
Metabolic Crosstalk between Liver and Brain: From Diseases to Mechanisms

Abstract
:
1. Introduction
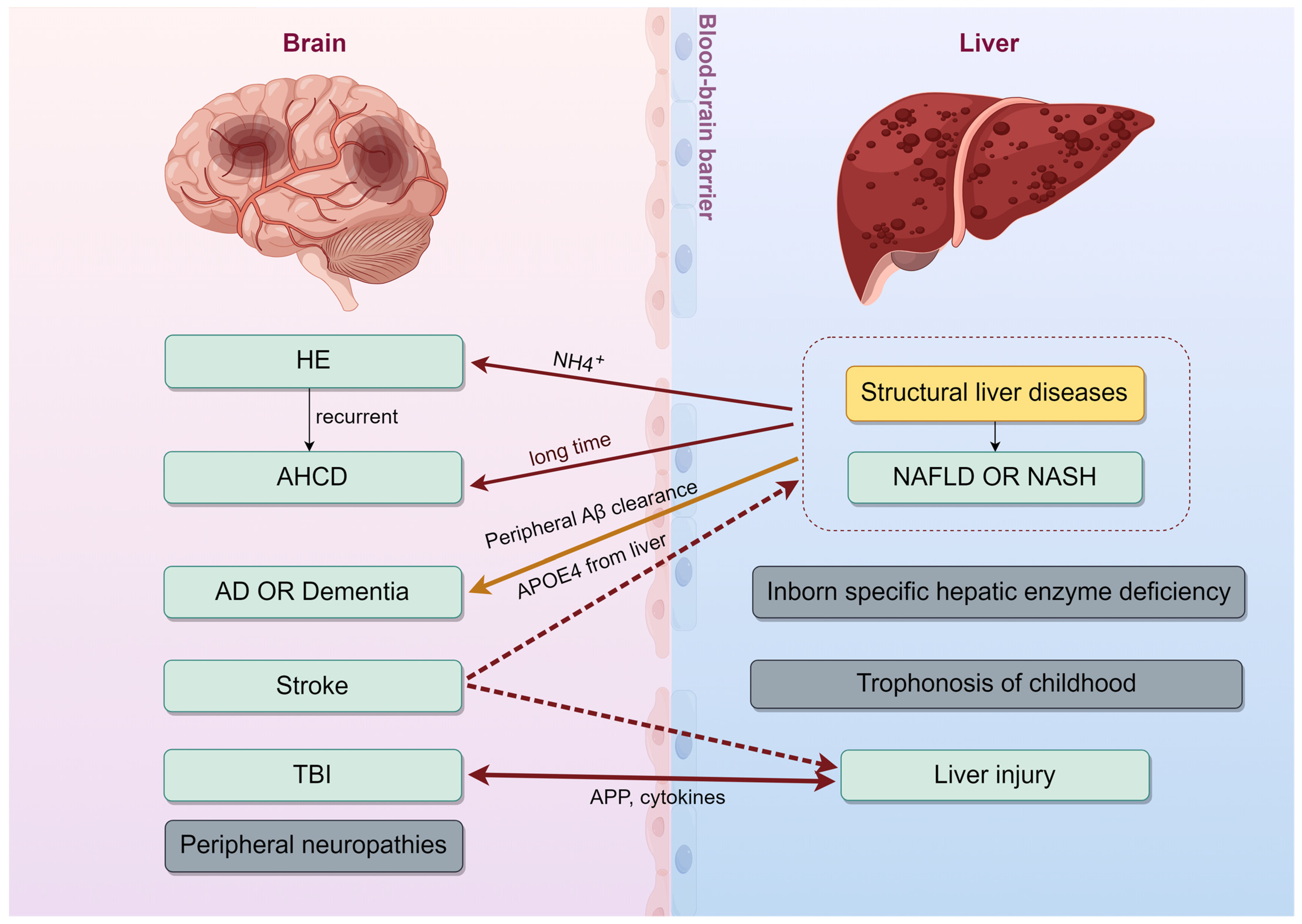
2. Interconnected Diseases of the Liver and Brain
2.1. Neurological Syndrome Linked to a Liver Disease
2.1.1. Hepatic Encephalopathy (HE)
2.1.2. Acquired Hepatocerebral Degeneration (AHCD)
2.1.3. Stroke
2.2. Neurodegenerative Diseases and the Liver
2.2.1. Liver’s Role in Neurodegeneration
2.2.2. Liver’s Clearance of Aβ
2.2.3. Liver-Derived APOE in AD Pathogenesis
2.3. Hepatic Responses to Cerebral Lesions
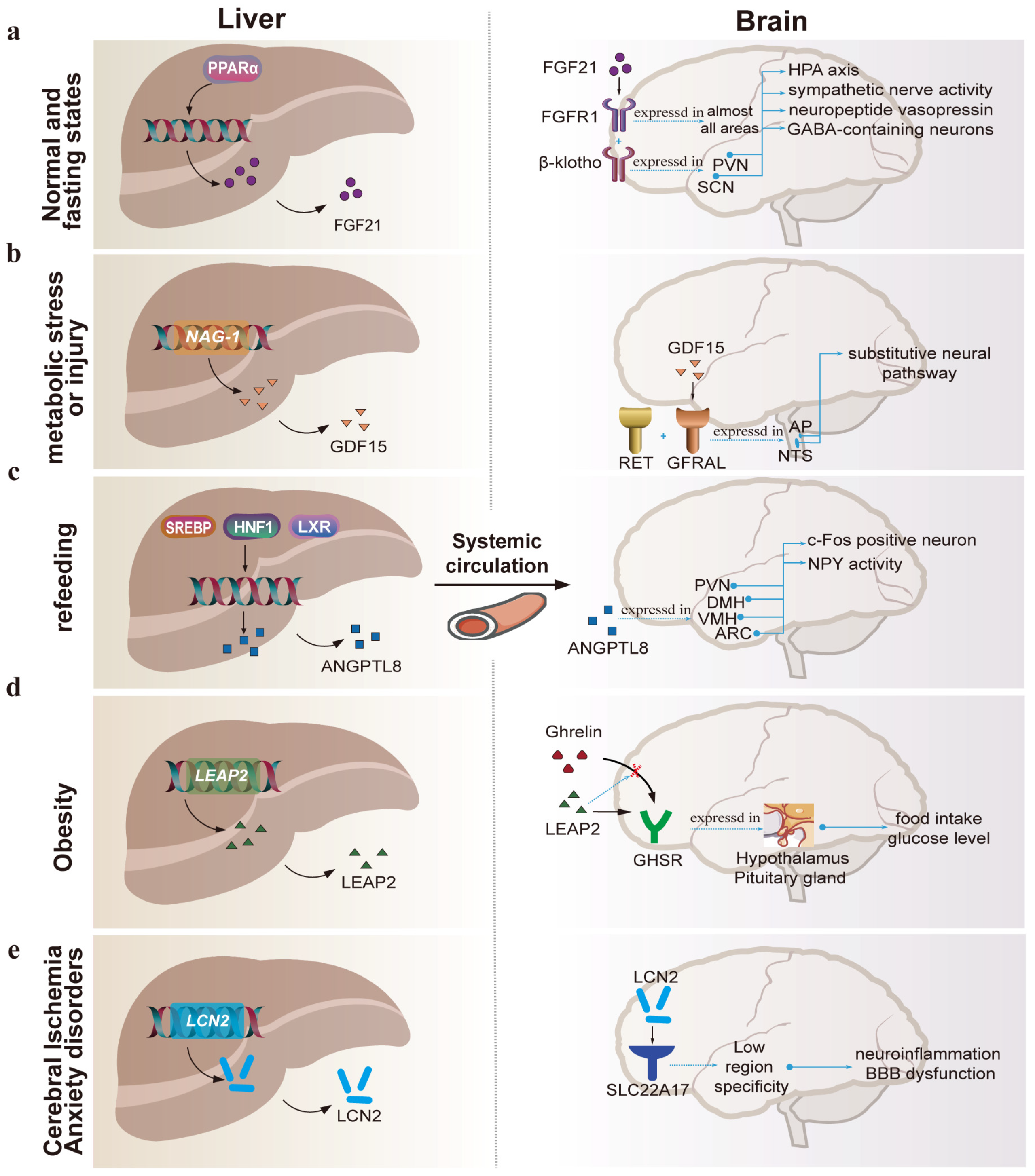
3. Hepatokines Which Act on the Brain
3.1. FGF21

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hepatokines | Molecular Weight | Concentration in Human Blood | Receptors | Central Site of Receptor Expression | Effects on the Brain | Diseases with Therapeutic Potential |
|---|---|---|---|---|---|---|
| Apolipoprotein E (APOE) | 34 kDa | 0.03~0.05 g/L [65] | Low density lipoprotein receptor(LDLR) family; LDL receptor-related protein 1(LRP1) | Low region specificity | Maintains cholesterol homeostasis of brain; liver-expressed apoE4 exacerbated brain Aβ deposition and cerebrovascular dysfunction, whereas apoE3 reduced it. | Alzheimer’s disease [33,34] |
| Fibroblast growth factor 21 (FGF21) | 19.5 kDa | 200~300 pg/mL [66] | FGF receptor (FGFR1); FGF co-receptor (β-klotho) | FGFR1 is spread throughout the nervous system, but co-receptor β-klotho is predominantly expressed in hypothalamus, hippocampal region, subiculum, and amygdala [55] | Regulates energy homeostasis, via activation of the hypothalamus–pituitary–adrenal axis. | Obesity; NAFLD; Diabetes mellitus [57] |
| Growth differentiation factor 15 (GDF15) | 24.8 kDa | 100~1200 pg/mL [47] | Glial-derived neurotropic factor receptor-a like (GFRAL); co-receptor rearranged during transfection (RET) | The area postrema (AP); the nucleus of solitary tract (NTS) [67] | Conveys peripheral metabolic messages to the brain where it activates substitutive neuronal pathways to adapt to shifting energy demands; reduces food intake and body mass. | Diabetes mellitus; Obesity; NAFLD [68,69] |
| Tsukushi (TSK) | 34 kDa | 18–49 ng/mL [70] | Not clear yet | Not clear yet | Functions as a liver-derived feedback hormone that attenuates energy expenditure by engaging in crosstalk with the CNS in hypermetabolic states. | Metabolic disease [49] |
| Angiopoietin-like protein 8 (ANGPTL8) | 22.5 kDa | ~300 pg/mL [71] | Leukocyte immunoglobulin-like receptor B3 (LILRB3) [72] | Low region specificity and vasculature (mainly) | Is involved in the regulation of appetite. | Anorexia; Diabetes mellitus; Obesity; NAFLD [73] |
| Insulin-like growth factor 1 (IGF-1) | 7.6 kDa | 82~487 ng/mL [74] | Insulin like growth factor 1 receptor (IGF1R) | Low region specificity | Mediates brain growth and development; functions as an anti-apoptotic agent by enhancing cell survival. | Disorders related to brain development [75]; Traumatic brain injury [76]; Age-Related Neurological Conditions [77,78] |
| Energy Homeostasis Associated gene (ENHO) (Adropin) | 5.0 kDa | 3.4~4.5 ng/mL [79] | Not clear yet [80] | Not clear yet | Regulates endothelial cells and maintains blood–brain barrier integrity. | Transient Ischemic Stroke [81,82]; Aging-related neuropathology [83] |
| Liver-enriched antimicrobial peptide-2 (LEAP2) | 23 kDa | 5~20 ng/mL [84] | Growth hormone secretagogue receptor (GHSR) | Hypothalamus, Pituitary gland | Endogenous antagonist of Ghrelin Receptor, thus preventing the effects of ghrelin; regulator of food intake, glucose level and body weight. | Obesity [85] |
| Lipocalin-2 (LCN2) | 22.6 kDa | 590 µg/L 1 | Solute carrier family 22 member 17 (SLC22A17) | Low region specificity | Induces neuroinflammation and blood–brain barrier dysfunction [86]; induces anxity-like behavior through Lcn2 receptors in the medial prefrontal cortex (mPFC). | Cerebral Ischemia [87]; Anxiety disorders [88]; Neurodegenerative diseases [89] |
3.2. GDF15
3.3. ANGPTL8
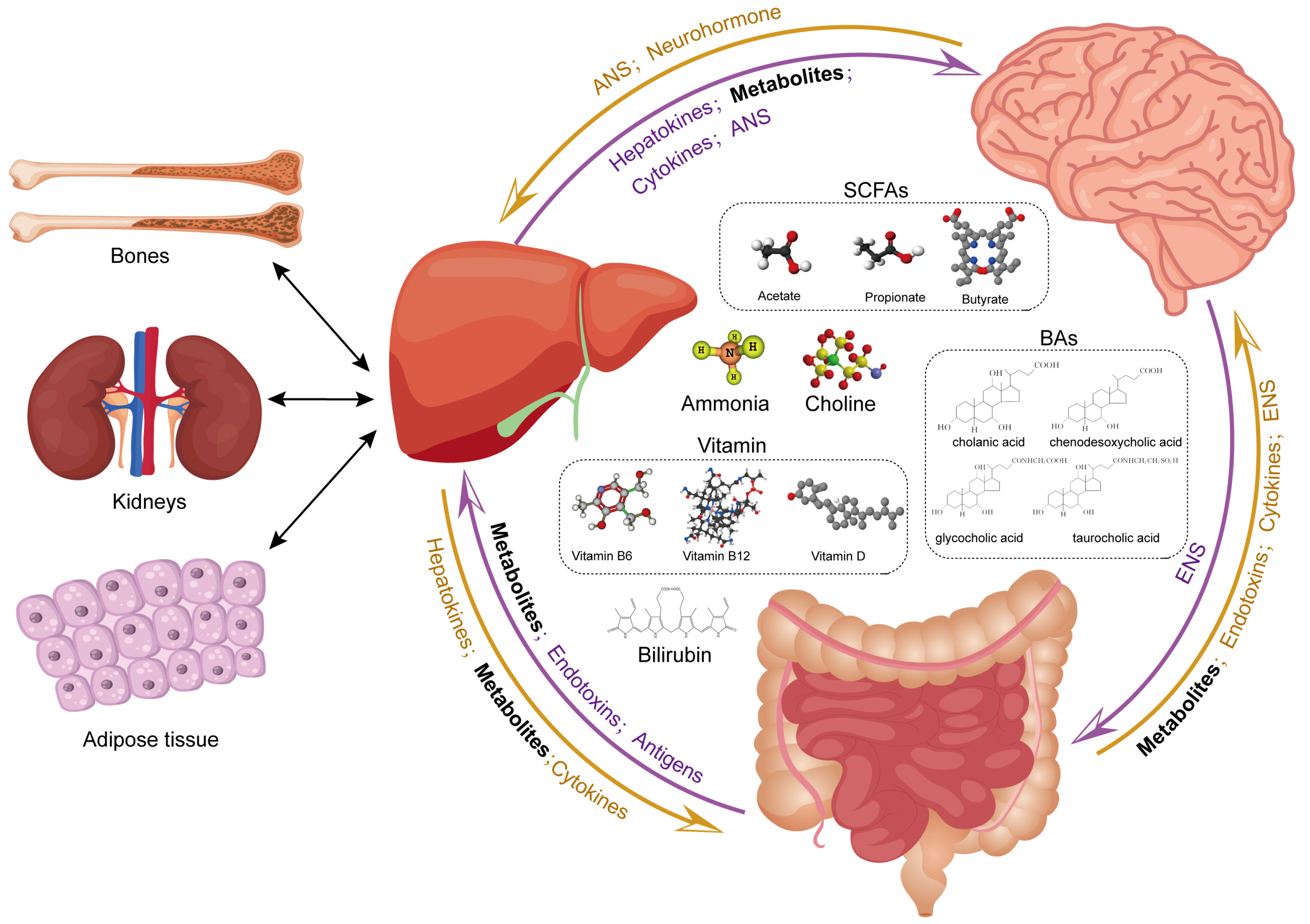
4. Metabolites from the Liver to the Brain
4.1. Bile Acids
4.2. Short-Chain Fatty Acids
4.3. Ammonia
4.4. Bilirubin
4.5. Vitamin
4.6. Choline
4.7. Liver–Brain Axis and Gut Metabolites: Possible Association
5. Neural Interfaces between Brain and Liver
5.1. Hypothalamic Nuclei Influence Liver Metabolism
5.1.1. Hypothalamic Nuclei and Liver
5.1.2. ARC and Liver Metabolism
5.1.3. PVN and Liver Metabolism
5.1.4. VMH and Liver Metabolism
5.2. Nerve Fiber Connections between the CNS and the Liver
5.2.1. Afferent Sensory Nerves
5.2.2. Efferent Nerve Pathways
5.2.3. Neurohormone and the Liver
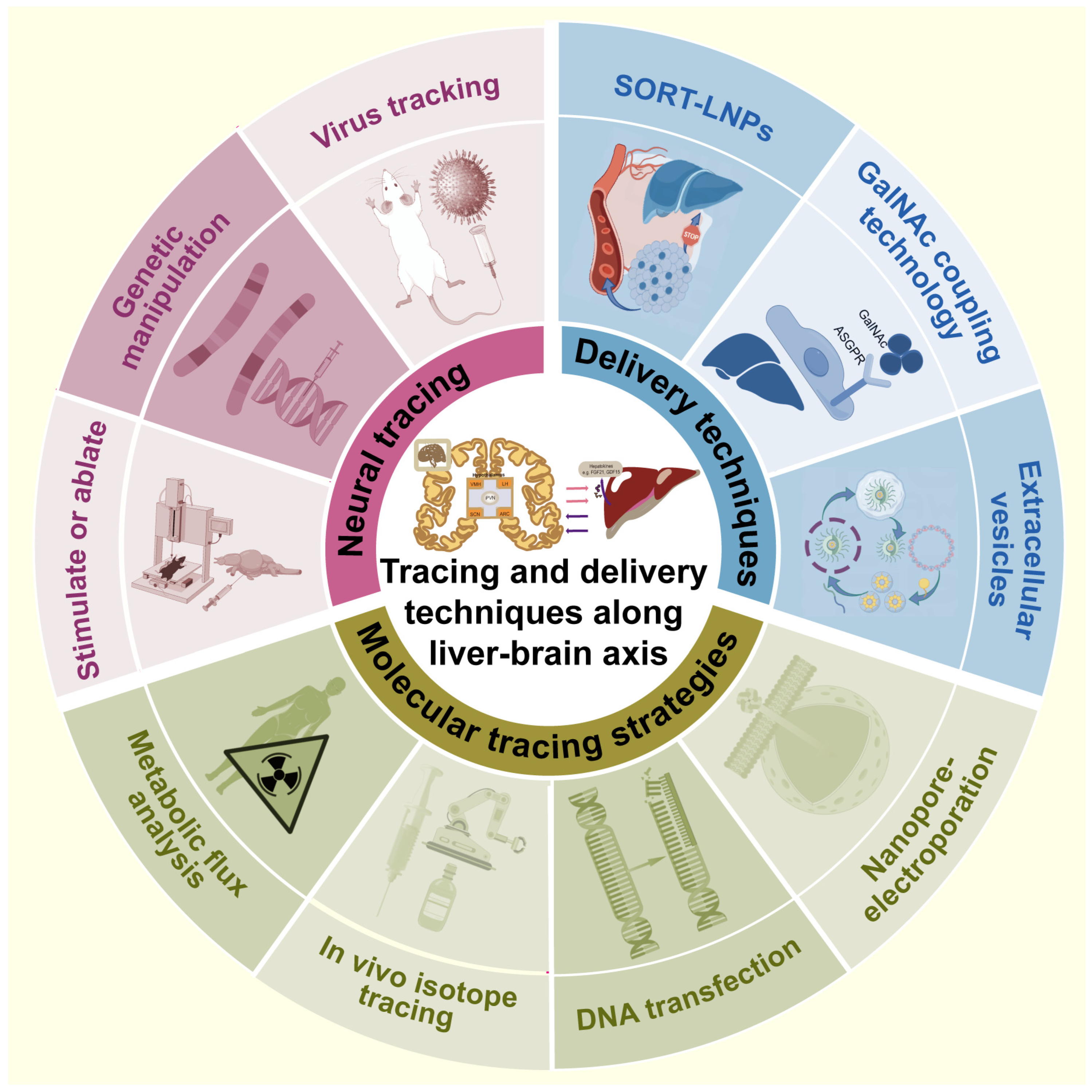
6. Advanced Techniques for Tracing and Transmitting along the Liver–Brain Axis
6.1. Molecular Tracing
6.2. Neural Tracing
6.3. Transmitting Techniques
7. Summary and Perspectives
- (1)
- While certain liver-derived factors are known to be recruited to the CNS, only a few of these circulating factors have been identified for their central receptors and central action. It is worth continuing to study the effects of liver factors on the central nervous system and the sites of action, and trying to find specific drugs that regulate the central metabolism.
- (2)
