
Interplay between mTOR and Purine Metabolism Enzymes and Its Relevant Role in Cancer
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Purine “De Novo” Synthesis and Mitochondria
2.1. Purinosome and Mitochondria
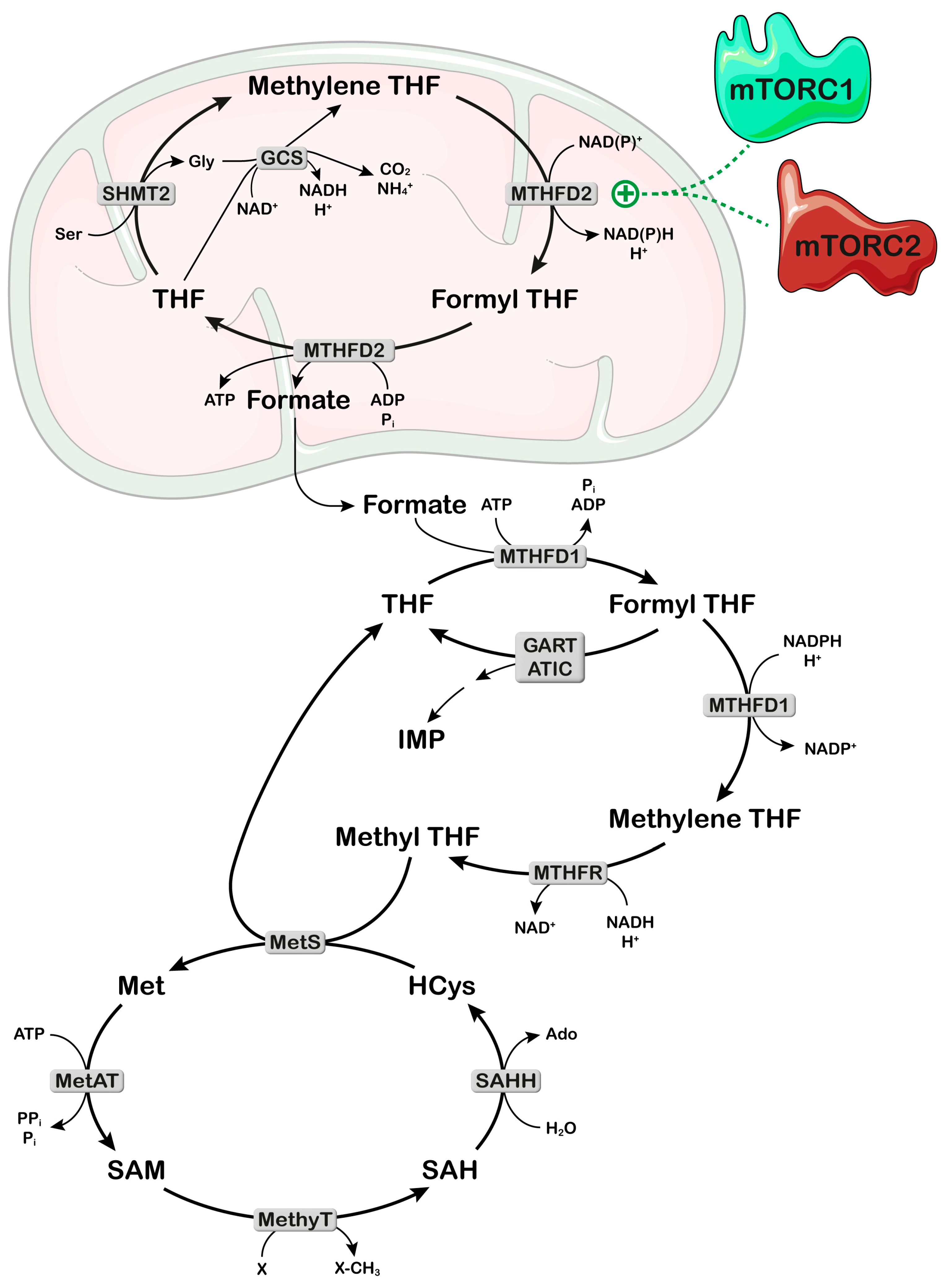
2.2. Mitochondria and One-Carbon Metabolism
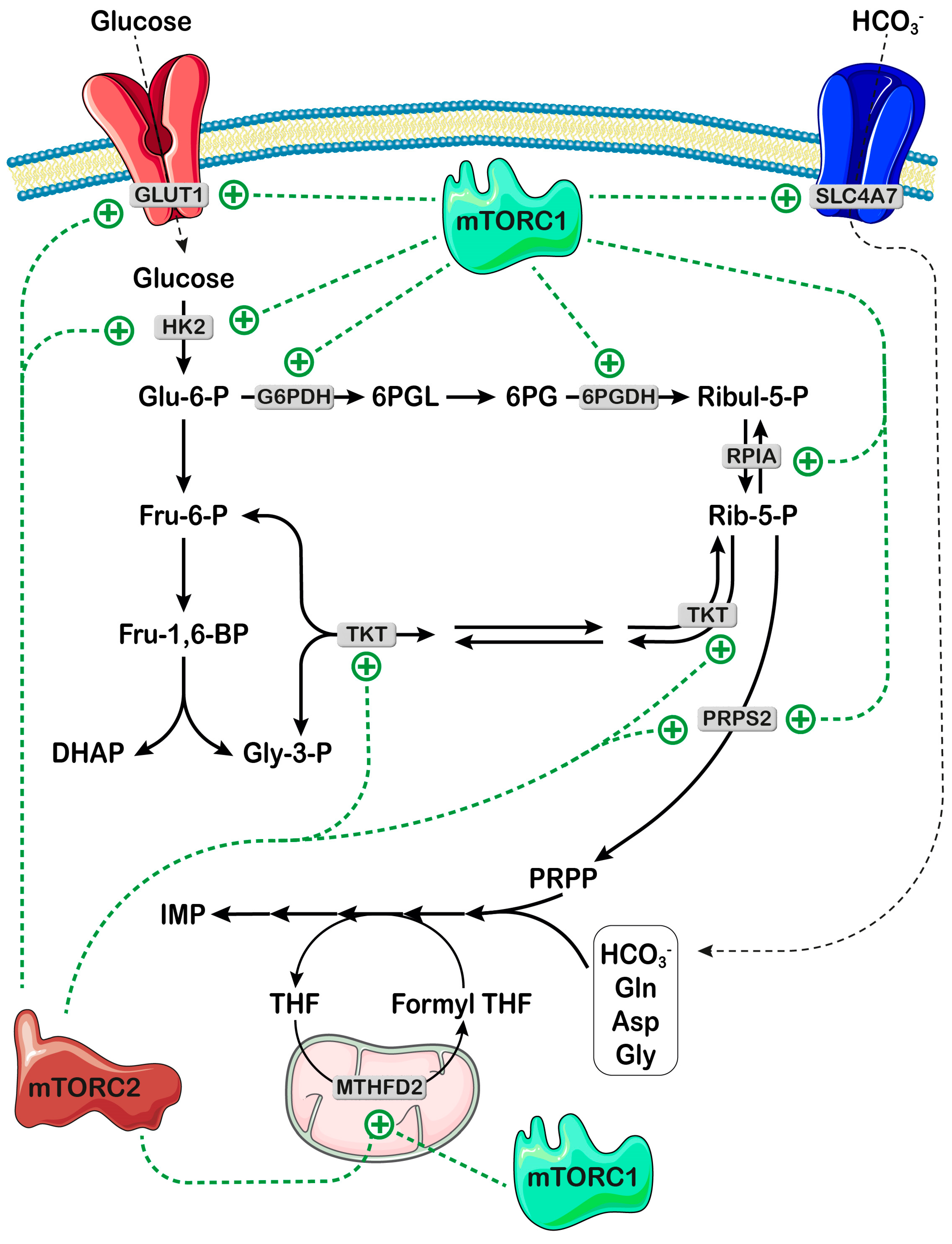
3. mTOR Signaling Controls Availability of Both Bicarbonate and PRPP for Nucleotide Synthesis
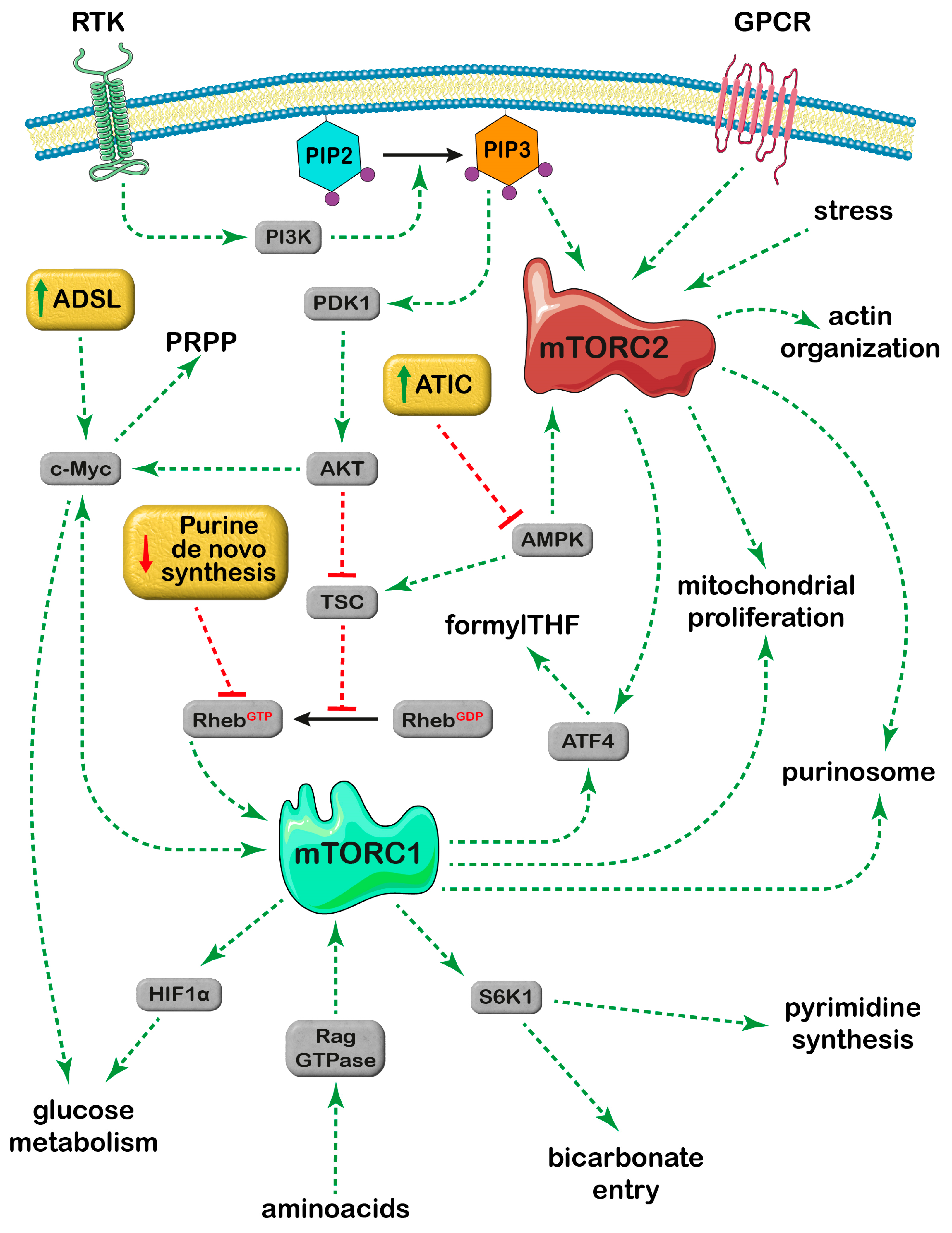
4. Effect of Purine Metabolizing Enzymes on the mTOR Signaling Pathways
4.1. ADSL and mTOR
4.2. ATIC and mTOR
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol. Biol. Evol. 2002, 19, 640–653. [Google Scholar] [CrossRef]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef]
- French, J.B.; Jones, S.A.; Deng, H.; Pedley, A.M.; Kim, D.; Chan, C.Y.; Hu, H.; Pugh, R.J.; Zhao, H.; Zhang, Y.; et al. Spatial colocalization and functional link of purinosomes with mitochondria. Science 2016, 351, 733–737. [Google Scholar] [CrossRef]
- Zhao, H.; Chiaro, C.R.; Zhang, L.; Smith, P.B.; Chan, C.Y.; Pedley, A.M.; Pugh, R.J.; French, J.B.; Patterson, A.D.; Benkovic, S.J. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 2015, 290, 6705–6713. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kondo, M.; Honda, S.; Iwahana, H.; Moritani, M.; Ii, S.; Yoshimoto, K.; Itakura, M. Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 1997, 272, 17719–17725. [Google Scholar] [CrossRef]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef]
- Shi, D.D.; Savani, M.R.; Abdullah, K.G.; McBrayer, S.K. Emerging roles of nucleotide metabolism in cancer. Trends Cancer 2023, 9, 624–635. [Google Scholar] [CrossRef]
- Camici, M.; Garcia-Gil, M.; Pesi, R.; Allegrini, S.; Tozzi, M.G. Purine-Metabolising Enzymes and Apoptosis in Cancer. Cancers 2019, 11, 1354. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Petrotto, E.; Tozzi, M.G. Emerging Role of Purine Metabolizing Enzymes in Brain Function and Tumors. Int. J. Mol. Sci. 2018, 19, 3598. [Google Scholar] [CrossRef]
