

Finding miRNA–RNA Network Biomarkers for Predicting Metastasis and Prognosis in Cancer


Abstract
:1. Introduction
2. Results and Discussion
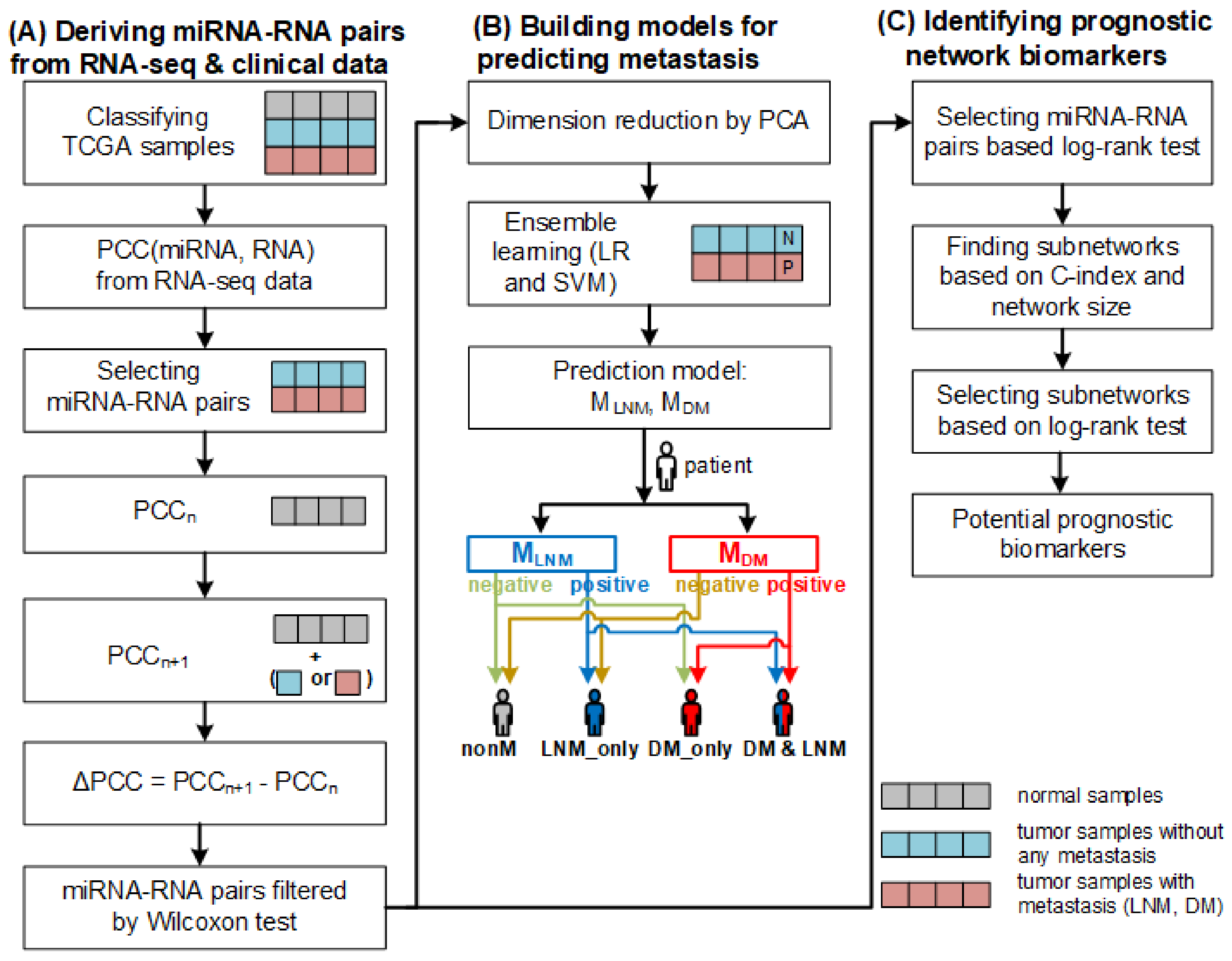
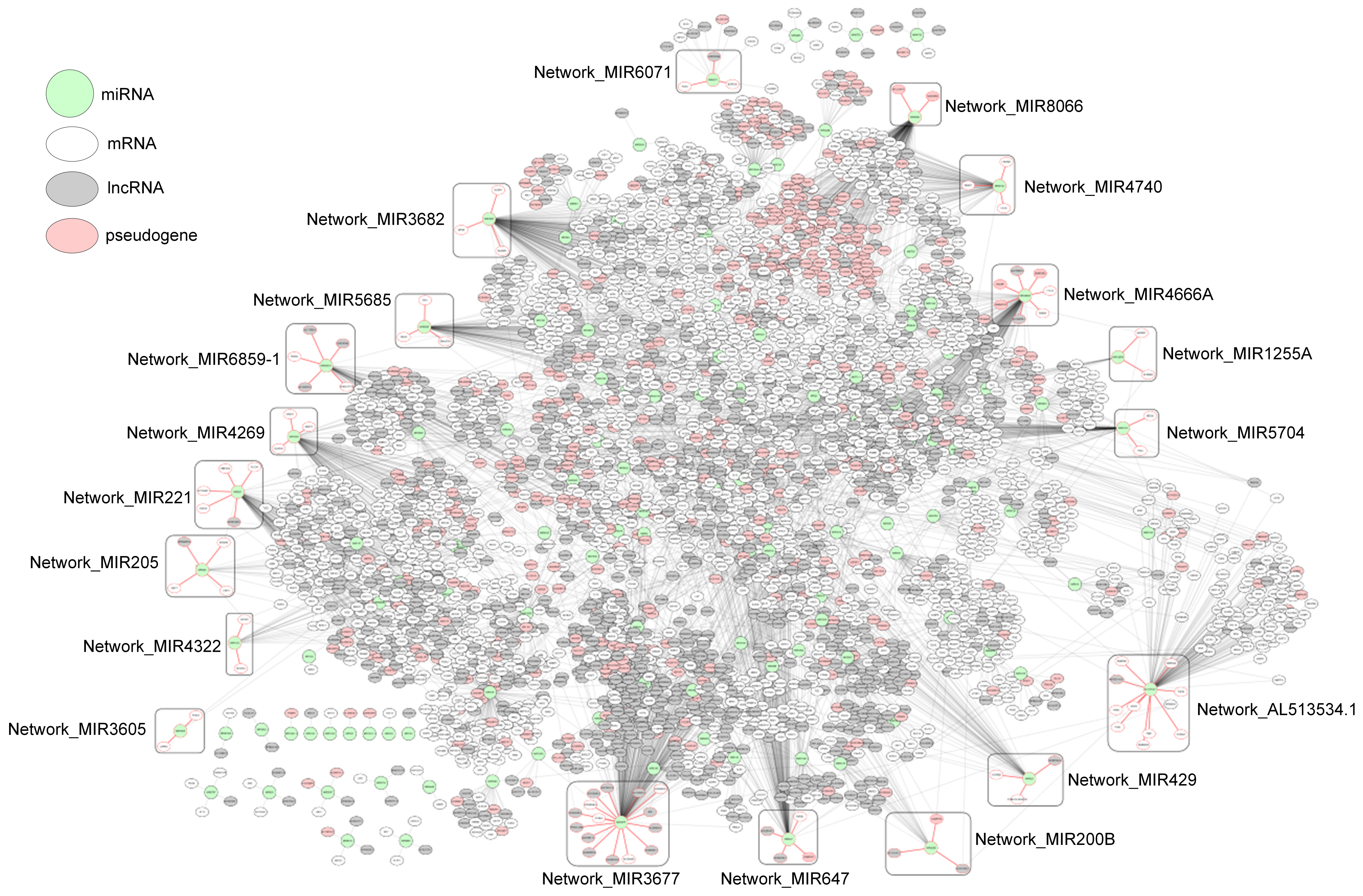
2.1. miRNA–RNA Pairs
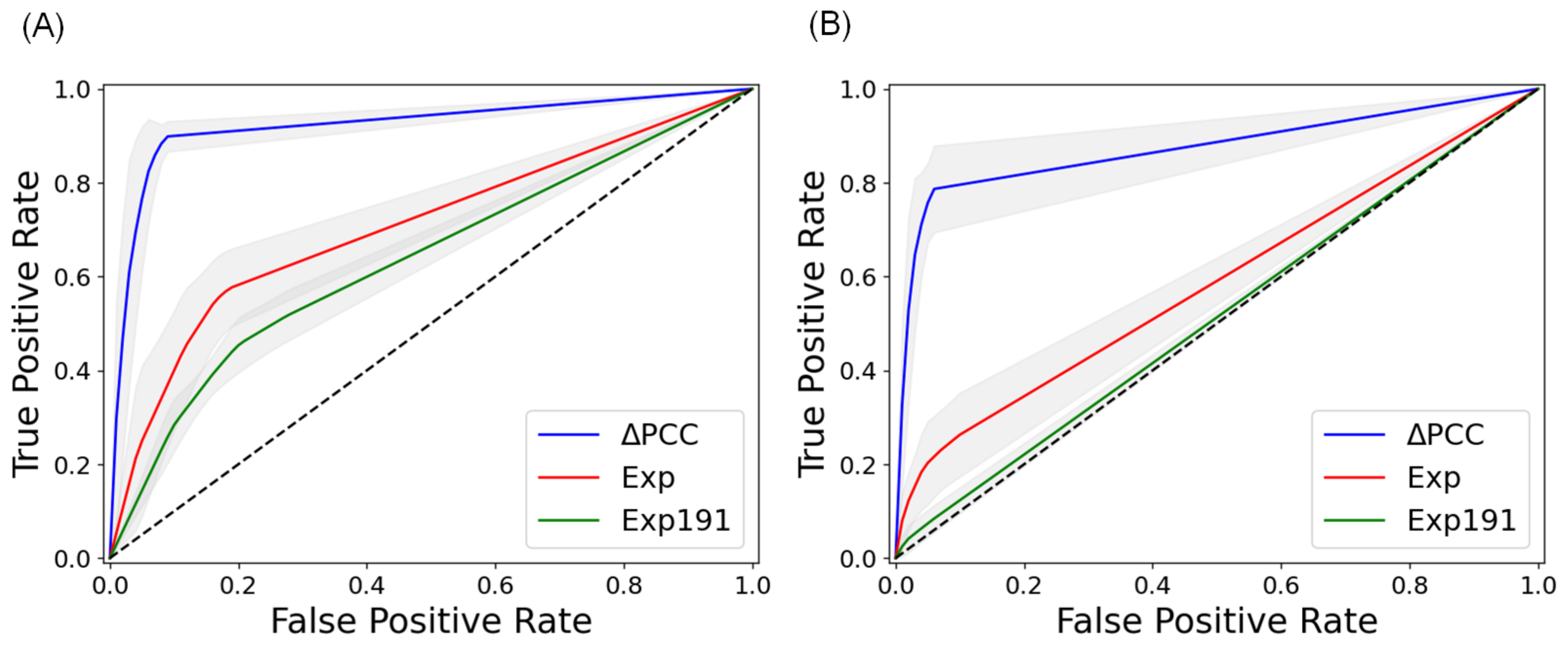
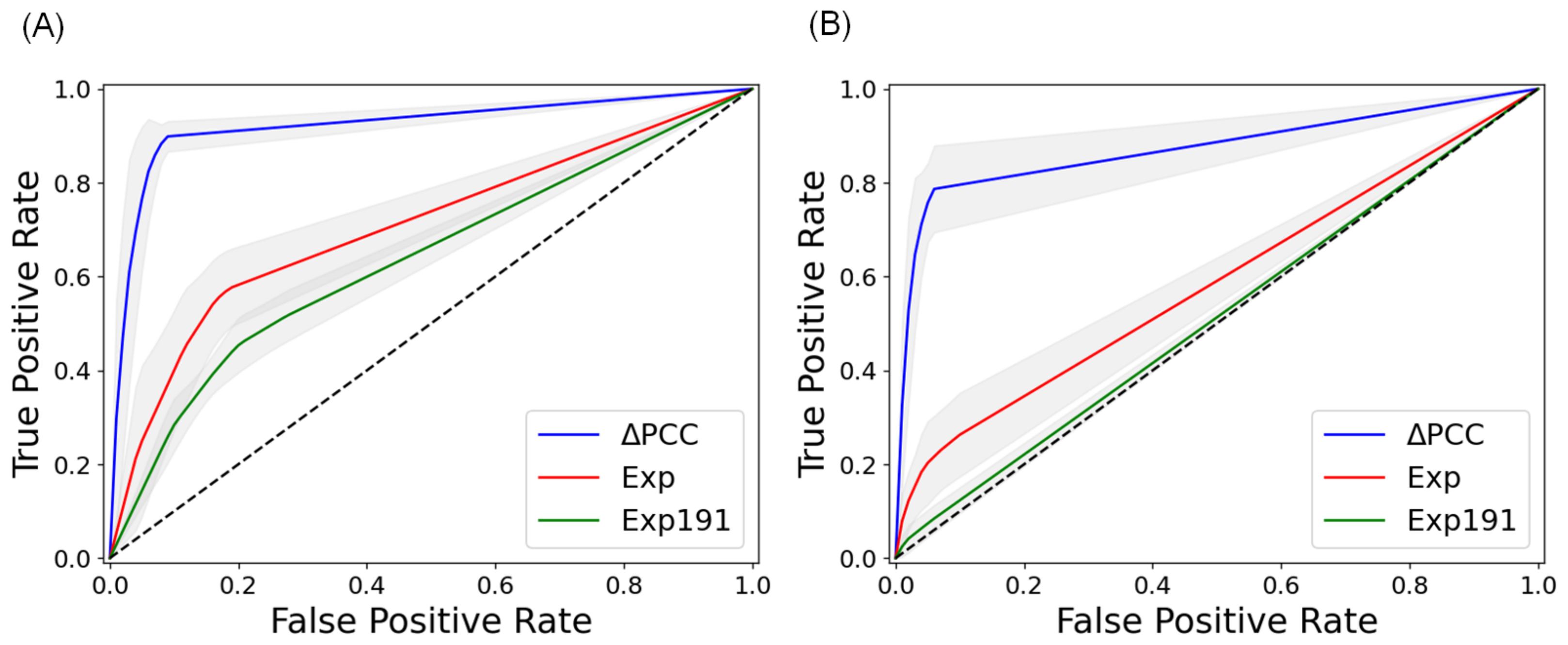
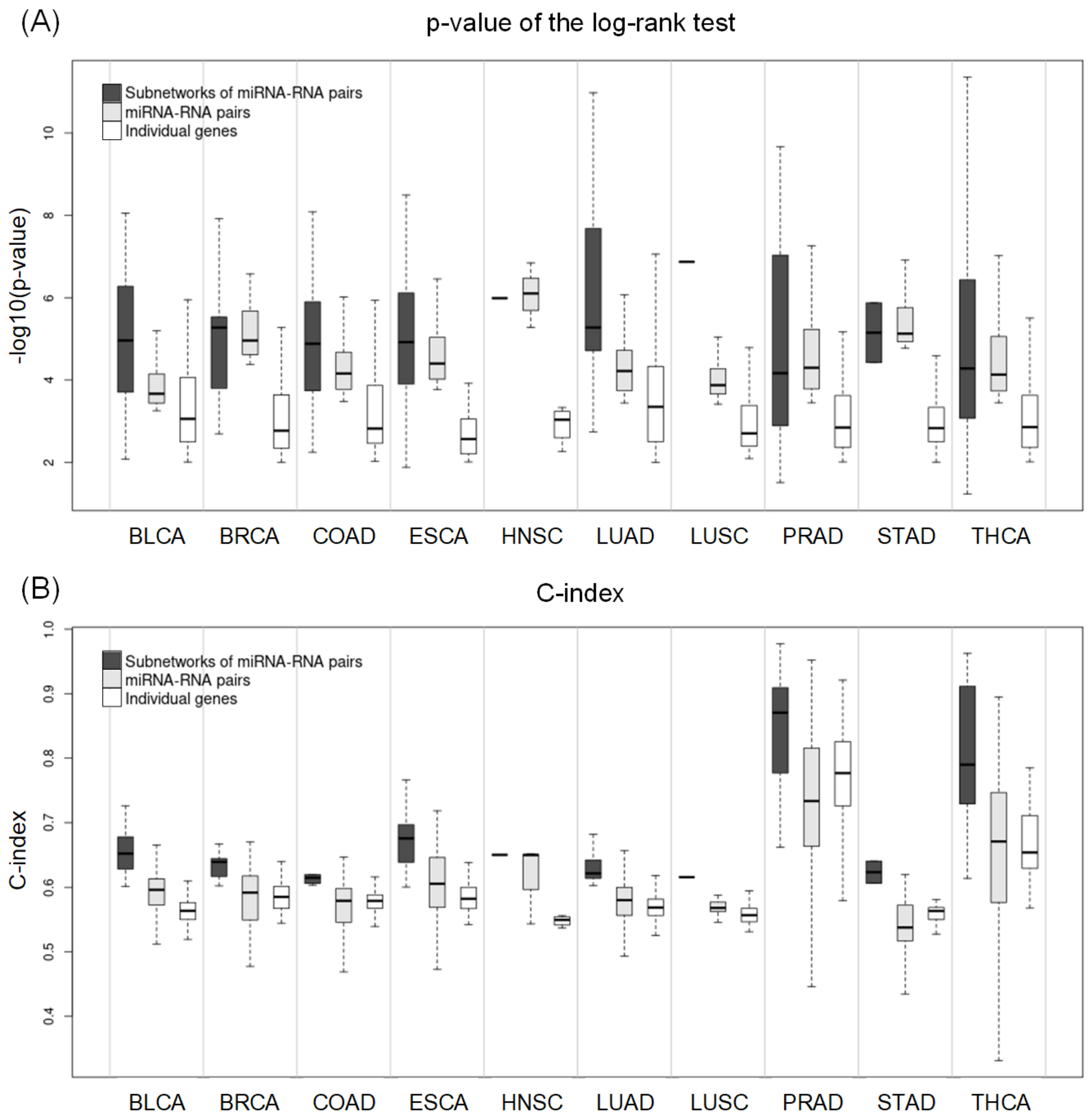
2.2. Prediction of Metastasis and Comparison with Other Methods
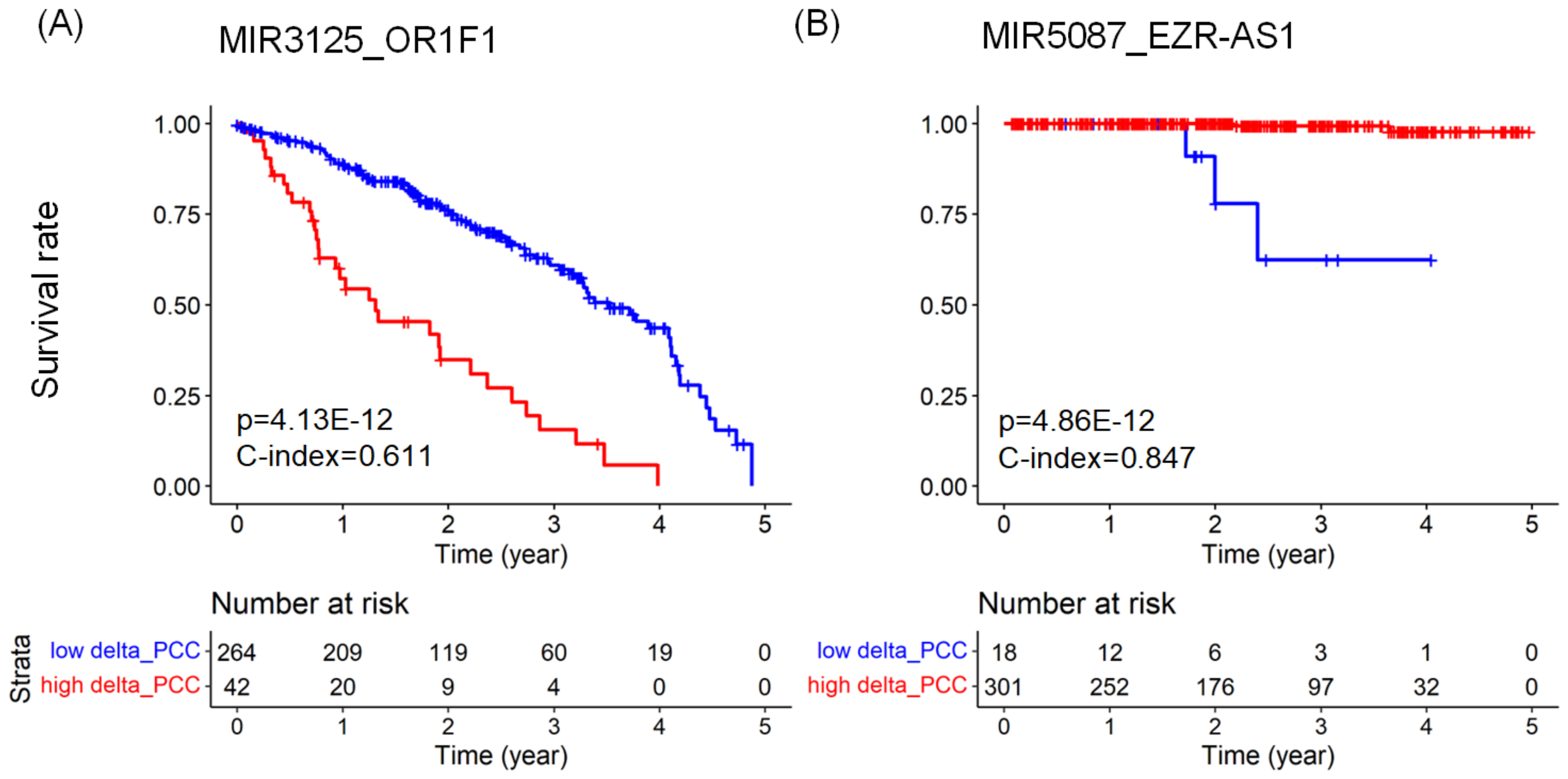
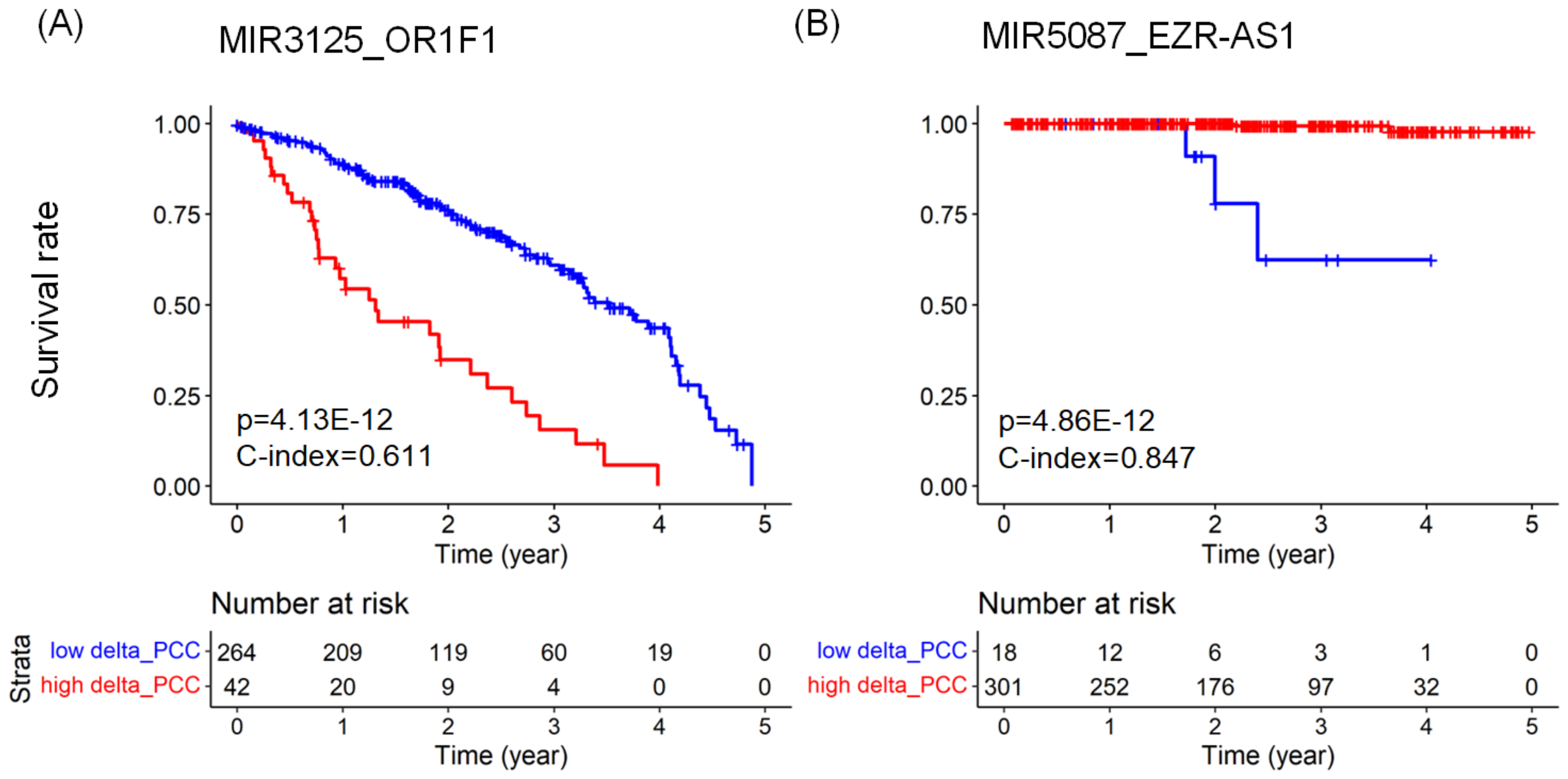
2.3. Predicting Prognosis and Potential Prognostic Biomarkers
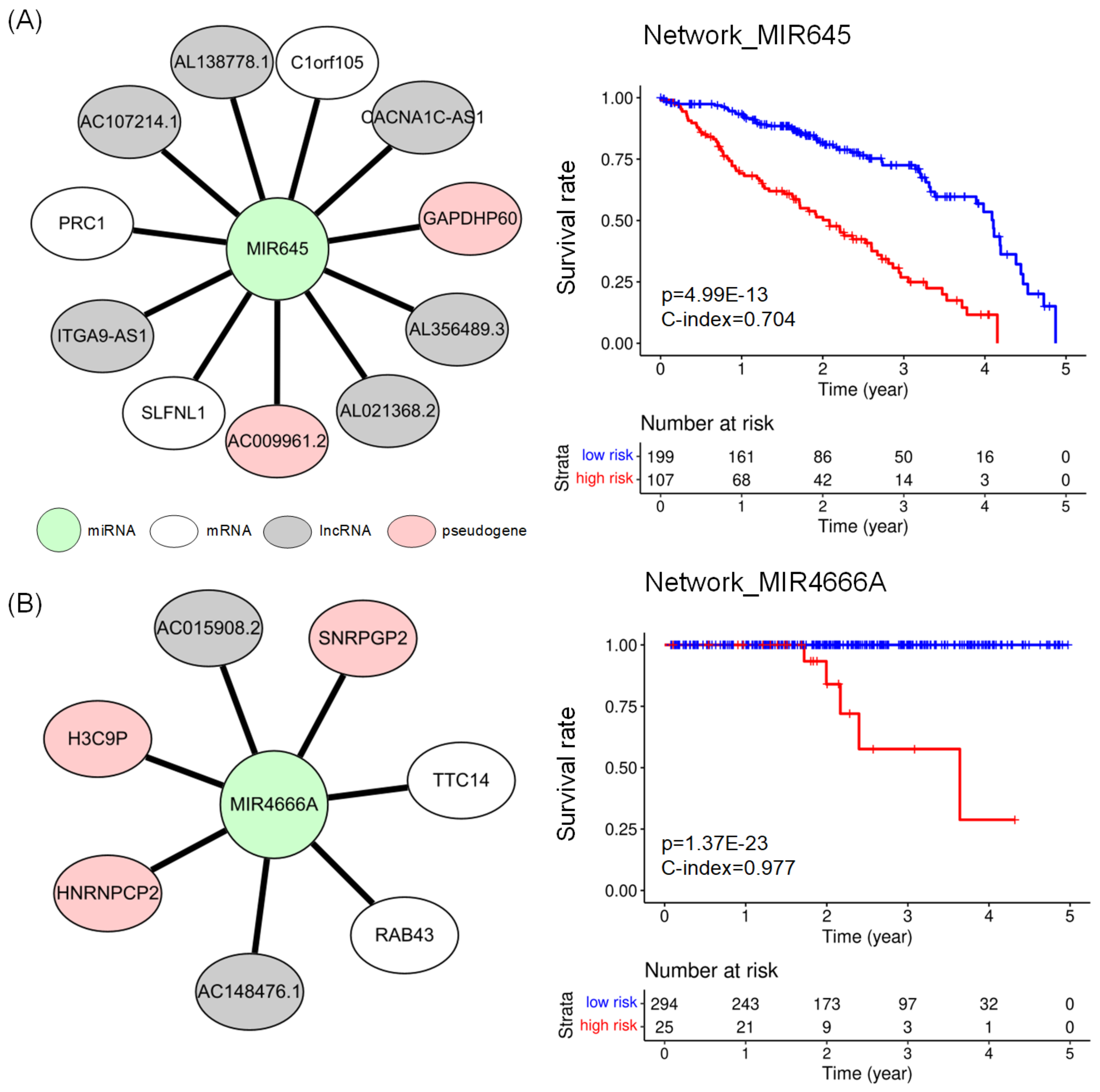
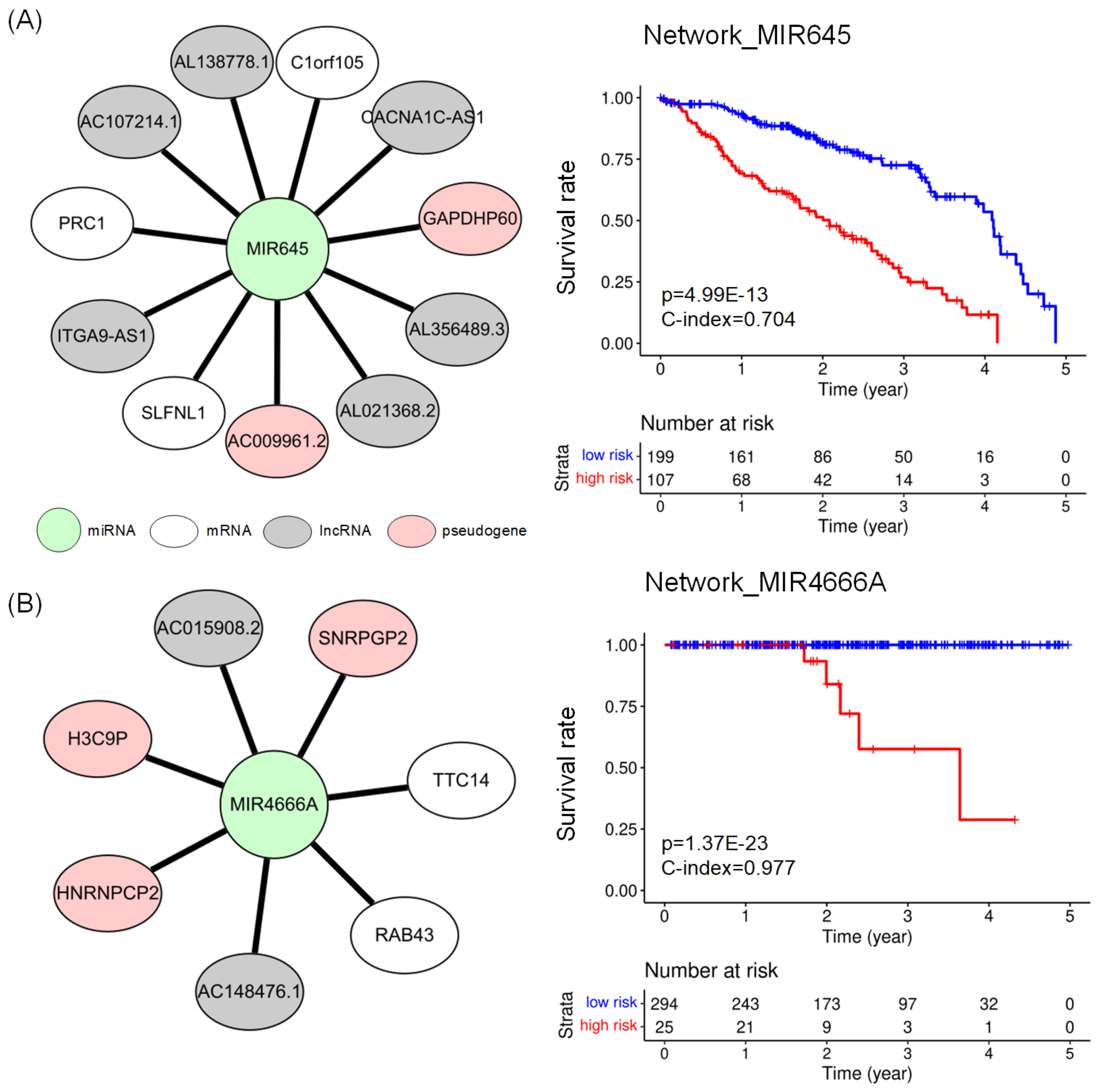
2.4. Subnetworks for the Cancer Prognosis
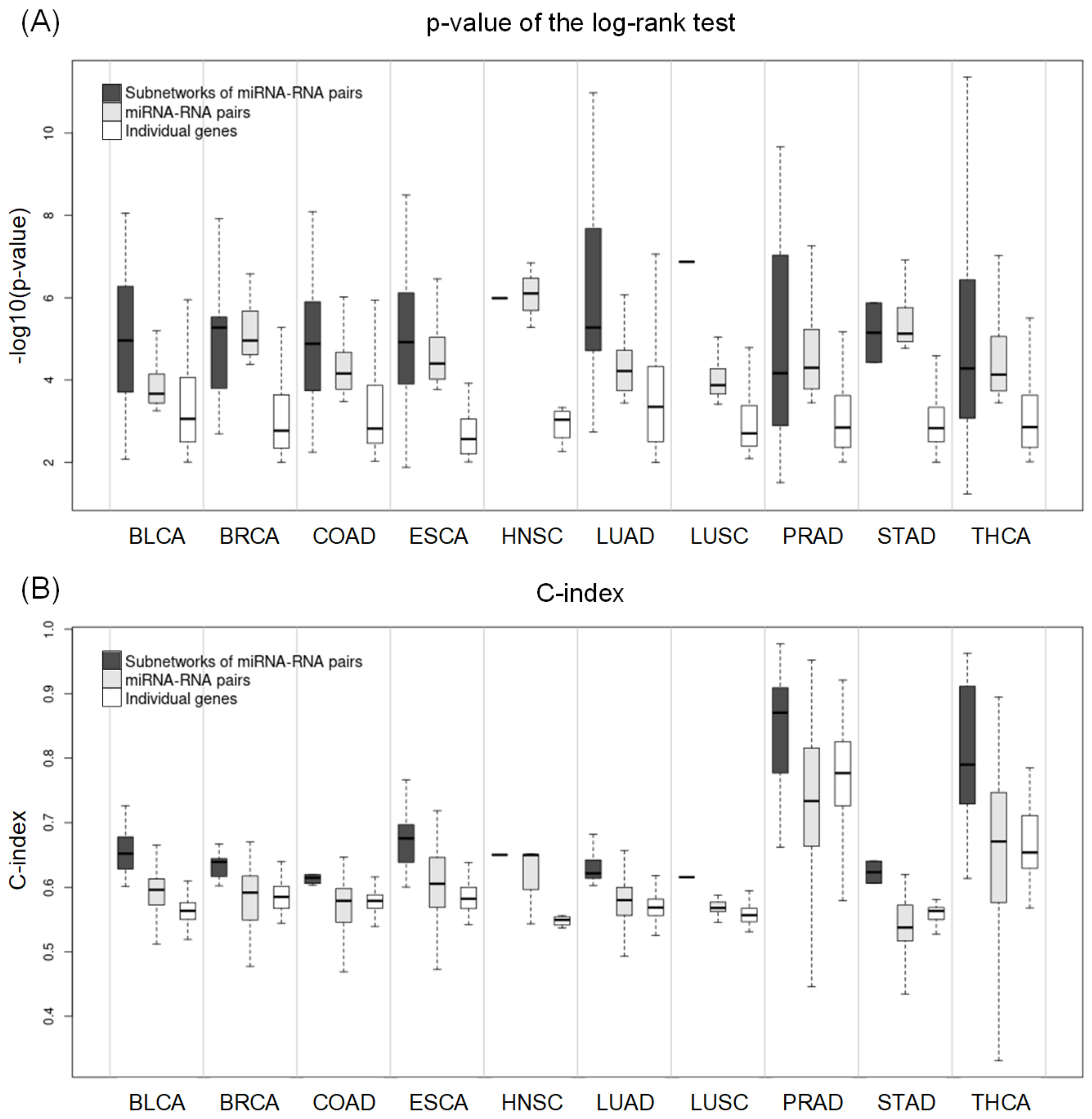
2.5. Comparing Potential Prognostic Biomarkers to Other Methods
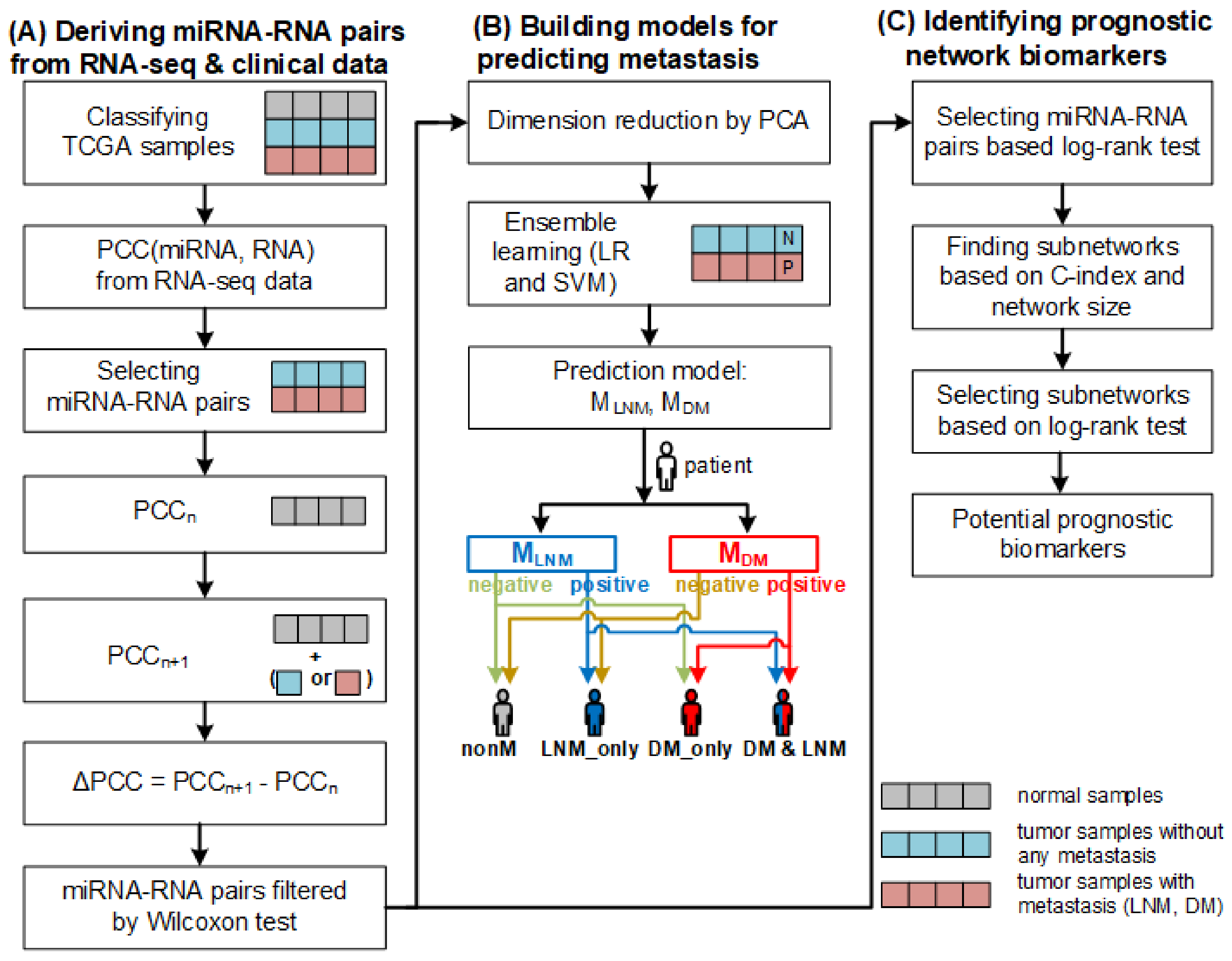
3. Materials and Methods
3.1. Data Collection and Preparation
- Samples with no metastasis (nonM): T stage of 1–4, N stage of 0, and M stage of 0
- Samples with lymph node metastasis only (LNM_only): T stage of 1–4, N stage of 1–3, and M stage of 0
- Samples with distant metastasis only (DM_only): T stage of 1–4, N stage of 0, and M stage of 1
- Samples with both lymph node metastasis and distant metastasis (LNM&DM): T stage of 1–4, N stage of 1–3, and M stage of 1
3.2. Deriving miRNA–RNA Interactions
3.3. Construction of Models for Predicting Metastasis
3.4. Finding Biomarkers for Predicting Prognosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TCGA | The cancer genome atlas |
| BLCA | Urothelial bladder carcinoma |
| BRCA | Breast invasive carcinoma |
| COAD | Colon adenocarcinoma |
| ESCA | Esophageal carcinoma |
| HNSC | Head-neck squamous cell carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| PRAD | Prostate adenocarcinoma |
| STAD | Stomach adenocarcinoma |
| THCA | Thyroid carcinoma |
| TNM | Tumor, node, metastasis |