- Intestinal flora metabolites mediate liver–brain interactions. The complexity of the gut microbiome is daunting. Most of the current research is an observational snapshot of the gut microbiota and has not explored in detail the dynamic evolution of microbial products in the liver and brain. How to determine the source of intestinal metabolites? What role does the liver play in these processes? What is the dynamic evolution of gut microbes in different tissues? These questions are left for future studies.
- (3)
- Though the central location of receptors for several liver factors has been identified, delivering drugs to specific brain regions and avoiding side effects are still a challenge. Future studies could explore discovering more hepatogenic molecules with central receptor and regulatory roles to develop highly selective agonists or antagonists for the neuroregulation of metabolism.
- (4)
- The hypothalamus–ANS–liver axis has been confirmed, with several hypothalamic nuclei engaging in hepatic metabolism through ANS outputs. However, the precise neuroanatomy and the transmission of metabolic information via nerve fibers in the liver require further refinement.
- (5)
- Cutting-edge techniques such as single-molecule tracking and cell type-specific transgenic methods will be instrumental in deciphering how the liver communicates with the brain. However, the throughput and resolution are low, and we call for the development of single-cell and single-organelle metabolomics technologies.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Castera, L.; Laouenan, C.; Vallet-Pichard, A.; Vidal-Trécan, T.; Manchon, P.; Paradis, V.; Roulot, D.; Gault, N.; Boitard, C.; Terris, B.; et al. High Prevalence of NASH and Advanced Fibrosis in Type 2 Diabetes: A Prospective Study of 330 Outpatients Undergoing Liver Biopsies for Elevated ALT, Using a Low Threshold. Diabetes Care 2023, 46, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Mark, H.E.; Villota-Rivas, M.; Palayew, A.; Carrieri, P.; Colombo, M.; Ekstedt, M.; Esmat, G.; George, J.; Marchesini, G.; et al. The Global NAFLD Policy Review and Preparedness Index: Are Countries Ready to Address This Silent Public Health Challenge? J. Hepatol. 2022, 76, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef] [PubMed]
- Wijdicks, E.F.M. Hepatic Encephalopathy. N. Engl. J. Med. 2016, 375, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Winnick, J.J.; Kraft, G.; Gregory, J.M.; Edgerton, D.S.; Williams, P.; Hajizadeh, I.A.; Kamal, M.Z.; Smith, M.; Farmer, B.; Scott, M.; et al. Hepatic Glycogen Can Regulate Hypoglycemic Counterregulation via a Liver-Brain Axis. J. Clin. Investig. 2016, 126, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Kiyohara, H.; Teratani, T.; Mikami, Y.; Kanai, T. Organ and Brain Crosstalk: The Liver-Brain Axis in Gastrointestinal, Liver, and Pancreatic Diseases. Neuropharmacology 2022, 205, 108915. [Google Scholar] [CrossRef]
- Penticoff, H.B.; Fortin, J.S. Toxic/Metabolic Diseases of the Nervous System. In Neurobiology of Brain Disorders, 2nd ed.; Zigmond, M.J., Wiley, C.A., Chesselet, M.-F., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 379–401. ISBN 978-0-323-85654-6. [Google Scholar]
- Bateman, J.R.; Roque, D.A. Teaching NeuroImages: Acquired Hepatocerebral Degeneration: An Underrecognized Complication of Advanced Liver Disease. Neurology 2016, 87, e144. [Google Scholar] [CrossRef]
- Gleason, A.; Hayhow, B.; Walterfang, M.; Evans, A.; Mocellin, R.; Gates, P.; Velakoulis, D. Neuropsychiatric Symptoms as the Presenting Feature of Acquired Hepatocerebral Degeneration. Aust. N. Z. J. Psychiatry 2014, 48, 959–960. [Google Scholar] [CrossRef]
- Malaquias, M.J.; Pinto, C.M.; Ramos, C.; Ferreira, S.; Gandara, J.; Almeida, A.; Cavaco, S.; Miranda, H.P.; Magalhães, M. Acquired Hepatocerebral Degeneration and Hepatic Encephalopathy: One or Two Entities? Eur. J. Neurol. 2020, 27, 2396–2404. [Google Scholar] [CrossRef]
- Au, M.; Smart, C.; Chinnaratha, M.A. A Rare Neurological Complication in Advanced Liver Disease: Acquired Hepatocerebral Degeneration. Am. J. Med. Sci. 2022, 363, e37–e38. [Google Scholar] [CrossRef] [PubMed]
- Rajoriya, N.; Brahmania, M.; Feld, J.J. Implications of Manganese in Chronic Acquired Hepatocerebral Degeneration. Ann. Hepatol. 2019, 18, 274–278. [Google Scholar] [CrossRef]
- Nascimento, H.; Malaquias, M.J.; Pinto, C.M.; Sá Silva, J.; Rochate, D.; Fraga, C.; Alves, J.E.; Ramos, C.; Gandara, J.; Ferreira, S.; et al. Trace Element Imbalances in Acquired Hepatocerebral Degeneration and Changes after Liver Transplant. Biology 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dai, L.; Zhang, Y.; Wang, A.; Li, H.; Wang, Y.; Meng, X.; Wu, S.; Wang, Y. Severity of Nonalcoholic Fatty Liver Disease and Risk of Future Ischemic Stroke Events. Stroke 2021, 52, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zha, M.; Lv, Q.; Xie, Y.; Yuan, K.; Zhang, X.; Liu, X. Non-Alcoholic Fatty Liver Disease and Stroke: A Mendelian Randomization Study. Eur. J. Neurol. 2022, 29, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Non-Alcoholic Fatty Liver Disease and Risk of Incident Acute Myocardial Infarction and Stroke: Findings from Matched Cohort Study of 18 Million European Adults. BMJ 2019, 367, l5367. [Google Scholar] [CrossRef]
- Canillas, L.; Soriano-Varela, A.; Rodríguez-Campello, A.; Giralt-Steinhauer, E.; Cuadrado-Godia, E.; Broquetas, T. High Prevalence of Non-Alcoholic Fatty Liver Disease in Patients with a First Episode of Acute Ischemic Stroke. Impact on Disability and Death. Front. Endocrinol. 2022, 13, 1003878. [Google Scholar] [CrossRef]
