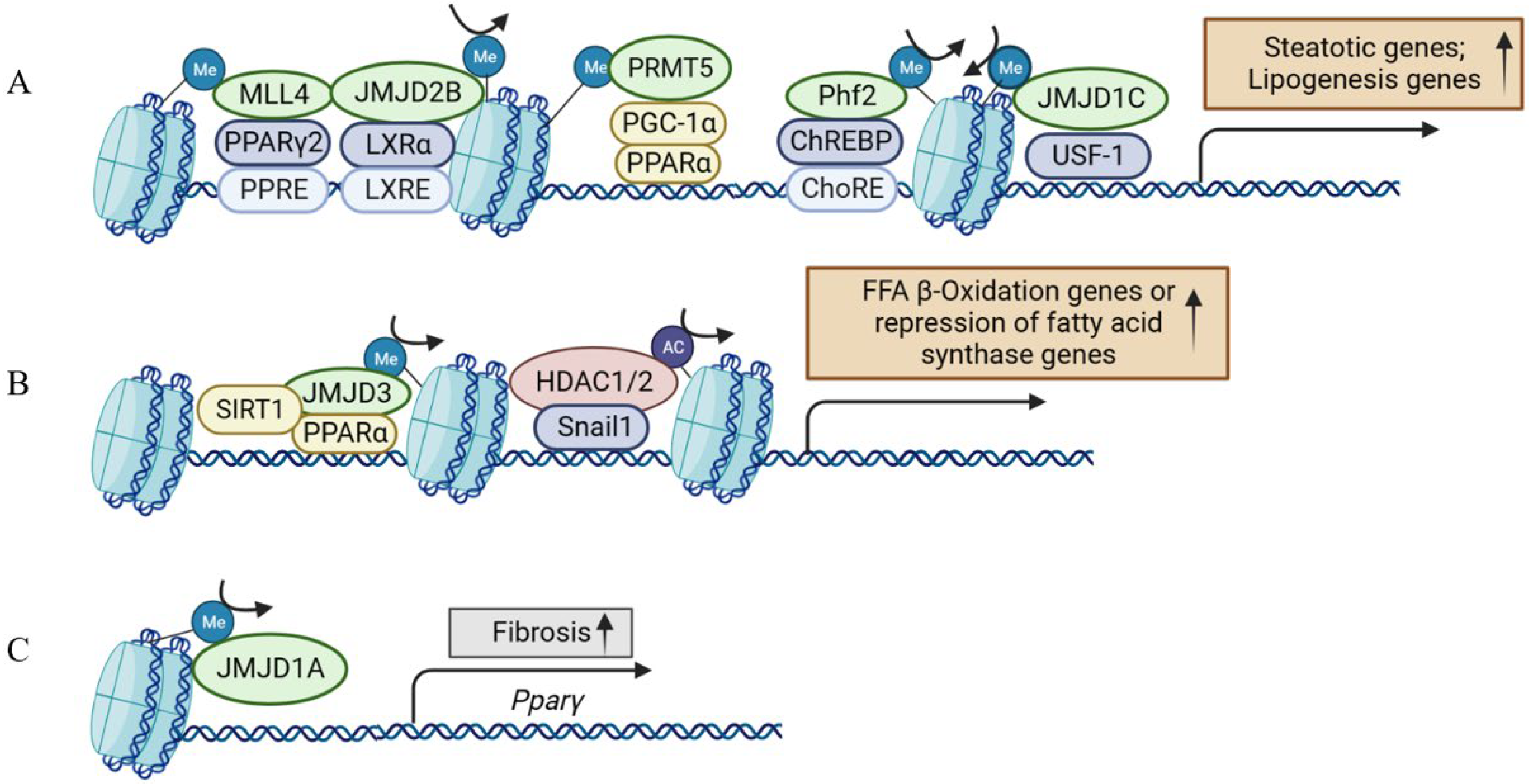
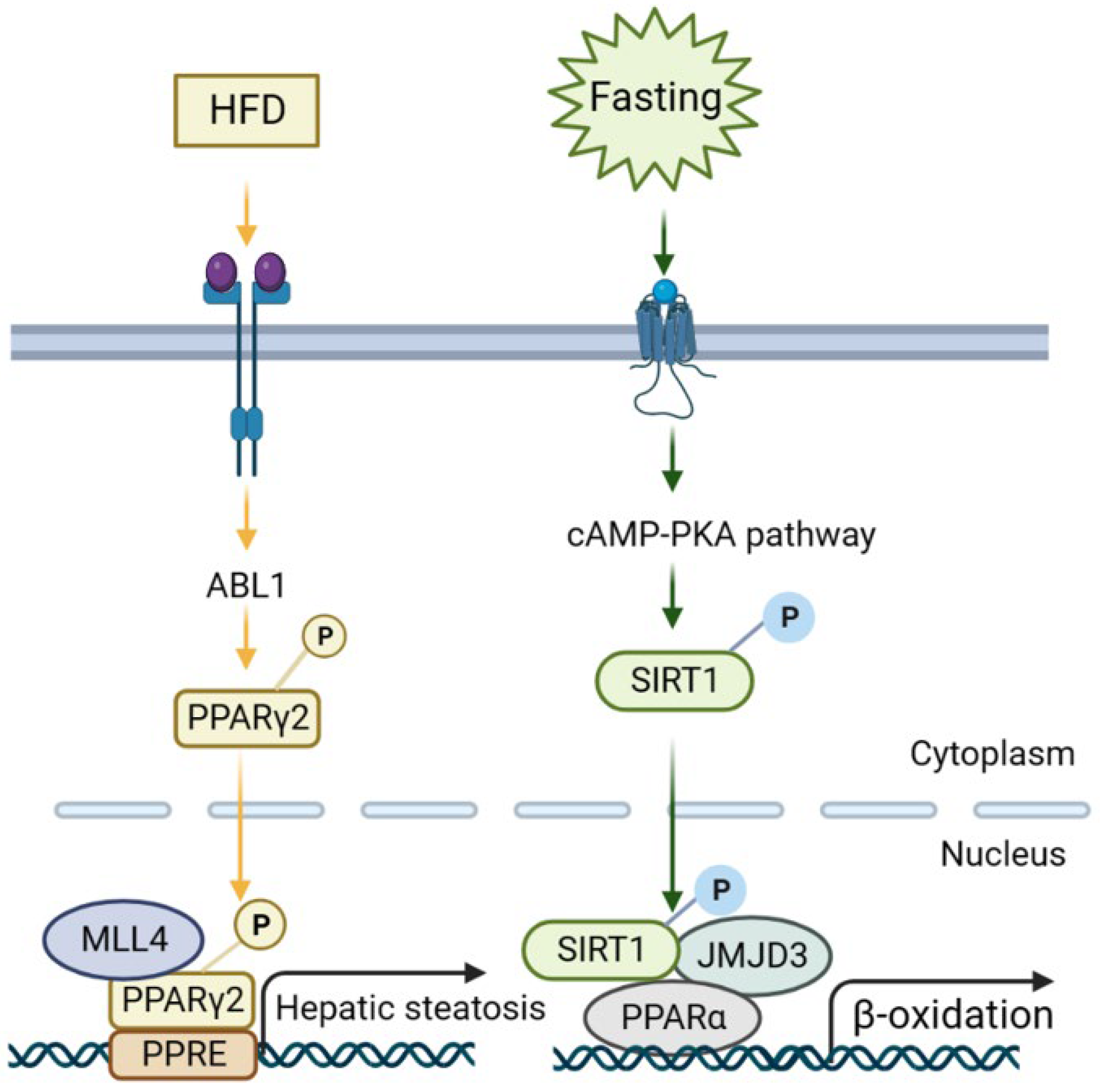
Early evidence suggested histone H3 lysine 4 (H3K4) methyltransferase MLL2 could influence the metabolism via random ENU mutagenesis. A study revealed that a germline Mll2 mutation led to insulin resistance and impaired glucose tolerance in mice [35]. In another study, HFD feeding led to the activation of ABL1 kinase, which phosphorylated PPARγ2 and enhanced the MLL4–PPARγ2 interaction. Consequently, overnutrition enhanced the recruitment of MLL4 to the promoter of PPARγ2-regulated steatosis target genes. This, in turn, increased the H3K4 methylation and transcriptional activation. Thus, the interaction between MLL4 and PPARγ2 proteins played a role in the development of fatty liver in HFD-fed mice [36,37] (Figure 3 and Figure 4). Moreover, the activation of hepatic stellate cells (HSCs), a key event in the transition from NAFLD to NASH, is also regulated by histone methylation. In activated HSCs, ASH1, another HMT for H3K4 methylation, directly bound to the regulatory genomic regions of alpha smooth muscle actin (α-SMA), collagen I, tissue inhibitor of metalloproteinase-1 (TIMP1), and transforming growth factor beta 1 (TGF-β1) to facilitate H3K4 methylation and transcriptional activation. Conversely, inhibiting ASH1 led to the downregulation of these fibrogenic gene expressions [38,39].
