
A Narrative Review on the Pathophysiology of Preeclampsia

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. The Etiology of Preeclampsia
2.2. Abnormal Placentation
2.2.1. Immune Response
2.2.2. Glycosylation and the Expression of Galectins
2.2.3. Genetic Factors
2.2.4. Apoptosis
2.2.5. Dyslipidemia
2.2.6. Role of Ion Channels in Trophoblast Invasion and Vascular Remodeling
2.2.7. Syncytiotrophoblast-Derived Extracellular Vesicles
2.3. Maternal Systemic Response
2.3.1. Pro-Angiogenic and Anti-Angiogenic Factors
2.3.2. Endothelial Dysfunction
2.3.3. Oxidative Stress
2.3.4. Endothelial Cell Activation
2.3.5. The Role of Adipokines
2.3.6. Systemic Inflammation
2.3.7. The Role of Ferroptosis
2.3.8. Genetic and Epigenetic Factors

{kind=link}
{kind=link}
{kind=link}
| Genetic/Epigenetic Factor | Description | Effect | Key Gene/Factor |
|---|---|---|---|
| MTHFR polymorphisms | Hypomethylation of MTHFR promoter increases homocysteine levels and reduces enzyme activity [73,75]. | Contributes to endothelial dysfunction. | MTHFR |
| miR-155 upregulation | Induced by inflammatory stimuli (TNF-α, LPS); regulates genes involved in angiogenesis [74]. | Leads to reduced vascularization and placental insufficiency. | miR-155, CYR61 |
| FLT1 gene variants | Polymorphisms (e.g., rs4769613, rs12050029, rs149427560) affect FLT1 expression [76]. | Increases sFlt-1 production, disrupting vascular remodeling. | FLT1, sFlt-1 |
| miR-155 promoter polymorphism | rs767649 polymorphism enhances miR-155 expression [105]. | Heightens inflammation and gene regulation disruption. | miR-155 |
| Complement system SNPs | SNPs in the C3 gene (e.g., rs2287845) affect gene expression through epigenetic regulation [106]. | Contributes to inflammatory and immune responses. | C3 |
| TNF-α gene polymorphisms | Polymorphisms at -308 and -238 positions increase TNF-α expression [107]. | Promotes inflammation and endothelial dysfunction. | TNF-α |
| sFlt-1 alternative splicing | Hypoxia induces alternative splicing of the sFlt-1 gene [32]. | Increases soluble sFlt-1, reducing angiogenesis and causing endothelial dysfunction. | sFlt-1 |
| MEG3 downregulation | lncRNA MEG3 is underexpressed in preeclamptic placentas [108]. | Decreases trophoblast migration and invasion, increases apoptosis. | MEG3 |
| NOS3 and GUCY1A3 polymorphisms | Polymorphisms affecting NO production and signaling [111]. | Leads to reduced NO bioavailability and endothelial dysfunction. | NOS3, GUCY1A3 |
2.3.9. Other Associated Mechanisms
2.3.10. The Role of Mitochondrial Dysfunction
2.3.11. The Role of Platelet Activation
2.3.12. The Role of the Renin-Angiotensin-Aldosterone System
2.3.13. The Role of Oxidative Stress
2.3.14. The Role of Micronutrient Deficiencies and Inositol Messengers
2.3.15. The Role of HO-1/CO and CSE/H2S Pathways
2.3.16. The Role of Environmental Factors and Endocrine Disruptors
2.3.17. The Role of Estrogens and Androgens
3. Discussion
3.1. Main Findings
3.2. Clinical Interpretation
3.3. Potential Therapeutic Targets
3.4. Research Interpretation and Future Directions
4. Materials and Methods
4.1. Eligibility Criteria
4.2. Study Selection
4.3. Data Extraction and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres-Torres, J.; Villafan-Bernal, J.R.; Martinez-Portilla, R.J.; Hidalgo-Carrera, J.A.; Estrada-Gutierrez, G.; Adalid-Martinez-Cisneros, R.; Rojas-Zepeda, L.; Acevedo-Gallegos, S.; Camarena-Cabrera, D.M.; Cruz-Martínez, M.Y.; et al. Performance of machine-learning approach for prediction of pre-eclampsia in a middle-income country. Ultrasound Obstet. Gynecol. 2024, 63, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, E.; Rolnik, D.L.; Zhou, W.; Estrada-Gutierrez, G.; Koga, K.; Francisco, R.P.V.; Whitehead, C.; Hyett, J.; da Silva Costa, F.; Nicolaides, K.; et al. Pre-eclampsia. Nat. Rev. Dis. Prim. 2023, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237–e260. [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S.; et al. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2018, 13, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Mooij, R.; Kapanga, R.R.; Mwampagatwa, I.H.; Mgalega, G.C.; van Dillen, J.; Stekelenburg, J.; de Kok, B.C. Beyond severe acute maternal morbidity: A mixed-methods study on the long-term consequences of (severe pre-)eclampsia in rural Tanzania. Trop. Med. Int. Health 2021, 26, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Davidesko, S.; Nahum Sacks, K.; Friger, M.; Haim, A.; Sheiner, E. Prenatal exposure to preeclampsia as a risk factor for long-term endocrine morbidity of the offspring. Hypertens. Pregnancy 2021, 40, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kedar Sade, E.; Wainstock, T.; Tsumi, E.; Sheiner, E. Prenatal Exposure to Preeclampsia and Long-Term Ophthalmic Morbidity of the Offspring. J. Clin. Med. 2020, 9, 1271. [Google Scholar] [CrossRef]
- Kessous, R.; Shoham-Vardi, I.; Pariente, G.; Sergienko, R.; Sheiner, E. Long-term maternal atherosclerotic morbidity in women with pre-eclampsia. Heart 2015, 101, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.S.; Davey, M.A.; Mol, B.W.; Rolnik, D.L. The evolution of the diagnostic criteria of preeclampsia-eclampsia. Am. J. Obstet. Gynecol. 2022, 226, S835–S843. [Google Scholar] [CrossRef]
