
Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis


Abstract
1. Introduction
2. Mitochondria Anatomy, Role, and Functions
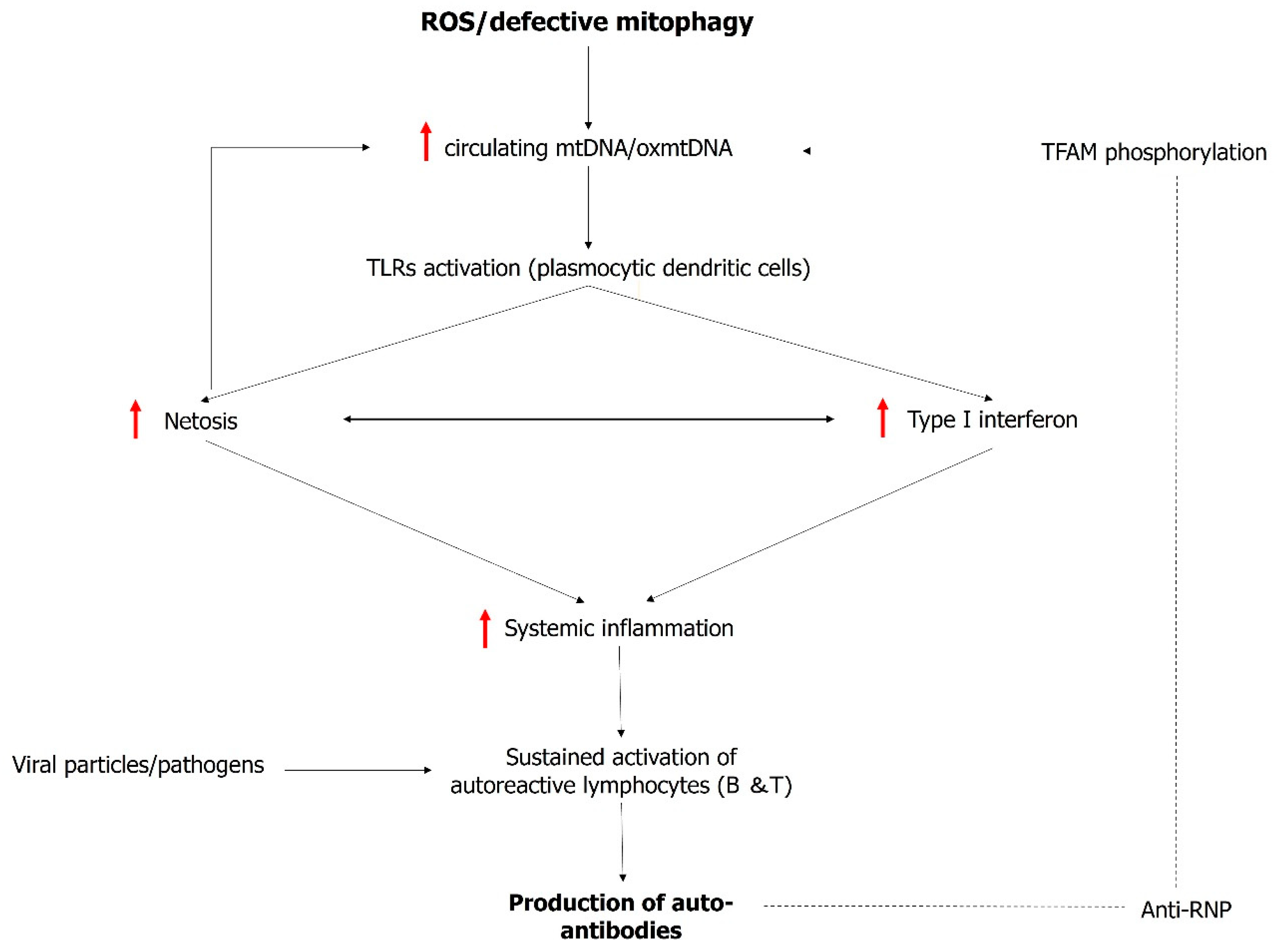
3. Mitochondrial Dysfunction: A Trigger and Amplifier of Type I Interferon
4. Mitochondrial Dysfunction in Lupus Nephritis
5. Mitochondria as Therapeutic Targets in SLE
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Cruz, D.P.; Khamashta, M.A.; Hughes, G.R. Systemic lupus erythematosus. Lancet 2007, 369, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.R.W.; Drenkard, C.; Falasinnu, T.; Hoi, A.; Mak, A.; Kow, N.Y.; Svenungsson, E.; Peterson, J.; Clarke, A.E.; Ramsey-Goldman, R. Global epidemiology of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 515–532. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Tsao, B.P. Current topics in human SLE genetics. Springer Semin. Immunopathol. 2006, 28, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.H. T cells and B cells in lupus nephritis. Semin. Nephrol. 2007, 27, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.S. Lupus nephritis. J. Am. Soc. Nephrol. 1999, 10, 413–424. [Google Scholar] [CrossRef]
- Bono, L.; Cameron, J.S.; Hicks, J.A. The very long-term prognosis and complications of lupus nephritis and its treatment. QJM 1999, 92, 211–218. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.K.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Teng, Y.K.O.; Ginzler, E.M.; Arriens, C.; Caster, D.J.; Romero-Diaz, J.; Gibson, K.; Kaplan, J.; Lisk, L.; Navarra, S.; et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): A double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2070–2080. [Google Scholar] [CrossRef]
- Rovin, B.; Furie, R. B-cell depletion and response in a randomized, controlled trial of obinutuzumab for proliferative lupus nephritis. Ann Rheum Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef]
- Halfon, M.; Bachelet, D.; Hanouna, G.; Dema, B.; Pellefigues, C.; Manchon, P.; Laouenan, C.; Charles, N.; Daugas, E. CD62L on blood basophils: A first pre-treatment predictor of remission in severe lupus nephritis. Nephrol. Dial. Transplant. 2021, 36, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.; Boki, K.A.; Katsorida, M.E.; Drosos, A.A.; Skopouli, F.N.; Boletis, J.N.; Moutsopoulos, H.M. Remission, relapse, and re-remission of proliferative lupus nephritis treated with cyclophosphamide. Kidney Int. 2000, 57, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gong, M.; Park, Y.P.; Elshikha, A.S.; Choi, S.C.; Brown, J.; Kanda, N.; Yeh, W.-I.; Peters, L.; Titov, A.A.; et al. Lupus susceptibility gene Esrrg modulates regulatory T cells through mitochondrial metabolism. JCI Insight 2021, 6, e143540. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.N.; Fernandez, D.R.; Talaber, G.; Oaks, Z.; Haas, M.; Madaio, M.P.; Lai, Z.W.; Miklossy, G.; Singh, R.R.; Chudakov, D.M.; et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis represents a target for treatment in, S.L.E. Ann. Rheum. Dis. 2014, 73, 1888–1897. [Google Scholar] [CrossRef]
- Ma, L.; Roach, T.; Morel, L. Immunometabolic alterations in lupus: Where do they come from and where do we go from there? Curr. Opin. Immunol. 2022, 78, 102245. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Choi, S.C.; Zeumer-Spataro, L.; Scindia, Y.; Moser, E.K.; Morel, L. Metabolic regulation of follicular helper T cell differentiation in a mouse model of lupus. Immunol. Lett. 2022, 247, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.C.; Titov, A.A.; Abboud, G.; Seay, H.R.; Brusko, T.M.; Roopenian, D.C.; Salek-Ardakani, S.; Morel, L. Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat. Commun. 2018, 9, 4369. [Google Scholar] [CrossRef] [PubMed]
- Doke, T.; Susztak, K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. 2022, 32, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Miyazono, Y.; Hirashima, S.; Ishihara, N.; Kusukawa, J.; Nakamura, K.I.; Ohta, K. Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission-independent manner. Sci. Rep. 2018, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Habbane, M.; Montoya, J.; Rhouda, T.; Sbaoui, Y.; Radallah, D.; Emperador, S. Human Mitochondrial DNA: Particularities and Diseases. Biomedicines 2021, 9, 1364. [Google Scholar] [CrossRef]
- Spelbrink, J.N. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life 2010, 62, 19–32. [Google Scholar] [CrossRef]
- Rensch, T.; Villar, D.; Horvath, J.; Odom, D.T.; Flicek, P. Mitochondrial heteroplasmy in vertebrates using ChIP-sequencing data. Genome Biol. 2016, 17, 139. [Google Scholar] [CrossRef]
- Tan, T.; Zimmermann, M.; Reichert, A.S. Controlling quality and amount of mitochondria by mitophagy: Insights into the role of ubiquitination and deubiquitination. Biol. Chem. 2016, 397, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Atici, A.E.; Crother, T.R.; Noval Rivas, M. Mitochondrial quality control in health and cardiovascular diseases. Front. Cell Dev. Biol. 2023, 11, 1290046. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef]
- Guo, H.J.; Rahimi, N.; Tadi, P. Biochemistry, Ubiquitination; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Pattingre, S.; Turtoi, A. BAG Family Members as Mitophagy Regulators in Mammals. Cells 2022, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Hua, F.; Fang, P.; Li, C.; Deng, F.; Chen, S.; Ying, J.; Wang, X. Regulation of Mitophagy by Sirtuin Family Proteins: A Vital Role in Aging and Age-Related Diseases. Front. Aging Neurosci. 2022, 14, 845330. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, J.; Gao, C.; Wu, M.; Du, Q.; Tsoi, B.; Wang, Q.; Yang, D.; Shen, J. Nitration of Drp1 provokes mitophagy activation mediating neuronal injury in experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2019, 143, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Talaber, G.; Miklossy, G.; Oaks, Z.; Liu, Y.; Tooze, S.A.; Chudakov, D.M.; Banki, K.; Perl, A. HRES-1/Rab4 promotes the formation of LC3(+) autophagosomes and the accumulation of mitochondria during autophagy. PLoS ONE 2014, 9, e84392. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Tummala, R.; Streicher, K.; Nogueira da Costa, A.; Brohawn, P.Z. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 11286. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Wittmann, M.; Vital, E.M. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat. Rev. Rheumatol. 2022, 18, 575–590. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef]
- Skopelja-Gardner, S.; Tai, J.; Sun, X.; Tanaka, L.; Kuchenbecker, J.A.; Snyder, J.M.; Kubes, P.; Mustelin, T.; Elkon, K.B. Acute skin exposure to ultraviolet light triggers neutrophil-mediated kidney inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019097118. [Google Scholar] [CrossRef]
- Castellano, G.; Cafiero, C.; Divella, C.; Sallustio, F.; Gigante, M.; Pontrelli, P.; De Palma, G.; Rossini, M.; Grandaliano, G.; Gesualdo, L. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthritis Res Ther. 2015, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Zhao, Y.; Pandya, K.; Yap, G.S. Growth differentiation factor-15 is an IFN-gamma regulated mediator of infection-induced weight loss and the hepatic FGF21 response. Brain Behav. Immun. 2024, 116, 24–33. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.A.; van Vollenhoven, R.F.; Kalunian, K.; Navarra, S.; Romero-Diaz, J.; Werth, V.P.; Huang, X.; Clark, G.; Carroll, H.; Meyers, A.; et al. Trial of Anti-BDCA2 Antibody Litifilimab for Systemic Lupus Erythematosus. N. Engl. J. Med. 2022, 387, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Vassilopoulos, D.; Boletis, J.; Boumpas, D.T. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmun. Rev. 2019, 18, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 484. [Google Scholar] [CrossRef]
- Fortner, K.A.; Blanco, L.P.; Buskiewicz, I.; Huang, N.; Gibson, P.C.; Cook, D.L.; Pedersen, H.L.; Yuen, P.S.; Murphy, M.; Perl, A.; et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci. Med. 2020, 7, e000387. [Google Scholar] [CrossRef]
- Melki, I.; Allaeys, I.; Tessandier, N.; Levesque, T.; Cloutier, N.; Laroche, A.; Vernoux, N.; Becker, Y.; Benk-Fortin, H.; Zufferey, A.; et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci. Transl. Med. 2021, 13, eaav5928. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Schoeniger, A.; Edlich, F. Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.N.; Talaber, G.; Perl, A. Metabolic regulation of organelle homeostasis in lupus T cells. Clin. Immunol. 2012, 144, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Katsuyama, E.; Satyam, A.; Li, H.; Rubio, J.; Jung, S.; Andrzejewski, S.; Becherer, J.D.; Tsokos, M.G.; Abdi, R.; et al. CD38 reduces mitochondrial fitness and cytotoxic T cell response against viral infection in lupus patients by suppressing mitophagy. Sci. Adv. 2022, 8, eabo4271. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Cardenas, J.; de Jesus, A.A.; Baisch, J.; Walters, L.; Blanck, J.P.; Balasubramanian, P.; Stagnar, C.; Ohouo, M.; Hong, S.; et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human, S.L.E. Cell 2021, 184, 4464–4479. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Jiao, C.; Ma, C.; Zhang, Y.; Hao, X.; Zhou, G.; Fu, J.; Qiu, X.; Li, H.; Yang, W.; et al. circMTND5 Participates in Renal Mitochondrial Injury and Fibrosis by Sponging MIR6812 in Lupus Nephritis. Oxid. Med. Cell. Longev. 2022, 2022, 2769487. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, H.; Miao, X.; Xu, J.; Yang, R.; Zhao, L.; Liu, J.; Yang, L.; Gao, F.; Zhang, W.; et al. Nestin protects podocyte from injury in lupus nephritis by mitophagy and oxidative stress. Cell Death Dis. 2020, 11, 319. [Google Scholar] [CrossRef]