- Zheng, H.; Cai, A.; Shu, Q.; Niu, Y.; Xu, P.; Li, C.; Lin, L.; Gao, H. Tissue-Specific Metabolomics Analysis Identifies the Liver as a Major Organ of Metabolic Disorders in Amyloid Precursor Protein/Presenilin 1 Mice of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 1218–1227. [Google Scholar] [CrossRef]
- Lu, Y.; Pike, J.R.; Hoogeveen, R.C.; Walker, K.A.; Raffield, L.M.; Selvin, E.; Avery, C.L.; Engel, S.M.; Mielke, M.M.; Garcia, T.; et al. Liver Integrity and the Risk of Alzheimer’s Disease and Related Dementias. Alzheimers Dement. 2024, 20, 1913–1922. [Google Scholar] [CrossRef]
- Giuffrè, M.; Merli, N.; Pugliatti, M.; Moretti, R. The Metabolic Impact of Nonalcoholic Fatty Liver Disease on Cognitive Dysfunction: A Comprehensive Clinical and Pathophysiological Review. Int. J. Mol. Sci. 2024, 25, 3337. [Google Scholar] [CrossRef]
- Xiao, T.; van Kleef, L.A.; Ikram, M.K.; de Knegt, R.J.; Ikram, M.A. Association of Nonalcoholic Fatty Liver Disease and Fibrosis with Incident Dementia and Cognition: The Rotterdam Study. Neurology 2022, 99, e565–e573. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Pike, J.R.; Hoogeveen, R.; Walker, K.; Raffield, L.; Selvin, E.; Avery, C.; Engel, S.; Mielke, M.M.; Garcia, T.; et al. Nonalcoholic Fatty Liver Disease and Longitudinal Change in Imaging and Plasma Biomarkers of Alzheimer Disease and Vascular Pathology. Neurology 2024, 102, e209203. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhang, X.; Xu, Z.; Li, L.; Mo, X.; Peng, Z.; Shan, Z.; Yan, H.; Xu, J.; Liu, L. Peripheral Amyloid-β Clearance Mediates Cognitive Impairment in Non-Alcoholic Fatty Liver Disease. EBioMedicine 2024, 102, 105079. [Google Scholar] [CrossRef]
- Kim, D.-G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.-A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-Alcoholic Fatty Liver Disease Induces Signs of Alzheimer’s Disease (AD) in Wild-Type Mice and Accelerates Pathological Signs of AD in an AD Model. J. Neuroinflammation 2016, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.-Y.; Tian, D.-Y.; Chen, S.-H.; Ren, J.-R.; Sun, H.-L.; Xu, M.-Y.; Tan, C.-R.; Fan, D.-Y.; Jian, J.-M.; et al. Physiological β-Amyloid Clearance by the Liver and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2023, 145, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; O’Donnell, A.; Davis-Plourde, K.; Zelber-Sagi, S.; Ghosh, S.; DeCarli, C.S.; Thibault, E.G.; Sperling, R.A.; Johnson, K.A.; Beiser, A.S.; et al. Non-Alcoholic Fatty Liver Disease, Liver Fibrosis, and Regional Amyloid-β and Tau Pathology in Middle-Aged Adults: The Framingham Study. J. Alzheimers Dis. 2022, 86, 1371–1383. [Google Scholar] [CrossRef]
- Huang, Z.; Lin, H.W.K.; Zhang, Q.; Zong, X. Targeting Alzheimer’s Disease: The Critical Crosstalk between the Liver and Brain. Nutrients 2022, 14, 4298. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological Amyloid-Beta Clearance in the Periphery and Its Therapeutic Potential for Alzheimer’s Disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Qosa, H.; Abuasal, B.S.; Romero, I.A.; Weksler, B.; Couraud, P.-O.; Keller, J.N.; Kaddoumi, A. Differences in Amyloid-β Clearance across Mouse and Human Blood-Brain Barrier Models: Kinetic Analysis and Mechanistic Modeling. Neuropharmacology 2014, 79, 668–678. [Google Scholar] [CrossRef]
- Haney, M.S.; Pálovics, R.; Munson, C.N.; Long, C.; Johansson, P.K.; Yip, O.; Dong, W.; Rawat, E.; West, E.; Schlachetzki, J.C.M.; et al. APOE4/4 Is Linked to Damaging Lipid Droplets in Alzheimer’s Disease Microglia. Nature 2024, 628, 154–161. [Google Scholar] [CrossRef]
- Liu, C.-C.; Wang, N.; Chen, Y.; Inoue, Y.; Shue, F.; Ren, Y.; Wang, M.; Qiao, W.; Ikezu, T.C.; Li, Z.; et al. Cell-Autonomous Effects of APOE4 in Restricting Microglial Response in Brain Homeostasis and Alzheimer’s Disease. Nat. Immunol. 2023, 24, 1854–1866. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhao, J.; Fu, Y.; Inoue, Y.; Ren, Y.; Chen, Y.; Doss, S.V.; Shue, F.; Jeevaratnam, S.; Bastea, L.; et al. Peripheral apoE4 Enhances Alzheimer’s Pathology and Impairs Cognition by Compromising Cerebrovascular Function. Nat. Neurosci. 2022, 25, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Takechi, R.; Hackett, M.J.; Francis, R.; Bynevelt, M.; Celliers, L.M.; Nesbit, M.; Mamsa, S.; Arfuso, F.; Das, S.; et al. Synthesis of Human Amyloid Restricted to Liver Results in an Alzheimer Disease-like Neurodegenerative Phenotype. PLoS Biol. 2021, 19, e3001358. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Menon, D.K.; Manley, G.T.; Abrams, M.; Åkerlund, C.; Andelic, N.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. Traumatic Brain Injury: Progress and Challenges in Prevention, Clinical Care, and Research. Lancet Neurol. 2022, 21, 1004–1060. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the Global Incidence of Traumatic Brain Injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Tian, Y.; Jiang, R.; Wang, Y.; Nie, M.; Li, X.; He, Y.; Liu, X.; Zhao, R.; Zhang, J. Susceptibility to Hepatotoxic Drug-Induced Liver Injury Increased After Traumatic Brain Injury in Mice. J. Neurotrauma 2024, 41, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Dong, J.; Wu, Y.; Zhu, M.; Xiong, W.; Li, H.; Zhao, Y.; Hammock, B.D.; Zhu, X. Enhancement of the Liver’s Neuroprotective Role Ameliorates Traumatic Brain Injury Pathology. Proc. Natl. Acad. Sci. USA 2023, 120, e2301360120. [Google Scholar] [CrossRef]