- Rorman, E.; Freud, A.; Wainstock, T.; Sheiner, E. Maternal preeclampsia and long-term infectious morbidity in the offspring—A population based cohort analysis. Pregnancy Hypertens. 2020, 21, 30–34. [Google Scholar] [CrossRef]
- Nahum Sacks, K.; Friger, M.; Shoham-Vardi, I.; Sergienko, R.; Spiegel, E.; Landau, D.; Sheiner, E. Long-term neuropsychiatric morbidity in children exposed prenatally to preeclampsia. Early Hum. Dev. 2019, 130, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Nahum Sacks, K.; Friger, M.; Shoham-Vardi, I.; Spiegel, E.; Sergienko, R.; Landau, D.; Sheiner, E. Prenatal exposure to preeclampsia as an independent risk factor for long-term cardiovascular morbidity of the offspring. Pregnancy Hypertens. 2018, 13, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torres, J.; Espino, Y.S.S.; Villafan-Bernal, J.R.; Orozco-Guzman, L.E.; Solis-Paredes, J.M.; Estrada-Gutierrez, G.; Martinez-Cisneros, R.A.; Mateu-Rogell, P.; Acevedo-Gallegos, S.; Martinez-Portilla, R.J. Effects of maternal characteristics and medical history on first trimester biomarkers for preeclampsia. Front. Med. 2023, 10, 1050923. [Google Scholar] [CrossRef] [PubMed]
- Magee, L.A.; Nicolaides, K.H.; von Dadelszen, P. Preeclampsia. N. Engl. J. Med. 2022, 386, 1817–1832. [Google Scholar] [CrossRef]
- Myatt, L. The prediction of preeclampsia: The way forward. Am. J. Obstet. Gynecol. 2022, 226, S1102–S1107.e8. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Romero, R.; Yeo, L.; Gomez-Lopez, N.; Chaemsaithong, P.; Jaovisidha, A.; Gotsch, F.; Erez, O. The etiology of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S844–S866. [Google Scholar] [CrossRef]
- Carter, A.M. Evolution of placental function in mammals: The molecular basis of gas and nutrient transfer, hormone secretion, and immune responses. Physiol. Rev. 2012, 92, 1543–1576. [Google Scholar] [CrossRef] [PubMed]
- Kornacki, J.; Olejniczak, O.; Sibiak, R.; Gutaj, P.; Wender-Ożegowska, E. Pathophysiology of Pre-Eclampsia-Two Theories of the Development of the Disease. Int. J. Mol. Sci. 2023, 25, 307. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Pre-eclampsia, the placenta and the maternal systemic inflammatory response—A review. Placenta 2003, 24 (Suppl. A), S21–S27. [Google Scholar] [CrossRef]
- Kamrani, A.; Alipourfard, I.; Ahmadi-Khiavi, H.; Yousefi, M.; Rostamzadeh, D.; Izadi, M.; Ahmadi, M. The role of epigenetic changes in preeclampsia. Biofactors 2019, 45, 712–724. [Google Scholar] [CrossRef]
- Hansen, A.T.; Bernth Jensen, J.M.; Hvas, A.M.; Christiansen, M. The genetic component of preeclampsia: A whole-exome sequencing study. PLoS ONE 2018, 13, e0197217. [Google Scholar] [CrossRef] [PubMed]
- Laresgoiti-Servitje, E. A leading role for the immune system in the pathophysiology of preeclampsia. J. Leukoc. Biol. 2013, 94, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Greer, I.A. Pregnancy complications and maternal cardiovascular risk: Opportunities for intervention and screening? BMJ 2002, 325, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Soares, M.J. Modeling Trophoblast Cell-Guided Uterine Spiral Artery Transformation in the Rat. Int. J. Mol. Sci. 2022, 23, 2947. [Google Scholar] [CrossRef] [PubMed]
- Knöfler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.; James, J. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell. Mol. Life Sci. 2019, 76, 3479–3496. [Google Scholar] [CrossRef]
- Da Cunha Castro, E.C.; Popek, E. Abnormalities of placenta implantation. APMIS 2018, 126, 613–620. [Google Scholar] [CrossRef]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am. J. Obstet. Gynecol. 2022, 226, S1019–S1034. [Google Scholar] [CrossRef]
- Cornelius, D.C. Preeclampsia: From Inflammation to Immunoregulation. Clin. Med. Insights Blood Disord. 2018, 11, 1179545X17752325. [Google Scholar] [CrossRef]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef]
- Xu, X.; Pan, J.R.; Zhang, Y.Z. CoQ10 alleviate preeclampsia symptoms by enhancing the function of mitochondria in the placenta of pregnant rats with preeclampsia. Hypertens. Pregnancy 2019, 38, 217–222. [Google Scholar] [CrossRef]
- Teran, E.; Hernández, I.; Tana, L.; Teran, S.; Galaviz-Hernandez, C.; Sosa-Macías, M.; Molina, G.; Calle, A. Mitochondria and Coenzyme Q10 in the Pathogenesis of Preeclampsia. Front. Physiol. 2018, 9, 1561. [Google Scholar] [CrossRef]
- Vishnyakova, P.; Elchaninov, A.; Fatkhudinov, T.; Sukhikh, G. Role of the Monocyte-Macrophage System in Normal Pregnancy and Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3695. [Google Scholar] [CrossRef]
- Raguema, N.; Moustadraf, S.; Bertagnolli, M. Immune and Apoptosis Mechanisms Regulating Placental Development and Vascularization in Preeclampsia. Front. Physiol. 2020, 11, 98. [Google Scholar] [CrossRef]
- Faas, M.M.; de Vos, P. Uterine NK cells and macrophages in pregnancy. Placenta 2017, 56, 44–52. [Google Scholar] [CrossRef]
- Figueiredo, A.S.; Schumacher, A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology 2016, 148, 13–21. [Google Scholar] [CrossRef]
