

The Brain–Heart Network of Syncope


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Early Recognition of Syncope and Hints to Its Mechanism
Types of Syncope
3. The Basic Anatomy of Syncope
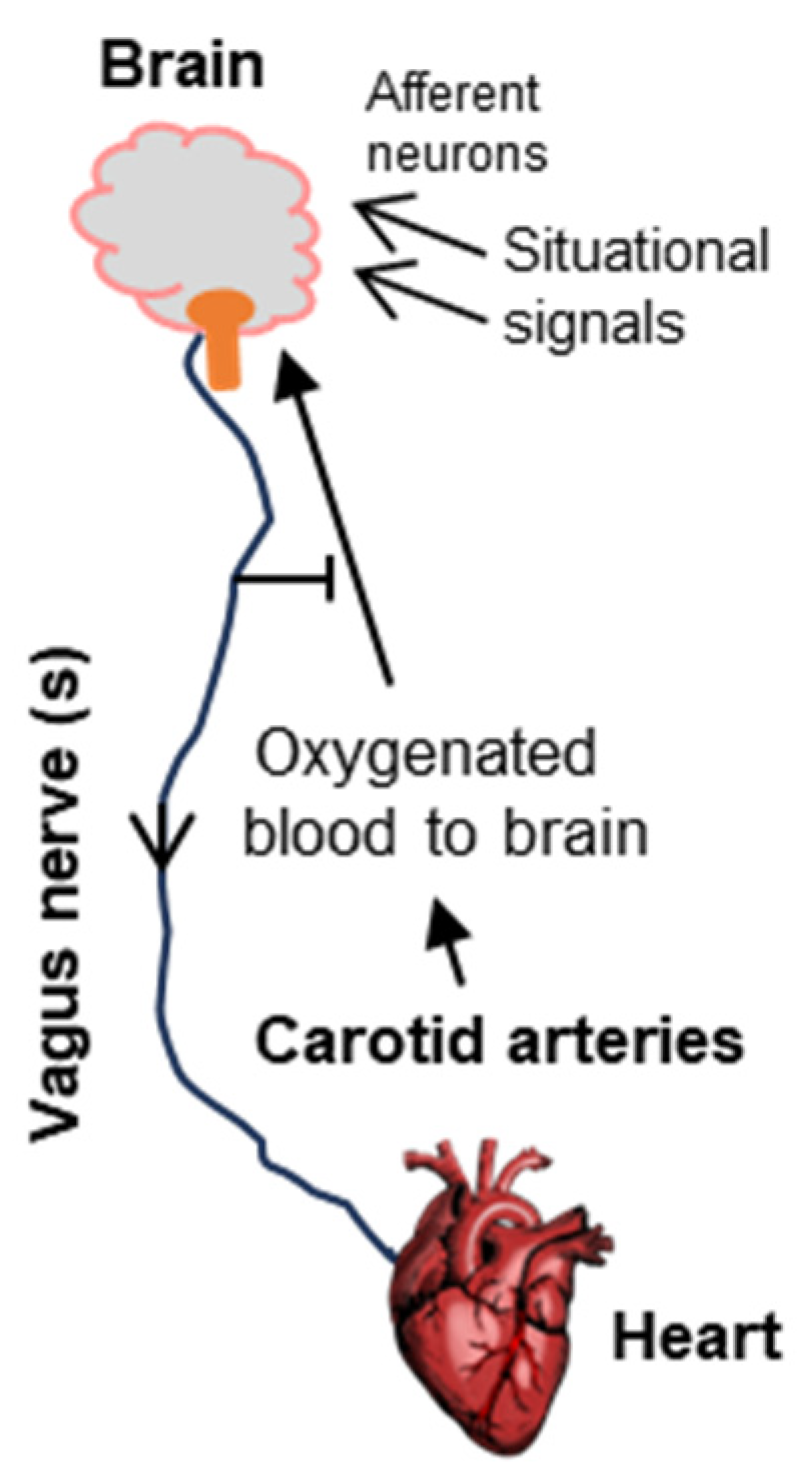
3.1. The Vagus Nerve
3.2. The GPCR Feature of A1/A2A Receptors and NPY2R
3.3. The Brain and the Heart
4. Molecular Signaling in Syncope
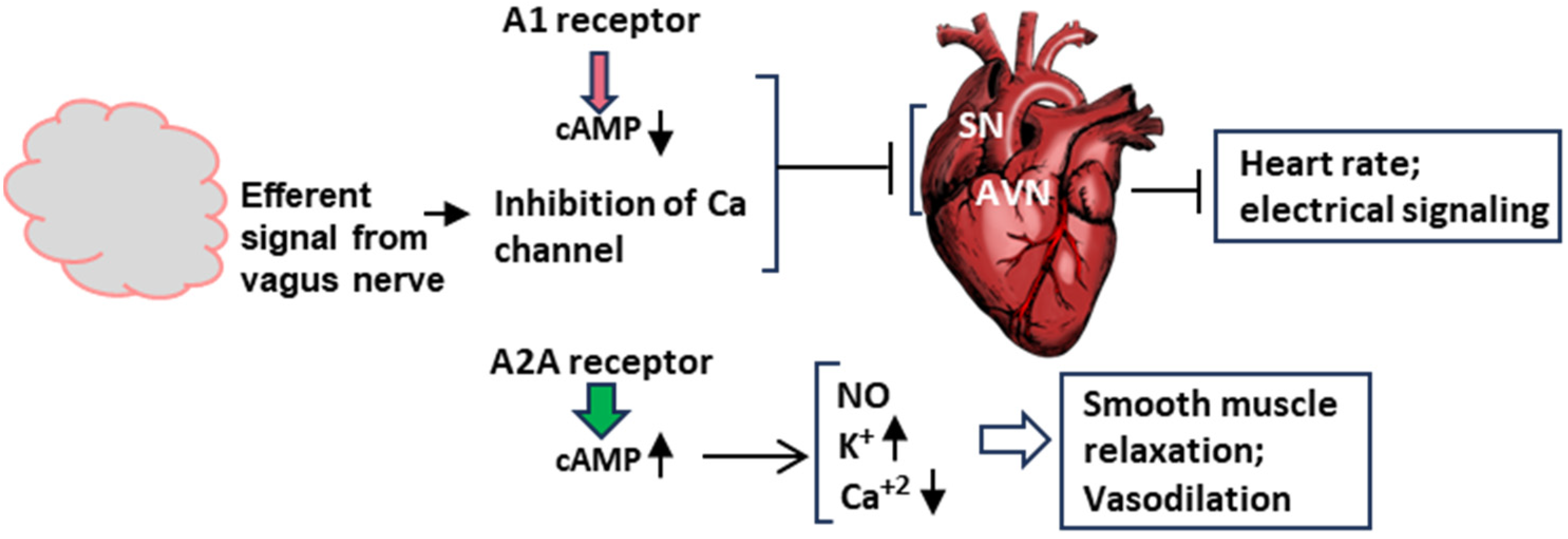
4.1. Adenosinergic Signaling
4.2. Molecular Signaling Pathways of Syncope at the Organ Level
5. Potential Interventions Based on Known Signaling Pathways
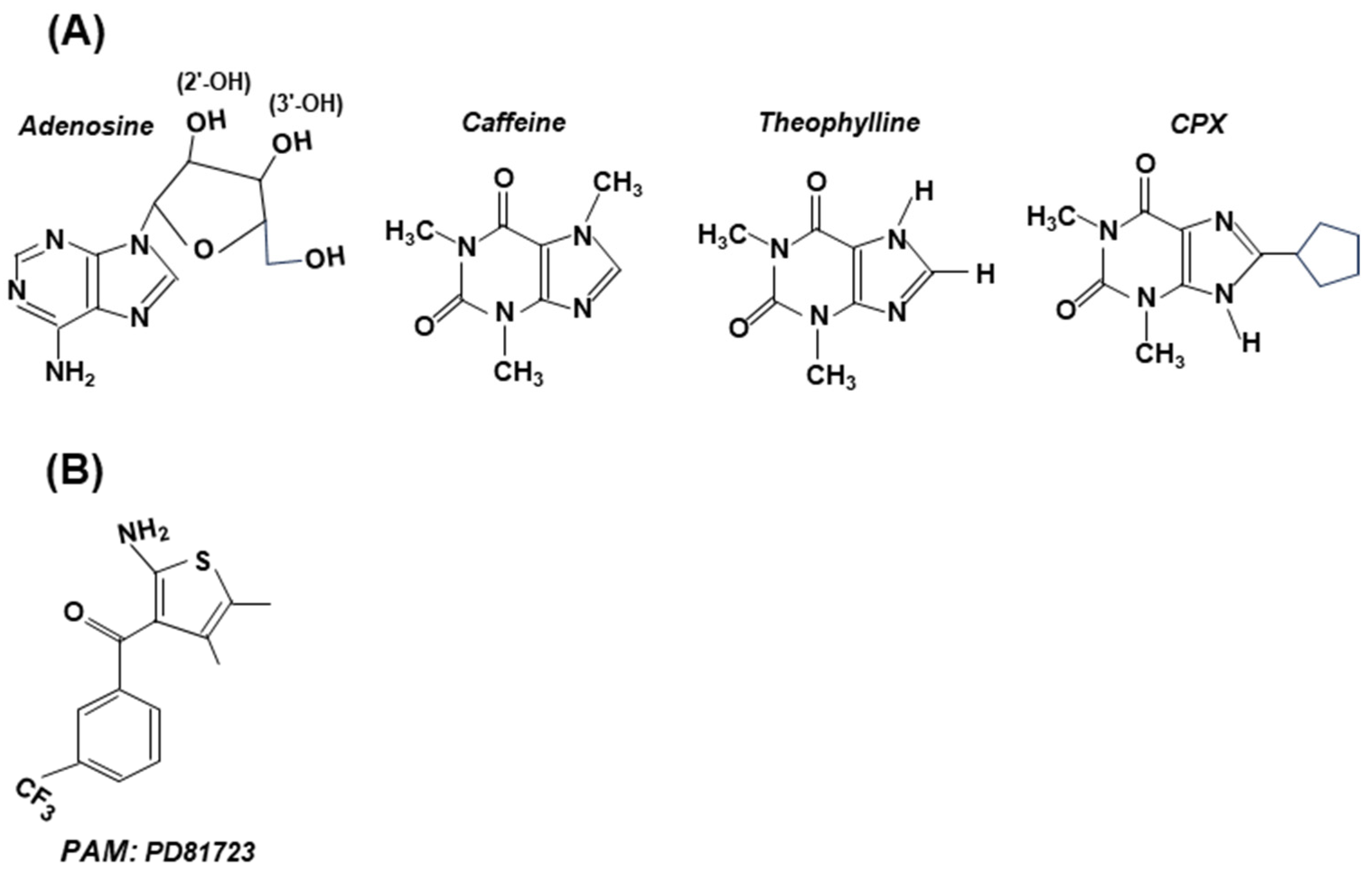
5.1. Intervention via the GPCRs of Adenosine Receptors
5.2. Intervention via the GPCR of Vagus Nerve NPY2R
6. Summary and Conclusions
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lovelace, J.W.; Ma, J.; Yadav, S.; Chhabria, K.; Shen, H.; Pang, Z.; Qi, T.; Sehgal, R.; Zhang, Y.; Bali, T.; et al. Vagal sensory neurons mediate the Bezold-Jarisch reflex and induce syncope. Nature 2023, 623, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, W.N. Syncope. N. Engl. J. Med. 2000, 343, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, J.G.; Thijs, R.D.; Benditt, D.G.; Wieling, W. A guide to disorders causing transient loss of consciousness: Focus on syncope. Nat. Rev. Neurol. 2009, 5, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Wieling, W.; Thijs, R.D.; van Dijk, N.; Wilde, A.A.M.; Benditt, D.G.; van Dijk, J.G. Symptoms and signs of syncope: A review of the link between physiology and clinical clues. Brain 2009, 132, 2630–2642. [Google Scholar] [CrossRef] [PubMed]
- McBride, D.W.; Reis, C.; Frank, W.; Klebe, D.W.; Zhang, J.H.; Applegate, R., 2nd; Tang, J. An experimental model of vasovagal syncope induces cerebral hypoperfusion and fainting-like behavior in awake rats. PLoS ONE 2016, 11, e0163280. [Google Scholar] [CrossRef]
- Lim, G.B. Explaining how a cardiac reflex causes syncope. Nat. Rev. Cardiol. 2024, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Guieu, R.; Deharo, J.-C.; Ruf, J.; Mottola, G.; Kipson, N.; Bruzzese, L.; Gerolami, V.; Franceschi, F.; Ungar, A.; Tomaino, M.; et al. Adenosine and clinical forms of neurally-mediated syncope. J. Am. Coll. Cardiol. 2015, 66, 204–205. [Google Scholar] [CrossRef]