| GDC | Genomic data commons |
| CPM | Counts per million |
| TMM | Trimmed mean of M values |
| PCC | Pearson correlation coefficient |
| PCA | Principal component analysis |
| PC | Principal component |
| SVM | Support vector machine |
| RBF | Radial basis function |
| LR | Logistic regression |
| HR | Hazard ratio |
| AUC | Area under the curve |
| HPA | Human protein atlas |
Appendix A
Appendix B
Appendix C
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widakowich, C.; de Castro, G., Jr.; de Azambuja, E.; Dinh, P.; Awada, A. Review: Side effects of approved molecular targeted therapies in solid cancers. Oncologist 2007, 12, 1443–1455. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Kurzrock, R. Toxicity of targeted therapy: Implications for response and impact of genetic polymorphisms. Cancer Treat. Rev. 2014, 40, 883–891. [Google Scholar] [CrossRef]
- Verma, M. Personalized medicine and cancer. J. Pers. Med. 2012, 2, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Lee, W.; Han, K. Predicting lymph node metastasis and prognosis of individual cancer patients based on miRNA-mediated RNA interactions. BMC Med. Genom. 2022, 15, 87. [Google Scholar] [CrossRef]
- Lee, S.; Lee, W.; Ren, S.; Park, B.; Han, K. Constructing Integrative ceRNA Networks and Finding Prognostic Biomarkers in Renal Cell Carcinoma. IEEE/ACM Trans. Comp. Biol. Bioinform. 2022. ahead of print. [Google Scholar] [CrossRef]
- Gaidatzis, D.; van Nimwegen, E.; Hausser, J.; Zavolan, M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinform. 2007, 8, 69. [Google Scholar]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language. Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huang, C.; Li, Q.; Wu, X. Construction and Comprehensive Analysis for Dysregulated Long Non-Coding RNA (lncRNA)-Associated Competing Endogenous RNA (ceRNA) Network in Gastric Cancer. Med. Sci. Monit. 2018, 24, 33–49. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Jiang, C.; Huang, Z.; Xu, D.; Zheng, S. Comprehensive analysis of dysregulated lncRNAs, miRNAs and mRNAs with associated ceRNA network in esophageal squamous cell carcinoma. Gene 2019, 696, 206–218. [Google Scholar] [CrossRef]