- Kirkgöz, K.; Vogtmann, R.; Xie, Y.; Zhao, F.; Riedel, A.; Adam, L.M.; Freitag, N.; Harms, C.; Garcia, M.G.; Plösch, T.; et al. Placental glycosylation senses the anti-angiogenic milieu induced by human sFLT1 during pregnancy. J. Reprod. Immunol. 2024, 164, 104284. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, F.; Freitag, N.; Borowski, S.; Wang, Y.; Harms, C.; Pang, P.C.; Desforges, J.; Wen, T.; Schwedhelm, E.; et al. Maternal-derived galectin-1 shapes the placenta niche through Sda terminal glycosylation: Implication for preeclampsia. PNAS Nexus 2023, 2, pgad247. [Google Scholar] [CrossRef]
- Blois, S.M.; Dveksler, G.; Vasta, G.R.; Freitag, N.; Blanchard, V.; Barrientos, G. Pregnancy Galectinology: Insights into a Complex Network of Glycan Binding Proteins. Front. Immunol. 2019, 10, 1166. [Google Scholar] [CrossRef]
- Kasture, V.; Sahay, A.; Joshi, S. Cell death mechanisms and their roles in pregnancy related disorders. Adv. Protein Chem. Struct. Biol. 2021, 126, 195–225. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yoshimoto, C.; Matsubara, S.; Shigetomi, H.; Imanaka, S. An integral role of mitochondrial function in the pathophysiology of preeclampsia. Mol. Biol. Rep. 2024, 51, 330. [Google Scholar] [CrossRef]
- Lu, Y.; Jia, Z.; Su, S.; Han, L.; Meng, L.; Tang, G.; Wang, J.; Zhang, C.; Xie, X.; Zhang, Y.; et al. Establishment of trimester-specific reference intervals of serum lipids and the associations with pregnancy complications and adverse perinatal outcomes: A population-based prospective study. Ann. Med. 2021, 53, 1632–1641. [Google Scholar] [CrossRef]
- Melhem, H.; Kallol, S.; Huang, X.; Lüthi, M.; Ontsouka, C.E.; Keogh, A.; Stroka, D.; Thormann, W.; Schneider, H.; Albrecht, C. Placental secretion of apolipoprotein A1 and E: The anti-atherogenic impact of the placenta. Sci. Rep. 2019, 9, 6225. [Google Scholar] [CrossRef]
- Adank, M.C.; Benschop, L.; Peterbroers, K.R.; Smak Gregoor, A.M.; Kors, A.W.; Mulder, M.T.; Schalekamp-Timmermans, S.; Roeters Van Lennep, J.E.; Steegers, E.A.P. Is maternal lipid profile in early pregnancy associated with pregnancy complications and blood pressure in pregnancy and long term postpartum? Am. J. Obstet. Gynecol. 2019, 221, 150.e1–150.e13. [Google Scholar] [CrossRef]
- Staff, A.C.; Dechend, R.; Pijnenborg, R. Learning from the placenta: Acute atherosis and vascular remodeling in preeclampsia-novel aspects for atherosclerosis and future cardiovascular health. Hypertension 2010, 56, 1026–1034. [Google Scholar] [CrossRef]
- Staff, A.C.; Johnsen, G.M.; Dechend, R.; Redman, C.W.G. Preeclampsia and uteroplacental acute atherosis: Immune and inflammatory factors. J. Reprod. Immunol. 2014, 101–102, 120–126. [Google Scholar] [CrossRef]
- Baczyk, D.; Kingdom, J.C.; Uhlén, P. Calcium signaling in placenta. Cell Calcium 2011, 49, 350–356. [Google Scholar] [CrossRef]
- Lorigo, M.; Oliveira, N.; Cairrao, E. Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells 2020, 9, 1956. [Google Scholar] [CrossRef]
- Tannetta, D.; Masliukaite, I.; Vatish, M.; Redman, C.; Sargent, I. Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J. Reprod. Immunol. 2017, 119, 98–106. [Google Scholar] [CrossRef]
- Ma, K.; Jin, H.; Hu, R.; Xiong, Y.; Zhou, S.; Ting, P.; Cheng, Y.; Yang, Y.; Yang, P.; Li, X. A proteomic analysis of placental trophoblastic cells in preeclampsia-eclampsia. Cell Biochem. Biophys. 2014, 69, 247–258. [Google Scholar] [CrossRef]
- Motta-Mejia, C.; Kandzija, N.; Zhang, W.; Mhlomi, V.; Cerdeira, A.S.; Burdujan, A.; Tannetta, D.; Dragovic, R.; Sargent, I.L.; Redman, C.W.; et al. Placental Vesicles Carry Active Endothelial Nitric Oxide Synthase and Their Activity is Reduced in Preeclampsia. Hypertension 2017, 70, 372–381. [Google Scholar] [CrossRef]
- Boisramé-Helms, J.; Meziani, F.; Sananès, N.; Boisramé, T.; Langer, B.; Schneider, F.; Ragot, T.; Andriantsitohaina, R.; Tesse, A. Detrimental arterial inflammatory effect of microparticles circulating in preeclamptic women: Ex vivo evaluation in human arteries. Fundam. Clin. Pharmacol. 2015, 29, 450–461. [Google Scholar] [CrossRef]
- Chiarello, D.I.; Salsoso, R.; Toledo, F.; Mate, A.; Vázquez, C.M.; Sobrevia, L. Foetoplacental communication via extracellular vesicles in normal pregnancy and preeclampsia. Mol. Asp. Med. 2018, 60, 69–80. [Google Scholar] [CrossRef]
- Thilaganathan, B.; Kalafat, E. Cardiovascular System in Preeclampsia and Beyond. Hypertension 2019, 73, 522–531. [Google Scholar] [CrossRef]
- Yagel, S.; Cohen, S.M.; Goldman-Wohl, D. An integrated model of preeclampsia: A multifaceted syndrome of the maternal cardiovascular-placental-fetal array. Am. J. Obstet. Gynecol. 2022, 226, S963–S972. [Google Scholar] [CrossRef]
- Han, C.; Huang, P.; Lyu, M.; Dong, J. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants 2020, 9, 1139. [Google Scholar] [CrossRef]
- Bueno-Pereira, T.O.; Bertozzi-Matheus, M.; Zampieri, G.M.; Abbade, J.F.; Cavalli, R.C.; Nunes, P.R.; Sandrim, V.C. Markers of Endothelial Dysfunction Are Attenuated by Resveratrol in Preeclampsia. Antioxidants 2022, 11, 2111. [Google Scholar] [CrossRef]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef]
- San Juan-Reyes, S.; Gómez-Oliván, L.M.; Islas-Flores, H.; Dublán-García, O. Oxidative stress in pregnancy complicated by preeclampsia. Arch. Biochem. Biophys. 2020, 681, 108255. [Google Scholar] [CrossRef]
- Freire, V.A.F.; Melo, A.D.; Santos, H.L.; Barros-Pinheiro, M. Evaluation of oxidative stress markers in subtypes of preeclampsia: A systematic review and meta-analysis. Placenta 2023, 132, 55–67. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2022, 226, S907–S927. [Google Scholar] [CrossRef]
- Kornacki, J.; Wirstlein, P.; Wender-Ozegowska, E. Markers of Endothelial Injury and Dysfunction in Early- and Late-Onset Preeclampsia. Life 2020, 10, 239. [Google Scholar] [CrossRef]
- Pankiewicz, K.; Issat, T. Understanding the Role of Chemerin in the Pathophysiology of Pre-Eclampsia. Antioxidants 2023, 12, 830. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, Y.; Fan, P.; Yang, L.; Liu, X. Role of leptin in the pathophysiology of preeclampsia. Placenta 2023, 142, 128–134. [Google Scholar] [CrossRef]
- Yu, M.; Yang, Y.; Huang, C.; Ge, L.; Xue, L.; Xiao, Z.; Xiao, T.; Zhao, H.; Ren, P.; Zhang, J.V. Chemerin: A Functional Adipokine in Reproductive Health and Diseases. Biomedicines 2022, 10, 1910. [Google Scholar] [CrossRef]
- Shah, D.A.; Khalil, R.A. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem. Pharmacol. 2015, 95, 211–226. [Google Scholar] [CrossRef]
- Ribeiro, V.R.; Romao-Veiga, M.; Romagnoli, G.G.; Matias, M.L.; Nunes, P.R.; Borges, V.T.M.; Peracoli, J.C.; Peracoli, M.T.S. Association between cytokine profile and transcription factors produced by T-cell subsets in early- and late-onset pre-eclampsia. Immunology 2017, 152, 163–173. [Google Scholar] [CrossRef]
- Ng, S.W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. [Google Scholar] [CrossRef]
- Chen, Z.; Gan, J.; Zhang, M.; Du, Y.; Zhao, H. Ferroptosis and Its Emerging Role in Pre-Eclampsia. Antioxidants 2022, 11, 1282. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Mohammadpour-Gharehbagh, A.; Salimi, S.; Keshavarzi, F.; Saeidian, F.; Mousavi, M.; Teimoori, B.; Esmaeilipour, M.; Mokhtari, M. Genetic variants in 3′-UTRs of MTHFR in the pregnancies complicated with preeclampsia and bioinformatics analysis. J. Cell. Biochem. 2018, 119, 773–781. [Google Scholar] [CrossRef]
- Ayoub, S.E.; Shaker, O.G.; Abdelwahed, M.Y.; Ahmed, N.A.; Abdelhameed, H.G.; Bosilah, A.H.; Mohammed, S.R. Association of MicroRNA-155rs767649 Polymorphism with Susceptibility to Preeclampsia. Int. J. Mol. Cell. Med. 2019, 8, 247–257. [Google Scholar] [CrossRef]
- Previtera, F.; Restaino, S.; Romano, G.; Vizzielli, G.; Neri, A.; Scalzotto, E.; Vetrugno, L.; Montessoro, B.; Mioni, R.; Driul, L. Gene Polymorphism in Five Target Genes of Immunosuppressive Therapy and Risk of Development of Preeclampsia. Healthcare 2021, 9, 821. [Google Scholar] [CrossRef]
- Ohwaki, A.; Nishizawa, H.; Kato, A.; Kato, T.; Miyazaki, J.; Yoshizawa, H.; Noda, Y.; Sakabe, Y.; Ichikawa, R.; Sekiya, T.; et al. Placental Genetic Variants in the Upstream Region of the FLT1 Gene in Pre-eclampsia. J. Reprod. Infertil. 2020, 21, 240–246. [Google Scholar] [CrossRef]
- Agbani, E.O.; Skeith, L.; Lee, A. Preeclampsia: Platelet procoagulant membrane dynamics and critical biomarkers. Res. Pract. Thromb. Haemost. 2023, 7, 100075. [Google Scholar] [CrossRef]
- Hu, X.Q.; Zhang, L. Mitochondrial Dysfunction in the Pathogenesis of Preeclampsia. Curr. Hypertens. Rep. 2022, 24, 157–172. [Google Scholar] [CrossRef]
- Gathiram, P.; Moodley, J. The Role of the Renin-Angiotensin-Aldosterone System in Preeclampsia: A Review. Curr. Hypertens. Rep. 2020, 22, 89. [Google Scholar] [CrossRef]
- Ahmed, A. Molecular mechanisms and therapeutic implications of the carbon monoxide/hmox1 and the hydrogen sulfide/CSE pathways in the prevention of pre-eclampsia and fetal growth restriction. Pregnancy Hypertens. 2014, 4, 243–244. [Google Scholar] [CrossRef]
- Testai, L.; Brancaleone, V.; Flori, L.; Montanaro, R.; Calderone, V. Modulation of EndMT by Hydrogen Sulfide in the Prevention of Cardiovascular Fibrosis. Antioxidants 2021, 10, 910. [Google Scholar] [CrossRef]
- Anto, E.O.; Ofori Boadu, W.I.; Addai-Mensah, O.; Wiafe, Y.A.; Owiredu, W.K.; Obirikorang, C.; Annani-Akollor, M.E.; Adua, E.; Appiah, M.; Opoku, S.; et al. Association between micronutrients, oxidative stress biomarkers and angiogenic growth mediators in early and late-onset preeclamptic Ghanaian women. SAGE Open Med. 2023, 11, 20503121231175759. [Google Scholar] [CrossRef]
- Kunjara, S.; McLean, P.; Rademacher, L.; Rademacher, T.W.; Fascilla, F.; Bettocchi, S.; Scioscia, M. Putative Key Role of Inositol Messengers in Endothelial Cells in Preeclampsia. Int. J. Endocrinol. 2016, 2016, 7695648. [Google Scholar] [CrossRef]
- Piani, F.; Tossetta, G.; Fantone, S.; Agostinis, C.; Di Simone, N.; Mandalà, M.; Bulla, R.; Marzioni, D.; Borghi, C. First Trimester CD93 as a Novel Marker of Preeclampsia and Its Complications: A Pilot Study. High Blood Press. Cardiovasc. Prev. 2023, 30, 591–594. [Google Scholar] [CrossRef]
- Smith-Jackson, K.; Hentschke, M.R.; Poli-de-Figueiredo, C.E.; Pinheiro da Costa, B.E.; Kurlak, L.O.; Broughton Pipkin, F.; Czajka, A.; Mistry, H.D. Placental expression of eNOS, iNOS and the major protein components of caveolae in women with pre-eclampsia. Placenta 2015, 36, 607–610. [Google Scholar] [CrossRef]