- Mullen, N.J.; Singh, P.K. Nucleotide metabolism: A pan-cancer metabolic dependency. Nat. Rev. Cancer 2023, 23, 275–294. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Kondo, M.; Yamaoka, T.; Honda, S.; Miwa, Y.; Katashima, R.; Moritani, M.; Yoshimoto, K.; Hayashi, Y.; Itakura, M. The rate of cell growth is regulated by purine biosynthesis via ATP production and G(1) to S phase transition. J. Biochem. 2000, 128, 57–64. [Google Scholar] [CrossRef]
- Liu, C.; Knudsen, G.M.; Pedley, A.M.; He, J.; Johnson, J.L.; Yaron, T.M.; Cantley, L.C.; Benkovic, S.J. Mapping Post-Translational Modifications of de Novo Purine Biosynthetic Enzymes: Implications for Pathway Regulation. J. Proteome Res. 2019, 18, 2078–2087. [Google Scholar] [CrossRef]
- Deng, Y.; Gam, J.; French, J.B.; Zhao, H.; An, S.; Benkovic, S.J. Mapping protein-protein proximity in the purinosome. J. Biol. Chem. 2012, 287, 36201–36207. [Google Scholar] [CrossRef]
- French, J.B.; Zhao, H.; An, S.; Niessen, S.; Deng, Y.; Cravatt, B.F.; Benkovic, S.J. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. USA 2013, 110, 2528–2533. [Google Scholar] [CrossRef]
- Chan, C.Y.; Pedley, A.M.; Kim, D.; Xia, C.; Zhuang, X.; Benkovic, S.J. Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 13009–13014. [Google Scholar] [CrossRef]
- Doigneaux, C.; Pedley, A.M.; Mistry, I.N.; Papayova, M.; Benkovic, S.J.; Tavassoli, A. Hypoxia drives the assembly of the multienzyme purinosome complex. J. Biol. Chem. 2020, 295, 9551–9566. [Google Scholar] [CrossRef]
- Chou, M.C.; Wang, Y.H.; Chen, F.Y.; Kung, C.Y.; Wu, K.P.; Kuo, J.C.; Chan, S.J.; Cheng, M.L.; Lin, C.Y.; Chou, Y.C.; et al. PAICS ubiquitination recruits UBAP2 to trigger phase separation for purinosome assembly. Mol. Cell 2023, 83, 4123–4140.e4112. [Google Scholar] [CrossRef]
- Fang, Y.; French, J.; Zhao, H.; Benkovic, S. G-protein-coupled receptor regulation of de novo purine biosynthesis: A novel druggable mechanism. Biotechnol. Genet. Eng. Rev. 2013, 29, 31–48. [Google Scholar] [CrossRef]
- Venerando, A.; Ruzzene, M.; Pinna, L.A. Casein kinase: The triple meaning of a misnomer. Biochem. J. 2014, 460, 141–156. [Google Scholar] [CrossRef]
- Ali, E.S.; Sahu, U.; Villa, E.; O’Hara, B.P.; Gao, P.; Beaudet, C.; Wood, A.W.; Asara, J.M.; Ben-Sahra, I. ERK2 Phosphorylates PFAS to Mediate Posttranslational Control of De Novo Purine Synthesis. Mol. Cell 2020, 78, 1178–1191.e1176. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Pedley, A.M.; Karras, G.I.; Zhang, X.; Lindquist, S.; Benkovic, S.J. Role of HSP90 in the Regulation of de Novo Purine Biosynthesis. Biochemistry 2018, 57, 3217–3221. [Google Scholar] [CrossRef]