- Bai, Y.; Long, J.; Liu, Z.; Lin, J.; Huang, H.; Wang, D.; Yang, X.; Miao, F.; Mao, Y.; Sang, X.; et al. Comprehensive analysis of a ceRNA network reveals potential prognostic cytoplasmic lncRNAs involved in HCC progression. J. Cell. Physiol. 2019, 234, 18837–18848. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chang, Z.; Wu, C.; Zhu, Y.; Li, K.; Xu, Y. Identification of potential cancer-related pseudogenes in lung adenocarcinoma based on ceRNA hypothesis. Oncotarget 2017, 8, 59036–59047. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, Y.; Feng, L.; Li, F.; Sun, Z.; Wu, T.; Shi, X.; Li, J.; Li, X. Comprehensive characterization of lncRNA-mRNA related ceRNA network across 12 major cancers. Oncotarget 2016, 7, 64148–64167. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Bian, Y.; Zhang, Q.; Hu, J.; Li, L.; Yang, M.; Qian, H.; Yu, L.; Liu, B.; Qian, X. Construction and analysis of dysregulated lncRNA-associated ceRNA network in colorectal cancer. J. Cell. Biochem. 2019, 120, 9250–9263. [Google Scholar] [CrossRef]
- Jiang, R.; Zhao, C.; Gao, B.; Xu, J.; Song, W.; Shi, P. Mixomics analysis of breast cancer: Long non-coding RNA linc01561 acts as ceRNA involved in the progression of breast cancer. Int. J. Biochem. Cell Biol. 2018, 102, 1–9. [Google Scholar] [CrossRef]
- Chen, L. Network biomarker for quantifying regular state of a biological system, and dynamic network biomarker for quantifying critical state of a biological system. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine 2020, Seoul, Republic of Korea, 16–19 December 2020. [Google Scholar]