- Caldeira-Dias, M.; Viana-Mattioli, S.; de Souza Rangel Machado, J.; Carlström, M.; de Carvalho Cavalli, R.; Sandrim, V.C. Resveratrol and grape juice: Effects on redox status and nitric oxide production of endothelial cells in in vitro preeclampsia model. Pregnancy Hypertens. 2021, 23, 205–210. [Google Scholar] [CrossRef]
- Piani, F.; Agnoletti, D.; Baracchi, A.; Scarduelli, S.; Verde, C.; Tossetta, G.; Montaguti, E.; Simonazzi, G.; Degli Esposti, D.; Borghi, C. Serum uric acid to creatinine ratio and risk of preeclampsia and adverse pregnancy outcomes. J. Hypertens. 2023, 41, 1333–1338. [Google Scholar] [CrossRef]
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxidative Med. Cell. Longev. 2019, 2019, 8238727. [Google Scholar] [CrossRef]
- Vaka, R.; Deer, E.; LaMarca, B. Is Mitochondrial Oxidative Stress a Viable Therapeutic Target in Preeclampsia? Antioxidants 2022, 11, 210. [Google Scholar] [CrossRef]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, C.; Zhang, K.; Lan, X.; Chen, X.; Zang, W.; Wang, Z.; Guan, F.; Zhu, C.; Yang, X.; et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free. Radic. Biol. Med. 2019, 131, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.G.; Jardim, N.S.; Quines, C.B.; Nogueira, C.W. Diphenyl diselenide regulates Nrf2/Keap-1 signaling pathway and counteracts hepatic oxidative stress induced by bisphenol A in male mice. Environ. Res. 2018, 164, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, L.; Zhang, G.; Zhang, Z.; Guo, W. Oxidative stress activated by Keap-1/Nrf2 signaling pathway in pathogenesis of preeclampsia. Int. J. Clin. Exp. Pathol. 2020, 13, 382–392. [Google Scholar] [PubMed]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 Signaling in Preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef] [PubMed]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef]
- Ben Dhaou, C.; Mandi, K.; Frye, M.; Acheampong, A.; Radi, A.; De Becker, B.; Antoine, M.; Baeyens, N.; Wittamer, V.; Parmentier, M. Chemerin regulates normal angiogenesis and hypoxia-driven neovascularization. Angiogenesis 2022, 25, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadeh, M.; Norouzian, M.; Arabpour, F.; Naderi, N. Regulatory T-cells and preeclampsia: An overview of literature. Expert Rev. Clin. Immunol. 2016, 12, 209–227. [Google Scholar] [CrossRef]
- Boij, R.; Mjösberg, J.; Svensson-Arvelund, J.; Hjorth, M.; Berg, G.; Matthiesen, L.; Jenmalm, M.C.; Ernerudh, J. Regulatory T-cell Subpopulations in Severe or Early-onset Preeclampsia. Am. J. Reprod. Immunol. 2015, 74, 368–378. [Google Scholar] [CrossRef]
- Steinborn, A.; Schmitt, E.; Kisielewicz, A.; Rechenberg, S.; Seissler, N.; Mahnke, K.; Schaier, M.; Zeier, M.; Sohn, C. Pregnancy-associated diseases are characterized by the composition of the systemic regulatory T cell (Treg) pool with distinct subsets of Tregs. Clin. Exp. Immunol. 2012, 167, 84–98. [Google Scholar] [CrossRef]
- Soares, M.J.; Iqbal, K.; Kozai, K. Hypoxia and Placental Development. Birth Defects Res. 2017, 109, 1309–1329. [Google Scholar] [CrossRef] [PubMed]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef]
- Peng, X.; Lin, Y.; Li, J.; Liu, M.; Wang, J.; Li, X.; Liu, J.; Jia, X.; Jing, Z.; Huang, Z.; et al. Evaluation of Glutathione Peroxidase 4 role in Preeclampsia. Sci. Rep. 2016, 6, 33300. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Bounds, K.R.; Chiasson, V.L.; Pan, L.J.; Gupta, S.; Chatterjee, P. MicroRNAs: New Players in the Pathobiology of Preeclampsia. Front. Cardiovasc. Med. 2017, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Lokki, A.I.; Kaartokallio, T.; Holmberg, V.; Onkamo, P.; Koskinen, L.L.E.; Saavalainen, P.; Heinonen, S.; Kajantie, E.; Kere, J.; Kivinen, K.; et al. Analysis of Complement C3 Gene Reveals Susceptibility to Severe Preeclampsia. Front. Immunol. 2017, 8, 589. [Google Scholar] [CrossRef]
- Lin, C.W.; Chen, C.H.; Wu, M.H.; Chang, F.M.; Kang, L. Polymorphisms within the Tumor Necrosis Factor-Alpha Gene Is Associated with Preeclampsia in Taiwanese Han Populations. Biomedicines 2023, 11, 862. [Google Scholar] [CrossRef]
- Wang, R.; Zou, L. Downregulation of LncRNA-MEG3 promotes HTR8/SVneo cells apoptosis and attenuates its migration by repressing Notch1 signal in preeclampsia. Reproduction 2020, 160, 21–29. [Google Scholar] [CrossRef]
- Zhang, Y.; Zou, Y.; Wang, W.; Zuo, Q.; Jiang, Z.; Sun, M.; De, W.; Sun, L. Down-regulated long non-coding RNA MEG3 and its effect on promoting apoptosis and suppressing migration of trophoblast cells. J. Cell. Biochem. 2015, 116, 542–550. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, P.; Shi, Y.; Cui, B.; Meng, J. Long noncoding RNA maternally expressed gene 3 improves trophoblast dysfunction and inflammation in preeclampsia through the Wnt/β-Catenin/nod-like receptor pyrin domain-containing 3 axis. Front. Mol. Biosci. 2022, 9, 1022450. [Google Scholar] [CrossRef]
- Pereira, D.A.; Luizon, M.R.; Palei, A.C.; Tanus-Santos, J.E.; Cavalli, R.C.; Sandrim, V.C. Functional polymorphisms of NOS3 and GUCY1A3 affect both nitric oxide formation and association with hypertensive disorders of pregnancy. Front. Genet. 2024, 15, 1293082. [Google Scholar] [CrossRef] [PubMed]