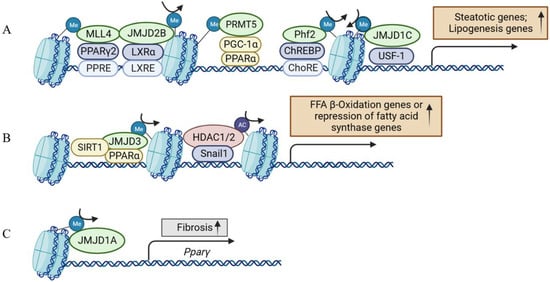
Figure 3.
Histone-modifying enzymes and transcriptional regulators are concordantly involved in the multiple aspects of NAFLD development. (A) Histone-modifying enzymes involved in the process of hepatic steatosis. (B) Histone-modifying enzymes involved in the process of fatty acid β-oxidation or the repression of fatty acid synthase. (C) Histone-modifying enzymes involved in the process of hepatic fibrosis. The epigenetic regulation in (A,B) involves hepatocytes, and (C) involves hepatic stellate cells in addition to hepatocytes. MLL4, mixed lineage leukemia 4; PPARγ, peroxisome proliferator-activated receptor γ; PPRE, PPAR response element; JMJD2B, Jumonji domain-containing protein 2B; LXRα, liver X receptors α; LXRE, LXR response element; PRMT5, protein arginine methyltransferase 5; PGC-1α, peroxisome proliferator-activated receptor γ coactivator 1 alpha; PPARα, peroxisome proliferator-activated receptor α; Phf2, Plant Homeodomain Finger 2; ChREBP, carbohydrate-responsive element-binding protein; ChoRE, carbohydrate responsive element; JMJD1C, Jumonji domain-containing protein 1C; USF-1, upstream stimulatory factor 1; SIRT1, Sirtuin 1; HDAC1/2, histone deacetylases 1/2; JMJD1A, Jumonji domain-containing protein 1A.
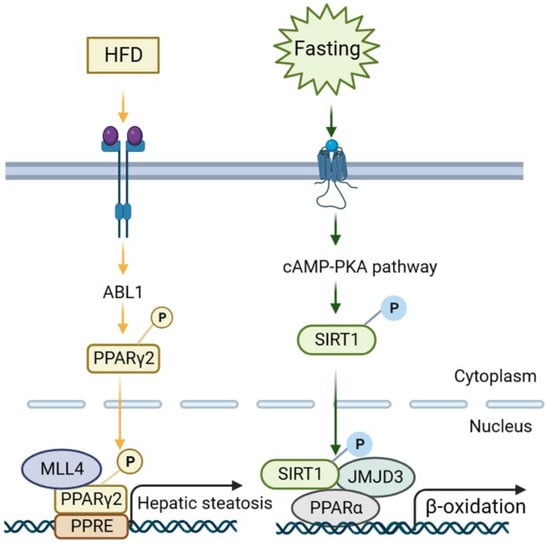
Figure 4.
Representative signaling mechanisms of NAFLD regulation via histone-modifying enzymes. (1) Under HFD feeding, insulin and glucose signaling activates ABL1 kinase, which then phosphorylates PPARγ2, and the PPARγ2-MLL4 complex forms to promote hepatic steatotic target genes. (2) Fasting initiates cAMP/PKA signaling, resulting in the phosphorylation of SIRT1 and the formation of a JMJD3-SIRT1-PPARα complex in hepatocytes to increase the expression of its own gene and SIRT1-targeted β-oxidation via H3K27me3 demethylation.
Protein arginine methyltransferase 5 (PRMT5) affects gene expression by methylating the arginine residues on histones, including H4R3, H3R8, and H2AR3. A previous study showed PRMT5 promoted the development of hepatic steatosis under a high-fat diet by facilitating the suppression of transcription regulators in mitochondrial biogenesis such as PPARα [40] (Figure 3A). PPARα functions as a pivotal transcription factor governing processes like fatty acid uptake, mitochondrial and peroxisomal fatty acid oxidation, and ketogenesis in the liver [41,42]. Mechanistically, PRMT5 was upregulated in the liver upon HFD. Conversely, the silencing or deletion of PRMT5 led to diminished AKT phosphorylation while increasing the expression of PPARα and PGC-1. This, in turn, elevated mitochondrial and peroxisomal fatty acid oxidation, demonstrating a propensity to slow down the development of fatty liver, although the mechanism with which PRMT5 regulates AKT phosphorylation remains unclear [40,43].
Methylations on K27 or 9 of histone H3 (H3K27 or H3K9 methylation) are typically linked to transcriptional repression. Polycomb repressive complex 2 (PRC2), a multicomponent protein complex, is the only methyltransferase for H3K27 and conserved from fungi to mammals. EZH2, the enzymatic subunit of PRC2, catalyzes the mono-, di-, and trimethylation of H3K27, which plays an important role in cell proliferation and differentiation. It has been reported that the EZH2 protein level was downregulated in the rat liver of a diet-induced NAFLD model and the fatty acid-induced insulin-resistant HepG2 model, although lacking a mechanism [44]. On the other hand, many studies have pointed toward out that EZH2/PRC2 activity or H3K27me3 may facilitate the disease progression to HCC via the repression of multiple tumor-suppressive microRNAs [45]. For example, it was reported that miR-200c was repressed by chromatin H3K27me3, and EZH2 depletion upregulated miR-200c and inhibited the growth of Huh7 in vitro and in vivo [46].
Unlike H3K27, there are multiple H3K9 methyltransferases, such as SUV39H2/KMT1B and the dimeric G9a, and they exhibit differential catalytic activities and target genes [47]. In methionine- and choline-deficient (MCD) diet-fed mice, SUV39H2 promoted hepatic steatosis by downregulating SIRT1, a NAD+-dependent histone deacetylase executing a protective role in the liver (more details in Section 2.4) [48]. Conversely, Suv39h2 deletion alleviated diet-induced NASH in mice [49]. As H3K9 methylation is usually mechanistically associated with transcriptional repression, these effects may or may not be direct modulations and require further investigation. Nonetheless, the presented evidence underscores the role of histone methylation in the pathogenesis of NAFLD.
Potential Targets and Compounds Modulating Histone Methyltransferases in NAFLD
As NAFLD is a multifactorial chronic disease, treatment methods and approaches are still scarce despite the increased attention it has received. Since epigenetic modifications and NAFLD are closely related, altering histone modifications holds the potential to offer new opportunities in the treatment of NAFLD.
Targeting EZH2 has been intensively studied for its potential therapeutic implications in the treatment of NAFLD [50]. A fundamental event in the pathogenesis of hepatic fibrosis is the activation of quiescent HSC and their subsequent transformation into myofibroblasts. EZH2 has been reported to promote this transformation by suppressing the expression of PPARγ [51]. In addition, EZH2 was found to be inhibited by SIRT1, the protective NAD+-dependent histone deacetylase, and EZH2 inhibition is required for the protective effect of SIRT1 activation in myofibroblasts [52]. Furthermore, the inhibition of EZH2 decreased fibrogenic gene transcription in the TGF-β1-treated HSCs [53]. Indeed, DZNep, an HMT inhibitor, and GSK-503, a specific EZH2 inhibitor, prevent the progression of liver fibrosis in vivo by decreasing H3K27 methylation [44,50,53,54]. In addition, the herbal prescription Yang-Gan-Wan (YGW) and its active ingredients, rosmarinic acid (RA) and baicalin (BC), showed the potential to treat liver fibrosis by de-repressing Pparγ in an epigenetic-dependent way, which suppressed the expression of EZH2 and reduced H3K27 di-methylation [55].
In addition, the expression of histone H3K9 methyltransferase G9a and the DNA methyltransferase DNMT1 was found to be upregulated in human cirrhotic liver and during mouse HSC activation. Using a dual chemical inhibitor of G9a and DNMT1 CM272, the authors showed that the inhibition of them simultaneously disrupted the profibrogenic metabolic reprogramming of HSC induced by TGF-β1 and inhibited liver fibrogenesis in vivo. Thus, the dual targeting of G9a and DNMT1 may provide a potential therapeutic approach for the treatment of liver fibrosis [56]. Targeting the complex epigenetic mechanisms involved in fibrogenesis with innovative molecules like CM272 may pave the way for better therapies.
2.2. Histone Demethylation in NAFLD and its Therapeutic Implications
In addition to HMTs, HDMs have been shown to be involved in NAFLD development. The histone demethylase Plant Homeodomain Finger 2 (PHF2) can erase the H3K9me2 mark. Mice with adenovirus overexpressing Phf2 in the liver showed that increased levels of DAG and TG were protected from insulin resistance and inflammation [57] (Figure 3A). The reason was that the overexpression of Phf2 could increase the level of SCD1, which catalyzes the desaturation of saturated fatty acids (SFA) to monounsaturated fatty acids (MuFA), and MuFA can prevent lipotoxicity. Consequently, PHF2 could prevent the progression to NASH with inflammation and fibrosis [57]. In addition, the lysine (K)-specific demethylase KDM7A belongs to the PHF2/PHF8 family of the Jumonji C (JmjC) domain-containing demethylase (JMJD demethylase) and has two identifiable domains: a PHD and a JmjC domain [58]. KDM7A overexpression could erase the H3K9me2 and H3K27me2 repressive markers on the DGAT2 promoter, thereby increasing the expression of DGAT2 and TG accumulation, which, finally, induced hepatic steatosis [59]. As SCD1 and DGAT2 enzymes are potential targets for the treatment of NAFLD and clinical trials are ongoing, PHF2 and KDM7A could provide potential therapeutic targets in treating NAFLD.
The KDM4 family is made up of four isoforms, KDM4A to D (also called JMJD2A to D). KDM4A, B, and C encode their respective proteins containing one JmjC, one JmjN, two PHD domains, and two Tudor domains. KDM4D is different from the other three isoforms, because it lacks both the PHD and Tudor domains [60,61]. KDM4D catalyzed the di-demethylation and tri-demethylation of H3K9, which stimulated TLR4 expression and triggered hepatic fibrogenesis by activating the NF-κB pathway. Meanwhile, KDM4D was significantly upregulated during HSC activation [60,62,63]. Unlike KDM4D, the other three isoforms were downregulated in HSC activation and facilitated the transcription of miR-29 together with SREBP2 to antagonize liver fibrosis [64]. In addition, KDM4B/JMJD2B could upregulate PPARγ2 and its target genes related to lipid droplet formation and fatty acid uptake by removing H3K9 methylation to promote hepatic steatosis [65]. A later study demonstrated that KDM4B plays a pivotal role in liver X receptor alpha (LXRα)-mediated lipogenesis [66], which provided another mechanism for KDM4B in hepatic steatosis (Figure 3A).
In addition to KDM4s, the role of the KDM3 subfamily in the liver has also been studied. KDM3A, KDM3B, and KDM3C (JMJD1A–C) are roughly 50% identical at the amino acid level and are all capable of removing dimethyl and monomethyl marks from H3K9 and H4R3 and nonhistone proteins, to a lesser extent. PPARγ expression was epigenetically regulated by KDM3A/JMJD1A during HSC activation (Figure 3C). KDM3A knockdown led to the elevated expression of fibrosis markers in HSCs and a mouse liver fibrosis model [67]. KDM3C/JMJD1C facilitated nutrient signaling to elevate the triglyceride levels in the liver and plasma by promoting the expression of lipogenesis genes [68]. Mechanistically, USF-1 recruited JMJD1C into multiple lipogenic genes in the fed state to demethylate H3K9me2 and increase chromatin accessibility (Figure 3A). JMJD1C was phosphorylated at T505 by the mTOR complex, which enhanced the direct interaction between USF-1 and JMJD1C and transduced the nutrient signal [68]. Therefore, a potential treatment approach for hepatosteatosis and IR is to target JMJD1C phosphorylation by mTOR, a critical lipogenic insulin signaling cascade [68]. KDM6B/JMJD3, a H3K27 demethylase, is another notable HDM in liver metabolism and NAFLD [69]. Under fasting conditions, JMJD3 and SIRT1 worked synergistically to activate fatty acid oxidation genes, such as Fgf21, Cpt1a, and Mcad. The liver-specific downregulation of JMJD3 reduced fatty acid oxidation and led to hepatic steatosis [70] (Figure 3B).
Potential Targets and Compounds Modulating Histone Demethylase in NAFLD
Small molecules that activate JMJD3 or promote the interaction of JMJD3 with SIRT1 specifically decreased the lipid levels, which may provide a therapeutic approach to treat obesity and hepatosteatosis [70]. Furthermore, fasting conditions also induced Fibroblast Growth Factor-21 (FGF21) signaling, which required JMJD3 to activate hepatic autophagy and lipid degradation through upregulating the global autophagy network genes [71] (Figure 4). This study also demonstrated that FGF21 administration to alleviate fatty liver in HFD mice was mediated by JMJD3 [71]. Thus, targeting the histone demethylase JMJD3 could be a potential treatment for NAFLD.
GSK2879552 is an LSD1 inhibitor that showed beneficial effect to inhibit FASN expression and ameliorate hepatic steatosis in mice. Mechanistically, the transcription factor Slug was upregulated in hepatocytes by insulin in fed state, which recruited the histone K3K9 demethylase LSD1 to Fasn promoter and promote FASN expression. So GSK2879552 blocked lipogenesis activated by Slug-LSD1 pathway and may be a useful therapeutic rationale for the treatment of NAFLD [72]. In addition, Gomisin N (GN) is a phytochemical from Schisandra chinensis, exhibiting hepatoprotective, anti-cancer, and anti-inflammatory properties [73]. In one study, the administration of GN was found to downregulate the expression of PPARγ2 and JMJD2B in the liver of HFD-induced obese mice [65], which may contribute to the alleviation of HFD-induced hepatic steatosis. GN was also found to reduce the tunicamycin-induced hepatic ER stress and TG accumulation in mice [74]. Thus, GN might be helpful for the treatment of NAFLD, although it may not work mainly through histone demethylation.
2.3. Histone Acetylation in NAFLD and Its Therapeutic Implications
Histone acetylation is a post-translational modification and mostly occurs at specific lysine residues in the N-terminal tails of the histone H3 and H4. Histone acetylation is always associated with chromatin opening and transcriptional activation. Histone acetylation is usually quite dynamic and jointly determined by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which add or remove the acetyl group on particular histone lysine residues.
Histone acetylation may affect the expression of individual critical genes. In HepG2, H3 and H4 acetylation at fatty acid synthase gene FASN promoter was transiently increased upon insulin stimulation in a manner of cross-regulation with ChREBP, although the HAT was not identified [75]. In addition, it has been demonstrated that liver-specific knockdown of nuclear receptor subfamily 2, group F, member 6 (NR2F6) alleviated obesity-associated hepatosteatosis and MCD diet-induced NASH through downregulating CD36 expression in mouse models [76]. NR2F6 bound directly to CD36 promoter in hepatocytes, recruited nuclear receptor coactivator 1 (SRC-1), a component of p300/CBP HAT complex, and promoted H3 acetylation on CD36 promoter [76]. Interestingly, NR2F6 expression was increased in the livers of NAFLD patients and reduced by metformin treatment in obese mice [76]. Therefore, NR2F6 antagonists might offer a therapeutic approach for treating NAFLD through histone acetylation.
In addition, histone acetylation may be involved in the regulation of multiple genes/pathways simultaneously. Homozygous knock-in of a serine-to-alanine mutation at Ser196 (S196A) in LXRα to abolish the phosphorylation could affect the hepatic H3K27 acetylome and transcriptome during the progression of NAFLD. For example, the H3K27Ac at the Ces1f gene locus and the expression of Ces1f were high in the liver of LXRα-S196A mice comparing with WT mice when fed high-fat-high-cholesterol (HFHC) diet. Ces1f is a member of the carboxylesterase 1 family that controls hepatic lipid mobilization. Meanwhile, the H3K27Ac and expression of inflammation and fibrosis related genes including Spp1 and Col1e1 were reduced in S196A. So, LXRα-S196A could induce liver steatosis but prevent cholesterol accumulation, inflammation and fibrosis, thereby slowing the development from simple hepatic steatosis to NASH [77].
Potential Targets and Compounds Modulating Histone Acetylase in NAFLD
A few studies further suggested that histone acetylation can be a potential target for NAFLD. The active phosphorylated form of FTY720/fingolimod, a prodrug treating multiple sclerosis, could reduce FASN expression by histone acetylation alteration, inhibit ceramide production and hepatic steatosis in diet-induced NAFLD mice [78]. Tannic acid (TA), a HAT inhibitor, inhibited lipid accumulation in vivo and reduced the mRNA expression of genes associated with lipogenesis. Mechanistically, TA eliminated the occupancy of p300 on the sterol regulatory elements (SREs) in the promoters of FASN and ATP-citrate lyase (ACLY) genes, thereby decreasing acetylation of H3K9 and H3K36 [79].
The biguanide medicine metformin is the most popular anti-diabetic medication for the treatment of type 2 diabetes (T2D), which relieves hyperglycemia by reducing hepatic gluconeogenesis and improving insulin sensitivity [80,81,82]. It was reported that metformin activated AMPK, which directly phosphorylated and activated HAT1, promoted histone acetylation, and upregulated genes in mitochondria biogenesis [83]. Another study demonstrated that metformin promoted the phosphorylation of CBP at Ser436, which resulted in the dissociation of the CREB-CBP-TORC2 complex and downregulated the expression of the genes encoding gluconeogenic enzymes. Thus, metformin dramatically reduced the blood glucose level [84]. In addition, metformin increased hepatic protein levels of SIRT1 and GCN5 to inhibit hepatic gluconeogenesis [85].
2.4. Histone Deacetylation in NAFLD and Its Therapeutic Implications
The histone deacetylases (HDACs) family includes four distinct classes, namely class I, II, III and IV. HDAC1-3 and HDAC8 constitute Class I HDACs. Class II includes HDAC 4, 5, 6, 7, 9, and 10. On the other hand, class III HDAC enzymes, also known as sirtuins or silent information regulators (SIRT1-7), rely on NAD+ as a cofactor. Class IV HDAC exclusively consist of HDAC11 [86].
HDACs promote a dense chromatin structure and inhibit transcription by deacetylating lysine residues. In a diet-induced NAFLD and HCC mouse model, SREBP1 directly upregulated the expression of HDAC8, which worked with EZH2 concordantly through H4 deacetylation and H3K27 trimethylation to repress Wnt antagonists, thereby activating the Wnt pathway [87,88]. Snail1, a zinc-finger transcription factor, was reported to repress the expression of FASN through recruiting HDAC1/2 to deacetylate H3K9 and H3K27 at FASN promoter. In HFD-fed mice, Snail1 overexpression in the liver decreased the insulin-stimulated lipogenesis in hepatocytes and attenuated the fatty liver. Conversely, disruption of the insulin-snail1 pathway may lead to NAFLD (Figure 3B) [89]. HDAC6 was also reported to deacetylate the transcription factor FOXO1. S100 calcium binding protein A11 (S100A11) was upregulated in NAFLD liver, and then blocked the interaction between HDAC6 and FOXO1 to stimulate lipogenesis and liver steatosis [90]. Here the role of HDAC6 is beneficial along the line of NAFLD prevention.
SIRT1 is a NAD+ coenzyme-dependent histone deacetylase. Various studies have shown that SIRT1 can influence NAFLD through a variety of pathways. On one hand, SIRT1 is a unique upstream regulator of LKB1/AMPK sensing energy signaling [91]. Under fasting condition, the SREBP-1c acetylation level in mouse liver was consistently reduced and its interaction with SIRT1 was increased. The SREBP-1c-SIRT1 interaction was decreased after feeding, while SREBP-1c acetylation went up and better promoted lipogenesis [92]. In HFD model, liver-specific Sirt1 knockout impaired PPARα/PGC-1α signaling and reduced fatty acid oxidation, thereby resulting in increased hepatic steatosis. This provided compelling evidence for a significant association between SIRT1 and hepatic fatty acid metabolism [93]. In diet-induced and genetically obese mice, pharmacological SIRT1 activators suppressed the hepatic lipid and cholesterol levels as well as liver steatosis [94]. In addition, SIRT1 could also inhibit NF-κB activity to reduce the inflammatory response, which is a powerful defender against pathologic conditions like fatty liver [95,96]. Thus, interventions stimulating SIRT1 activity could potentially offer therapeutic benefits for the management of hepatic diseases and metabolic syndrome associated with obesity [93].
Like SIRT1, SIRT6 has been implicated in the negative regulation of lipid metabolism and inflammation. Liver-specific Sirt6 knockout mice exhibited a tendency to increase hepatic steatosis, inflammation and insulin resistance under high-fat and high-fructose (HFHF) diet through upregulated BTB domain and CNC homolog 1 (Bach1), a nuclear repressor of Nrf2 [97]. In a similar study, liver-specific Sirt6 deletion led to fatty liver through reduced β-oxidation and enhanced glycolysis, lipogenesis and TG synthesis [98]. Mechanistically, SIRT1 interacts with FOXO3a and NRF1 on SIRT6 promoter to positively regulate SIRT6 expression [98]. Indeed, multiple studies support the idea that SIRT6 promotes β-oxidation in a fasting state. In one study, SIRT6 regulated hepatic PPARα activity in vivo via deacetylation of the cofactor NCOA2 at K780 [99]. In parallel, SIRT6 could increase the activity of long-chain acyl-CoA synthase 5 (ACSL5) by deacetylating K98, K361 and K367 and promote fatty acid β-oxidation, thereby increasing the cellular lipid utilization and ultimately resisting the NAFLD process [100]. In addition, SIRT6 engaged in an interaction with acetyltransferase GCN5 and increased its activity, thereby suppressing hepatic gluconeogenesis. Therefore, hepatic SIRT6 activation may be therapeutically useful in the prevention of IR and NAFLD [101]. Another study demonstrated that SIRT6 overexpression in liver reduced steatosis, inflammation, and fibrosis caused by a HFHF diet, indicating the SIRT6 activator may be a promising therapeutic direction for treating NASH by reducing oxidative stress and inflammation [97]. Together, it is valuable to further explore the therapeutic agonists of SIRT1 and SIRT6 for the treatment of NAFLD.
Potential Targets and Compounds Modulating Histone Deacetylase in NAFLD
HDAC chemical inhibitors have been developed to treat cancer, and many of them have also been tested in mice models of NAFLD or obesity. The treatment of mice with valproic acid (VPA), a class I and II HDAC inhibitor, could decrease collagen deposition and HSC activation in the CCl4 model [102]. VPA is expected to have potential in preventing the further progression of liver fibrosis. Suberoylanilide hydroxamic acid (SAHA), another HDAC inhibitor, was shown to reduce liver fibrosis in rats through the suppression of TGF-β1 signaling [103]. In addition, sodium butyrate (NaB) increased H3K9Ac on the PPARα promoter, enhanced fatty acid oxidation, and inhibited the NF-κB inflammatory pathway, thereby alleviating NAFLD in the rat HFD model [104]. Thus, HDAC inhibitors may hold promise for the treatment of NAFLD.
Resveratrol (RSV), a natural polyphenol, not only exhibits anti-inflammatory and antioxidative characteristics but also activates SIRT1 [105]. RSV alleviated HFD-induced hepatic steatosis in mice liver and reduced lipid droplet accumulation in a SIRT1/ATF6-dependent manner [106]. In addition to the studies on mice models, there have been clinical studies demonstrating resveratrol has the potential to improve NAFLD in patients [107,108]. Patients receiving a daily dose of 300 mg resveratrol for 3 months had lower ALT and aspartate transaminase (AST) levels, higher lipid metabolism, and less inflammation [108]. On the other hand, it was also reported that patient treated with 3000 mg of resveratrol daily for 8 weeks did not show an improvement in their ALT and AST levels, hepatic steatosis, and insulin resistance [109]. Therefore, the dose and long-term effects of RSV require more study. Furthermore, SRT1720 is a specific SIRT1 activator that prevents diet-induced obesity and insulin resistance through enhancing fatty acid oxidation in the liver, muscle, and brown adipose tissue in mice [110]. SRT1720 has a significant protective effect against NAFLD in monosodium glutamate (MSG) mice. Treatment with SRT1720 reduced the expression of markers of oxidative stress, as well as inflammatory cytokines [111].
Interestingly, Olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor approved for cancer treatment, could increase hepatic SIRT1 activity/expression in mice [112] and showed the potential to treat fatty liver disorders. Although upregulating SIRT1 is only one aspect of the multifaceted mechanism of Olaparib, mice receiving Olaparib for 5 weeks showed significantly attenuated liver injury, inflammation, and fibrosis in a MCD diet model [112]. The monoterpenic phenol carvacrol (CVL), which exists in a variety of essential oils from the Labiatae family plants, also upregulates SIRT1. It has prospective hepatoprotective and neuroprotective effects [113]. Combined CVL and rosiglitazone treatment in HFD-fed mice improves the symptoms of diabetes mellitus, such as the reduction in the ALT/AST, plasma glucose, and insulin levels [114].
In addition to SIRT1 and SIRT6, SIRT3 is a mitochondrial sirtuin that has been investigated widely, which is essential for the maintenance of mitochondrial functions [115]. Protocatechuic acid (PCA), also known as 3,4-dihydroxybenzoic acid, is a natural phenolic compound in various food plants. Although it also has antioxidant and anti-inflammatory properties [116,117], PCA bound and upregulated the SIRT3 protein to prevent liver damage and steatosis in HFD-fed mice, likely by regulating the acetylation and degradation of Acyl-CoA synthetase family member 3 (ACSF3) and fatty acid metabolism [118].
2.5. Histone Ubiquitination and Phosphorylation in NAFLD
Histone ubiquitination is the process by which the ubiquitin molecule is specifically conjugated to histones on lysine residues in the presence of a family of enzymes, such as when activating, binding, and degrading ubiquitin. The ubiquitination of histones plays a role in changing the conformation of chromatin and recruiting and activating downstream readers or chromatin regulator proteins. For example, the overexpression of RNF20 effectively inhibited IL-6, TNFα, and VEGFA to prevent TGF-β-induced hepatic fibrosis via the ubiquitination of H2BK120 (H2BK120ub) [119]. Unlike acetylation and methylation, histone H3 serine 10 (H3S10) phosphorylation is a marker for mitotic chromatin and affected by the counteractions between kinases and phosphatases. Histone phosphorylation often cross-talks with other histone modifications and may regulate the chromatin status upon DNA damage and other stresses. For example, it was reported that ChREBP bound to ChoRE to upregulate the expression of FASN through the histone acetylation, methylation, and phosphorylation of histone H3 serine 10 (H3S10) [120], promoting hepatic steatosis. There are relatively scarce reports on histone ubiquitination and phosphorylation in NAFLD, and more studies may be in the queue.
3. Conclusions and Future Perspectives
NAFLD is a chronic and progressive hepatic disorder characterized by the increase of an excessive amount of fat in the liver. This accumulation of fat induces stress and damage to hepatocytes, resulting in inflammation and fibrosis. If left untreated, these pathological processes can ultimately lead to the development of liver injury, cirrhosis, hepatocellular carcinoma, and, ultimately, mortality [121]. Epigenetic mechanisms, specifically histone modifications here, are dynamic and can be reversibly regulated by a variety of external cues, such as nutrient signals. The role and mechanisms of histone modifications and the related enzymes in NAFLD have been investigated, and some of the interesting findings are summarized here (briefly listed in Table 1). Some of the useful compounds affecting these histone modifications are briefly listed in Table 2. Although they may not be directly useful in the treatment of patients, they provide specific tools for target validation in cell and animal models.

Table 1.
Summary of histone modifications and the corresponding mechanisms involved in NAFLD.
Table 2.
Summary of histone modification-modulating compounds tested in NAFLD models.
One of the noticeable features is that multiple histone modifications can be concordantly regulated and coordinately promote or inhibit specific gene networks. A few key cases are summarized in Figure 3 and may happen in different types of cells critical in NAFLD, such as hepatocytes and HSCs. Another feature worth mentioning is that many histone modifications are regulated by nutrient availability and status. Two cases are shown in Figure 4 to support this notion. More broadly, cofactors for histone modification enzymes include acetyl-CoA for acetyltransferases, S-adenosyl methionine for methyltransferases, and NAD+ for Class III HDACs. The concentrations of these cofactors are comprehensively affected by energy metabolism and other physiological/pathological conditions. In this way, histone modifications and other epigenetic marks may capture and reflect the integrated state of external inputs.
As histone modification plays an important role in the development of NAFLD and may be reversely regulated, there is increasing interest in the development of novel therapies focusing on modulating epigenetic variations [57]. Histone-modifying enzymes may provide targets for NAFLD therapy. However, NAFLD is a complex disease associated with multiple metabolic disorders, and its potential side effects still need to be considered. Therefore, although there is interest in the development of histone modification enzyme inhibitors as the treatment for NAFLD, more research needs to be done to examine the possible side effects, to discover target-selective inhibitors, and carefully assess their effectiveness in patients. Targeting the complex epigenetic mechanisms in NAFLD with dual-inhibitory molecules may also be tried. The prospective unexplored potential in histone modifications remains to be investigated and released in the future.
Author Contributions
Conceptualization, W.Q.; writing—original draft preparation, Y.S.; writing—review and editing, W.Q.; visualization, Y.S.; funding acquisition, W.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NNSF of China grant number 32070609 and 32270648 to W.Q. And The APC was funded by 32070609.
Informed Consent Statement
This study did not require ethical approval or consent from patients.
Conflicts of Interest
The authors declare no potential conflict of interest.
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Giral, P.; Khan, J.F.; Rosenbaum, D.; Housset, C.; Poynard, T.; Ratziu, V. Fatty liver is an independent predictor of early carotid atherosclerosis. J. Hepatol. 2016, 65, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 195–203. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef]
- Mucinski, J.M.; Manrique-Acevedo, C.; Kasumov, T.; Garrett, T.J.; Gaballah, A.; Parks, E.J. Relationships between Very Low-Density Lipoproteins-Ceramides, -Diacylglycerols, and -Triacylglycerols in Insulin-Resistant Men. Lipids 2020, 55, 387–393. [Google Scholar] [CrossRef]
- Huby, T.; Gautier, E.L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 2022, 22, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Harrison, S.A.; Abdelmalek, M.F.; Anstee, Q.M.; Bedossa, P.; Castera, L.; Dimick-Santos, L.; Friedman, S.L.; Greene, K.; Kleiner, D.E.; et al. Case definitions for inclusion and analysis of endpoints in clinical trials for nonalcoholic steatohepatitis through the lens of regulatory science. Hepatology 2018, 67, 2001–2012. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Yilma, M.; Saxena, V.; Mehta, N. Models to Predict Development or Recurence of Hepatocellular Carcinoma (HCC) in Patients with Advanced Hepatic Fibrosis. Curr. Gastroenterol. Rep. 2022, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.N., Jr.; Harrison, S.A. Lifestyle and Dietary Interventions in the Management of Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, J.; Sanyal, A.J. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 575–594. [Google Scholar] [CrossRef]
- Volynets, V.; Küper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef]
- Membrez, M.; Blancher, F.; Jaquet, M.; Bibiloni, R.; Cani, P.D.; Burcelin, R.G.; Corthesy, I.; Macé, K.; Chou, C.J. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008, 22, 2416–2426. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Investig. 2012, 122, 4130–4144. [Google Scholar] [CrossRef]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes. 2010, 34, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Balcar, L.; Scheiner, B.; Urheu, M.; Weinberger, P.; Paternostro, R.; Simbrunner, B.; Semmler, G.; Willheim, C.; Pinter, M.; Ferenci, P.; et al. The impact of transmembrane 6 superfamily 2 (TM6SF2) rs58542926 on liver-related events in patients with advanced chronic liver disease. Dig. Liver Dis. 2023, 55, 1072–1080. [Google Scholar] [CrossRef]
- Franzago, M.; Pilenzi, L.; Di Rado, S.; Vitacolonna, E.; Stuppia, L. The epigenetic aging, obesity, and lifestyle. Front. Cell Dev. Biol. 2022, 10, 985274. [Google Scholar] [CrossRef]
- Lee, J.H.; Friso, S.; Choi, S.W. Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Nutrients 2014, 6, 3303–3325. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Jang, H.J.; Kim, S.; Choi, S.S.; Khim, K.W.; Eom, H.J.; Hyun, J.; Shin, K.J.; Chae, Y.C.; Kim, H.; et al. Hepatic MIR20B promotes nonalcoholic fatty liver disease by suppressing PPARA. Elife 2021, 10, e70472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Werner, C.T.; Stewart, A.F.; Fulton, S.L.; Zhong, P.; Farrelly, L.A.; Smith, A.C.W.; Ramakrishnan, A.; Lyu, Y.; Bastle, R.M.; et al. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 2020, 368, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Goldsworthy, M.; Absalom, N.L.; Schröter, D.; Matthews, H.C.; Bogani, D.; Moir, L.; Long, A.; Church, C.; Hugill, A.; Anstee, Q.M.; et al. Mutations in Mll2, an H3K4 methyltransferase, result in insulin resistance and impaired glucose tolerance in mice. PLoS ONE 2013, 8, e61870. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef]
- Kim, J.; Lee, B.; Kim, D.H.; Yeon, J.G.; Lee, J.; Park, Y.; Lee, Y.; Lee, S.K.; Lee, S.; Lee, J.W. UBE3A Suppresses Overnutrition-Induced Expression of the Steatosis Target Genes of MLL4 by Degrading MLL4. Hepatology 2019, 69, 1122–1134. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef]
- Atta, H.; El-Rehany, M.; Hammam, O.; Abdel-Ghany, H.; Ramzy, M.; Roderfeld, M.; Roeb, E.; Al-Hendy, A.; Raheim, S.A.; Allam, H.; et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: Role of ASH1 and EZH2 methyltransferases repression. PLoS ONE 2014, 9, e112384. [Google Scholar] [CrossRef]
- Huang, L.; Liu, J.; Zhang, X.O.; Sibley, K.; Najjar, S.M.; Lee, M.M.; Wu, Q. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem. 2018, 293, 10884–10894. [Google Scholar] [CrossRef]
- Motojima, K.; Passilly, P.; Peters, J.M.; Gonzalez, F.J.; Latruffe, N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J. Biol. Chem. 1998, 273, 16710–16714. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Verwilligen, R.A.F.; Van Eck, M.; Hoekstra, M. PRMT5 inhibition induces pro-inflammatory macrophage polarization and increased hepatic triglyceride levels without affecting atherosclerosis in mice. J. Cell. Mol. Med. 2023, 27, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Vella, S.; Gnani, D.; Crudele, A.; Ceccarelli, S.; De Stefanis, C.; Gaspari, S.; Nobili, V.; Locatelli, F.; Marquez, V.E.; Rota, R.; et al. EZH2 down-regulation exacerbates lipid accumulation and inflammation in in vitro and in vivo NAFLD. Int. J. Mol. Sci. 2013, 14, 24154–24168. [Google Scholar] [CrossRef]
- Au, S.L.; Wong, C.C.; Lee, J.M.; Fan, D.N.; Tsang, F.H.; Ng, I.O.; Wong, C.M. Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef]
- Xu, L.; Lin, J.; Deng, W.; Luo, W.; Huang, Y.; Liu, C.Q.; Zhang, F.P.; Qin, Y.F.; Wong, P.P.; Liu, C. EZH2 facilitates BMI1-dependent hepatocarcinogenesis through epigenetically silencing microRNA-200c. Oncogenesis 2020, 9, 101. [Google Scholar] [CrossRef]
- Brower-Toland, B.; Riddle, N.C.; Jiang, H.; Huisinga, K.L.; Elgin, S.C. Multiple SET methyltransferases are required to maintain normal heterochromatin domains in the genome of Drosophila melanogaster. Genetics 2009, 181, 1303–1319. [Google Scholar] [CrossRef]
- Shao, J.; Li, L.; Xu, H.; Yang, L.; Bian, Y.; Fang, M.; Xu, Y. Suv39h2 deficiency ameliorates diet-induced steatosis in mice. Biochem. Biophys. Res. Commun. 2017, 485, 658–664. [Google Scholar] [CrossRef]
- Fan, Z.; Li, L.; Li, M.; Zhang, X.; Hao, C.; Yu, L.; Zeng, S.; Xu, H.; Fang, M.; Shen, A.; et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology 2017, 65, 1904–1919. [Google Scholar] [CrossRef]
- Lim, H.J.; Kim, M. EZH2 as a Potential Target for NAFLD Therapy. Int. J. Mol. Sci. 2020, 21, 8617. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Z.; Tang, F.; Zhao, Y.; Feng, D.; Li, Y.; Hu, Y.; Wang, C.; Zhou, J.; Tian, X.; et al. Carnosol-mediated Sirtuin 1 activation inhibits Enhancer of Zeste Homolog 2 to attenuate liver fibrosis. Pharmacol. Res. 2018, 128, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Martin-Mateos, R.; De Assuncao, T.M.; Arab, J.P.; Jalan-Sakrikar, N.; Yaqoob, U.; Greuter, T.; Verma, V.K.; Mathison, A.J.; Cao, S.; Lomberk, G.; et al. Enhancer of Zeste Homologue 2 Inhibition Attenuates TGF-β Dependent Hepatic Stellate Cell Activation and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 197–209. [Google Scholar] [CrossRef]
- Zeybel, M.; Luli, S.; Sabater, L.; Hardy, T.; Oakley, F.; Leslie, J.; Page, A.; Moran Salvador, E.; Sharkey, V.; Tsukamoto, H.; et al. A Proof-of-Concept for Epigenetic Therapy of Tissue Fibrosis: Inhibition of Liver Fibrosis Progression by 3-Deazaneplanocin A. Mol. Ther. 2017, 25, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.D.; Chiang, Y.M.; Higashiyama, R.; Asahina, K.; Mann, D.A.; Mann, J.; Wang, C.C.; Tsukamoto, H. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor γ in hepatic stellate cells for their antifibrotic effect. Hepatology 2012, 55, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L.; et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2021, 70, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Alves-Guerra, M.C.; Esteves, P.; Prip-Buus, C.; Bertrand-Michel, J.; Guillou, H.; Chang, C.J.; Vander Wal, M.N.; Canonne-Hergaux, F.; Mathurin, P.; et al. The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat. Commun. 2018, 9, 2092. [Google Scholar] [CrossRef]
- Huang, C.; Xiang, Y.; Wang, Y.; Li, X.; Xu, L.; Zhu, Z.; Zhang, T.; Zhu, Q.; Zhang, K.; Jing, N.; et al. Dual-specificity histone demethylase KIAA1718 (KDM7A) regulates neural differentiation through FGF4. Cell Res. 2010, 20, 154–165. [Google Scholar] [CrossRef]
- Kim, J.H.; Nagappan, A.; Jung, D.Y.; Suh, N.; Jung, M.H. Histone Demethylase KDM7A Contributes to the Development of Hepatic Steatosis by Targeting Diacylglycerol Acyltransferase 2. Int. J. Mol. Sci. 2021, 22, 11085. [Google Scholar] [CrossRef]
- Dong, F.; Jiang, S.; Li, J.; Wang, Y.; Zhu, L.; Huang, Y.; Jiang, X.; Hu, X.; Zhou, Q.; Zhang, Z.; et al. The histone demethylase KDM4D promotes hepatic fibrogenesis by modulating Toll-like receptor 4 signaling pathway. EBioMedicine 2019, 39, 472–483. [Google Scholar] [CrossRef]
- Chen, Z.; Zang, J.; Whetstine, J.; Hong, X.; Davrazou, F.; Kutateladze, T.G.; Simpson, M.; Mao, Q.; Pan, C.H.; Dai, S.; et al. Structural insights into histone demethylation by JMJD2 family members. Cell 2006, 125, 691–702. [Google Scholar] [CrossRef]
- Li, M.; Deng, Y.; Zhuo, M.; Zhou, H.; Kong, X.; Xia, X.; Su, Z.; Chen, Q.; Guo, P.; Mo, P.; et al. Demethylase-independent function of JMJD2D as a novel antagonist of p53 to promote Liver Cancer initiation and progression. Theranostics 2020, 10, 8863–8879. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, M.; Zhuo, M.; Guo, P.; Chen, Q.; Mo, P.; Li, W.; Yu, C. Histone demethylase JMJD2D promotes the self-renewal of liver cancer stem-like cells by enhancing EpCAM and Sox9 expression. J. Biol. Chem. 2021, 296, 100121. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Wu, J.; Fan, Z.; Chen, B.; Wu, T.; Xu, Y. The histone demethylase Kdm4 suppresses activation of hepatic stellate cell by inducing MiR-29 transcription. Biochem. Biophys. Res. Commun. 2019, 514, 16–23. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Kim, J.H.; Jung, D.Y.; Kim, H.R.; Jung, M.H. Histone H3K9 Demethylase JMJD2B Plays a Role in LXRα-Dependent Lipogenesis. Int. J. Mol. Sci. 2020, 21, 8313. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Viscarra, J.A.; Wang, Y.; Nguyen, H.P.; Choi, Y.G.; Sul, H.S. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat. Commun. 2020, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xiang, C.; Zhong, F.; Zhang, Y.; Wang, L.; Zhao, Y.; Wang, J.; Ding, C.; Jin, L.; He, F.; et al. Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics 2021, 11, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.; Kim, Y.C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, H.; Jiang, L.; Shang, Q.; Yin, L.; Lin, J.D.; Wu, W.S.; Rui, L. Hepatic Slug epigenetically promotes liver lipogenesis, fatty liver disease, and type 2 diabetes. J. Clin. Investig. 2020, 130, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.R.; Kim, J.H.; Kim, J.H.; Jung, M.H. Protective effects of gomisin N against hepatic steatosis through AMPK activation. Biochem. Biophys. Res. Commun. 2017, 482, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Yun, Y.R.; Kim, S.H.; Kim, J.H.; Jung, M.H. Protective Effect of Gomisin N against Endoplasmic Reticulum Stress-Induced Hepatic Steatosis. Biol. Pharm. Bull. 2016, 39, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Cai, C.; Yao, J.; Zhou, Y.; Yu, H.; Shen, W. Histone modifications in FASN modulated by sterol regulatory element-binding protein 1c and carbohydrate responsive-element binding protein under insulin stimulation are related to NAFLD. Biochem. Biophys. Res. Commun. 2017, 483, 409–417. [Google Scholar] [CrossRef]
- Zhou, B.; Jia, L.; Zhang, Z.; Xiang, L.; Yuan, Y.; Zheng, P.; Liu, B.; Ren, X.; Bian, H.; Xie, L.; et al. The Nuclear Orphan Receptor NR2F6 Promotes Hepatic Steatosis through Upregulation of Fatty Acid Transporter CD36. Adv. Sci. 2020, 7, 2002273. [Google Scholar] [CrossRef]
- Becares, N.; Gage, M.C.; Voisin, M.; Shrestha, E.; Martin-Gutierrez, L.; Liang, N.; Louie, R.; Pourcet, B.; Pello, O.M.; Luong, T.V.; et al. Impaired LXRα Phosphorylation Attenuates Progression of Fatty Liver Disease. Cell Rep. 2019, 26, 984–995.e6. [Google Scholar] [CrossRef]
- Rohrbach, T.D.; Asgharpour, A.; Maczis, M.A.; Montefusco, D.; Cowart, L.A.; Bedossa, P.; Sanyal, A.J.; Spiegel, S. FTY720/fingolimod decreases hepatic steatosis and expression of fatty acid synthase in diet-induced nonalcoholic fatty liver disease in mice. J. Lipid Res. 2019, 60, 1311–1322. [Google Scholar] [CrossRef]
- Chung, M.Y.; Song, J.H.; Lee, J.; Shin, E.J.; Park, J.H.; Lee, S.H.; Hwang, J.T.; Choi, H.K. Tannic acid, a novel histone acetyltransferase inhibitor, prevents non-alcoholic fatty liver disease both in vivo and in vitro model. Mol. Metab. 2019, 19, 34–48. [Google Scholar] [CrossRef]
- Sharma, M.; Nazareth, I.; Petersen, I. Trends in incidence, prevalence and prescribing in type 2 diabetes mellitus between 2000 and 2013 in primary care: A retrospective cohort study. BMJ Open 2016, 6, e010210. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef]
- Caton, P.W.; Nayuni, N.K.; Kieswich, J.; Khan, N.Q.; Yaqoob, M.M.; Corder, R. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5. J. Endocrinol. 2010, 205, 97–106. [Google Scholar] [CrossRef]