- Zhou, J.; Lu, X.; Chang, W.; Wan, C.; Lu, X.; Zhang, C.; Cao, S. PLUS: Predicting Cancer Metastasis Potential Based on Positive and Unlabeled Learning. PLoS Comput. Biol. 2022, 18, e1009956. [Google Scholar] [CrossRef]
- Poliseno, L.; Marranci, A.; Pandolfi, P.P. Pseudogenes in Human Cancer. Front. Med. 2015, 2, 68. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Lu, X.; Yuan, L. LncRNA: A link between RNA and cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1097–1109. [Google Scholar] [CrossRef]
- Kang, J.; Tang, Q.; He, J.; Li, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA Interactome Repository with Redefined Confidence Scoring System and Improved Accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-X.; Liu, Z.; Deng, F.; Wang, D.-D.; Li, X.-W.; Tian, T.; Zhang, J.; Tang, J.-H. MiR-145: A Potential Biomarker of Cancer Migration and Invasion. Am. J. Transl. Res. 2019, 11, 6739–6753. [Google Scholar] [PubMed]
- Zhu, B.; Tian, T.; Zhao, M. MiR-645 Promotes Proliferation and Migration of Non-Small Cell Lung Cancer Cells by Targeting TP53I11. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6150–6156. [Google Scholar] [PubMed]
- Liu, W.; Li, Y.; Feng, S.; Guan, Y.; Cao, Y. MicroRNA-760 Inhibits Cell Viability and Migration through down-Regulating BST2 in Gastric Cancer. J. Biochem. 2020, 168, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Wang, Y.; Duan, Q.-H.; Liu, S.-S.; Liu, G.-J. MicroRNA-760 Acts as a Tumor Suppressor in Gastric Cancer Development via Inhibiting G-Protein-Coupled Receptor Kinase Interacting Protein-1 Transcription. World J. Gastroenterol. 2019, 25, 6619–6633. [Google Scholar] [CrossRef]
- Sha, H.-H.; Wang, D.-D.; Chen, D.; Liu, S.-W.; Wang, Z.; Yan, D.-L.; Dong, S.-C.; Feng, J.-F. MiR-138: A Promising Therapeutic Target for Cancer. Tumour Biol. 2017, 39, 1010428317697575. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, 6352. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. Available online: https://portal.gdc.cancer.gov/ (accessed on 31 May 2022). [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A Scaling Normalization Method for Differential Expression Analysis of RNA-Seq Data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Wilcoxon, F. Individual comparisons by ranking methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Puccini, A.; Seebe, A.; Xiu, J.; Goldberg, R.M.; Soldato, D.; Grothey, A.; Shields, A.F.; Salem, M.E.; Battaglin, F.; Berger, M.D.; et al. Molecular differences between lymph nodes and distant metastases compared with primaries in colorectal cancer patients. NPJ Precis. Oncol. 2021, 5, 95. [Google Scholar] [CrossRef]
- Wang, G.; Hao, J.; Ma, J.; Jiang, H. A Comparative Assessment of Ensemble Learning for Credit Scoring. Expert Syst. Appl. 2011, 38, 223–230. [Google Scholar] [CrossRef]
- Wolpert, D.H. Stacked generalization. Neural Netw. 1992, 5, 241–259. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. The Cox Model. In Statistics for Biology and Health; Springer: New York, NY, USA, 2000; pp. 39–77. [Google Scholar]
- Davidson-Pilon, C. Lifelines: Survival analysis in Python. J. Open Source Softw. 2019, 4, 1317. [Google Scholar] [CrossRef] [Green Version]
- Dardis, C.; Dardis, M.C. Package ‘survMisc’. Available online: https://CRAN.R-project.org/package=survMisc (accessed on 1 November 2022).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | #nonM samples | #LNM_only samples | #DM_only samples | #LNM&DM samples | #normal samples | #total samples |
|---|---|---|---|---|---|---|
| BLCA | 118 | 44 | 0 | 7 | 19 | 188 |
| BRCA | 450 | 449 | 1 | 18 | 113 | 1031 |
| COAD | 228 | 105 | 9 | 55 | 41 | 438 |
| ESCA | 56 | 64 | 2 | 6 | 11 | 139 |
| HNSC | 81 | 98 | 0 | 1 | 44 | 224 |
| LUAD | 219 | 124 | 11 | 11 | 59 | 424 |
| LUSC | 258 | 149 | 3 | 3 | 49 | 462 |
| PRAD | 316 | 75 | 1 | 1 | 52 | 445 |
| STAD | 103 | 210 | 2 | 23 | 32 | 370 |