- Pease, M.; Mittal, A.; Merkaj, S.; Okonkwo, D.O.; Gonzalez-Martinez, J.A.; Elmer, J.; Liou, W.-S.; Pingue, V.; Hammond, F.M.; Abramovici, S.; et al. Early Seizure Prophylaxis in Mild and Moderate Traumatic Brain Injury: A Systematic Review and Meta-Analysis. JAMA Neurol. 2024, 81, 507–514. [Google Scholar] [CrossRef]
- Anthony, D.C.; Couch, Y.; Losey, P.; Evans, M.C. The Systemic Response to Brain Injury and Disease. Brain. Behav. Immun. 2012, 26, 534–540. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and Metabolism: Deciphering Communication from the Liver. Mol. Metab. 2021, 44, 101138. [Google Scholar] [CrossRef]
- Iroz, A.; Couty, J.-P.; Postic, C. Hepatokines: Unlocking the Multi-Organ Network in Metabolic Diseases. Diabetologia 2015, 58, 1699–1703. [Google Scholar] [CrossRef]
- Stefan, N.; Schick, F.; Birkenfeld, A.L.; Häring, H.-U.; White, M.F. The Role of Hepatokines in NAFLD. Cell Metab. 2023, 35, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, S.M.; Saudek, V.; O’Rahilly, S. GDF15: A Hormone Conveying Somatic Distress to the Brain. Endocr. Rev. 2020, 41, bnaa007. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, M.; Zhang, S.; Dong, Z.; Wang, Y.; Zhang, W.; Liu, C. Angptl8 Mediates Food-Driven Resetting of Hepatic Circadian Clock in Mice. Nat. Commun. 2019, 10, 3518. [Google Scholar] [CrossRef] [PubMed]
- Hultman, K.; Scarlett, J.M.; Baquero, A.F.; Cornea, A.; Zhang, Y.; Salinas, C.B.G.; Brown, J.; Morton, G.J.; Whalen, E.J.; Grove, K.L.; et al. The Central Fibroblast Growth Factor Receptor/Beta Klotho System: Comprehensive Mapping in Mus Musculus and Comparisons to Nonhuman Primate and Human Samples Using an Automated in Situ Hybridization Platform. J. Comp. Neurol. 2019, 527, 2069–2085. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.-N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL Is the Receptor for GDF15 and the Ligand Promotes Weight Loss in Mice and Nonhuman Primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yuan, J.; Zhang, C.; Wang, L.; Liu, Y.; Song, L.; Zhong, W.; Chen, X.; Dong, J. Neuropeptide Y-Positive Neurons in the Dorsomedial Hypothalamus Are Involved in the Anorexic Effect of Angptl8. Front. Mol. Neurosci. 2018, 11, 451. [Google Scholar] [CrossRef]
- Wang, Q.; Sharma, V.P.; Shen, H.; Xiao, Y.; Zhu, Q.; Xiong, X.; Guo, L.; Jiang, L.; Ohta, K.; Li, S.; et al. The Hepatokine Tsukushi Gates Energy Expenditure via Brown Fat Sympathetic Innervation. Nat. Metab. 2019, 1, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake during Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Régnier, M.; Fouché, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef]
- Qiu, H.; Song, E.; Hu, Y.; Li, T.; Ku, K.C.; Wang, C.; Cheung, B.M.Y.; Cheong, L.Y.; Wang, Q.; Wu, X.; et al. Hepatocyte-Secreted Autotaxin Exacerbates Nonalcoholic Fatty Liver Disease Through Autocrine Inhibition of the PPARα/FGF21 Axis. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 1003–1023. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21 and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.e3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, M.; Tan, X.; Wang, X.; Yang, X.; Xiao, J.; Li, X.; Wang, F. Negative Correlation between Cerebrospinal Fluid FGF21 Levels and BDI Scores in Male Chinese Subjects. Psychiatry Res. 2017, 252, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Bono, B.S.; Koziel Ly, N.K.; Miller, P.A.; Williams-Ikhenoba, J.; Dumiaty, Y.; Chee, M.J. Spatial Distribution of Beta-Klotho mRNA in the Mouse Hypothalamus, Hippocampal Region, Subiculum, and Amygdala. J. Comp. Neurol. 2022, 530, 1634–1657. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; de Groot, M.H.M.; Owen, B.M.; Lee, S.; Gautron, L.; Lawrence, H.L.; Ding, X.; Elmquist, J.K.; Takahashi, J.S.; Mangelsdorf, D.J.; et al. FGF21 Regulates Metabolism and Circadian Behavior by Acting on the Nervous System. Nat. Med. 2013, 19, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.L.; Xu, A. FGF21 Maintains Glucose Homeostasis by Mediating the Cross Talk between Liver and Brain during Prolonged Fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.M.; Ding, X.; Morgan, D.A.; Coate, K.C.; Bookout, A.L.; Rahmouni, K.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 Acts Centrally to Induce Sympathetic Nerve Activity, Energy Expenditure, and Weight Loss. Cell Metab. 2014, 20, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Owen, B.M.; Bookout, A.L.; Ding, X.; Lin, V.Y.; Atkin, S.D.; Gautron, L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF21 Contributes to Neuroendocrine Control of Female Reproduction. Nat. Med. 2013, 19, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Pena-Leon, V.; Folgueira, C.; Barja-Fernández, S.; Pérez-Lois, R.; Da Silva Lima, N.; Martin, M.; Heras, V.; Martinez-Martinez, S.; Valero, P.; Iglesias, C.; et al. Prolonged Breastfeeding Protects from Obesity by Hypothalamic Action of Hepatic FGF21. Nat. Metab. 2022, 4, 901–917. [Google Scholar] [CrossRef]
- Von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286.e6. [Google Scholar] [CrossRef]
- Flippo, K.H.; Trammell, S.A.J.; Gillum, M.P.; Aklan, I.; Perez, M.B.; Yavuz, Y.; Smith, N.K.; Jensen-Cody, S.O.; Zhou, B.; Claflin, K.E.; et al. FGF21 Suppresses Alcohol Consumption through an Amygdalo-Striatal Circuit. Cell Metab. 2022, 34, 317–328.e6. [Google Scholar] [CrossRef]
