
Exploring the Gut Microbiota–Muscle Axis in Duchenne Muscular Dystrophy
 , , , and
, , , and 
Abstract
:1. The Bidirectional Gut–Muscle Axis
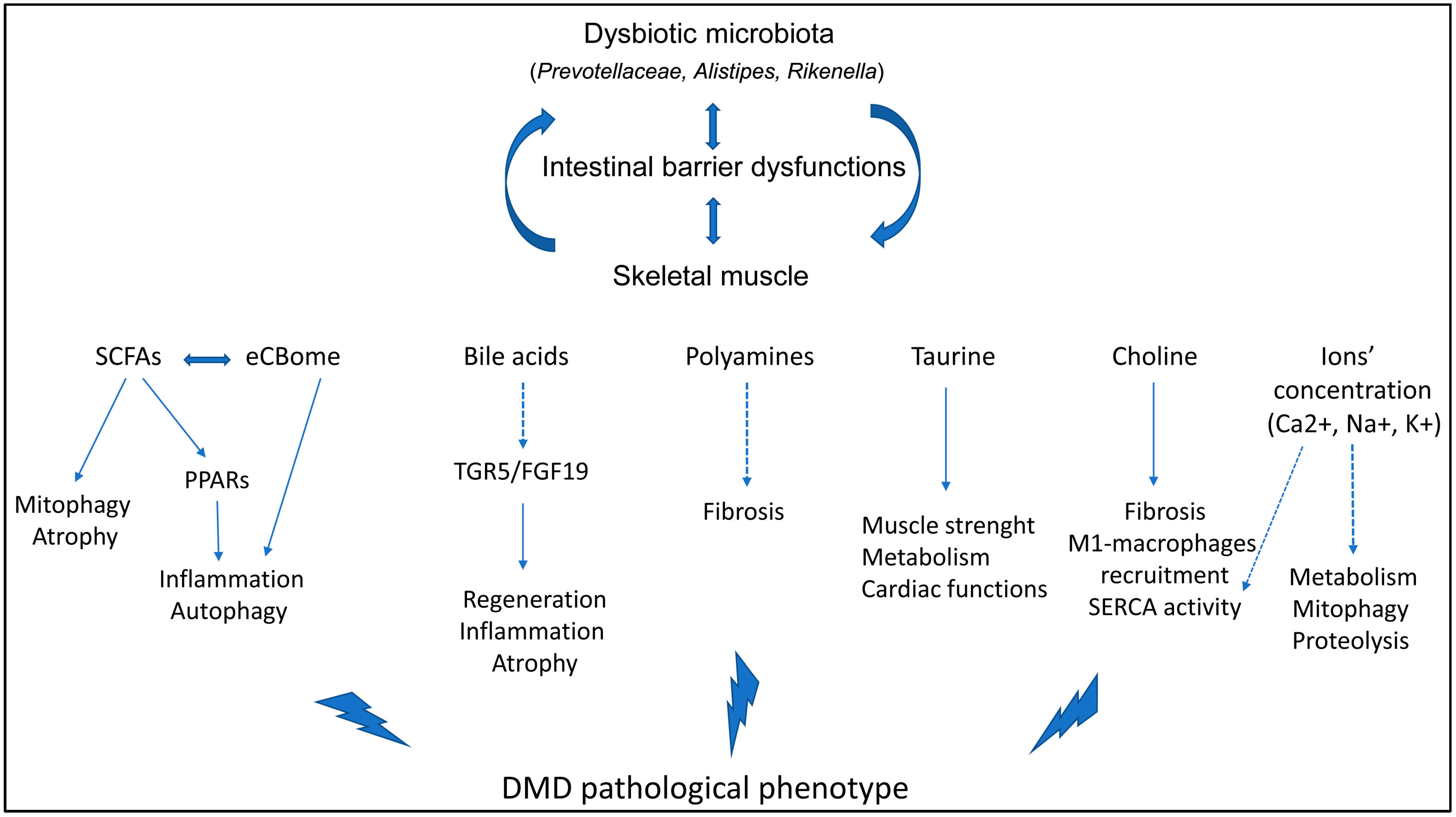
2. Interplay of Factors in Regulating Skeletal Muscle Physiology and Immune Balance
2.1. The Peroxisome Proliferator-Activated Receptors (PPARs)
2.2. Gut-Derived Metabolites
2.3. Short-Chain Fatty Acids (SCFAs)
2.4. Choline Derivatives
2.5. Polyamines
2.6. Tryptophan (Trp)
2.7. Bile Acids
2.8. Taurine
3. Therapies Targeting the Dysbiotic Microbiota
4. The Pathogenesis of Duchenne Muscular Dystrophy
5. Ions’ Homeostasis
5.1. Calcium
5.2. Sodium
5.3. Potassium
6. Sarcoplasmatic Reticulum (SR)
7. The Involvement of the Gut Microbiota in DMD
8. Microbiota Interactors and Microbiota-Derived Metabolites Modulate the DMD Phenotype
8.1. PPARs Inhibition
8.2. The Endocannabinoid System
8.3. Choline Inhibition
8.4. Polyamines’ Inhibition
8.5. Taurine Inhibition
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H.; et al. The gut microbiota influences skeletal muscle mass and function in mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Chen, W.D.; Wang, Y.D. Gut Microbiota: An Integral Moderator in Health and Disease. Front. Microbiol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jin, B.; Fan, Z. Mechanisms Involved in Gut Microbiota Regulation of Skeletal Muscle. Oxidative Med. Cell. Longev. 2022, 2022, 2151191. [Google Scholar] [CrossRef] [PubMed]
- Marullo, A.L.; O’Halloran, K.D. Microbes, metabolites and muscle: Is the gut-muscle axis a plausible therapeutic target in Duchenne muscular dystrophy? Exp. Physiol. 2023, 108, 1132–1143. [Google Scholar] [CrossRef]
- Guo, A.; Li, K.; Tian, H.C.; Fan, Z.; Chen, Q.N.; Yang, Y.F.; Yu, J.; Wu, Y.X.; Xiao, Q. FGF19 protects skeletal muscle against obesity-induced muscle atrophy, metabolic derangement and abnormal irisin levels via the AMPK/SIRT-1/PGC-alpha pathway. J. Cell. Mol. Med. 2021, 25, 3585–3600. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, Q.; Xu, T.; Yuan, Q.; Hu, Q.; Hu, N.; Sun, W.; Bai, Y.; Liu, L.; Feng, J.; et al. Fndc5/irisin deficiency leads to dysbiosis of gut microbiota contributing to the depressive-like behaviors in mice. Brain Res. 2023, 1819, 148537. [Google Scholar] [CrossRef] [PubMed]
- Nay, K.; Jollet, M.; Goustard, B.; Baati, N.; Vernus, B.; Pontones, M.; Lefeuvre-Orfila, L.; Bendavid, C.; Rue, O.; Mariadassou, M.; et al. Gut bacteria are critical for optimal muscle function: A potential link with glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E158–E171. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wei, Q.; Wang, H.; Kim, D.M.; Balderas, M.; Wu, G.; Lawler, J.; Safe, S.; Guo, S.; Devaraj, S.; et al. Protective Effects of Ghrelin on Fasting-Induced Muscle Atrophy in Aging Mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 621–630. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ishizu, Y.; Honda, T.; Ito, T.; Imai, N.; Nakamura, M.; Kawashima, H.; Kitaura, Y.; Ishigami, M.; Fujishiro, M. Patients with low muscle mass have characteristic microbiome with low potential for amino acid synthesis in chronic liver disease. Sci. Rep. 2022, 12, 3674. [Google Scholar] [CrossRef]
- Haran, J.P.; Bucci, V.; Dutta, P.; Ward, D.; McCormick, B. The nursing home elder microbiome stability and associations with age, frailty, nutrition and physical location. J. Med. Microbiol. 2018, 67, 40–51. [Google Scholar] [CrossRef] [PubMed]
- van Tongeren, S.P.; Slaets, J.P.; Harmsen, H.J.; Welling, G.W. Fecal microbiota composition and frailty. Appl. Environ. Microbiol. 2005, 71, 6438–6442. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut 2018, 67, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Ponziani, F.R.; Calvani, R.; Marini, F.; Biancolillo, A.; Coelho-Junior, H.J.; Gervasoni, J.; Primiano, A.; Putignani, L.; Del Chierico, F.; et al. Gut Microbial, Inflammatory and Metabolic Signatures in Older People with Physical Frailty and Sarcopenia: Results from the BIOSPHERE Study. Nutrients 2019, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, L.; Yang, Z.; Wang, D.; Li, T.; Yang, F.; Li, Z.; Bai, X.; Wang, Y. Gut-muscle axis and sepsis-induced myopathy: The potential role of gut microbiota. Biomed. Pharmacother. 2023, 163, 114837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chu, J.; Feng, S.; Guo, C.; Xue, B.; He, K.; Li, L. Immunological mechanisms of inflammatory diseases caused by gut microbiota dysbiosis: A review. Biomed. Pharmacother. 2023, 164, 114985. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.; Lim, Y.P.; Lim, W.S.; Chambers, E.S.; Frost, G.; Wong, S.H.; Ali, Y. Gut-muscle crosstalk. A perspective on influence of microbes on muscle function. Front. Med. 2022, 9, 1065365. [Google Scholar] [CrossRef] [PubMed]
- Gizard, F.; Fernandez, A.; De Vadder, F. Interactions between gut microbiota and skeletal muscle. Nutr. Metab. Insights 2020, 13, 1178638820980490. [Google Scholar] [CrossRef] [PubMed]
- Giron, M.; Thomas, M.; Dardevet, D.; Chassard, C.; Savary-Auzeloux, I. Gut microbes and muscle function: Can probiotics make our muscles stronger? J. Cachexia Sarcopenia Muscle 2022, 13, 1460–1476. [Google Scholar] [CrossRef]
- Messaritakis, I.; Vogiatzoglou, K.; Tsantaki, K.; Ntretaki, A.; Sfakianaki, M.; Koulouridi, A.; Tsiaoussis, J.; Mavroudis, D.; Souglakos, J. The Prognostic Value of the Detection of Microbial Translocation in the Blood of Colorectal Cancer Patients. Cancers 2020, 12, 1058. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Douek, D.C. Microbial translocation across the GI tract. Annu. Rev. Immunol. 2012, 30, 149–173. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef]
- Manickam, R.; Duszka, K.; Wahli, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. Int. J. Mol. Sci. 2020, 21, 8056. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Pena, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vazquez-Carrera, M. PPARbeta/delta: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef]
- Miura, P.; Chakkalakal, J.V.; Boudreault, L.; Belanger, G.; Hebert, R.L.; Renaud, J.M.; Jasmin, B.J. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum. Mol. Genet. 2009, 18, 4640–4649. [Google Scholar] [CrossRef] [PubMed]
- Manickam, R.; Oh, H.Y.P.; Tan, C.K.; Paramalingam, E.; Wahli, W. Metronidazole Causes Skeletal Muscle Atrophy and Modulates Muscle Chronometabolism. Int. J. Mol. Sci. 2018, 19, 2418. [Google Scholar] [CrossRef] [PubMed]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Perandini, L.A.; Chimin, P.; Lutkemeyer, D.D.S.; Camara, N.O.S. Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: Can physical exercise restore the satellite cell niche? FEBS J. 2018, 285, 1973–1984. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Wang, L.; Huangfu, M.; Li, H. The impact of microbiota-derived short-chain fatty acids on macrophage activities in disease: Mechanisms and therapeutic potentials. Biomed. Pharmacother. 2023, 165, 115276. [Google Scholar] [CrossRef]
- Wang, J.; Chen, W.D.; Wang, Y.D. The Relationship Between Gut Microbiota and Inflammatory Diseases: The Role of Macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Mancabelli, L.; Tagliaferri, S.; Nouvenne, A.; Milani, C.; Del Rio, D.; Lauretani, F.; Maggio, M.G.; Ventura, M.; Meschi, T. The Gut-Muscle Axis in Older Subjects with Low Muscle Mass and Performance: A Proof of Concept Study Exploring Fecal Microbiota Composition and Function with Shotgun Metagenomics Sequencing. Int. J. Mol. Sci. 2020, 21, 8946. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.; Murphy, K.G.; Frost, G.; Chambers, E.S. Short-chain fatty acids as potential regulators of skeletal muscle metabolism and function. Nat. Metab. 2020, 2, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Lustgarten, M.S. The Role of the Gut Microbiome on Skeletal Muscle Mass and Physical Function: 2019 Update. Front. Physiol. 2019, 10, 1435. [Google Scholar] [CrossRef] [PubMed]
- da Costa, K.A.; Badea, M.; Fischer, L.M.; Zeisel, S.H. Elevated serum creatine phosphokinase in choline-deficient humans: Mechanistic studies in C2C12 mouse myoblasts. Am. J. Clin. Nutr. 2004, 80, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Frohman, M.A.; Blusztajn, J.K. Generation of choline for acetylcholine synthesis by phospholipase D isoforms. BMC Neurosci. 2001, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Cisterna, B.A.; Vargas, A.A.; Puebla, C.; Fernandez, P.; Escamilla, R.; Lagos, C.F.; Matus, M.F.; Vilos, C.; Cea, L.A.; Barnafi, E.; et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat. Commun. 2020, 11, 1073. [Google Scholar] [CrossRef]
- Sagar, N.A.; Tarafdar, S.; Agarwal, S.; Tarafdar, A.; Sharma, S. Polyamines: Functions, Metabolism, and Role in Human Disease Management. Med. Sci. 2021, 9, 44. [Google Scholar] [CrossRef]
- Galasso, L.; Cappella, A.; Mule, A.; Castelli, L.; Ciorciari, A.; Stacchiotti, A.; Montaruli, A. Polyamines and Physical Activity in Musculoskeletal Diseases: A Potential Therapeutic Challenge. Int. J. Mol. Sci. 2023, 24, 9798. [Google Scholar] [CrossRef]
- Tabbaa, M.; Ruz Gomez, T.; Campelj, D.G.; Gregorevic, P.; Hayes, A.; Goodman, C.A. The regulation of polyamine pathway proteins in models of skeletal muscle hypertrophy and atrophy: A potential role for mTORC1. Am. J. Physiol. Cell Physiol. 2021, 320, C987–C999. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mao, T.; Wang, Y.; Qi, X.; Zhao, W.; Chen, H.; Zhang, C.; Li, X. Effect of Gut Microbiota-Mediated Tryptophan Metabolism on Inflammaging in Frailty and Sarcopenia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2024, 79, glae044. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Nakamura, N.; Nakamura, H.; Mizutani, T.; Kaneda, Y.; Yamaguchi, K.; Matsumoto, T.; Kitagawa, J.; Kanemura, N.; Shiraki, M.; et al. Low Levels of Serum Tryptophan Underlie Skeletal Muscle Atrophy. Nutrients 2020, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yu, J.; Li, Y.; Yang, F.; Yu, H.; Xue, M.; Zhang, F.; Jiang, X.; Ji, X.; Bao, Z. Depletion of gut microbiota induces skeletal muscle atrophy by FXR-FGF15/19 signalling. Ann. Med. 2021, 53, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Mancin, L.; Wu, G.D.; Paoli, A. Gut microbiota-bile acid-skeletal muscle axis. Trends Microbiol. 2023, 31, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kuboyama, A.; Mita, M.; Murata, S.; Shimizu, M.; Inoue, J.; Mori, K.; Sato, R. The exercise-inducible bile acid receptor Tgr5 improves skeletal muscle function in mice. J. Biol. Chem. 2018, 293, 10322–10332. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Hara, N.; Sugimoto, R.; Mifuji-Moroka, R.; Tanaka, H.; Eguchi, A.; Iwasa, M.; Hasegawa, H.; Iwata, K.; Takei, Y.; et al. The Associations between Circulating Bile Acids and the Muscle Volume in Patients with Non-alcoholic Fatty Liver Disease (NAFLD). Intern. Med. 2017, 56, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.A.; Durieux, A.C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, Y.; Hu, S.; Chen, Y.; Shen, Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020, 11, 349. [Google Scholar] [CrossRef]
- Qiu, Y.; Yu, J.; Ji, X.; Yu, H.; Xue, M.; Zhang, F.; Li, Y.; Bao, Z. Ileal FXR-FGF15/19 signaling activation improves skeletal muscle loss in aged mice. Mech. Ageing Dev. 2022, 202, 111630. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yoshikawa, N.; Inui, T.; Miyazaki, N.; Schaffer, S.W.; Azuma, J. Tissue depletion of taurine accelerates skeletal muscle senescence and leads to early death in mice. PLoS ONE 2014, 9, e107409. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, H.; Pagano, E.; Paris, D.; Panza, E.; Cuozzo, M.; Moriello, C.; Piscitelli, F.; Abolghasemi, A.; Gazzerro, E.; Silvestri, C.; et al. Targeting gut dysbiosis against inflammation and impaired autophagy in Duchenne muscular dystrophy. EMBO Mol. Med. 2023, 15, e16225. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.; Shen, L.; Cheng, R.; Wang, P.; Wen, L.; Sun, Y.; Wang, Q.; Chen, J.; Lin, S.; Liao, Y.; et al. Faecal microbiota transplantation from young rats attenuates age-related sarcopenia revealed by multiomics analysis. J. Cachexia Sarcopenia Muscle 2023, 14, 2168–2183. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, C.; Zhang, F.; Liu, Y.; Guo, Y.; Yao, Q. The gut microbiota attenuates muscle wasting by regulating energy metabolism in chemotherapy-induced malnutrition rats. Cancer Chemother. Pharmacol. 2020, 85, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Beck, R.; Schakman, O.; Martin, J.C.; De Backer, F.; Sohet, F.M.; Dewulf, E.M.; Pachikian, B.D.; Neyrinck, A.M.; Thissen, J.P.; et al. Restoring specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS ONE 2012, 7, e37971. [Google Scholar] [CrossRef]
- Horiba, T.; Katsukawa, M.; Mita, M.; Sato, R. Dietary obacunone supplementation stimulates muscle hypertrophy, and suppresses hyperglycemia and obesity through the TGR5 and PPARgamma pathway. Biochem. Biophys. Res. Commun. 2015, 463, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Wei, C.C.; Huang, C.C.; Chen, W.L.; Huang, H.Y. The Beneficial Effects of Lactobacillus plantarum PS128 on High-Intensity, Exercise-Induced Oxidative Stress, Inflammation, and Performance in Triathletes. Nutrients 2019, 11, 353. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Yang, X.; Zheng, L.; Wang, Z.; Wu, L.; Jiang, J.; Yang, T.; Ma, L.; Fu, Z. Lactobacillus and Bifidobacterium Improves Physiological Function and Cognitive Ability in Aged Mice by the Regulation of Gut Microbiota. Mol. Nutr. Food Res. 2019, 63, e1900603. [Google Scholar] [CrossRef]
- Kariyawasam, D.; D’Silva, A.; Mowat, D.; Russell, J.; Sampaio, H.; Jones, K.; Taylor, P.; Farrar, M. Incidence of Duchenne muscular dystrophy in the modern era; an Australian study. Eur. J. Hum. Genet. 2022, 30, 1398–1404. [Google Scholar] [CrossRef]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Bez Batti Angulski, A.; Hosny, N.; Cohen, H.; Martin, A.A.; Hahn, D.; Bauer, J.; Metzger, J.M. Duchenne muscular dystrophy: Disease mechanism and therapeutic strategies. Front. Physiol. 2023, 14, 1183101. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Belosludtsev, K.N. Ion Channels of the Sarcolemma and Intracellular Organelles in Duchenne Muscular Dystrophy: A Role in the Dysregulation of Ion Homeostasis and a Possible Target for Therapy. Int. J. Mol. Sci. 2023, 24, 2229. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef] [PubMed]
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2020, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Sakurai, H. Orai1-STIM1 Regulates Increased Ca(2+) Mobilization, Leading to Contractile Duchenne Muscular Dystrophy Phenotypes in Patient-Derived Induced Pluripotent Stem Cells. Biomedicines 2021, 9, 1589. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Castaneda, M.; Michelucci, A.; Zhao, N.; Malik, S.; Dirksen, R.T. Postdevelopmental knockout of Orai1 improves muscle pathology in a mouse model of Duchenne muscular dystrophy. J. Gen. Physiol. 2022, 154, e202213081. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Creismeas, A.; Gazaille, C.; Bourdon, A.; Lallemand, M.A.; Francois, V.; Allais, M.; Ledevin, M.; Larcher, T.; Toumaniantz, G.; Lafoux, A.; et al. TRPC3, but not TRPC1, as a good therapeutic target for standalone or complementary treatment of DMD. J. Transl. Med. 2021, 19, 519. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.L.; Shin, J.Y.; Jeffreys, W.P.; Wang, N.; Lukban, C.A.; Moorer, M.C.; Velarde, E.; Hanselman, O.A.; Kwon, S.; Kannan, S.; et al. Pharmacological TRPC6 inhibition improves survival and muscle function in mice with Duchenne muscular dystrophy. JCI Insight 2022, 7, e158906. [Google Scholar] [CrossRef] [PubMed]
- Wasala, N.B.; Yue, Y.; Lostal, W.; Wasala, L.P.; Niranjan, N.; Hajjar, R.J.; Babu, G.J.; Duan, D. Single SERCA2a Therapy Ameliorated Dilated Cardiomyopathy for 18 Months in a Mouse Model of Duchenne Muscular Dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Law, M.L.; Prins, K.W.; Olander, M.E.; Metzger, J.M. Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca(2+) cycling in vivo. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1544–H1552. [Google Scholar] [CrossRef] [PubMed]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, A.; Madiraju, S.; Dhindsa, B.S.; Hassen, G.W.; Rochling, F.A. Gigantic Stomach: A Rare Manifestation of Duchenne Muscular Dystrophy. Cureus 2019, 11, e4609. [Google Scholar] [CrossRef] [PubMed]
- Jollet, M.; Mariadassou, M.; Rue, O.; Pessemesse, L.; Ollendorff, V.; Ramdani, S.; Vernus, B.; Bonnieu, A.; Bertrand-Gaday, C.; Goustard, B.; et al. Insight into the Role of Gut Microbiota in Duchenne Muscular Dystrophy: An Age-Related Study in Mdx Mice. Am. J. Pathol. 2024, 194, 264–279. [Google Scholar] [CrossRef]
- Farini, A.; Tripodi, L.; Villa, C.; Strati, F.; Facoetti, A.; Baselli, G.; Troisi, J.; Landolfi, A.; Lonati, C.; Molinaro, D.; et al. Microbiota dysbiosis influences immune system and muscle pathophysiology of dystrophin-deficient mice. EMBO Mol. Med. 2023, 15, e16244. [Google Scholar] [CrossRef]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert. Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Sitzia, C.; Farini, A.; Colleoni, F.; Fortunato, F.; Razini, P.; Erratico, S.; Tavelli, A.; Fabrizi, F.; Belicchi, M.; Meregalli, M.; et al. Improvement of endurance of DMD animal model using natural polyphenols. BioMed Res. Int. 2015, 2015, 680615. [Google Scholar] [CrossRef] [PubMed]
- Banfi, S.; D’Antona, G.; Ruocco, C.; Meregalli, M.; Belicchi, M.; Bella, P.; Erratico, S.; Donato, E.; Rossi, F.; Bifari, F.; et al. Supplementation with a selective amino acid formula ameliorates muscular dystrophy in mdx mice. Sci. Rep. 2018, 8, 14659. [Google Scholar] [CrossRef]
- Tripodi, L.; Molinaro, D.; Fortunato, F.; Mella, C.; Cassani, B.; Torrente, Y.; Farini, A. Immunoproteasome Inhibition Ameliorates Aged Dystrophic Mouse Muscle Environment. Int. J. Mol. Sci. 2022, 23, 4657. [Google Scholar] [CrossRef]
- Sitzia, C.; Meregalli, M.; Belicchi, M.; Farini, A.; Arosio, M.; Bestetti, D.; Villa, C.; Valenti, L.; Brambilla, P.; Torrente, Y. Preliminary Evidences of Safety and Efficacy of Flavonoids- and Omega 3-Based Compound for Muscular Dystrophies Treatment: A Randomized Double-Blind Placebo Controlled Pilot Clinical Trial. Front. Neurol. 2019, 10, 755. [Google Scholar] [CrossRef]
- Iljazovic, A.; Roy, U.; Galvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef]
- Cha, K.H.; Yang, J.S.; Kim, K.A.; Yoon, K.Y.; Song, D.G.; Erdene-Ochir, E.; Kang, K.; Pan, C.H.; Ko, G. Improvement in host metabolic homeostasis and alteration in gut microbiota in mice on the high-fat diet: A comparison of calcium supplements. Food Res. Int. 2020, 136, 109495. [Google Scholar] [CrossRef]
- Bueno Junior, C.R.; Pantaleao, L.C.; Voltarelli, V.A.; Bozi, L.H.; Brum, P.C.; Zatz, M. Combined effect of AMPK/PPAR agonists and exercise training in mdx mice functional performance. PLoS ONE 2012, 7, e45699. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. Review of the Endocannabinoid System. Biol. Psychiatry. Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Heyman, E.; Gamelin, F.X.; Aucouturier, J.; Di Marzo, V. The role of the endocannabinoid system in skeletal muscle and metabolic adaptations to exercise: Potential implications for the treatment of obesity. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2012, 13, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Schonke, M.; Martinez-Tellez, B.; Rensen, P.C. Role of the endocannabinoid system in the regulation of the skeletal muscle response to exercise. Curr. Opin. Pharmacol. 2020, 52, 52–60. [Google Scholar] [CrossRef]
- Silvestri, C.; Ligresti, A.; Di Marzo, V. Peripheral effects of the endocannabinoid system in energy homeostasis: Adipose tissue, liver and skeletal muscle. Rev. Endocr. Metab. Disord. 2011, 12, 153–162. [Google Scholar] [CrossRef]
- Manca, C.; Boubertakh, B.; Leblanc, N.; Deschenes, T.; Lacroix, S.; Martin, C.; Houde, A.; Veilleux, A.; Flamand, N.; Muccioli, G.G.; et al. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J. Lipid Res. 2020, 61, 70–85. [Google Scholar] [CrossRef]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Cuddihey, H.; MacNaughton, W.K.; Sharkey, K.A. Role of the Endocannabinoid System in the Regulation of Intestinal Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 947–963. [Google Scholar] [CrossRef]
- Alves, M.; Caldow, M.K.; Trieu, J.; Naim, T.; Montgomery, M.K.; Watt, M.J.; Lynch, G.S.; Koopman, R. Choline administration attenuates aspects of the dystrophic pathology in mdx mice. Clin. Nutr. Exp. 2019, 24, 83–91. [Google Scholar] [CrossRef]
- Rae, C.; Scott, R.B.; Thompson, C.H.; Dixon, R.M.; Dumughn, I.; Kemp, G.J.; Male, A.; Pike, M.; Styles, P.; Radda, G.K. Brain biochemistry in Duchenne muscular dystrophy: A 1H magnetic resonance and neuropsychological study. J. Neurol. Sci. 1998, 160, 148–157. [Google Scholar] [CrossRef]
- Rae, C.; Griffin, J.L.; Blair, D.H.; Bothwell, J.H.; Bubb, W.A.; Maitland, A.; Head, S. Abnormalities in brain biochemistry associated with lack of dystrophin: Studies of the mdx mouse. Neuromuscul. Disord. 2002, 12, 121–129. [Google Scholar] [CrossRef]
- Kemaladewi, D.U.; Benjamin, J.S.; Hyatt, E.; Ivakine, E.A.; Cohn, R.D. Increased polyamines as protective disease modifiers in congenital muscular dystrophy. Hum. Mol. Genet. 2018, 27, 1905–1912. [Google Scholar] [CrossRef]
- Mele, A.; Mantuano, P.; De Bellis, M.; Rana, F.; Sanarica, F.; Conte, E.; Morgese, M.G.; Bove, M.; Rolland, J.F.; Capogrosso, R.F.; et al. A long-term treatment with taurine prevents cardiac dysfunction in mdx mice. Transl. Res. J. Lab. Clin. Med. 2019, 204, 82–99. [Google Scholar] [CrossRef]
- Ren, X.; Xu, H.; Barker, R.G.; Lamb, G.D.; Murphy, R.M. Elevated MMP2 abundance and activity in mdx mice are alleviated by prenatal taurine supplementation. Am. J. Physiol. Cell Physiol. 2020, 318, C1083–C1091. [Google Scholar] [CrossRef]
- Terrill, J.R.; Grounds, M.D.; Arthur, P.G. Taurine deficiency, synthesis and transport in the mdx mouse model for Duchenne Muscular Dystrophy. Int. J. Biochem. Cell Biol. 2015, 66, 141–148. [Google Scholar] [CrossRef]
- Merckx, C.; De Paepe, B. The Role of Taurine in Skeletal Muscle Functioning and Its Potential as a Supportive Treatment for Duchenne Muscular Dystrophy. Metabolites 2022, 12, 193. [Google Scholar] [CrossRef]

{kind=link}
| Protein Intermediates or Metabolites/Sources | Role | Effects on the Skeletal Muscle | References |
|---|---|---|---|
| Peroxisome proliferator-activated receptors (PPARs)/Gene expression | α: cellular uptake, energy homeostasis, and inflammation. | Expression of adipogenic genes, fatty acid metabolism, atrophy, inflammation, and myofiber type switching. | [24,25,26] |
| β/δ: energy expenditure, tissue regeneration and repair, and inflammation. | |||
| γ: energy homeostasis, adipogenesis, triglyceride storage, and deposition of fat. | |||
| Short-chain fatty acids (SCFAs)/Bacterial fermentation of non-digestible carbohydrates | Anti-inflammatory effects through GPR43 binding and the modulation of HDAC activity and cytokine, and PGE2 synthesis. | Regulation of lipid and carbohydrate expression, protein metabolism, and blood flow; and anti-inflammatory properties. | [18,20,31,34] |
| Choline (and its derivatives)/Dietary sources | Liver and neural metabolisms, and metabolism of membrane constituents. | Integrity of skeletal muscle cells and synthesis of acetylcholine (ACh). | [36,37] |
| Polyamines/Decarboxylation of amino acids | Cell growth, metabolism, and development; and antioxidant, anti-inflammatory, and anti-apoptotic effects. | Regulation of atrophy and muscle fiber size via mTORC1. | [39,40,41] |
| Tryptophan (Trp) and its metabolites/Dietary sources and gut microbiota | Inflammation in the gastrointestinal tract, nervous system, and muscles. | Age-related frailty and sarcopenia, and atrophy. | [42,43] |
| Bile acids (BAs)/Cholesterol | Digestion, absorption of dietary lipids and fat-soluble vitamins; lipid and glucose metabolisms; and systemic inflammation. | Regulation of skeletal muscle mass and function; and the involvement in atrophy and sarcopenia. | [6,44,46,47,48,49,51] |
| Taurine (2-aminoethanesulfonic acid)/Dietary sources | Osmotic pressure of different tissues and oxidative stress; and cytoprotective and anti-aging activities. | Myofiber necrosis, protein folding, and mitochondrial activity. | [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostosi, D.; Molinaro, M.; Saccone, S.; Torrente, Y.; Villa, C.; Farini, A. Exploring the Gut Microbiota–Muscle Axis in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 5589. https://doi.org/10.3390/ijms25115589
Mostosi D, Molinaro M, Saccone S, Torrente Y, Villa C, Farini A. Exploring the Gut Microbiota–Muscle Axis in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(11):5589. https://doi.org/10.3390/ijms25115589
Chicago/Turabian StyleMostosi, Debora, Monica Molinaro, Sabrina Saccone, Yvan Torrente, Chiara Villa, and Andrea Farini. 2024. "Exploring the Gut Microbiota–Muscle Axis in Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 11: 5589. https://doi.org/10.3390/ijms25115589
APA StyleMostosi, D., Molinaro, M., Saccone, S., Torrente, Y., Villa, C., & Farini, A. (2024). Exploring the Gut Microbiota–Muscle Axis in Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 25(11), 5589. https://doi.org/10.3390/ijms25115589