| THCA | 145 | 127 | 3 | 4 | 59 | 338 |
| Cancer | #miRNAs | #lncRNAs | #mRNAs | #pseudogenes |
|---|---|---|---|---|
| BLCA | 143 | 9612 | 18,038 | 4994 |
| BRCA | 150 | 10,070 | 18,035 | 5380 |
| COAD | 144 | 8477 | 17,515 | 5102 |
| ESCA | 418 | 12,588 | 18,658 | 8713 |
| HNSC | 88 | 8563 | 17,912 | 4493 |
| LUAD | 182 | 10,291 | 18,037 | 5845 |
| LUSC | 147 | 10,206 | 18,152 | 5507 |
| PRAD | 126 | 8764 | 17,731 | 4686 |
| STAD | 345 | 12,472 | 18,657 | 8700 |
| THCA | 140 | 8256 | 17,487 | 4610 |
| Cancer | #miRNA–RNA Pairs after PCC Filtering | #miRNA–RNA Pairs after Wilcoxon Test | #PCs after PCA |
|---|---|---|---|
| LNM | |||
| BLCA | 169,439 | 9501 | 45 |
| BRCA | 170,673 | 3619 | 166 |
| COAD | 312,968 | 3970 | 162 |
| ESCA | 706,722 | 27,312 | 65 |
| HNSC | 82,959 | 2281 | 58 |
| LUAD | 320,323 | 13,891 | 137 |
| LUSC | 43,340 | 1296 | 83 |
| PRAD | 78,722 | 5036 | 150 |
| STAD | 230,038 | 8136 | 120 |
| THCA | 124,722 | 12,738 | 102 |
| DM | |||
| BRCA | 572,862 | 19,634 | 134 |
| COAD | 273,660 | 4968 | 112 |
| LUAD | 863,846 | 20,632 | 55 |
| STAD | 1,222,396 | 43,240 | 58 |
| Cancer | LNM | DM | ||||
|---|---|---|---|---|---|---|
| PCC 1 | Exp 2 | Exp191 3 | PCC 1 | Exp 2 | Exp191 3 | |
| BLCA | 0.938 | 0.668 | 0.541 | - | - | - |
| BRCA | 0.732 | 0.626 | 0.550 | 0.907 | 0.605 | 0.500 |
| COAD | 0.936 | 0.713 | 0.637 | 0.889 | 0.580 | 0.512 |
| ESCA | 0.961 | 0.670 | 0.501 | - | - | - |
| HNSC | 0.924 | 0.727 | 0.520 | - | - | - |
| LUAD | 0.787 | 0.636 | 0.557 | 0.733 | 0.613 | 0.500 |
| LUSC | 0.840 | 0.598 | 0.498 | - | - | - |
| PRAD | 0.815 | 0.655 | 0.534 | - | - | - |
| STAD | 0.897 | 0.596 | 0.507 | 0.853 | 0.661 | 0.498 |
| THCA | 0.802 | 0.675 | 0.638 | - | - | - |
| Cancer | miRNA–RNA Pair | Type of RNA | HR | p-Value | C-Index |
|---|---|---|---|---|---|
| BLCA | MIR6793_CST4 | mRNA | 0.164 | 0.639 | |
| BRCA | MIR186_AP1S1 | mRNA | 3.820 | 0.642 | |