- Song, P.; Zechner, C.; Hernandez, G.; Cánovas, J.; Xie, Y.; Sondhi, V.; Wagner, M.; Stadlbauer, V.; Horvath, A.; Leber, B.; et al. The Hormone FGF21 Stimulates Water Drinking in Response to Ketogenic Diet and Alcohol. Cell Metab. 2018, 27, 1338–1347.e4. [Google Scholar] [CrossRef]
- Sofat, R.; Cooper, J.A.; Kumari, M.; Casas, J.P.; Mitchell, J.P.; Acharya, J.; Thom, S.; Hughes, A.D.; Humphries, S.E.; Hingorani, A.D. Circulating Apolipoprotein E Concentration and Cardiovascular Disease Risk: Meta-Analysis of Results from Three Studies. PLoS Med. 2016, 13, e1002146. [Google Scholar] [CrossRef]
- Solomon, T.P.J.; Carter, S.; Haus, J.M.; Karstoft, K.; von Holstein-Rathlou, S.; Nielsen, M.S.; Gillum, M.P. Plasma FGF21 Concentrations Are Regulated by Glucose Independently of Insulin and GLP-1 in Lean, Healthy Humans. PeerJ 2022, 10, e12755. [Google Scholar] [CrossRef]
- Villanueva, M.T. GDF15 Tells the Brain to Lose Weight. Nat. Rev. Drug Discov. 2017, 16, 827. [Google Scholar] [CrossRef]
- Wang, D.; Day, E.A.; Townsend, L.K.; Djordjevic, D.; Jørgensen, S.B.; Steinberg, G.R. GDF15: Emerging Biology and Therapeutic Applications for Obesity and Cardiometabolic Disease. Nat. Rev. Endocrinol. 2021, 17, 592–607. [Google Scholar] [CrossRef]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.-D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e12. [Google Scholar] [CrossRef]
- Furuhashi, M.; Higashiura, Y.; Sakai, A.; Koyama, M.; Tanaka, M.; Saitoh, S.; Shimamoto, K.; Ohnishi, H. Plasma Tsukushi Concentration Is Associated with High Levels of Insulin and FGF21 and Low Level of Total Cholesterol in a General Population without Medication. Metabolites 2022, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Espes, D.; Lau, J.; Carlsson, P.-O. Increased Circulating Levels of Betatrophin in Individuals with Long-Standing Type 1 Diabetes. Diabetologia 2014, 57, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wei, J.; Zhang, Y.; Wang, Z.; Tang, J.; Tang, J.; Gao, Y.; Zhang, X.; Li, Y.; Liu, Y.; et al. ANGPTL8 Is a Negative Regulator in Pathological Cardiac Hypertrophy. Cell Death Dis. 2022, 13, 621. [Google Scholar] [CrossRef]
- Guo, C.; Wang, C.; Deng, X.; He, J.; Yang, L.; Yuan, G. ANGPTL8 in Metabolic Homeostasis: More Friend than Foe? Open Biol. 2021, 11, 210106. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, N.; Wolf, P.; Piedvache, C.; Trabado, S.; Verdelet, T.; Cornu, C.; Souberbielle, J.-C.; Chanson, P. Reference Values for IGF-I Serum Concentration in an Adult Population: Use of the VARIETE Cohort for Two New Immunoassays. Endocr. Connect. 2021, 10, 1027. [Google Scholar] [CrossRef]
- Potenzieri, A.; Uccella, S.; Preiti, D.; Pisoni, M.; Rosati, S.; Lavarello, C.; Bartolucci, M.; Debellis, D.; Catalano, F.; Petretto, A.; et al. Early IGF-1 Receptor Inhibition in Mice Mimics Preterm Human Brain Disorders and Reveals a Therapeutic Target. Sci. Adv. 2024, 10, eadk8123. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Q.; Wang, W.; Wang, Y.; Fang, K.; Wan, Q.; Li, H.; Wu, T. Potential Use of Bioactive Nanofibrous Dural Substitutes with Controlled Release of IGF-1 for Neuroprotection after Traumatic Brain Injury. Nanoscale 2022, 14, 18217–18230. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Li, F.; Kang, D.; Anderson, T.; Pitcher, T.; Dalrymple-Alford, J.; Shorten, P.; Singh-Mallah, G. Cyclic Glycine-Proline (cGP) Normalises Insulin-Like Growth Factor-1 (IGF-1) Function: Clinical Significance in the Ageing Brain and in Age-Related Neurological Conditions. Molecules 2023, 28, 1021. [Google Scholar] [CrossRef]
- Cao, Z.; Min, J.; Tan, Q.; Si, K.; Yang, H.; Xu, C. Circulating Insulin-like Growth Factor-1 and Brain Health: Evidence from 369,711 Participants in the UK Biobank. Alzheimers Res. Ther. 2023, 15, 140. [Google Scholar] [CrossRef]
- Demïrdöğen, F.; Akdağ, T.; Gündüz, Z.B.; Odabaş, F.Ö. Investigation of Serum Adropin Levels and Its Relationship with Hypothalamic Atrophy in Patients with Multiple Sclerosis. Mult. Scler. Relat. Disord. 2022, 66, 103948. [Google Scholar] [CrossRef] [PubMed]
- Mushala, B.A.S.; Scott, I. Adropin: A Hepatokine Modulator of Vascular Function and Cardiac Fuel Metabolism. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H238–H244. [Google Scholar] [CrossRef]
- Gunraj, R.E.; Yang, C.; Liu, L.; Larochelle, J.; Candelario-Jalil, E. Protective Roles of Adropin in Neurological Disease. Am. J. Physiol. Cell Physiol. 2023, 324, C674–C678. [Google Scholar] [CrossRef]
- Yang, C.; Liu, L.; Lavayen, B.P.; Larochelle, J.; Gunraj, R.E.; Butler, A.A.; Candelario-Jalil, E. Therapeutic Benefits of Adropin in Aged Mice After Transient Ischemic Stroke via Reduction of Blood-Brain Barrier Damage. Stroke 2023, 54, 234–244. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghoshal, S.; Girardet, C.; DeMars, K.M.; Yang, C.; Niehoff, M.L.; Nguyen, A.D.; Jayanth, P.; Hoelscher, B.A.; Xu, F.; et al. Adropin Correlates with Aging-Related Neuropathology in Humans and Improves Cognitive Function in Aging Mice. npj Aging Mech. Dis. 2021, 7, 1–17. [Google Scholar] [CrossRef]
- Mani, B.K.; Puzziferri, N.; He, Z.; Rodriguez, J.A.; Osborne-Lawrence, S.; Metzger, N.P.; Chhina, N.; Gaylinn, B.; Thorner, M.O.; Thomas, E.L.; et al. LEAP2 Changes with Body Mass and Food Intake in Humans and Mice. J. Clin. Investig. 2019, 129, 3909–3923. [Google Scholar] [CrossRef]