- Anto, E.O.; Coall, D.A.; Asiamah, E.A.; Afriyie, O.O.; Addai-Mensah, O.; Wiafe, Y.A.; Owiredu, W.; Obirikorang, C.; Annani-Akollor, M.E.; Titiloye, N.A.; et al. Placental lesions and differential expression of pro- and anti-angiogenic growth mediators and oxidative DNA damage marker in placentae of Ghanaian suboptimal and optimal health status pregnant women who later developed preeclampsia. PLoS ONE 2022, 17, e0265717. [Google Scholar] [CrossRef] [PubMed]
- Haram, K.; Mortensen, J.H.; Myking, O.; Magann, E.F.; Morrison, J.C. The Role of Oxidative Stress, Adhesion Molecules and Antioxidants in Preeclampsia. Curr. Hypertens. Rev. 2019, 15, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.K.; Bijalwan, V.; Yadav, S.; Sarkar, K.; Das, S.; Singh, D.P. Susceptibility of male reproductive system to bisphenol A, an endocrine disruptor: Updates from epidemiological and experimental evidence. J. Biochem. Mol. Toxicol. 2023, 37, e23292. [Google Scholar] [CrossRef]
- Hirke, A.; Varghese, B.; Varade, S.; Adela, R. Exposure to endocrine-disrupting chemicals and risk of gestational hypertension and preeclampsia: A systematic review and meta-analysis. Environ. Pollut. 2023, 317, 120828. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, A.G.C.; Coutinho, V.L.; Meyer, A.; Lisboa, P.C.; de Moura, E.G. Human exposure to bisphenol A (BPA) through medical-hospital devices: A systematic review. Environ. Toxicol. Pharmacol. 2023, 97, 104040. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Das, M.K.; Duttaroy, A.K. Plastics derived endocrine-disrupting compounds and their effects on early development. Birth Defects Res. 2020, 112, 1308–1325. [Google Scholar] [CrossRef] [PubMed]
- Peretz, J.; Vrooman, L.; Ricke, W.A.; Hunt, P.A.; Ehrlich, S.; Hauser, R.; Padmanabhan, V.; Taylor, H.S.; Swan, S.H.; VandeVoort, C.A.; et al. Bisphenol a and reproductive health: Update of experimental and human evidence, 2007–2013. Environ. Health Perspect. 2014, 122, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef]
- Fernandes, L.M.; Lorigo, M.; Cairrao, E. Relationship between Androgens and Vascular and Placental Function during Pre-eclampsia. Curr. Issues Mol. Biol. 2024, 46, 1668–1693. [Google Scholar] [CrossRef]
- Kumar, S.; Gordon, G.H.; Abbott, D.H.; Mishra, J.S. Androgens in maternal vascular and placental function: Implications for preeclampsia pathogenesis. Reproduction 2018, 156, R155–R167. [Google Scholar] [CrossRef] [PubMed]
- Salustiano, E.M.; De Pinho, J.C.; Provost, K.; Ruano, R.; Zugaib, M. Maternal serum hormonal factors in the pathogenesis of preeclampsia. Obstet. Gynecol. Surv. 2013, 68, 141–150. [Google Scholar] [CrossRef]
- Kohli, S.; Singh, K.K.; Gupta, A.; Markmeyer, P.; Lochmann, F.; Gupta, D.; Rana, R.; Elwakiel, A.; Huebner, H.; Ruebner, M.; et al. Placental thromboinflammation impairs embryonic survival by reducing placental thrombomodulin expression. Blood 2021, 137, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Negi, R.; Pande, D.; Karki, K.; Kumar, A.; Khanna, R.S.; Khanna, H.D. Association of oxidative DNA damage, protein oxidation and antioxidant function with oxidative stress induced cellular injury in pre-eclamptic/eclamptic mothers during fetal circulation. Chem. Biol. Interact. 2014, 208, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Sandrim, V.C.; Luizon, M.R.; Pilan, E.; Caldeira-Dias, M.; Coeli-Lacchini, F.B.; Kors, G.; Berndt, I.; Lacchini, R.; Cavalli, R.C. Interaction Between NOS3 and HMOX1 on Antihypertensive Drug Responsiveness in Preeclampsia. Rev. Bras. Ginecol. Obstet. 2020, 42, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Boudreau, A.; Ahmed, S.; Atlas, E. In Vitro Effects of Bisphenol A β-D-Glucuronide (BPA-G) on Adipogenesis in Human and Murine Preadipocytes. Environ. Health Perspect. 2015, 123, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Just, A.C.; Williams, P.L.; Smith, K.W.; Calafat, A.M.; Hauser, R. Personal care product use and urinary phthalate metabolite and paraben concentrations during pregnancy among women from a fertility clinic. J. Expo. Sci. Environ. Epidemiol. 2014, 24, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Tschugguel, W.; Dietrich, W.; Zhegu, Z.; Stonek, F.; Kolbus, A.; Huber, J.C. Differential regulation of proteasome-dependent estrogen receptor alpha and beta turnover in cultured human uterine artery endothelial cells. J. Clin. Endocrinol. Metab. 2003, 88, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.D.; Pepe, G.J. Estrogen regulation of placental angiogenesis and fetal ovarian development during primate pregnancy. Int. J. Dev. Biol. 2010, 54, 397–408. [Google Scholar] [CrossRef]
- McCrohon, J.A.; Jessup, W.; Handelsman, D.J.; Celermajer, D.S. Androgen exposure increases human monocyte adhesion to vascular endothelium and endothelial cell expression of vascular cell adhesion molecule-1. Circulation 1999, 99, 2317–2322. [Google Scholar] [CrossRef]
- Aggarwal, R.; Jain, A.K.; Mittal, P.; Kohli, M.; Jawanjal, P.; Rath, G. Association of pro- and anti-inflammatory cytokines in preeclampsia. J. Clin. Lab. Anal. 2019, 33, e22834. [Google Scholar] [CrossRef] [PubMed]