| COAD | MIR4538_SLAMF1 | mRNA | 3.294 | 0.630 | |
| ESCA | MIR4755_CCDC18-AS1 | lncRNA | 5.298 | 0.681 | |
| HNSC | MIR4537_EMC3-AS1 | pseudogene | 0.256 | 0.651 | |
| LUAD | MIR3125_OR1F1 | mRNA | 3.868 | 0.611 | |
| LUSC | MIR6071_SFTA3 | lncRNA | 0.408 | 0.579 | |
| PRAD | MIR5087_EZR-AS1 | lncRNA | 0.022 | 0.847 | |
| STAD | MIR6757_AC104619.3 | pseudogene | 5.724 | 0.537 | |
| THCA | MIR4664_AL353138.1 | lncRNA | 0.014 | 0.863 |
| Cancer | Network | #edges | HR | p-Value | C-Index |
|---|---|---|---|---|---|
| BLCA | network_MIR145 | 15 | 7.476 | 0.710 | |
| BRCA | network_MIR378J | 3 | 3.357 | 0.638 | |
| COAD | network_MIR4538 | 15 | 3.491 | 0.689 | |
| ESCA | network_MIR4644 | 15 | 6.312 | 0.788 | |
| HNSC | network_MIR8058 | 2 | 4.146 | 0.650 | |
| LUAD | network_MIR645 | 11 | 3.628 | 0.704 | |
| LUSC | network_MIR6071 | 15 | 2.400 | 0.615 | |
| PRAD | network_MIR4666A | 7 | 0.977 | ||
| STAD | network_MIR760 | 5 | 2.325 | 0.640 | |
| THCA | network_MIR138-1 | 2 | 46.806 | 0.789 |
| Cancer | Type of Feature | Number of Features | p-Value | C-Index |
|---|---|---|---|---|
| BLCA | networks | 32 | 0.7264 | |
| miRNA–RNA pairs | 514 | 0.6656 | ||
| individual genes | 297 | 0.6100 | ||
| BRCA | networks | 14 | 0.6673 | |
| miRNA–RNA pairs | 93 | 0.6701 | ||
| individual genes | 52 | 0.6396 | ||
| COAD | networks | 10 | 0.6895 | |
| miRNA–RNA pairs | 190 | 0.6470 | ||
| individual genes | 100 | 0.6192 | ||
| ESCA | networks | 34 | 0.7888 | |
| miRNA–RNA pairs | 311 | 0.7185 | ||
| individual genes | 98 | 0.6384 | ||
| HNSC | networks | 1 | 0.6502 | |
| miRNA–RNA pairs | 3 | 0.6516 | ||
| individual genes | 4 | 0.5562 | ||
| LUAD | networks | 39 | 0.7154 | |
| miRNA–RNA pairs | 632 | 0.6565 | ||
| individual genes | 342 | 0.6242 | ||
| LUSC | networks | 1 | 0.6157 | |
| miRNA–RNA pairs | 53 | 0.5875 | ||
| individual genes | 42 | 0.5943 | ||
| PRAD | networks | 19 | 0.9773 | |