- Ge, X.; Yang, H.; Bednarek, M.A.; Galon-Tilleman, H.; Chen, P.; Chen, M.; Lichtman, J.S.; Wang, Y.; Dalmas, O.; Yin, Y.; et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018, 27, 461–469.e6. [Google Scholar] [CrossRef]
- Mondal, A.; Bose, D.; Saha, P.; Sarkar, S.; Seth, R.; Kimono, D.; Albadrani, M.; Nagarkatti, M.; Nagarkatti, P.; Chatterjee, S. Lipocalin 2 Induces Neuroinflammation and Blood-Brain Barrier Dysfunction through Liver-Brain Axis in Murine Model of Nonalcoholic Steatohepatitis. J. Neuroinflammation 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Li, W.; Shi, J.; Yu, Z.; Garcia-Gabilondo, M.; Held, A.; Huang, L.; Deng, W.; Ning, M.; Ji, X.; Rosell, A.; et al. SLC22A17 as a Cell Death-Linked Regulator of Tight Junctions in Cerebral Ischemia. Stroke 2024, 55, 1650–1659. [Google Scholar] [CrossRef]
- Yan, L.; Yang, F.; Wang, Y.; Shi, L.; Wang, M.; Yang, D.; Wang, W.; Jia, Y.; So, K.-F.; Zhang, L. Stress Increases Hepatic Release of Lipocalin 2 Which Contributes to Anxiety-like Behavior in Mice. Nat. Commun. 2024, 15, 3034. [Google Scholar] [CrossRef]
- Jung, B.-K.; Ryu, K.-Y. Lipocalin-2: A Therapeutic Target to Overcome Neurodegenerative Diseases by Regulating Reactive Astrogliosis. Exp. Mol. Med. 2023, 55, 2138–2146. [Google Scholar] [CrossRef]
- Baek, S.J.; Eling, T. Growth Differentiation Factor 15 (GDF15): A Survival Protein with Therapeutic Potential in Metabolic Diseases. Pharmacol. Ther. 2019, 198, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Walker, K.; Min, X.; Hale, C.; Tran, T.; Komorowski, R.; Yang, J.; Davda, J.; Nuanmanee, N.; Kemp, D.; et al. Long-Acting MIC-1/GDF15 Molecules to Treat Obesity: Evidence from Mice to Monkeys. Sci. Transl. Med. 2017, 9, eaan8732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, C.; Chen, J.; Sang, T.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. Overexpression of NAG-1/GDF15 Prevents Hepatic Steatosis through Inhibiting Oxidative Stress-Mediated dsDNA Release and AIM2 Inflammasome Activation. Redox. Biol. 2022, 52, 102322. [Google Scholar] [CrossRef]
- Patel, S.; Haider, A.; Alvarez-Guaita, A.; Bidault, G.; El-Sayed Moustafa, J.S.; Guiu-Jurado, E.; Tadross, J.A.; Warner, J.; Harrison, J.; Virtue, S.; et al. Combined Genetic Deletion of GDF15 and FGF21 Has Modest Effects on Body Weight, Hepatic Steatosis and Insulin Resistance in High Fat Fed Mice. Mol. Metab. 2022, 65, 101589. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-Homeostatic Body Weight Regulation through a Brainstem-Restricted Receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef]
- Yang, L.; Chang, C.-C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL Is the Receptor for GDF15 and Is Required for the Anti-Obesity Effects of the Ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The Metabolic Effects of GDF15 Are Mediated by the Orphan Receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Insights Into Mechanisms of GDF15 and Receptor GFRAL: Therapeutic Targets. Trends Endocrinol. Metab. 2020, 31, 939–951. [Google Scholar] [CrossRef]
- Muniyan, S.; Pothuraju, R.; Seshacharyulu, P.; Batra, S.K. Macrophage Inhibitory Cytokine-1 in Cancer: Beyond the Cellular Phenotype. Cancer Lett. 2022, 536, 215664. [Google Scholar] [CrossRef]
- Patel, S.; Alvarez-Guaita, A.; Melvin, A.; Rimmington, D.; Dattilo, A.; Miedzybrodzka, E.L.; Cimino, I.; Maurin, A.-C.; Roberts, G.P.; Meek, C.L.; et al. GDF15 Provides an Endocrine Signal of Nutritional Stress in Mice and Humans. Cell Metab. 2019, 29, 707–718.e8. [Google Scholar] [CrossRef]
- Luan, H.H.; Wang, A.; Hilliard, B.K.; Carvalho, F.; Rosen, C.E.; Ahasic, A.M.; Herzog, E.L.; Kang, I.; Pisani, M.A.; Yu, S.; et al. GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 2019, 178, 1231–1244.e11. [Google Scholar] [CrossRef] [PubMed]
- Vatner, D.F.; Goedeke, L.; Camporez, J.-P.G.; Lyu, K.; Nasiri, A.R.; Zhang, D.; Bhanot, S.; Murray, S.F.; Still, C.D.; Gerhard, G.S.; et al. Angptl8 Antisense Oligonucleotide Improves Adipose Lipid Metabolism and Prevents Diet-Induced NAFLD and Hepatic Insulin Resistance in Rodents. Diabetologia 2018, 61, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Qamar, A.; Qu, L.; Qasim, A.N.; Mehta, N.N.; Reilly, M.P.; Rader, D.J. Differential Association of Plasma Angiopoietin-like Proteins 3 and 4 with Lipid and Metabolic Traits. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1057–1063. [Google Scholar] [CrossRef]
- Larsson, M.; Allan, C.M.; Heizer, P.J.; Tu, Y.; Sandoval, N.P.; Jung, R.S.; Walzem, R.L.; Beigneux, A.P.; Young, S.G.; Fong, L.G. Impaired Thermogenesis and Sharp Increases in Plasma Triglyceride Levels in GPIHBP1-Deficient Mice during Cold Exposure. J. Lipid Res. 2018, 59, 706–713. [Google Scholar] [CrossRef]
- Dang, F.; Wu, R.; Wang, P.; Wu, Y.; Azam, M.S.; Xu, Q.; Chen, Y.; Liu, Y. Fasting and Feeding Signals Control the Oscillatory Expression of Angptl8 to Modulate Lipid Metabolism. Sci. Rep. 2016, 6, 36926. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Ghosh, A.; Al-Khairi, I.; Madiraju, S.R.M.; Abubaker, J.; Prentki, M. The Multi-Faces of Angptl8 in Health and Disease: Novel Functions beyond Lipoprotein Lipase Modulation. Prog. Lipid Res. 2020, 80, 101067. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Trauner, M. Gut-Liver Axis: Pathophysiological Concepts and Clinical Implications. Cell Metab. 2022, 34, 1700–1718. [Google Scholar] [CrossRef]