- Mosqueda-Garcia, R.; Furlan, R.; Tank, J.; Fernandez-Violante, R. The elusive pathophysiology of neurally mediated syncope. Circulation 2000, 102, 2898–2906. [Google Scholar] [CrossRef]
- Mark, A.L. The Bezold-Jarisch reflex revisited: Clinical implications of inhibitory reflexes originating in the heart. J. Am. Coll. Cardiol. 1983, 1, 90–102. [Google Scholar] [CrossRef]
- Chang, R.B.; Strochlic, D.E.; Williams, E.K.; Umans, B.D.; Liberles, S.D. Vagal sensory neuron subtypes that differentially control breathing. Cell 2015, 161, 622–633. [Google Scholar] [CrossRef]
- Habecker, B.A.; Anderson, M.E.; Birren, S.J.; Fukuda, K.; Herring, N.; Hoover, D.B.; Kanazawa, H.; Paterson, D.J.; Ripplinger, C.M.; Habecker, B.A. Molecular and cellular neurocardiology: Development, and cellular and molecular adaptations to heart disease. J. Physiol. 2016, 594, 3853–3875. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, H.G. Albert von Bezold and nervous control of the heart. Clin. Cardiol. 2005, 28, 158–159. [Google Scholar] [CrossRef]
- da Silva, R.M.F.L. Syncope: Epidemiology, etiology, and prognosis. Front. Physiol. 2014, 5, 471. [Google Scholar] [CrossRef]
- Stavrakis, S.; Po, S. Ganglionated plexi ablation: Physiology and clinical applications. Arrhythm. Electrophysiol. Rev. 2017, 6, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Pachon, J.C.M.; Pachon, E.I.M.; Pachon, M.Z.C.; Lobo, T.J.; Pachon, J.C.M.; Santillana, T.G.P. Catheter ablation of severe neurally meditated reflex (neurocardiogenic or vasovagal) syncope: Cardioneuroablation long-term results. Europace 2011, 13, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zheng, L.; Qiao, Y.; Shi, R.; Hou, B.; Wu, L.; Guo, J.; Zhang, S.; Yao, Y. Catheter ablation as a treatment for vasovagal syncope: Long-term outcome of endocardial autonomic modification of the left atrium. J. Am. Heart Assoc. 2016, 5, e003471. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Silberstein, S.D. Vagus nerve and vagus nerve stimulation, a comprehensive review: Part I. Headache 2016, 56, 71–78. [Google Scholar] [CrossRef]