| miRNA–RNA pairs | 156 | 0.9519 | ||
| individual genes | 66 | 0.9210 | ||
| STAD | networks | 2 | 0.6406 | |
| miRNA–RNA pairs | 77 | 0.6196 | ||
| individual genes | 39 | 0.6135 | ||
| THCA | networks | 31 | 0.9627 | |
| miRNA–RNA pairs | 293 | 0.8950 | ||
| individual genes | 78 | 0.7854 |
| Cancer | Prognostic Networks Found in Our Study | Prognostic Genes in HPA | ||||
|---|---|---|---|---|---|---|
| Center miRNA | p-Value | C-Index | Gene | p-Value | C-Index | |
| BLCA | MIR4539 | 0.7264 | GARS1 | 0.6226 | ||
| BRCA | MIR4489 | 0.6673 | PGK1 | 0.6580 | ||
| COAD | MIR4538 | 0.6895 | PRKAR2A | 0.6411 | ||
| ESCA | MIR4644 | 0.7888 | - | - | - | |
| HNSC | MIR8058 | 0.6502 | IGHV3-13 | 0.6133 | ||
| LUAD | MIR624 | 0.7154 | DKK1 | 0.6480 | ||
| LUSC | MIR6071 | 0.6157 | NT5E | 0.5987 | ||
| PRAD | MIR466A | 0.9773 | SESN1 | 0.9029 | ||
| STAD | MIR760 | 0.6406 | ZBTB7A | 0.6135 | ||
| THCA | MIR4442 | 0.9627 | SNAI1 | 0.8500 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Cho, M.; Park, B.; Han, K. Finding miRNA–RNA Network Biomarkers for Predicting Metastasis and Prognosis in Cancer. Int. J. Mol. Sci. 2023, 24, 5052. https://doi.org/10.3390/ijms24055052
Lee S, Cho M, Park B, Han K. Finding miRNA–RNA Network Biomarkers for Predicting Metastasis and Prognosis in Cancer. International Journal of Molecular Sciences. 2023; 24(5):5052. https://doi.org/10.3390/ijms24055052
Chicago/Turabian StyleLee, Seokwoo, Myounghoon Cho, Byungkyu Park, and Kyungsook Han. 2023. "Finding miRNA–RNA Network Biomarkers for Predicting Metastasis and Prognosis in Cancer" International Journal of Molecular Sciences 24, no. 5: 5052. https://doi.org/10.3390/ijms24055052
APA StyleLee, S., Cho, M., Park, B., & Han, K. (2023). Finding miRNA–RNA Network Biomarkers for Predicting Metastasis and Prognosis in Cancer. International Journal of Molecular Sciences, 24(5), 5052. https://doi.org/10.3390/ijms24055052