- Lee, H.T.; Lin, C.S.; Pan, S.C.; Chen, W.S.; Tsai, C.Y.; Wei, Y.H. The Role of Plasma Cell-Free Mitochondrial DNA and Nuclear DNA in Systemic Lupus Erythematosus. Front. Biosci. 2022, 27, 333. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Fenton, K. The effect of cell death in the initiation of lupus nephritis. Clin. Exp. Immunol. 2015, 179, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Kaplan, M.J. Lupus neutrophils: ‘NET’ gain in understanding lupus pathogenesis. Curr. Opin. Rheumatol. 2012, 24, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Truszewska, A.; Wirkowska, A.; Gala, K.; Truszewski, P.; Krzemien-Ojak, L.; Perkowska-Ptasinska, A.; Mucha, K.; Pączek, L.; Foroncewicz, B. Cell-free DNA profiling in patients with lupus nephritis. Lupus 2020, 29, 1759–1772. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lin, C.S.; Chen, W.S.; Liao, H.T.; Tsai, C.Y.; Wei, Y.H. Leukocyte mitochondrial DNA alteration in systemic lupus erythematosus and its relevance to the susceptibility to lupus nephritis. Int. J. Mol. Sci. 2012, 13, 8853–8868. [Google Scholar] [CrossRef] [PubMed]
- Mambo, E.; Gao, X.; Cohen, Y.; Guo, Z.; Talalay, P.; Sidransky, D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homoplasmic mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Wang, H.; Shen, M.; Ma, Y.; Lan, L.; Jiang, X.; Cen, X.; Guo, G.; Zhou, Q.; Yuan, M.; Chen, J.; et al. Novel mitophagy inducer alleviates lupus nephritis by reducing myeloid cell activation and autoantigen presentation. Kidney Int. 2024, 105, 759–774. [Google Scholar] [CrossRef]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus With Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Standard Therapy for Lupus Nephritis | |||
|---|---|---|---|
| Drugs | Mechanism of action | Indication | Comments |
| Cyclophosphamide with corticosteroid * | Alkylating agent: reduces number of lymphocytes (both B and T cells) | Induction phase | Two regimens: Eurolupus protocol: IV cyclophosphamide 500 mg every 2 weeks for a total of six doses (3 months). NIH protocol: IV cyclophosphamide 500–1000 mg/m2 once a month for 6 months. |
| Mycophenolate mofetil with corticosteroid * | Inhibits the synthesis of guanine nucleotides | Both induction and maintenance phase | MMF 2–3 g per day, divided into two doses (1–1.5 g twice daily) for induction phase. MMF 1–2 g per day, divided into two doses (0.5–1 g twice daily) for maintenance phase. |
| Belimumab with SOC | Monoclonal antibody that inhibits the BAFF pathway | Both induction and maintenance phase | Use as a corticoid-sparing agent for articular SLE. Duration of treatment: two years (BLISS-LN study). |
| Non-Standard Therapy for Lupus Nephritis | |||
| Drugs | Mechanism of action | Indication | Comments |
| Rituximab | Monoclonal antibody that targets CD20 protein, which is expressed on the surface of pre-B and mature B lymphocytes | Induction phase or as rescue therapy | Could be used as monotherapy for inducing remission in pure Lupus nephritis Class V. Could be used in conjunction with low dose of MMF for induction remission in active LN (Rituxilup protocol). |
| Voclosporin with MMF and low dose of corticoid | Calcineurin inhibitor: inhibition of T cell activation | Both induction and maintenance phase | Duration of treatment up to three years (Aurora 2 study). |
| Obinutuzumab with MMF and low dose of corticoid | Monoclonal antibody that targets CD20 protein. More potent than rituximab and notably enhances antibody-dependent cellular cytotoxicity and cellular apoptosis | Induction phase | Obinutuzumab 1000 mg at day 1, then weeks 2, 24, and 26. |
| Tacrolimus with low dose of MMF and lose dose of corticoid | Calcineurin inhibitor: inhibition of T cell activation | Induction and maintenance phase | Part of the multitarget therapy. MMF 1–1.5 g twice daily. Tacrolimus: 2–4 mg daily, adjusted based on blood levels and clinical response. |
| Drug | Mechanism of Action on Mitochondria | Efficacy in SLE |
|---|---|---|
| Metformin | Inhibits the mitochondrial enzyme complex: mitochondrial glycerophosphate dehydrogenase | Increased renal function and reduced glomerular inflammation in a murine lupus model. No effect on lupus flare in a small, randomized control study. |
| Sirolimus | Restoration of mitophagy by interaction with HRES-1/Rab4 expression | Decrease in BILAG and in total dose of corticoid in patients with persistent SLE activity in a single-arm trial. |
| MitoQ | Mitochondrial antioxidant | Reduced NET formation, serum IFN-I, reduced immune complex deposit in kidneys in a murine model. |
| Idebenone | Analogue of coenzyme Q10 | Decreased glomerular inflammation and fibrosis and decreased NET formation in murine model. |
| UMI-77 | Inducer of mitophagy by interacting with the BAK/BAX pathway | Reduced glomerular inflammation, notably by decreasing T cell infiltration in the kidney in a murine model of LN. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halfon, M.; Tankeu, A.T.; Ribi, C. Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis. Int. J. Mol. Sci. 2024, 25, 6162. https://doi.org/10.3390/ijms25116162
Halfon M, Tankeu AT, Ribi C. Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis. International Journal of Molecular Sciences. 2024; 25(11):6162. https://doi.org/10.3390/ijms25116162
Chicago/Turabian StyleHalfon, Matthieu, Aurel T. Tankeu, and Camillo Ribi. 2024. "Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis" International Journal of Molecular Sciences 25, no. 11: 6162. https://doi.org/10.3390/ijms25116162
APA StyleHalfon, M., Tankeu, A. T., & Ribi, C. (2024). Mitochondrial Dysfunction in Systemic Lupus Erythematosus with a Focus on Lupus Nephritis. International Journal of Molecular Sciences, 25(11), 6162. https://doi.org/10.3390/ijms25116162