- Yuan, H.; Silberstein, S.D. Vagus nerve and vagus nerve stimulation, a comprehensive review: Part II. Headache 2016, 56, 258–266. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Functional and chemical anatomy of the afferent vagal system. Auton. Neurosci. 2000, 85, 1–17. [Google Scholar] [CrossRef]
- Prescott, S.L.; Liberles, S.D. Internal senses of the vagus nerve. Neuron 2022, 110, 579–599. [Google Scholar] [CrossRef]
- Nudell, V.; Wang, Y.; Pang, Z.; Lal, N.K.; Huang, M.; Shaabani, N.; Kanim, W.; Teijaro, J.; Maximov, A.; Ye, L. HYBRiD: Hydrogel-reinforced DISCO for clearing mammalian bodies. Nat. Methods 2022, 19, 479–485. [Google Scholar] [CrossRef]
- Parker, E.; Van Heek, M.; Stamford, A. Neuropeptide Y receptors as targets for anti-obesity drug development: Perspective and current status. Eur J. Pharmacol. 2022, 440, 173–187. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduction Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Kang, H.; Park, C.; Choi, Y.K.; Bae, J.; Kwon, S.; Kim, J.; Choi, C.; Seok, C.; Im, W.; Choi, H.-J. Structural basis for Y2 receptor-mediated neuropeptide Y and peptide YY signaling. Structure 2023, 31, 44–57. [Google Scholar] [CrossRef]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Gaitanaris, G.A. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef]
- Lempert, T. The eye movements of syncope. Neurology 1996, 46, 1086–1088. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Yao, Z.; van Velthoven, C.T.J.; Nguyen, T.N.; Goldy, J.; Sedeno-Cortes, A.E.; Baftizadeh, F. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 2021, 184, 3222–3241.e26. [Google Scholar] [CrossRef]
- Beauchamp, A.; Yee, Y.; Darwin, B.C.; Raznahan, A.; Mars, R.B.; Lerch, J.P. Whole-brain comparison of rodent and human brains using spatial transcriptomics. eLife 2022, 11, e79418. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [PubMed]
- Flammang, D.; Pelleg, A.; Benditt, D.G. The adenosine triphospate (ATP) test for evaluation of syncope of unknown origin. J. Cardiovasc. Electrophysiol. 2005, 16, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Saadjian, A.Y.; Lévy, S.; Franceschi, F.D.; Zouher, I.; Paganelli, F.; Guieu, R.G.P. Role of endogenous adenosine as a modulator of syncope induced during tilt testing. Circulation 2002, 106, 569–574. [Google Scholar] [CrossRef]
- Guieu, R.; Fromonot, J.; Mottola, G.; Maille, B.; Marlinge, M.; Groppelli, A.; Conte, S.; Bechah, Y.; Lalevee, N.; Michelet, P.; et al. Adenosinergic system and neuroendocrine syncope: What is the link? Cells 2023, 12, 2027. [Google Scholar] [CrossRef]
- Guieu, R.; Degioanni, C.; Fromonot, J.; De Maria, L.; Ruf, J.; Deharo, J.C.; Brignole, M. Adenosine, adenosine receptors and neurohumoral syncope: From molecular basis to personalized treatment. Biomedicines 2022, 10, 1127. [Google Scholar] [CrossRef]
- Sutton, R. The role of adenosine in syncope. Int. J. Cardiol. 2022, 365, 47–48. [Google Scholar] [CrossRef]
- Brignole, M.; Groppelli, A.; Brambilla, R.; Caldara, G.L.; Torresani, E.; Parati, G.; Solari, D.; Ungar, A.; Rafanelli, M.; Deharo, J.C.; et al. Plasma adenosine and neurally mediated syncope: Ready for clinical use. Eurospace 2020, 22, 847–853. [Google Scholar] [CrossRef]
- Cohen, F.R.; Lazareno, S.; Birdsall, N.J. The affinity of adenosine for the high- and low-affinity states of the human adenosine A1 receptor. Eur. J. Pharmacol. 1996, 309, 111–114. [Google Scholar] [CrossRef]
- Pacini, E.S.A.; Sanders-Silveira, S.; Godinho, R.O. The extracellular cAMP-adenosine pathway in airway smooth muscle. J. Pharmacol. Exp. Ther. 2018, 366, 75–83. [Google Scholar] [CrossRef]
- Belardinelli, L.; Shryock, J.C.; Song, Y.; Wang, D.; Srinivas, M.; Mozzicato, S.; Joshi, B.V.; Jacobson, K.A.; Liang, B.T.; Dougherty, C.; et al. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995, 9, 359–365. [Google Scholar] [CrossRef]
- De Waard, M.; Hering, J.; Weiss, N.; Feltz, A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol. Sci. 2005, 26, 427–436. [Google Scholar] [CrossRef]
- Spiegel, A.M. Signal transduction by guanine nucleotide binding proteins. Mol. Cell. Endocrinol. 1987, 49, 1–16. [Google Scholar] [CrossRef]
- Trzaskowski, B.; Latek, D.; Yuan, S.; Ghoshdastider, U.; Debinski, A.; Filipek, S. Action of molecular switches in GPCRs--theoretical and experimental studies. Curr. Med. Chem. 2012, 19, 1090–1109. [Google Scholar] [CrossRef]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Ann. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef]
- Bjarnadóttir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schiöth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263–273. [Google Scholar] [CrossRef]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 2016, 577, 101–108. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 414–420. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147, S193–S201. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.-G. Historical and current adenosine receptor agonists in preclinical and clinical development. Front. Cell. Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Knutsen, L.J.S. P1 and P2 purine and pyrimidine receptor ligands. Handb. Exp. Pharmacol. 2001, 151, 129–175. [Google Scholar]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef]
- Kiesman, W.F.; Elzein, E.; Zablocki, J. A1 Adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharmacol. 2009, 193, 25–58. [Google Scholar] [CrossRef]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets–What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef]
- Kim, D.G.; Bynoe, M.S. A2A adenosine receptor modulates drug efflux transporter P-glycoprotein at the blood–brain barrier. J. Clin. Investig. 2016, 126, 1717–1733. [Google Scholar] [CrossRef]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug. Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Brignole, M.; Iori, M.; Strano, S.; Tomaino, M.; Rivasi, G.; Ungar, A.; Carretta, D.; Solari, D.; Napoli, P.; Deharo, J.C.; et al. Theophylline in patients with syncope without prodrome, normal heart, and normal electrocardiogram: A propensity-score matched study verified by implantable cardiac monitor. Europace 2022, 24, 1164–1170. [Google Scholar] [CrossRef]
- Bruns, R.F.; Fergus, J.H. Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990, 38, 939–949. [Google Scholar]
- Bruns, R.F.; Fergus, J.H.; Coughenour, L.L.; Courtland, G.G.; Pugsley, T.A.; Dodd, J.H.; Tinney, F.J. Structure-activity relationships for enhancement of adenosine A1 receptor binding by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990, 38, 950–958. [Google Scholar] [PubMed]
- Vecchio, E.A.; Baltos, J.A.; Nguyen, A.T.N.; Christopoulos, A.; White, P.J.; May, L.T. New paradigms in adenosine receptor pharmacology: Allostery, oligomerization and biased agonism. Br. J. Pharmacol. 2018, 175, 4036–4046. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Gupta, D.; D’Avila, A.; Morillo, C.A. Cardioneuroablation for vasovagal syncope and atrioventricular block: A step-by-step guide. J. Cardiovasc. Electrophysiol. 2022, 33, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Guler, T.E.; Bozyel, S.; Yalin, K.; Gopinathannair, R. Usefulness of post-procedural heart rate response to predict syncope recurrence or positive head up tilt table testing after cardioneuroablation. Europace 2020, 22, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Po, S.; Yao, Y. Cardioneuroablation for treating vasovagal syncope: Current status and future directions. Arrhythm. Electrophysiol. Rev. 2023, 12, e18. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Marquez, M.F.; Fierro, E.F.; Baez, J.J.; Rockbrand, L.P.; Gomez-Flores, J. Cardioinhibitory syncope: From pathophysiology to treatment-should we think on cardioneuroablation? J. Interv. Card. Electrophysiol. 2020, 59, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Iordache, P.D.; Mates, D.; Gunnarsson, B.; Eggertsson, H.P.; Sulem, P.; Guðmundsson, J.; Benónísdóttir, S.; Csiki, I.E.; Rascu, S. Profile of common prostate cancer risk variants in an unscreened Romanian population. J. Cell. Mol. Med. 2018, 22, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Forstner, A.J.; Awasthi, S.; Wolf, C.; Maron, E.; Erhardt, A.; Czamara, D.; Eriksson, E.; Lavebratt, C.; Allgulander, C.; Friedrich, N.; et al. Genome-wide association study of panic disorder reveals genetic overlap with neuroticism and depression. Mol. Psychiatry 2021, 26, 4179–4190. [Google Scholar] [CrossRef] [PubMed]
- Haenig, C.; Atias, N.; Taylor, A.K.; Mazza, A.; Schaefer, M.H.; Russ, J.; Riechers, S.-P.; Jain, S. Interactome mapping provides a network of neurodegenerative disease proteins and uncovers widespread protein aggregation in affected brains. Cell Rep. 2020, 32, 108050. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Yang, C.; Lee, S.H. Vagus nerves stimulation: Clinical implication and practical issue as a neuropsychiatric treatment. Clin. Psychopharmacol. Neurosci. 2024, 22, 13–22. [Google Scholar] [CrossRef]
- Panebianco, M.; Rigby, A.; Marson, A.G. Vagus nerve stimulation for focal seizures. Cochrane Database Syst. Rev. 2022, 7, CD002896. [Google Scholar] [CrossRef]
- Wheless, J.W.; Gienapp, A.J.; Ryvlin, P. Vagus nerve stimulation (VNS) therapy update. Epilepsy Behav. 2018, 88S, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Helmers, S.L.; Duh, M.S.; Guérin, A.; Sarda, S.P.; Samuelson, T.M.; Bunker, M.T.; Olin, B.D.; Jackson, S.D.; Faught, E. Clinical and economic impact of vagus nerve stimulation therapy in patients with drug-resistant epilepsy. Epilepsy Behav. 2017, 22, 370–375. [Google Scholar] [CrossRef]
- Carreno, F.R.; Frazer, A. Vagal nerve stimulation for treatment-resistant depression. Neurotherapeutics 2017, 14, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Chepurny, O.G.; Bonaccorso, R.L.; Leech, C.A.; Wöllert, T.; Langford, G.M.; Schwede, F.; Roth, C.L.; Doyle, R.P.; Holz, G.G. Chimeric peptide EP45 as a dual agonist at GLP-1 and NPY2R receptors. Sci. Rep. 2018, 8, 3749. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, S.; Bhattacharya, S.; Walsh, E.M.; Okoh, G.I.; Thompson, D. Computational peptide design cotargeting glucagon and glucagon-like peptide-1 receptors. J. Chem. Inf. Model. 2023, 63, 4934–4947. [Google Scholar] [CrossRef]
- Lazris, A.; Roth, A.R.; Haskell, H.; James, J. Efficient approach to the evaluation of syncope. Am. Fam. Physician. 2021, 104, 305–308. [Google Scholar]
- Qiu, L.; Chang, A.; Ma, R.; Strong, T.V.; Okun, M.S.; Foote, K.D.; Wexler, A.; Gunduz, A.; Miller, J.L.; Halpern, C.H. Neuromodulation for the treatment of Prader-Willi syndrome–A systematic review. Neurotherapeutics 2024, 21, e00339. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barik, S.; Riddell, T. The Brain–Heart Network of Syncope. Int. J. Mol. Sci. 2024, 25, 6959. https://doi.org/10.3390/ijms25136959
Barik S, Riddell T. The Brain–Heart Network of Syncope. International Journal of Molecular Sciences. 2024; 25(13):6959. https://doi.org/10.3390/ijms25136959
Chicago/Turabian StyleBarik, Sailen, and Thomas Riddell. 2024. "The Brain–Heart Network of Syncope" International Journal of Molecular Sciences 25, no. 13: 6959. https://doi.org/10.3390/ijms25136959
APA StyleBarik, S., & Riddell, T. (2024). The Brain–Heart Network of Syncope. International Journal of Molecular Sciences, 25(13), 6959. https://doi.org/10.3390/ijms25136959