- Pedley, A.M.; Boylan, J.P.; Chan, C.Y.; Kennedy, E.L.; Kyoung, M.; Benkovic, S.J. Purine biosynthetic enzymes assemble into liquid-like condensates dependent on the activity of chaperone protein HSP90. J. Biol. Chem. 2022, 298, 101845. [Google Scholar] [CrossRef]
- Chou, S.D.; Prince, T.; Gong, J.; Calderwood, S.K. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS ONE 2012, 7, e39679. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef]
- Loffler, M.; Fairbanks, L.D.; Zameitat, E.; Marinaki, A.M.; Simmonds, H.A. Pyrimidine pathways in health and disease. Trends Mol. Med. 2005, 11, 430–437. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Tedeschi, P.M.; Markert, E.K.; Gounder, M.; Lin, H.; Dvorzhinski, D.; Dolfi, S.C.; Chan, L.L.; Qiu, J.; DiPaola, R.S.; Hirshfield, K.M.; et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013, 4, e877. [Google Scholar] [CrossRef]
- Li, A.M.; Ye, J. Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165841. [Google Scholar] [CrossRef]
- Walling, J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Investig. New Drugs 2006, 24, 37–77. [Google Scholar] [CrossRef]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Li, R.; Wilson, K.F.; Cerione, R.A. Elucidation of an mTORC2-PKC-NRF2 pathway that sustains the ATF4 stress response and identification of Sirt5 as a key ATF4 effector. Cell Death Discov. 2022, 8, 357. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Nagahama, Y.; Shimoda, M.; Mao, G.; Singh, S.K.; Kozakai, Y.; Sun, X.; Motooka, D.; Nakamura, S.; Tanaka, H.; Satoh, T.; et al. Regnase-1 controls colon epithelial regeneration via regulation of mTOR and purine metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, 11036–11041. [Google Scholar] [CrossRef]
- Ali, E.S.; Liponska, A.; O’Hara, B.P.; Amici, D.R.; Torno, M.D.; Gao, P.; Asara, J.M.; Yap, M.F.; Mendillo, M.L.; Ben-Sahra, I. The mTORC1-SLC4A7 axis stimulates bicarbonate import to enhance de novo nucleotide synthesis. Mol. Cell 2022, 82, 3284–3298.e3287. [Google Scholar] [CrossRef]