- Steiner, C.; Holleboom, A.G.; Karuna, R.; Motazacker, M.M.; Kuivenhoven, J.A.; Frikke-Schmidt, R.; Tybjaerg-Hansen, A.; Rohrer, L.; Rentsch, K.M.; Eckardstein, A. von Lipoprotein Distribution and Serum Concentrations of 7α-Hydroxy-4-Cholesten-3-One and Bile Acids: Effects of Monogenic Disturbances in High-Density Lipoprotein Metabolism. Clin. Sci. 2012, 122, 385–396. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Jia, W.; Li, Y.; Cheung, K.C.P.; Zheng, X. Bile Acid Signaling in the Regulation of Whole Body Metabolic and Immunological Homeostasis. Sci. China Life Sci. 2024, 67, 865–878. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Corlianò, M.; Singaraja, R.R. Bile Acids: A Communication Channel in the Gut-Brain Axis. Neuromolecular. Med. 2021, 23, 99–117. [Google Scholar] [CrossRef]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the Alternative Bile Acid Synthetic Pathway for Metabolic Diseases. Protein Cell 2021, 12, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Yanguas-Casás, N.; Barreda-Manso, M.A.; Nieto-Sampedro, M.; Romero-Ramírez, L. TUDCA: An Agonist of the Bile Acid Receptor GPBAR1/TGR5 with Anti-Inflammatory Effects in Microglial Cells. J. Cell Physiol. 2017, 232, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Velázquez-Villegas, L.A.; Bresciani, N.; Sun, Y.; Huang, Q.; Fénelon, V.S.; Castellanos-Jankiewicz, A.; Zizzari, P.; Bruschetta, G.; Jin, S.; et al. Central Anorexigenic Actions of Bile Acids Are Mediated by TGR5. Nat. Metab. 2021, 3, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Jankiewicz, A.; Guzmán-Quevedo, O.; Fénelon, V.S.; Zizzari, P.; Quarta, C.; Bellocchio, L.; Tailleux, A.; Charton, J.; Fernandois, D.; Henricsson, M.; et al. Hypothalamic Bile Acid-TGR5 Signaling Protects from Obesity. Cell Metab. 2021, 33, 1483–1492.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Zhang, S.-Y.; Hong, Y.-Z.; Chen, Z.-G.; Long, Y.; Yuan, D.-H.; Zhao, J.-J.; Tang, S.-S.; Wang, H.; Hong, H. TGR5-Mediated Lateral Hypothalamus-dCA3-Dorsolateral Septum Circuit Regulates Depressive-like Behavior in Male Mice. Neuron 2024, 112, 1795–1814.e10. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lv, Y.-G.; Du, Y.-F.; Hu, M.; Reed, M.N.; Long, Y.; Suppiramaniam, V.; Hong, H.; Tang, S.-S. Inhibitory Effect of INT-777 on Lipopolysaccharide-Induced Cognitive Impairment, Neuroinflammation, Apoptosis, and Synaptic Dysfunction in Mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Aasly, J.; Bandmann, O. Ursocholanic Acid Rescues Mitochondrial Function in Common Forms of Familial Parkinson’s Disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Seeger, D.; Golovko, S.; Golovko, M.; Combs, C.K. Liver Bile Acid Changes in Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7451. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.; Frampton, G.; Tobin, R.; Dusio, G.; Smith, J.; Shin, H.; Newell-Rogers, K.; Grant, S.; DeMorrow, S. TGR5 Signaling Reduces Neuroinflammation during Hepatic Encephalopathy. J. Neurochem. 2015, 135, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic Acid in the Treatment of Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2016, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhao, L.; Zhao, M.; Bao, T.; Chen, T.; Zhao, A.; Zheng, X.; Gu, X.; Sun, T.; Guo, Y.; et al. Increased Intestinal Bile Acid Absorption Contributes to Age-Related Cognitive Impairment. Cell Rep. Med. 2024, 5, 101543. [Google Scholar] [CrossRef]
- Ma, Y.; Nenkov, M.; Chen, Y.; Press, A.T.; Kaemmerer, E.; Gassler, N. Fatty Acid Metabolism and Acyl-CoA Synthetases in the Liver-Gut Axis. World J. Hepatol. 2021, 13, 1512–1533. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.-H.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Goswami, C.; Iwasaki, Y.; Yada, T. Short-Chain Fatty Acids Suppress Food Intake by Activating Vagal Afferent Neurons. J. Nutr. Biochem. 2018, 57, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Complex Regulatory Effects of Gut Microbial Short-Chain Fatty Acids on Immune Tolerance and Autoimmunity. Cell Mol. Immunol. 2023, 20, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Xie, R.; Liu, X.; Chen, S.; Tang, H. Mechanisms of Short-Chain Fatty Acids Derived from Gut Microbiota in Alzheimer’s Disease. Aging Dis. 2022, 13, 1252–1266. [Google Scholar] [CrossRef]
- Fu, S.-P.; Liu, B.-R.; Wang, J.-F.; Xue, W.-J.; Liu, H.-M.; Zeng, Y.-L.; Huang, B.-X.; Li, S.-N.; Lv, Q.-K.; Wang, W.; et al. β-Hydroxybutyric Acid Inhibits Growth Hormone-Releasing Hormone Synthesis and Secretion through the GPR109A/Extracellular Signal-Regulated 1/2 Signalling Pathway in the Hypothalamus. J. Neuroendocrinol. 2015, 27, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis through Regulation of the Inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef]
- Dong, Y.; Cui, C. The Role of Short-Chain Fatty Acids in Central Nervous System Diseases. Mol. Cell. Biochem. 2022, 477, 2595–2607. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, D.; Weng, F.; Jin, Y.; He, L. Sodium Butyrate Ameliorates the Cognitive Impairment of Alzheimer’s Disease by Regulating the Metabolism of Astrocytes. Psychopharmacology 2022, 239, 215–227. [Google Scholar] [CrossRef]
- Wang, P.; Wu, P.-F.; Wang, H.-J.; Liao, F.; Wang, F.; Chen, J.-G. Gut Microbiome-Derived Ammonia Modulates Stress Vulnerability in the Host. Nat. Metab. 2023, 5, 1986–2001. [Google Scholar] [CrossRef]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea Cycle Disorders-Update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef]