- Bergman, L.; Torres-Vergara, P.; Penny, J.; Wikström, J.; Nelander, M.; Leon, J.; Tolcher, M.; Roberts, J.M.; Wikström, A.K.; Escudero, C. Investigating Maternal Brain Alterations in Preeclampsia: The Need for a Multidisciplinary Effort. Curr. Hypertens. Rep. 2019, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Cherkerzian, S.; Herlihy, M.; Hacker, M.R.; McElrath, T.F.; Cantonwine, D.E.; Fichorova, R.; Oken, E.; Meydani, S.N. Supplementation with antioxidant micronutrients in pregnant women with obesity: A randomized controlled trial. Int. J. Obes. 2024, 48, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Rumbold, A.R.; Crowther, C.A.; Haslam, R.R.; Dekker, G.A.; Robinson, J.S. Vitamins C and E and the risks of preeclampsia and perinatal complications. N. Engl. J. Med. 2006, 354, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Briley, A.L.; Seed, P.T.; Kelly, F.J.; Shennan, A.H. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet 2006, 367, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kalpdev, A.; Saha, S.C.; Dhawan, V. Vitamin C and E supplementation does not reduce the risk of superimposed PE in pregnancy. Hypertens. Pregnancy 2011, 30, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.D.; Costantine, M.M. The role of statins in the prevention of preeclampsia. Am. J. Obstet. Gynecol. 2022, 226, S1171–S1181. [Google Scholar] [CrossRef] [PubMed]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N. Engl. J. Med. 2017, 377, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Khalil, R.A. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H661–H681. [Google Scholar] [CrossRef]
- Eddy, A.C.; Bidwell, G.L., 3rd; George, E.M. Pro-angiogenic therapeutics for preeclampsia. Biol. Sex Differ. 2018, 9, 36. [Google Scholar] [CrossRef]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
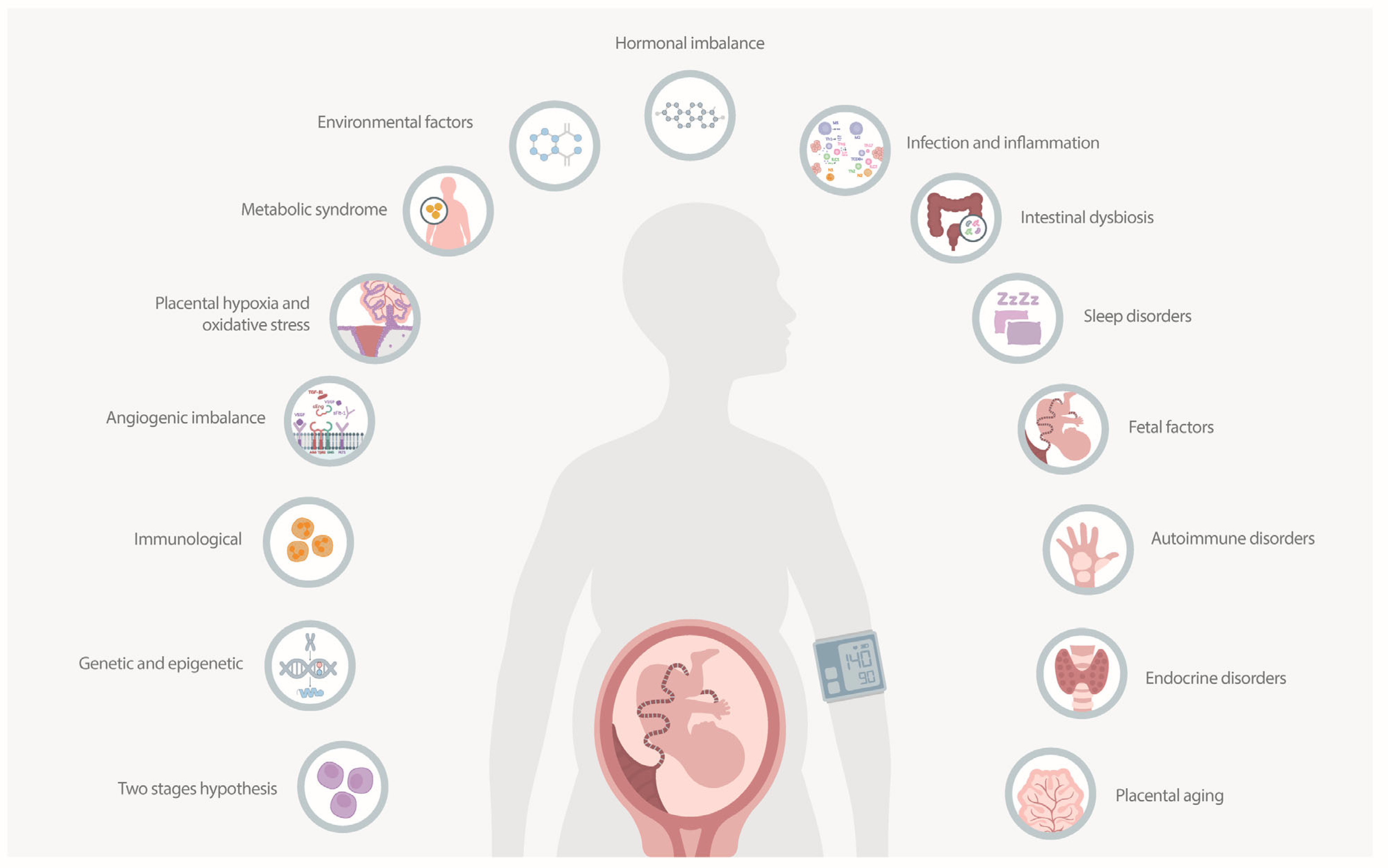
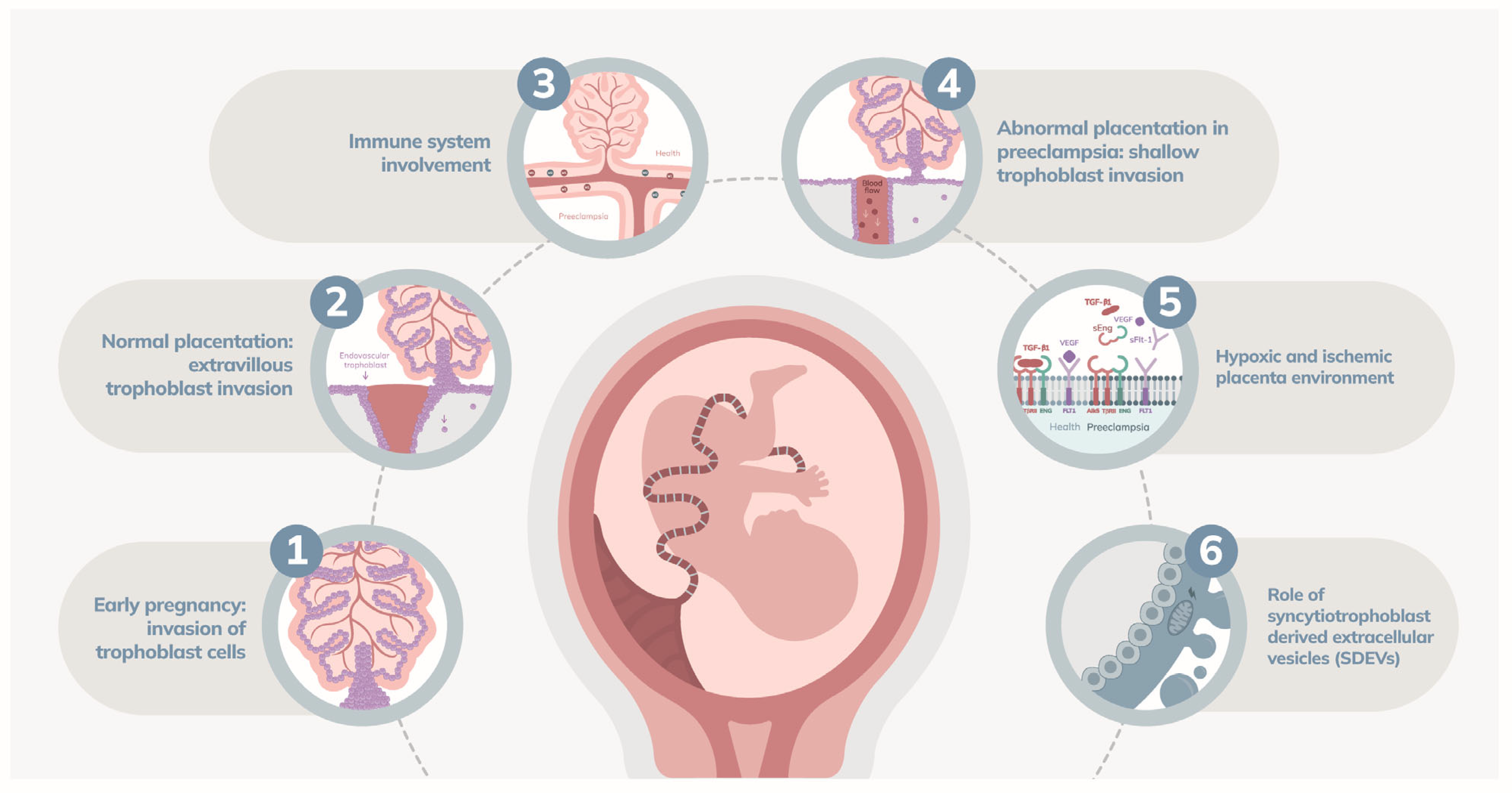

| Associated Mechanisms | Description |
|---|---|
| Abnormal placentation | Early pregnancy issues involving deficient invasion of extravillous trophoblasts and inadequate remodeling of spiral arteries, leading to reduced placental perfusion and hypoxia. Factors include genetic predisposition, reduced HLA-G expression, and dysfunctional interactions between uterine NK cells and trophoblasts [27,28,29]. |
| Maternal systemic response | Endothelial dysfunction caused by the hypoxic placenta-releasing factors like sFlt-1 and sEng, resulting in increased vascular permeability and heightened vascular reactivity. Clinical manifestations include hypertension and proteinuria due to an imbalance between vasodilators (nitric oxide) and vasoconstrictors (endothelin-1) [30]. |
| Systemic inflammation and oxidative stress | Oxidative stress from excess reactive oxygen species (ROS) production due to placental hypoxia, damaging endothelial cells. Systemic inflammation involves elevated pro-inflammatory cytokines (TNF-α, IL-6), activating inflammatory signaling pathways, and perpetuating endothelial damage and oxidative stress [31]. |
| Genetic and epigenetic factors | Genetic variants include polymorphisms in genes regulating endothelial function, inflammatory response, and angiogenesis (e.g., VEGF, ENG) that increase preeclampsia risk. Epigenetic modifications involve DNA methylation and histone modifications affecting key genes involved in blood pressure regulation and placental function [32]. |
| Other associated mechanisms | Mitochondrial dysfunction leads to excessive ROS production and oxidative damage, exacerbating endothelial dysfunction. Inflammatory pathways, such as NF-κB and JNK, increase pro-inflammatory cytokine production. RAAS alterations include overexpression of angiotensin II, contributing to hypertension [33,34]. |
| Mechanism | Description | Effect | Key Substance |
|---|---|---|---|
| Pro-angiogenic and anti-angiogenic factors | Inadequate trophoblast invasion and poor spiral artery remodeling trigger overproduction of sFlt-1 and sEng [59]. | Disruption of angiogenesis and endothelial repair, resulting in reduced NO production and clinical manifestations of PE [60]. sFlt-1 binds and neutralizes VEGF and PlGF, disrupting angiogenesis and endothelial repair, leading to endothelial dysfunction [30]. sEng inhibits TGF-β signaling, exacerbating endothelial damage [30,59]. | sFlt-1, sEng, VEGF, PlGF, TGF-β, CD93 |
| Endothelial dysfunction | Characterized by increased vascular permeability and reactivity, disrupted balance between vasodilators and vasoconstrictors [60] | Leads to vasoconstriction, increased blood pressure, and a pro-inflammatory state [59,60]. | NO, endothelin-1, ROS, TNF-α, IL-6 |
| Oxidative stress | Hypoxic placenta generates excess ROS through NADPH oxidase and xanthine oxidase activities [61]. | ROS damage cellular components, leading to endothelial cell apoptosis and further dysfunction [62,63]. | ROS, NADPH oxidase, xanthine oxidase |
| Endothelial cell activation | Increased expression of adhesion molecules by endothelial cells [64]. | Promotes leukocyte adhesion and transmigration, contributing to a pro-inflammatory and pro-thrombotic state [59,60]. | ICAM-1, VCAM-1 |
| The role of adipokines | Elevated levels of chemerin and leptin in PE [65]. | Contributes to endothelial dysfunction, metabolic dysregulation, and increased ROS production [66,67]. | Chemerin, leptin |
| Systemic inflammation | Characterized by elevated levels of pro-inflammatory cytokines [68]. | Promotes endothelial dysfunction, increased ROS, and chronic immune activation [69]. | TNF-α, IL-6, IL-17, Th1, Th17, Tregs, IL-10 |
| The role of ferroptosis | Iron-dependent cell death driven by lipid peroxidation and dysregulated iron metabolism [70]. | Contributes to placental dysfunction and exacerbates oxidative stress [71,72]. | GPX4, iron, PUFAs |
| Epigenetic factors | Hypomethylation and upregulation of specific genes like MTHFR and miR-155 [73]. | Altered gene expression leading to endothelial dysfunction and inflammation [74]. | MTHFR, miR-155 |
| Genetic factors | Polymorphisms in genes affecting angiogenesis and NO bioavailability [75] | Contributes to endothelial dysfunction and hypertension [75,76]. | sFlt-1, NOS3, GUCY1A3 |
| Other associated factors | Mitochondrial dysfunction, platelet activation, renin-angiotensin-aldosterone system, and adipokines [77,78,79]. | Contribute to oxidative stress, endothelial dysfunction, and inflammation [77,78,79]. | CoQ10, platelets, angiotensin II |
| Role of HO-1/CO and CSE/H2S pathways | Protective roles in preeclampsia by inhibiting the release of anti-angiogenic factors [80]. | Loss of HO activity contributes to pathogenesis, while H2S acts as a pro-angiogenic vasodilator [80,81]. | HO-1, CO, CSE, H2S |
| Micronutrient deficiencies and inositol messengers | Calcium and magnesium deficiencies modulate oxidative stress and angiogenic balance, while inositol messengers regulate the pyruvate dehydrogenase complex [82]. | Micronutrient deficiencies correlate with imbalanced AGMs and oxidative stress biomarkers, contributing to metabolic disturbances [82,83]. | Calcium, magnesium, IPG-P, IPG-A |
| Mechanism | Description | Effect | Key Factors |
|---|---|---|---|
| Mitochondrial dysfunction | Abnormalities in mitochondrial gene expression and lipid peroxidation [78]. | Increased oxidative stress and endothelial dysfunction. | Mitochondrial genes, CoQ10 |
| Platelet activation | Increased activation markers (e.g., P-selectin, CD63) and mean platelet volume [77]. | Contributes to prothrombotic state and inflammation. | P-selectin, CD63, platelets |
| RAAS dysregulation | Overexpression of angiotensin II and its vasoconstrictive effects [79]. | Leads to hypertension and endothelial dysfunction. | Angiotensin II, RAAS |
| Oxidative stress | Syncytiotrophoblast damage and local hypoxia increase ROS production [112,113]. | Leads to DNA damage, impaired angiogenesis, and adverse outcomes. | ROS, sFlt-1, PlGF, 8-OHdG |
| Micronutrient deficiencies | Deficiencies in calcium and magnesium [82]. | Modulates oxidative stress and angiogenic balance. | Calcium, magnesium |
| Inositol messengers | Regulation of the pyruvate dehydrogenase complex by IPG-P and IPG-A [83]. | Contributes to metabolic disturbances in preeclampsia. | IPG-P, IPG-A |
| HO-1/CO pathway | HO-1 and CO inhibit release of anti-angiogenic factors [80]. | Protective role by reducing sFlt-1 and sEng levels. | HO-1, CO |
| CSE/H2S pathway | H2S produced by CSE acts as a pro-angiogenic vasodilator [81]. | Reduced H2S leads to abnormal placentation and hypertension. | CSE, H2S |
| Environmental factors and endocrine disruptors | Exposure to compounds like BPA and phthalates disrupts hormone signaling and induces oxidative stress [114,115,116,117,118,119]. | Leads to impaired placental function and increased risk of preeclampsia. | BPA, phthalates, ESR1, PPARγ, TNF-α, IL-6, VEGF |
| Estrogen and androgen dysregulation | Disrupted estrogen and androgen signaling affects vascular function and angiogenic balance [120,121,122]. | Leads to endothelial dysfunction, impaired placental development, and inflammation. | ESR1, ESR2, eNOS, VEGF, PlGF, AR, NADPH oxidase, sFlt-1, sEng, PPARγ, TNF-α, IL-6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Torres, J.; Espino-y-Sosa, S.; Martinez-Portilla, R.; Borboa-Olivares, H.; Estrada-Gutierrez, G.; Acevedo-Gallegos, S.; Ruiz-Ramirez, E.; Velasco-Espin, M.; Cerda-Flores, P.; Ramirez-Gonzalez, A.; et al. A Narrative Review on the Pathophysiology of Preeclampsia. Int. J. Mol. Sci. 2024, 25, 7569. https://doi.org/10.3390/ijms25147569
Torres-Torres J, Espino-y-Sosa S, Martinez-Portilla R, Borboa-Olivares H, Estrada-Gutierrez G, Acevedo-Gallegos S, Ruiz-Ramirez E, Velasco-Espin M, Cerda-Flores P, Ramirez-Gonzalez A, et al. A Narrative Review on the Pathophysiology of Preeclampsia. International Journal of Molecular Sciences. 2024; 25(14):7569. https://doi.org/10.3390/ijms25147569
Chicago/Turabian StyleTorres-Torres, Johnatan, Salvador Espino-y-Sosa, Raigam Martinez-Portilla, Hector Borboa-Olivares, Guadalupe Estrada-Gutierrez, Sandra Acevedo-Gallegos, Erika Ruiz-Ramirez, Martha Velasco-Espin, Pablo Cerda-Flores, Andrea Ramirez-Gonzalez, and et al. 2024. "A Narrative Review on the Pathophysiology of Preeclampsia" International Journal of Molecular Sciences 25, no. 14: 7569. https://doi.org/10.3390/ijms25147569
APA StyleTorres-Torres, J., Espino-y-Sosa, S., Martinez-Portilla, R., Borboa-Olivares, H., Estrada-Gutierrez, G., Acevedo-Gallegos, S., Ruiz-Ramirez, E., Velasco-Espin, M., Cerda-Flores, P., Ramirez-Gonzalez, A., & Rojas-Zepeda, L. (2024). A Narrative Review on the Pathophysiology of Preeclampsia. International Journal of Molecular Sciences, 25(14), 7569. https://doi.org/10.3390/ijms25147569