- Nosal, J.M.; Switzer, R.L.; Becker, M.A. Overexpression, purification, and characterization of recombinant human 5-phosphoribosyl-1-pyrophosphate synthetase isozymes I and II. J. Biol. Chem. 1993, 268, 10168–10175. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Q.; Zhang, P.; Tan, W.; Li, Y.; Xu, Z.; Ma, J.; Kupfer, G.M.; Pei, Y.; Song, Q.; et al. Direct stimulation of de novo nucleotide synthesis by O-GlcNAcylation. Nat. Chem. Biol. 2024, 20, 19–29. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Tan, L.; Lee, J.H.; Xia, Y.; Cai, Q.; Zheng, Y.; Wang, H.; Lorenzi, P.L.; Lu, Z. Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy Stress. Cancer Discov. 2018, 8, 94–107. [Google Scholar] [CrossRef]
- Becker, M.A.; Kim, M. Regulation of purine synthesis de novo in human fibroblasts by purine nucleotides and phosphoribosylpyrophosphate. J. Biol. Chem. 1987, 262, 14531–14537. [Google Scholar] [CrossRef]
- Walter, M.; Herr, P. Re-Discovery of Pyrimidine Salvage as Target in Cancer Therapy. Cells 2022, 11, 739. [Google Scholar] [CrossRef]
- Fridman, A.; Saha, A.; Chan, A.; Casteel, D.E.; Pilz, R.B.; Boss, G.R. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 2013, 454, 91–99. [Google Scholar] [CrossRef]
- Camici, M.; Micheli, V.; Ipata, P.L.; Tozzi, M.G. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem. Int. 2010, 56, 367–378. [Google Scholar] [CrossRef]
- Fu, R.; Sutcliffe, D.; Zhao, H.; Huang, X.; Schretlen, D.J.; Benkovic, S.; Jinnah, H.A. Clinical severity in Lesch-Nyhan disease: The role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 2015, 114, 55–61. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Buj, R.; Chen, C.W.; Dahl, E.S.; Leon, K.E.; Kuskovsky, R.; Maglakelidze, N.; Navaratnarajah, M.; Zhang, G.; Doan, M.T.; Jiang, H.; et al. Suppression of p16 Induces mTORC1-Mediated Nucleotide Metabolic Reprogramming. Cell Rep. 2019, 28, 1971–1980.e1978. [Google Scholar] [CrossRef]
- Saha, A.; Connelly, S.; Jiang, J.; Zhuang, S.; Amador, D.T.; Phan, T.; Pilz, R.B.; Boss, G.R. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol. Cell 2014, 55, 264–276. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef]
- Song, L.; Li, P.; Sun, H.; Ding, L.; Wang, J.; Li, B.; Zhou, B.S.; Feng, H.; Li, Y. PRPS2 mutations drive acute lymphoblastic leukemia relapse through influencing PRPS1/2 hexamer stability. Blood Sci. 2023, 5, 39–50. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Hughes-Hallett, J.; Timson, R.C.; Ilagan, E.; Yuan, M.; Asara, J.M.; Ben-Sahra, I.; Manning, B.D. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017, 21, 1331–1346. [Google Scholar] [CrossRef]
- Emmanuel, N.; Ragunathan, S.; Shan, Q.; Wang, F.; Giannakou, A.; Huser, N.; Jin, G.; Myers, J.; Abraham, R.T.; Unsal-Kacmaz, K. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 2017, 19, 2665–2680. [Google Scholar] [CrossRef]
- Valvezan, A.J.; McNamara, M.C.; Miller, S.K.; Torrence, M.E.; Asara, J.M.; Henske, E.P.; Manning, B.D. IMPDH inhibitors for antitumor therapy in tuberous sclerosis complex. JCI Insight 2020, 5, e135071. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638.e5. [Google Scholar] [CrossRef]
- Lam, H.C.; Nijmeh, J.; Henske, E.P. New developments in the genetics and pathogenesis of tumours in tuberous sclerosis complex. J. Pathol. 2017, 241, 219–225. [Google Scholar] [CrossRef]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Taha-Mehlitz, S.; Bianco, G.; Coto-Llerena, M.; Kancherla, V.; Bantug, G.R.; Gallon, J.; Ercan, C.; Panebianco, F.; Eppenberger-Castori, S.; von Strauss, M.; et al. Adenylosuccinate lyase is oncogenic in colorectal cancer by causing mitochondrial dysfunction and independent activation of NRF2 and mTOR-MYC-axis. Theranostics 2021, 11, 4011–4029. [Google Scholar] [CrossRef]
- Li, M.; Jin, C.; Xu, M.; Zhou, L.; Li, D.; Yin, Y. Bifunctional enzyme ATIC promotes propagation of hepatocellular carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Commun. Signal 2017, 15, 52. [Google Scholar] [CrossRef]
- Niu, N.; Zeng, J.; Ke, X.; Zheng, W.; Fu, C.; Lv, S.; Fu, J.; Yu, Y. ATIC facilitates cell growth and migration by upregulating Myc expression in lung adenocarcinoma. Oncol. Lett. 2022, 23, 131. [Google Scholar] [CrossRef]
- Camici, M.; Garcia-Gil, M.; Allegrini, S.; Pesi, R.; Bernardini, G.; Micheli, V.; Tozzi, M.G. Inborn Errors of Purine Salvage and Catabolism. Metabolites 2023, 13, 787. [Google Scholar] [CrossRef]
- Zurlo, G.; Liu, X.; Takada, M.; Fan, C.; Simon, J.M.; Ptacek, T.S.; Rodriguez, J.; von Kriegsheim, A.; Liu, J.; Locasale, J.W.; et al. Prolyl hydroxylase substrate adenylosuccinate lyase is an oncogenic driver in triple negative breast cancer. Nat. Commun. 2019, 10, 5177. [Google Scholar] [CrossRef]
- Park, H.; Ohshima, K.; Nojima, S.; Tahara, S.; Kurashige, M.; Hori, Y.; Okuzaki, D.; Wada, N.; Ikeda, J.I.; Morii, E. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab. Investig. 2018, 98, 449–461. [Google Scholar] [CrossRef]
- Terzuoli, L.; Carlucci, F.; Martino, A.D.; Frosi, B.; Porcelli, B.; Minacci, C.; Vernillo, R.; Baldi, L.; Marinello, E.; Pagani, R.; et al. Determination of p185 and adenylosuccinate lyase (ASL) activity in preneoplastic colon lesions and intestinal mucosa of human subjects. Clin. Biochem. 1998, 31, 523–528. [Google Scholar] [CrossRef]
- Valizadeh Osalo, M.; Hosseini, P.; Charkhian, H.; Soltanzadeh, H.; Goharkhany, S.; Tuncer, S.B. The prevalence of ADSL (rs3788579) and CYP1A2 (rs17861162) polymorphisms in female breast cancer patients in North-West Iran. Discov. Oncol. 2024, 15, 59. [Google Scholar] [CrossRef]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef]
- Yin, J.; Ge, X.; Ding, F.; He, L.; Song, K.; Shi, Z.; Ge, Z.; Zhang, J.; Ji, J.; Wang, X.; et al. Reactivating PTEN to impair glioma stem cells by inhibiting cytosolic iron-sulfur assembly. Sci. Transl. Med. 2024, 16, eadg5553. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Di Cristofano, A.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal structure of the PTEN tumor suppressor: Implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef]
- Camici, M.; Allegrini, S.; Tozzi, M.G. Interplay between adenylate metabolizing enzymes and AMP-activated protein kinase. FEBS J. 2018, 285, 3337–3352. [Google Scholar] [CrossRef]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. Bioessays 2001, 23, 1112–1119. [Google Scholar] [CrossRef]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef]
- Li, R.; Chen, G.; Dang, Y.; He, R.; Liu, A.; Ma, J.; Wang, C. Upregulation of ATIC in multiple myeloma tissues based on tissue microarray and gene microarrays. Int. J. Lab. Hematol. 2021, 43, 409–417. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, P.; Liu, J.; Chen, Z.; Ma, W.; Yuan, Y. ATIC inhibits autophagy in hepatocellular cancer through the AKT/FOXO3 pathway and serves as a prognostic signature for modeling patient survival. Int. J. Biol. Sci. 2021, 17, 4442–4458. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef]
- Lu, X.; Paliogiannis, P.; Calvisi, D.F.; Chen, X. Role of the Mammalian Target of Rapamycin Pathway in Liver Cancer: From Molecular Genetics to Targeted Therapies. Hepatology 2021, 73 (Suppl. S1), 49–61. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, M.; Liu, P.; Zhang, S.; Shang, R.; Qiao, Y.; Che, L.; Ribback, S.; Cigliano, A.; Evert, K.; et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019, 70, 1600–1613. [Google Scholar] [CrossRef]
- Ragupathi, A.; Kim, C.; Jacinto, E. The mTORC2 signaling network: Targets and cross-talks. Biochem. J. 2024, 481, 45–91. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Abad, E.; Samino, S.; Grodzicki, R.L.; Pagano, G.; Trifuoggi, M.; Graifer, D.; Potesil, D.; Zdrahal, Z.; Yanes, O.; Lyakhovich, A. Identification of metabolic changes leading to cancer susceptibility in Fanconi anemia cells. Cancer Lett. 2021, 503, 185–196. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegrini, S.; Camici, M.; Garcia-Gil, M.; Pesi, R.; Tozzi, M.G. Interplay between mTOR and Purine Metabolism Enzymes and Its Relevant Role in Cancer. Int. J. Mol. Sci. 2024, 25, 6735. https://doi.org/10.3390/ijms25126735
Allegrini S, Camici M, Garcia-Gil M, Pesi R, Tozzi MG. Interplay between mTOR and Purine Metabolism Enzymes and Its Relevant Role in Cancer. International Journal of Molecular Sciences. 2024; 25(12):6735. https://doi.org/10.3390/ijms25126735
Chicago/Turabian StyleAllegrini, Simone, Marcella Camici, Mercedes Garcia-Gil, Rossana Pesi, and Maria Grazia Tozzi. 2024. "Interplay between mTOR and Purine Metabolism Enzymes and Its Relevant Role in Cancer" International Journal of Molecular Sciences 25, no. 12: 6735. https://doi.org/10.3390/ijms25126735
APA StyleAllegrini, S., Camici, M., Garcia-Gil, M., Pesi, R., & Tozzi, M. G. (2024). Interplay between mTOR and Purine Metabolism Enzymes and Its Relevant Role in Cancer. International Journal of Molecular Sciences, 25(12), 6735. https://doi.org/10.3390/ijms25126735