- Anand, A.C.; Acharya, S.K. The Story of Ammonia in Liver Disease: An Unraveling Continuum. J. Clin. Exp. Hepatol. 2024, 14, 101361. [Google Scholar] [CrossRef]
- Katayama, K.; Kakita, N. Possible Pathogenetic Role of Ammonia in Liver Cirrhosis without Hyperammonemia of Venous Blood: The so-Called Latency Period of Abnormal Ammonia Metabolism. Hepatol. Res. 2024, 54, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Mookerjee, R.; Rackayova, V.; Rangroo Thrane, V.; Vairappan, B.; Ott, P.; Rose, C. Ammonia Toxicity: From Head to Toe? Metab. Brain Dis. 2017, 32, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; McLin, V.A.; Cudalbu, C. Ammonia Toxicity to the Brain. J. Inherit. Metab. Dis. 2013, 36, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Pan, F.; Huang, Q.; Xie, G.; Chao, X.; Wu, L.; Wang, J.; Cui, L.; Sun, T.; Li, M.; et al. Metabolic Phenotyping Reveals an Emerging Role of Ammonia Abnormality in Alzheimer’s Disease. Nat. Commun. 2024, 15, 3796. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Lozano, M.; Torres, M.; Horrillo, R.; Afonso, N.; Núñez, L.; Mestre, A.; Pérez, A.; Cid, J.; Costa, M.; et al. Effect of Plasma Exchange with Albumin Replacement on Albumin Functionality and Organ Dysfunction in Acute-on-Chronic Liver Failure. JHEP Rep. 2024, 6, 101017. [Google Scholar] [CrossRef]
- Amin, S.B.; Saluja, S.; Kler, N. Unbound Bilirubin and Acute Bilirubin Encephalopathy in Infants Born Late Preterm and Term with Significant Hyperbilirubinemia. J. Pediatr. 2024, 266, 113880. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Kumar, P.; Testai, F.D. Bilirubin Encephalopathy. Curr. Neurol. Neurosci. Rep. 2022, 22, 343–353. [Google Scholar] [CrossRef]
- Licata, A.; Zerbo, M.; Como, S.; Cammilleri, M.; Soresi, M.; Montalto, G.; Giannitrapani, L. The Role of Vitamin Deficiency in Liver Disease: To Supplement or Not Supplement? Nutrients 2021, 13, 4014. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acid Metabolism in Liver Pathobiology. Gene Expr. 2018, 18, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef]
- Ford, T.C.; Downey, L.A.; Simpson, T.; McPhee, G.; Oliver, C.; Stough, C. The Effect of a High-Dose Vitamin B Multivitamin Supplement on the Relationship between Brain Metabolism and Blood Biomarkers of Oxidative Stress: A Randomized Control Trial. Nutrients 2018, 10, 1860. [Google Scholar] [CrossRef]
- Ford, A.H.; Almeida, O.P. Effect of Vitamin B Supplementation on Cognitive Function in the Elderly: A Systematic Review and Meta-Analysis. Drugs Aging 2019, 36, 419–434. [Google Scholar] [CrossRef]
- Sato, K. Why Is Vitamin B6 Effective in Alleviating the Symptoms of Autism? Med. Hypotheses 2018, 115, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Field, D.T.; Cracknell, R.O.; Eastwood, J.R.; Scarfe, P.; Williams, C.M.; Zheng, Y.; Tavassoli, T. High-dose Vitamin B6 Supplementation Reduces Anxiety and Strengthens Visual Surround Suppression. Hum. Psychopharmacol. 2022, 37, e2852. [Google Scholar] [CrossRef]
- Wu, F.; Xu, K.; Liu, L.; Zhang, K.; Xia, L.; Zhang, M.; Teng, C.; Tong, H.; He, Y.; Xue, Y.; et al. Vitamin B12 Enhances Nerve Repair and Improves Functional Recovery after Traumatic Brain Injury by Inhibiting ER Stress-Induced Neuron Injury. Front. Pharmacol. 2019, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Baltrusch, S. The Role of Neurotropic B Vitamins in Nerve Regeneration. Biomed. Res. Int. 2021, 2021, 9968228. [Google Scholar] [CrossRef]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef]
- Portugal, C.C.; Socodato, R.; Canedo, T.; Silva, C.M.; Martins, T.; Coreixas, V.S.M.; Loiola, E.C.; Gess, B.; Röhr, D.; Santiago, A.R.; et al. Caveolin-1-Mediated Internalization of the Vitamin C Transporter SVCT2 in Microglia Triggers an Inflammatory Phenotype. Sci. Signal. 2017, 10, eaal2005. [Google Scholar] [CrossRef]
- Chen, L.-J.; Sha, S.; Stocker, H.; Brenner, H.; Schöttker, B. The Associations of Serum Vitamin D Status and Vitamin D Supplements Use with All-Cause Dementia, Alzheimer’s Disease, and Vascular Dementia: A UK Biobank Based Prospective Cohort Study. Am. J. Clin. Nutr. 2024, 119, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Hu, J.; Huo, X.; Miao, R.; Zhang, Y.; Ma, F. Effects of Vitamin D Supplementation on Cognitive Function and Blood Aβ-Related Biomarkers in Older Adults with Alzheimer’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.-H.; Hsu, C.-C.; Yu, B.-H.; Lo, Y.-R.; Hsu, Y.-Y.; Chen, M.-H.; Juang, J.-L. Vitamin D Supplementation Worsens Alzheimer’s Progression: Animal Model and Human Cohort Studies. Aging Cell 2022, 21, e13670. [Google Scholar] [CrossRef] [PubMed]
- Fontana, I.C.; Kumar, A.; Nordberg, A. The Role of Astrocytic A7 Nicotinic Acetylcholine Receptors in Alzheimer Disease. Nat. Rev. Neurol. 2023, 19, 278–288. [Google Scholar] [CrossRef]
- Wallace, T.C.; Blusztajn, J.K.; Caudill, M.A.; Klatt, K.C.; Natker, E.; Zeisel, S.H.; Zelman, K.M. Choline: The Underconsumed and Underappreciated Essential Nutrient. Nutr. Today 2018, 53, 240. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, X.; Liu, C.; Ang, A.F.; Massaro, J.; Devine, S.A.; Auerbach, S.H.; Blusztajn, J.K.; Au, R.; Jacques, P.F. Is Dietary Choline Intake Related to Dementia and Alzheimer’s Disease Risks? Results from the Framingham Heart Study. Am. J. Clin. Nutr. 2022, 116, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Judd, J.M.; Decker, A.; Winslow, W.; Sarette, P.; Villarreal Espinosa, O.; