
Hepatic NLRP3-Derived Hsp70 Binding to TLR4 Mediates MASLD to MASH Progression upon Inhibition of PP2A by Harmful Algal Bloom Toxin Microcystin, a Second Hit

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. NLRP3 Knockout (KO) Alleviated the Pathophysiology of MASLD to MASH Progression upon PP2A Inhibition
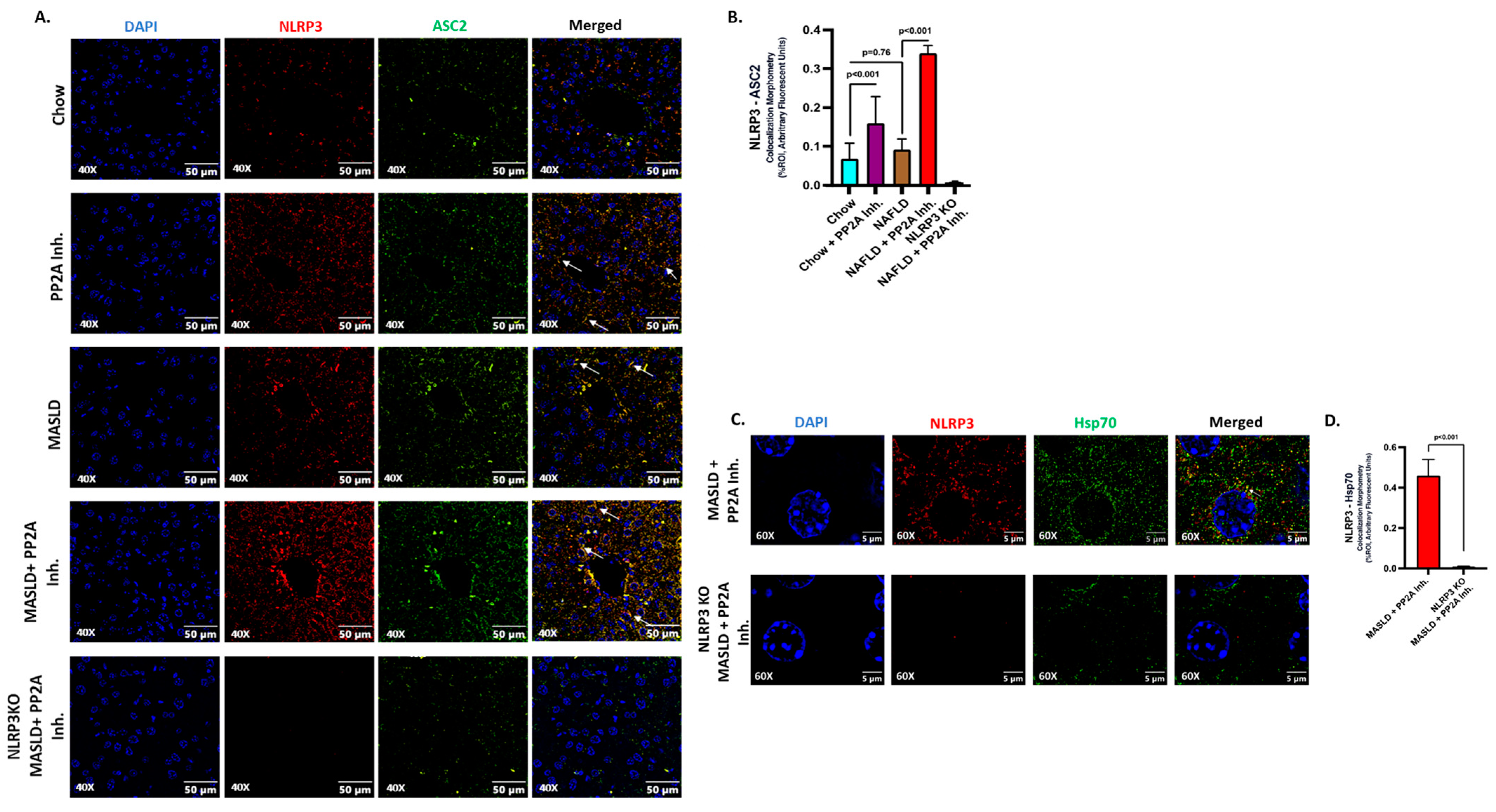
2.2. NLRP3 Drove the Inflammatory Events and Interacted with Hsp70 in Aggravating the Hepatic Injury
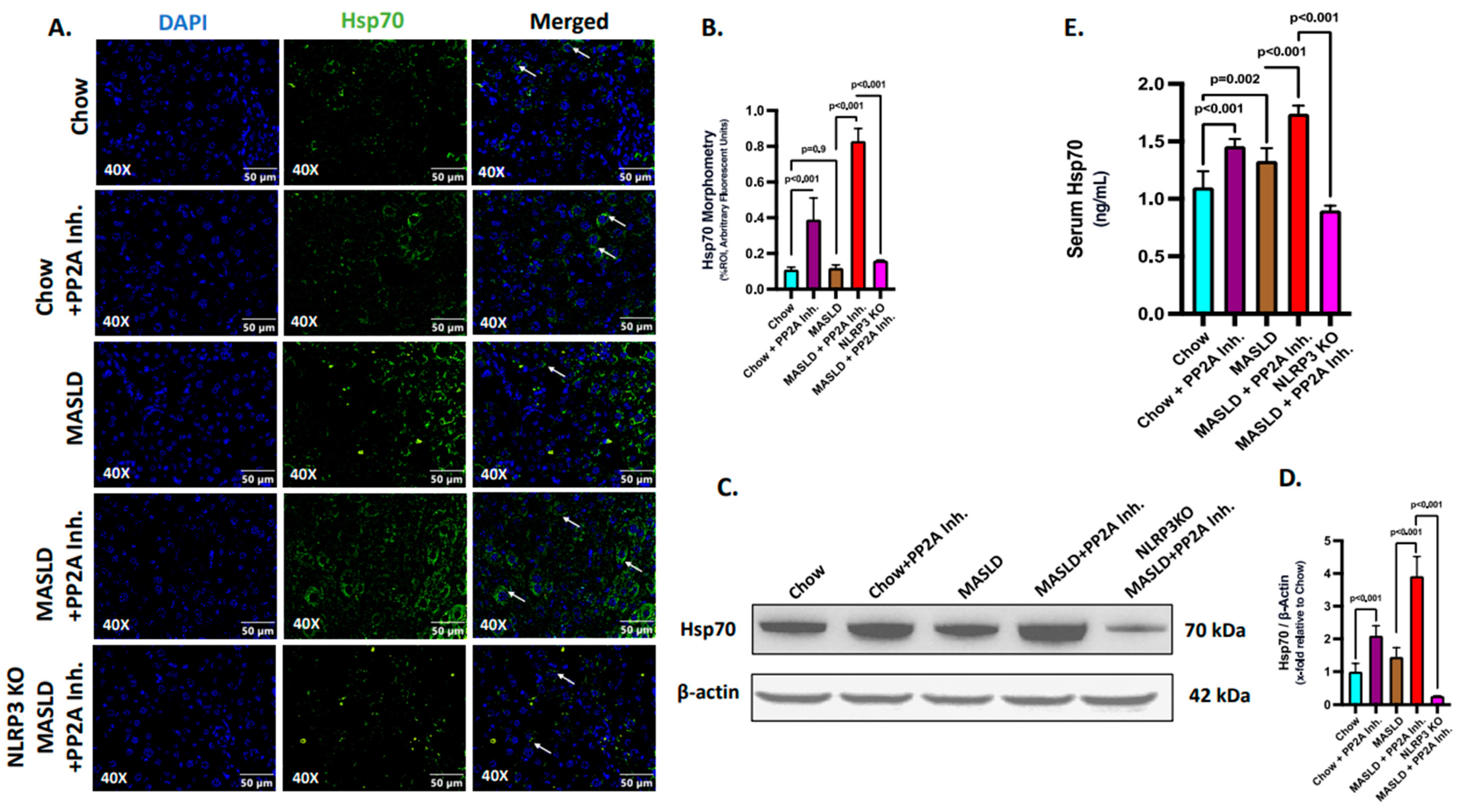
2.3. NLRP3 Was Key to the Production and Release of Hsp70 in the Liver
2.4. Hsp70 Served as a Ligand of TLR4 to Initiate the Inflammatory Cascade in Hepatocytes
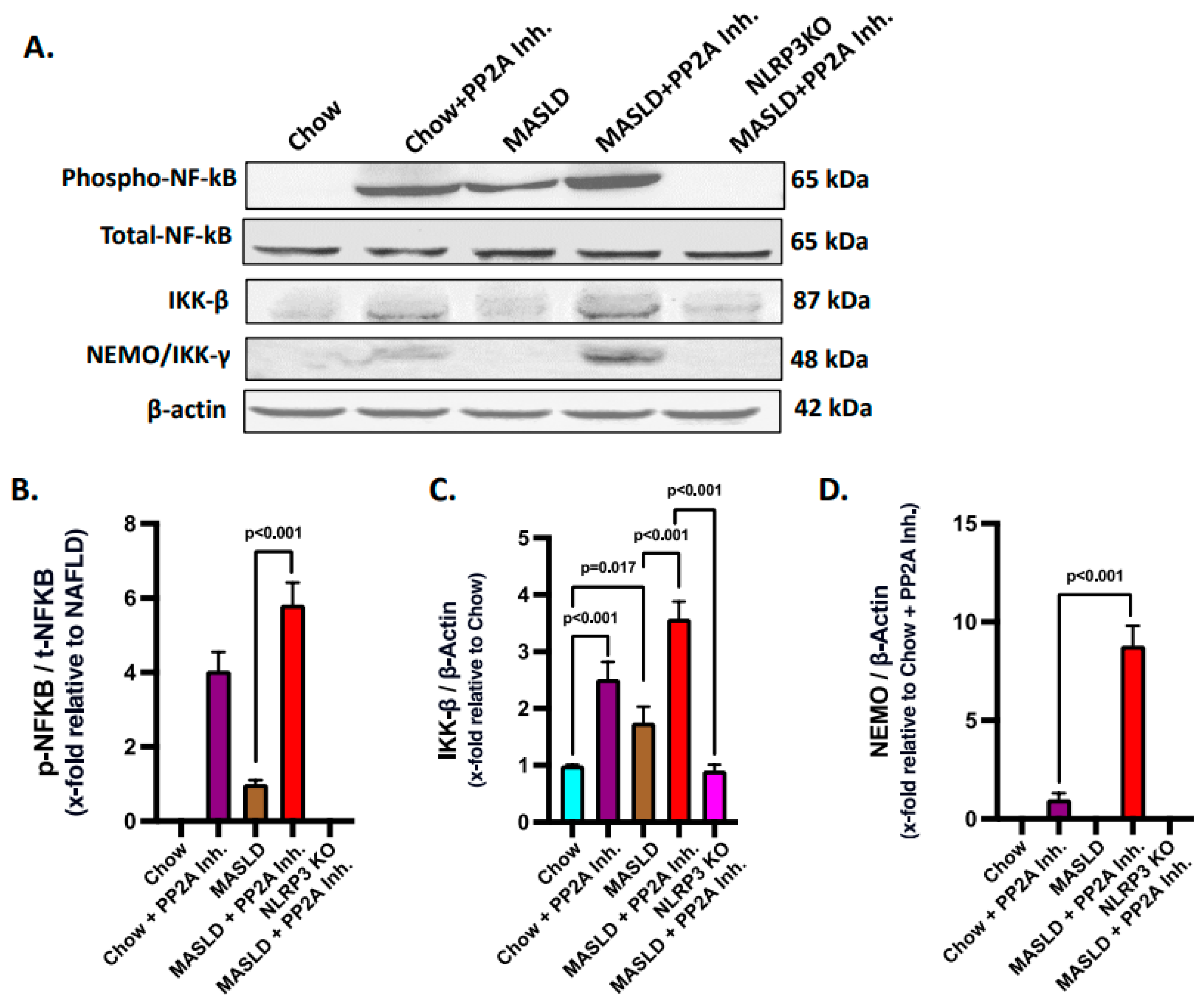
2.5. Hsp70–TLR4 Ligand Binding Regulated the NF-κB Phosphorylation to Manifest Hepatic Inflammation
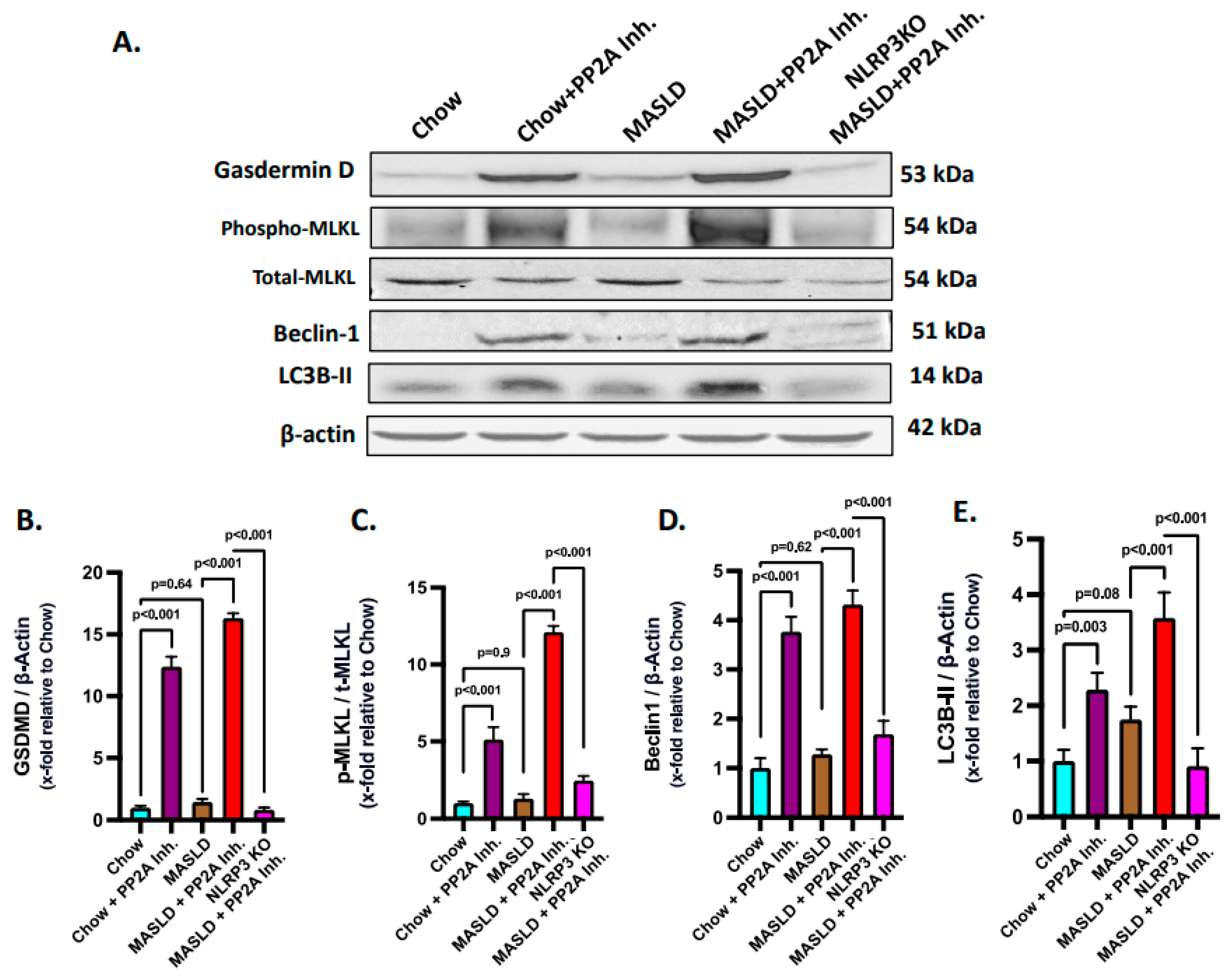
2.6. NLRP3–Hsp70–TLR4 Axis Triggered Cell Death and Survival Pathways in the Liver Following PP2A Inhibition by MC-LR
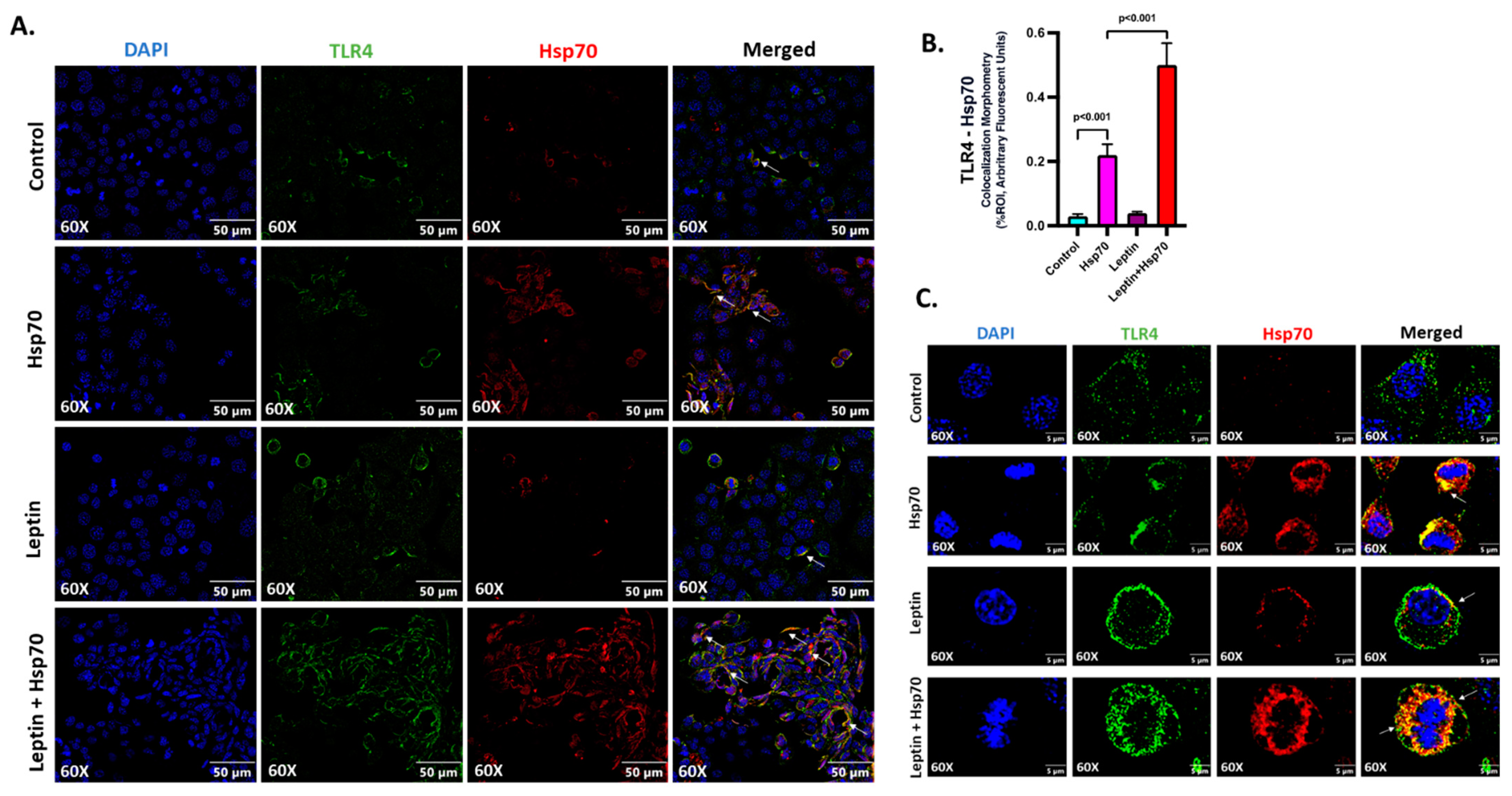
2.7. Hsp70–TLR4 Ligand Binding Augmented in Hepatocytes When Pre-Treated with Leptin
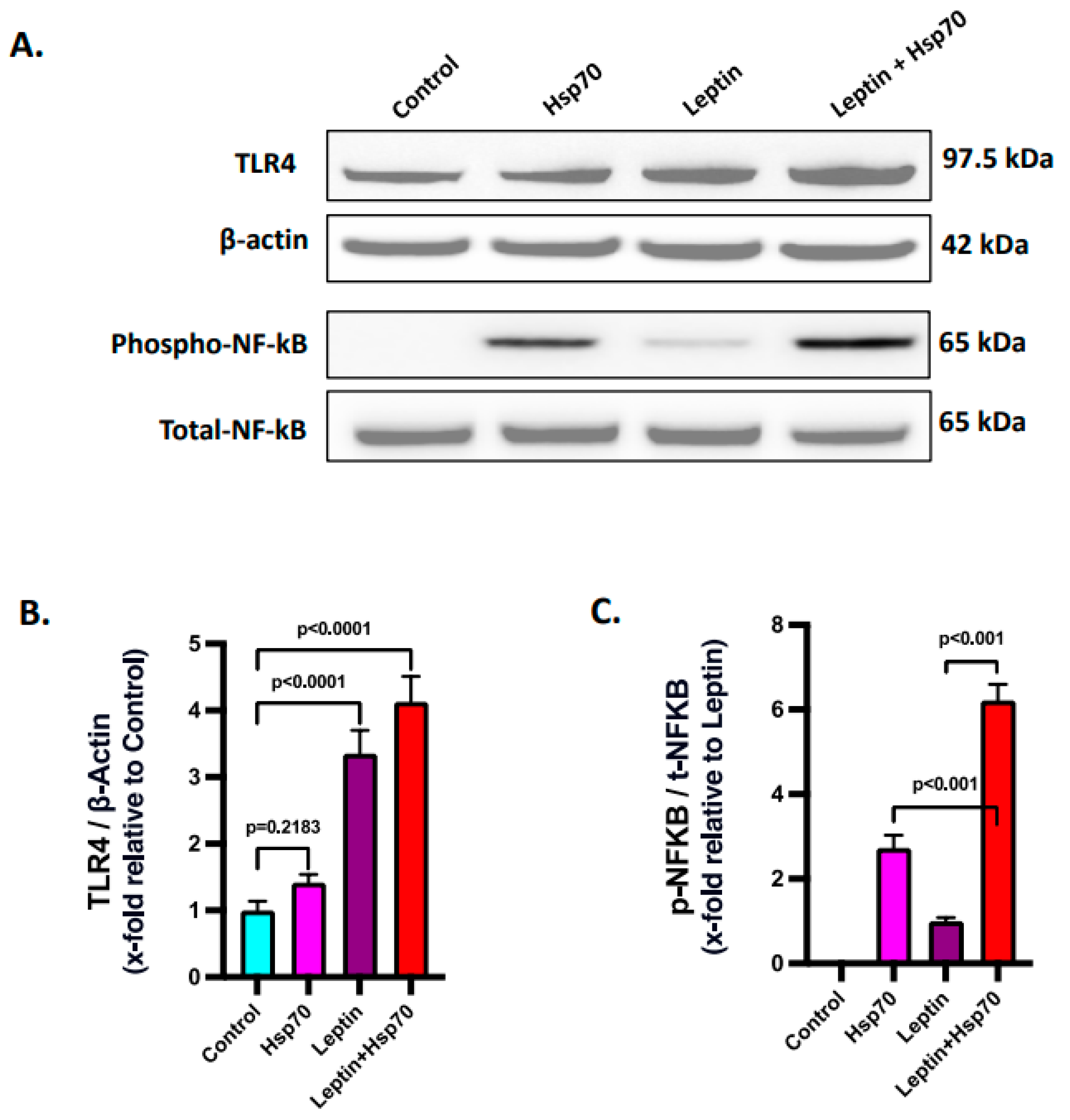
2.8. Leptin Elevated the Expression of TLR4 to Increase the Hsp70–TLR4 Ligand Binding and Subsequent NF-κB Phosphorylation in Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Experimental Models Used
- i.
- Chow: WT mice fed a chow diet only.
- ii.
- Chow + PP2A Inh.: WT mice fed a chow diet and exposed to PP2A inhibitor.
- iii.
- MASLD: WT mice fed an HFD (60% kCal) obtained from Research Diets (New Brunswick, NJ, USA) to induce MASLD.
- iv.
- MASLD + PP2A Inh.: WT mice fed a high-fat diet and exposed to PP2A inhibitor.
- v.
- NLRP3 KO MASLD + PP2A Inh.: NLRP3 knockout mice exposed to PP2A inhibitor and fed an HFD.
4.3. Cell Culture
4.4. Immunohistochemistry
4.5. Immunofluorescence
4.6. Quantitative Real-Time Polymerase Chain Reaction
4.7. Western Blot
4.8. ELISA
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Wong, V.W.; Ekstedt, M.; Wong, G.L.; Hagstrom, H. Changing epidemiology, global trends and implications for outcomes of NAFLD. J. Hepatol. 2023, 79, 842–852. [Google Scholar] [CrossRef]
- Arciello, M.; Gori, M.; Maggio, R.; Barbaro, B.; Tarocchi, M.; Galli, A.; Balsano, C. Environmental pollution: A tangible risk for NAFLD pathogenesis. Int. J. Mol. Sci. 2013, 14, 22052–22066. [Google Scholar] [CrossRef]
- Albadrani, M.; Seth, R.K.; Sarkar, S.; Kimono, D.; Mondal, A.; Bose, D.; Porter, D.E.; Scott, G.I.; Brooks, B.; Raychoudhury, S.; et al. Exogenous PP2A inhibitor exacerbates the progression of nonalcoholic fatty liver disease via NOX2-dependent activation of miR21. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G408–G428. [Google Scholar] [CrossRef]
- Seth, R.K.; Kumar, A.; Das, S.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci. 2013, 134, 291–303. [Google Scholar] [CrossRef]
- Al-Badrani, M.; Saha, P.; Mondal, A.; Seth, R.K.; Sarkar, S.; Kimono, D.; Bose, D.; Porter, D.E.; Scott, G.I.; Brooks, B.; et al. Early microcystin-LR exposure-linked inflammasome activation in mice causes development of fatty liver disease and insulin resistance. Environ. Toxicol. Pharmacol. 2020, 80, 103457. [Google Scholar] [CrossRef]
- Nagata, K.; Suzuki, H.; Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007, 32, 453–468. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Williams, C.D.; Ni, H.; Kumer, S.C.; Schmitt, T.; Kane, B.; Jaeschke, H. Microcystin-LR induced liver injury in mice and in primary human hepatocytes is caused by oncotic necrosis. Toxicon 2017, 125, 99–109. [Google Scholar] [CrossRef]
- Amado, L.L.; Monserrat, J.M. Oxidative stress generation by microcystins in aquatic animals: Why and how. Environ. Int. 2010, 36, 226–235. [Google Scholar] [CrossRef]
- Dawson, R.M. The toxicology of microcystins. Toxicon 1998, 36, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, C.; Fu, X.; Zhang, S.; Xin, Y.; Li, Y.; Xue, L.; Cheng, X.; Zhang, H. Microcystin-LR Induced Apoptosis in Rat Sertoli Cells via the Mitochondrial Caspase-Dependent Pathway: Role of Reactive Oxygen Species. Front. Physiol. 2016, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Ogura, Y.; Sutterwala, F.S.; Flavell, R.A. The inflammasome: First line of the immune response to cell stress. Cell 2006, 126, 659–662. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Zheng, C.; Zeng, H.; Lin, H.; Wang, J.; Feng, X.; Qiu, Z.; Chen, J.A.; Luo, J.; Luo, Y.; Huang, Y.; et al. Serum microcystin levels positively linked with risk of hepatocellular carcinoma: A case-control study in southwest China. Hepatology 2017, 66, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yan, Y.; Xie, L.; Wang, L.; He, Y.; Wan, X.; Xue, Q. Long-term environmental exposure to microcystins increases the risk of nonalcoholic fatty liver disease in humans: A combined fisher-based investigation and murine model study. Environ. Int. 2020, 138, 105648. [Google Scholar] [CrossRef]
- Sauve, S.; Desrosiers, M. A review of what is an emerging contaminant. Chem. Cent. J. 2014, 8, 15. [Google Scholar] [CrossRef]
- MacKintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef]
- Honkanen, R.E.; Zwiller, J.; Moore, R.E.; Daily, S.L.; Khatra, B.S.; Dukelow, M.; Boynton, A.L. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatases. J. Biol. Chem. 1990, 265, 19401–19404. [Google Scholar] [CrossRef]
- Xu, Y.; Xing, Y.; Chen, Y.; Chao, Y.; Lin, Z.; Fan, E.; Yu, J.W.; Strack, S.; Jeffrey, P.D.; Shi, Y. Structure of the protein phosphatase 2A holoenzyme. Cell 2006, 127, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Reynhout, S.; Janssens, V. Physiologic functions of PP2A: Lessons from genetically modified mice. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 31–50. [Google Scholar] [CrossRef]
- Xian, L.; Hou, S.; Huang, Z.; Tang, A.; Shi, P.; Wang, Q.; Song, A.; Jiang, S.; Lin, Z.; Guo, S.; et al. Liver-specific deletion of Ppp2calpha enhances glucose metabolism and insulin sensitivity. Aging 2015, 7, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, P.; Wu, X.; Yuan, T.; Su, Z.; Chen, S.; Zhou, Y.; Tao, W.A. Microcystin-LR Induces NLRP3 Inflammasome Activation via FOXO1 Phosphorylation, Resulting in Interleukin-1beta Secretion and Pyroptosis in Hepatocytes. Toxicol. Sci. 2021, 179, 53–69. [Google Scholar] [CrossRef]
- Maitiabula, G.; Tian, F.; Wang, P.; Zhang, L.; Gao, X.; Wan, S.; Sun, H.; Yang, J.; Zhang, Y.; Gao, T.; et al. Liver PP2A-Calpha Protects From Parenteral Nutrition-associated Hepatic Steatosis. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 669–692. [Google Scholar] [CrossRef]
- Wu, Y.; Song, P.; Xu, J.; Zhang, M.; Zou, M.H. Withdrawal: Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J. Biol. Chem. 2019, 294, 10741. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Sozio, M.S.; Shin, E.; Zhao, Z.; Xu, Y.; Ross, R.A.; Zeng, Y.; Crabb, D.W. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G1004–G1012. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Cai, C.Z.; Yu, M.L.; Feng, Z.M.; Zhang, Y.W.; Liu, P.H.; Zeng, H.; Yu, C.H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Sarkar, S.; Kimono, D.; Albadrani, M.; Seth, R.K.; Busbee, P.; Alghetaa, H.; Porter, D.E.; Scott, G.I.; Brooks, B.; Nagarkatti, M.; et al. Environmental microcystin targets the microbiome and increases the risk of intestinal inflammatory pathology via NOX2 in underlying murine model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 8742. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Spierings, J.; van Eden, W. Heat shock proteins and their immunomodulatory role in inflammatory arthritis. Rheumatology 2017, 56, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Moseley, P.L. Heat shock proteins and the inflammatory response. Ann. N. Y. Acad. Sci. 1998, 856, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Martine, P.; Rebe, C. Heat Shock Proteins and Inflammasomes. Int. J. Mol. Sci. 2019, 20, 4508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, N.; Peng, Y. Heat shock protein 70 promotes lipogenesis in HepG2 cells. Lipids Health Dis. 2018, 17, 73. [Google Scholar] [CrossRef]
- Rehati, A.; Abuduaini, B.; Liang, Z.; Chen, D.; He, F. Identification of heat shock protein family A member 5 (HSPA5) targets involved in nonalcoholic fatty liver disease. Genes Immun. 2023, 24, 124–129. [Google Scholar] [CrossRef]
- Sun, Y.; Meng, G.M.; Guo, Z.L.; Xu, L.H. Regulation of heat shock protein 27 phosphorylation during microcystin-LR-induced cytoskeletal reorganization in a human liver cell line. Toxicol. Lett. 2011, 207, 270–277. [Google Scholar] [CrossRef]
- Udono, H.; Srivastava, P.K. Heat shock protein 70-associated peptides elicit specific cancer immunity. J. Exp. Med. 1993, 178, 1391–1396. [Google Scholar] [CrossRef]
- Udono, H.; Srivastava, P.K. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J. Immunol. 1994, 152, 5398–5403. [Google Scholar] [CrossRef]
- Cosemans, G.; Merckx, C.; De Bleecker, J.L.; De Paepe, B. Inducible Heat Shock Protein 70 Levels in Patients and the mdx Mouse Affirm Regulation during Skeletal Muscle Regeneration in Muscular Dystrophy. Front. Biosci. Schol. Ed. 2022, 14, 19. [Google Scholar] [CrossRef]
- Boudesco, C.; Cause, S.; Jego, G.; Garrido, C. Hsp70: A Cancer Target Inside and Outside the Cell. Methods Mol. Biol. 2018, 1709, 371–396. [Google Scholar] [CrossRef]
- Martine, P.; Chevriaux, A.; Derangere, V.; Apetoh, L.; Garrido, C.; Ghiringhelli, F.; Rebe, C. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Hulina, A.; Grdic Rajkovic, M.; Jaksic Despot, D.; Jelic, D.; Dojder, A.; Cepelak, I.; Rumora, L. Extracellular Hsp70 induces inflammation and modulates LPS/LTA-stimulated inflammatory response in THP-1 cells. Cell Stress Chaperones 2018, 23, 373–384. [Google Scholar] [CrossRef]
- Ren, B.; Zou, G.; Huang, Y.; Xu, G.; Xu, F.; He, J.; Zhu, H.; Yu, P. Serum levels of HSP70 and other DAMP proteins can aid in patient diagnosis after traumatic injury. Cell Stress Chaperones 2016, 21, 677–686. [Google Scholar] [CrossRef]
- Fang, H.; Wu, Y.; Huang, X.; Wang, W.; Ang, B.; Cao, X.; Wan, T. Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J. Biol. Chem. 2011, 286, 30393–30400. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Yarla, N.; Haykal, T.; Nguyen, Y.V.; Cao, L.; Ferreira, M.; Holtzhausen, A.; Al-Rohil, R.; Salama, A.K.S.; Beasley, G.M.; et al. Tumor-intrinsic NLRP3-HSP70-TLR4 axis drives premetastatic niche development and hyperprogression during anti-PD-1 immunotherapy. Sci. Transl. Med. 2022, 14, eabq7019. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-κappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Engelmann, C.; Habtesion, A.; Hassan, M.; Kerbert, A.J.; Hammerich, L.; Novelli, S.; Fidaleo, M.; Philips, A.; Davies, N.; Ferreira-Gonzalez, S.; et al. Combination of G-CSF and a TLR4 inhibitor reduce inflammation and promote regeneration in a mouse model of ACLF. J. Hepatol. 2022, 77, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, C.; Wang, X.; Kim, K.; Bartolome, A.; Dongiovanni, P.; Yates, K.P.; Valenti, L.; Carrer, M.; Sadowski, T.; et al. Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci. Transl. Med. 2021, 13, eabe1692. [Google Scholar] [CrossRef] [PubMed]
- Di Naso, F.C.; Porto, R.R.; Fillmann, H.S.; Maggioni, L.; Padoin, A.V.; Ramos, R.J.; Mottin, C.C.; Bittencourt, A.; Marroni, N.A.; de Bittencourt, P.I., Jr. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity 2015, 23, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Shi, Y.; Shan, Z.; Yang, L.; Wang, X.; Shi, L. Bioaccumulation, oxidative stress and HSP70 expression in Cyprinus carpio L. exposed to microcystin-LR under laboratory conditions. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2012, 155, 483–490. [Google Scholar] [CrossRef]
- Ji, Y.; Lu, G.; Chen, G.; Huang, B.; Zhang, X.; Shen, K.; Wu, S. Microcystin-LR induces apoptosis via NF-κappaB/iNOS pathway in INS-1 cells. Int. J. Mol. Sci. 2011, 12, 4722–4734. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Lad, A.; Hunyadi, J.; Connolly, J.; Breidenbach, J.D.; Khalaf, F.K.; Dube, P.; Zhang, S.; Kleinhenz, A.L.; Baliu-Rodriguez, D.; Isailovic, D.; et al. Antioxidant Therapy Significantly Attenuates Hepatotoxicity following Low Dose Exposure to Microcystin-LR in a Murine Model of Diet-Induced Non-Alcoholic Fatty Liver Disease. Antioxidants 2022, 11, 1625. [Google Scholar] [CrossRef]
- Lad, A.; Su, R.C.; Breidenbach, J.D.; Stemmer, P.M.; Carruthers, N.J.; Sanchez, N.K.; Khalaf, F.K.; Zhang, S.; Kleinhenz, A.L.; Dube, P.; et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins 2019, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xue, Q.; Su, X.; Xie, L.; Yan, Y.; Wang, L.; Steinman, A.D. First Identification of the Toxicity of Microcystins on Pancreatic Islet Function in Humans and the Involved Potential Biomarkers. Environ. Sci. Technol. 2016, 50, 3137–3144. [Google Scholar] [CrossRef] [PubMed]
- Black, A.T.; Hayden, P.J.; Casillas, R.P.; Heck, D.E.; Gerecke, D.R.; Sinko, P.J.; Laskin, D.L.; Laskin, J.D. Regulation of Hsp27 and Hsp70 expression in human and mouse skin construct models by caveolae following exposure to the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol. Appl. Pharmacol. 2011, 253, 112–120. [Google Scholar] [CrossRef]
- Kuper, C.; Beck, F.X.; Neuhofer, W. Toll-like receptor 4 activates NF-κappaB and MAP kinase pathways to regulate expression of proinflammatory COX-2 in renal medullary collecting duct cells. Am. J. Physiol. Renal. Physiol. 2012, 302, F38–F46. [Google Scholar] [CrossRef]
- Galloway, E.; Shin, T.; Huber, N.; Eismann, T.; Kuboki, S.; Schuster, R.; Blanchard, J.; Wong, H.R.; Lentsch, A.B. Activation of hepatocytes by extracellular heat shock protein 72. Am. J. Physiol. Cell Physiol. 2008, 295, C514–C520. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, X.; Zhang, S.; Huang, H.; Wu, J.; Wang, Y.; Yuan, L.; Liu, C.; Zeng, X.; Cheng, X.; et al. Oxidative Stress Mediates Microcystin-LR-Induced Endoplasmic Reticulum Stress and Autophagy in KK-1 Cells and C57BL/6 Mice Ovaries. Front. Physiol. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ganini, D.; Tokar, E.J.; Kumar, A.; Das, S.; Corbett, J.; Kadiiska, M.B.; Waalkes, M.P.; Diehl, A.M.; Mason, R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol. 2013, 58, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; He, J.; Sun, Y.; Dong, X.; Yao, J.; Gu, H.; Liu, L. Leptin Induced TLR4 Expression via the JAK2-STAT3 Pathway in Obesity-Related Osteoarthritis. Oxid. Med. Cell Longev. 2021, 2021, 7385160. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Bose, D.; Saha, P.; Sarkar, S.; Seth, R.; Kimono, D.; Albadrani, M.; Nagarkatti, M.; Nagarkatti, P.; Chatterjee, S. Lipocalin 2 induces neuroinflammation and blood-brain barrier dysfunction through liver-brain axis in murine model of nonalcoholic steatohepatitis. J. Neuroinflamm. 2020, 17, 201. [Google Scholar] [CrossRef]
- Seth, R.K.; Das, S.; Dattaroy, D.; Chandrashekaran, V.; Alhasson, F.; Michelotti, G.; Nagarkatti, M.; Nagarkatti, P.; Diehl, A.M.; Bell, P.D.; et al. TRPV4 activation of endothelial nitric oxide synthase resists nonalcoholic fatty liver disease by blocking CYP2E1-mediated redox toxicity. Free Radic. Biol. Med. 2017, 102, 260–273. [Google Scholar] [CrossRef]
- Chandrashekaran, V.; Seth, R.K.; Dattaroy, D.; Alhasson, F.; Ziolenka, J.; Carson, J.; Berger, F.G.; Kalyanaraman, B.; Diehl, A.M.; Chatterjee, S. HMGB1-RAGE pathway drives peroxynitrite signaling-induced IBD-like inflammation in murine nonalcoholic fatty liver disease. Redox. Biol. 2017, 13, 8–19. [Google Scholar] [CrossRef]
- Alhasson, F.; Seth, R.K.; Sarkar, S.; Kimono, D.A.; Albadrani, M.S.; Dattaroy, D.; Chandrashekaran, V.; Scott, G.I.; Raychoudhury, S.; Nagarkatti, M.; et al. High circulatory leptin mediated NOX-2-peroxynitrite-miR21 axis activate mesangial cells and promotes renal inflammatory pathology in nonalcoholic fatty liver disease. Redox. Biol. 2018, 17, 1–15. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef]
- Yang, S.J.; Lim, Y. Resveratrol ameliorates hepatic metaflammation and inhibits NLRP3 inflammasome activation. Metabolism 2014, 63, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Saha, P.; Seth, R.K.; Mondal, A.; Bose, D.; Kimono, D.; Albadrani, M.; Mukherjee, A.; Porter, D.E.; Scott, G.I.; et al. Higher intestinal and circulatory lactate associated NOX2 activation leads to an ectopic fibrotic pathology following microcystin co-exposure in murine fatty liver disease. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2020, 238, 108854. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.A.; Edelman, I.; Culbertson, M.R.; Friedberg, E.C. Physiological levels of normal tRNA(CAGGln) can effect partial suppression of amber mutations in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1987, 84, 8031–8034. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef]
- Johnston, A.N.; Wang, Z. HSP70 promotes MLKL polymerization and necroptosis. Mol. Cell Oncol. 2020, 7, 1791561. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE((-/-)) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, N.; Qiu, T.; Sun, X. The Role of Autophagy and NLRP3 Inflammasome in Liver Fibrosis. Biomed. Res. Int. 2020, 2020, 7269150. [Google Scholar] [CrossRef]
- Yang, Q.; Shu, F.; Gong, J.; Ding, P.; Cheng, R.; Li, J.; Tong, R.; Ding, L.; Sun, H.; Huang, W.; et al. Sweroside ameliorates NAFLD in high-fat diet induced obese mice through the regulation of lipid metabolism and inflammatory response. J. Ethnopharmacol. 2020, 255, 112556. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wei, H.; Ren, C.; Yuan, S.; Fu, H.; Lv, Y.; Zhu, Y.; Zhang, T. Gene expression of Hsps in normal and abnormal embryonic development of mouse hindlimbs. Hum. Exp. Toxicol. 2015, 34, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Bouhy, D.; Juneja, M.; Katona, I.; Holmgren, A.; Asselbergh, B.; De Winter, V.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Libert, C.; et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 2018, 135, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Park, Y.J.; Tsuruta, Y.; Abraham, E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc. Natl. Acad. Sci. USA 2009, 106, 15819–15824. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer Sequence | Reverse Primer Sequence | Source |
|---|---|---|---|
| Hsp70 | CAGCGAGGCTGACAAGAAGAA | GGAGATGACCTCCTGGCACT | [56] |
| Hsp90 | TTGGTTACTTCCCCGTGCTG | GCCTTTTGCCGTAGGGTTTC | designed by us |
| Hsp60 | CATCGGAAGCCATTGGTCATAA | CGTGCTTAGAGCTTCTCCGTCA | [85] |
| Hsp25 | ACAGTGAAGACCAAGGAAGG | CTGGAGGGAGCGTGTATTT | designed by us |
| HspH1 | GACCCTCAAGGAGTTCCATATC | CTCTCGACTTCTCTCCATCTTTC | designed by us |
| HspB8 | AGACCCCTTTCGGGACTCA | GGCTGTCAAGTCGTCTGGAA | [86] |
| GAPDH | CGACTTCAACAGCAACTCCCACTCTTCC | TGGGTGGTCCAGGGTTTCTTACTCCTT | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, S.; Saha, P.; Bose, D.; Trivedi, A.; More, M.; Xiao, S.; Diehl, A.M.; Chatterjee, S. Hepatic NLRP3-Derived Hsp70 Binding to TLR4 Mediates MASLD to MASH Progression upon Inhibition of PP2A by Harmful Algal Bloom Toxin Microcystin, a Second Hit. Int. J. Mol. Sci. 2023, 24, 16354. https://doi.org/10.3390/ijms242216354
Roy S, Saha P, Bose D, Trivedi A, More M, Xiao S, Diehl AM, Chatterjee S. Hepatic NLRP3-Derived Hsp70 Binding to TLR4 Mediates MASLD to MASH Progression upon Inhibition of PP2A by Harmful Algal Bloom Toxin Microcystin, a Second Hit. International Journal of Molecular Sciences. 2023; 24(22):16354. https://doi.org/10.3390/ijms242216354
Chicago/Turabian StyleRoy, Subhajit, Punnag Saha, Dipro Bose, Ayushi Trivedi, Madhura More, Shuo Xiao, Anna Mae Diehl, and Saurabh Chatterjee. 2023. "Hepatic NLRP3-Derived Hsp70 Binding to TLR4 Mediates MASLD to MASH Progression upon Inhibition of PP2A by Harmful Algal Bloom Toxin Microcystin, a Second Hit" International Journal of Molecular Sciences 24, no. 22: 16354. https://doi.org/10.3390/ijms242216354
APA StyleRoy, S., Saha, P., Bose, D., Trivedi, A., More, M., Xiao, S., Diehl, A. M., & Chatterjee, S. (2023). Hepatic NLRP3-Derived Hsp70 Binding to TLR4 Mediates MASLD to MASH Progression upon Inhibition of PP2A by Harmful Algal Bloom Toxin Microcystin, a Second Hit. International Journal of Molecular Sciences, 24(22), 16354. https://doi.org/10.3390/ijms242216354