- Li, X.; Wu, X.Q.; Xu, T.; Li, X.F.; Yang, Y.; Li, W.X.; Huang, C.; Meng, X.M.; Li, J. Role of histone deacetylases(HDACs) in progression and reversal of liver fibrosis. Toxicol. Appl. Pharmacol. 2016, 306, 58–68. [Google Scholar] [CrossRef]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone Deacetylase HDAC8 Promotes Insulin Resistance and β-Catenin Activation in NAFLD-Associated Hepatocellular Carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef]
- Wang, L.T.; Chiou, S.S.; Chai, C.Y.; Hsi, E.; Wang, S.N.; Huang, S.K.; Hsu, S.H. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget 2017, 8, 7489–7501. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, L.; Sun, C.; Ireland, N.; Shah, Y.M.; Liu, Y.; Rui, L. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat. Commun. 2018, 9, 2751. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Li, C.; Zhu, T.; Gao, J.; Zhou, H.; Zheng, Y.; Chang, Q.; Wang, M.; Wu, J.; et al. S100A11 Promotes Liver Steatosis via FOXO1-Mediated Autophagy and Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 697–724. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Xu, Q.; Gao, H.; Peres-da-Silva, A.; Draper, D.W.; Fessler, M.B.; Purushotham, A.; Li, X. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol. Cell. Biol. 2010, 30, 4712–4721. [Google Scholar] [CrossRef] [PubMed]
- Escande, C.; Chini, C.C.; Nin, V.; Dykhouse, K.M.; Novak, C.M.; Levine, J.; van Deursen, J.; Gores, G.J.; Chen, J.; Lou, Z.; et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J. Clin. Investig. 2010, 120, 545–558. [Google Scholar] [CrossRef]
- Ka, S.O.; Bang, I.H.; Bae, E.J.; Park, B.H. Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J. 2017, 31, 3999–4010. [Google Scholar] [CrossRef]
- Kim, H.S.; Xiao, C.; Wang, R.H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic Beta-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e8. [Google Scholar] [CrossRef]
- Hou, T.; Tian, Y.; Cao, Z.; Zhang, J.; Feng, T.; Tao, W.; Sun, H.; Wen, H.; Lu, X.; Zhu, Q.; et al. Cytoplasmic SIRT6-mediated ACSL5 deacetylation impedes nonalcoholic fatty liver disease by facilitating hepatic fatty acid oxidation. Mol. Cell 2022, 82, 4099–4115.e9. [Google Scholar] [CrossRef]
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef]
- Mannaerts, I.; Nuytten, N.R.; Rogiers, V.; Vanderkerken, K.; van Grunsven, L.A.; Geerts, A. Chronic administration of valproic acid inhibits activation of mouse hepatic stellate cells in vitro and in vivo. Hepatology 2010, 51, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, L.; Jiao, F.Z.; Zhang, W.B.; Chen, Q.; Gong, Z.J. Histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates liver fibrosis by suppressing the transforming growth factor-β1 signal pathway. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Jia, Y.; Hong, J.; Sun, Q.; Gao, S.; Hu, Y.; Zhao, N.; Zhao, R. Sodium Butyrate Ameliorates High-Fat-Diet-Induced Non-alcoholic Fatty Liver Disease through Peroxisome Proliferator-Activated Receptor α-Mediated Activation of β Oxidation and Suppression of Inflammation. J. Agric. Food. Chem. 2018, 66, 7633–7642. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Zhou, R.; Yi, L.; Ye, X.; Zeng, X.; Liu, K.; Qin, Y.; Zhang, Q.; Mi, M. Resveratrol Ameliorates Lipid Droplet Accumulation in Liver Through a SIRT1/ ATF6-Dependent Mechanism. Cell. Physiol. Biochem. 2018, 51, 2397–2420. [Google Scholar] [CrossRef] [PubMed]
- Faghihzadeh, F.; Adibi, P.; Rafiei, R.; Hekmatdoost, A. Resveratrol supplementation improves inflammatory biomarkers in patients with nonalcoholic fatty liver disease. Nutr. Res. 2014, 34, 837–843. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, X.; Ran, L.; Wan, J.; Wang, X.; Qin, Y.; Shu, F.; Gao, Y.; Yuan, L.; Zhang, Q.; et al. Resveratrol improves insulin resistance, glucose and lipid metabolism in patients with non-alcoholic fatty liver disease: A randomized controlled trial. Dig. Liver Dis. 2015, 47, 226–232. [Google Scholar] [CrossRef]
- Chachay, V.S.; Macdonald, G.A.; Martin, J.H.; Whitehead, J.P.; O’Moore-Sullivan, T.M.; Lee, P.; Franklin, M.; Klein, K.; Taylor, P.J.; Ferguson, M.; et al. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2092–2103.e6. [Google Scholar] [CrossRef]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Usui, I.; Kanatani, Y.; Matsuya, Y.; Tsuneyama, K.; Fujisaka, S.; Bukhari, A.; Suzuki, H.; Senda, S.; Imanishi, S.; et al. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1179–E1186. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Varga, Z.V.; Gariani, K.; Ryu, D.; Cao, Z.; Holovac, E.; Park, O.; Zhou, Z.; et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J. Hepatol. 2017, 66, 589–600. [Google Scholar] [CrossRef]
- Baser, K.H. Biological and pharmacological activities of carvacrol and carvacrol bearing essential oils. Curr. Pharm. Des. 2008, 14, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Ezhumalai, M.; Radhiga, T.; Pugalendi, K.V. Antihyperglycemic effect of carvacrol in combination with rosiglitazone in high-fat diet-induced type 2 diabetic C57BL/6J mice. Mol. Cell. Biochem. 2014, 385, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Nagasawa, K.; Münch, C.; Xu, Y.; Satterstrom, K.; Jeong, S.; Hayes, S.D.; Jedrychowski, M.P.; Vyas, F.S.; Zaganjor, E.; et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016, 167, 985–1000.e21. [Google Scholar] [CrossRef]
- Sroka, Z.; Cisowski, W. Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food. Chem. Toxicol. 2003, 41, 753–758. [Google Scholar] [CrossRef]
- Min, S.W.; Ryu, S.N.; Kim, D.H. Anti-inflammatory effects of black rice, cyanidin-3-O-beta-D-glycoside, and its metabolites, cyanidin and protocatechuic acid. Int. Immunopharmacol. 2010, 10, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Kang, X.; Zhao, Y.; Wang, Z.; Wang, R.; Fu, R.; Li, Y.; Hu, Y.; Wang, Z.; Shan, W.; et al. Sirtuin 3-mediated deacetylation of acyl-CoA synthetase family member 3 by protocatechuic acid attenuates non-alcoholic fatty liver disease. Br. J. Pharmacol. 2020, 177, 4166–4180. [Google Scholar] [CrossRef]
- Chen, S.; Dai, X.; Li, H.; Gong, Y.; Zhao, Y.; Huang, H. Overexpression of ring finger protein 20 inhibits the progression of liver fibrosis via mediation of histone H2B lysine 120 ubiquitination. Hum. Cell 2021, 34, 759–770. [Google Scholar] [CrossRef]
- Cai, C.; Yu, H.; Huang, G.; Du, X.; Yu, X.; Zhou, Y.; Shen, W. Histone modifications in fatty acid synthase modulated by carbohydrate responsive element binding protein are associated with non-alcoholic fatty liver disease. Int. J. Mol. Med. 2018, 42, 1215–1228. [Google Scholar] [CrossRef]
- Zarei, M.; Pizarro-Delgado, J.; Barroso, E.; Palomer, X.; Vázquez-Carrera, M. Targeting FGF21 for the Treatment of Nonalcoholic Steatohepatitis. Trends Pharmacol. Sci. 2020, 41, 199–208. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).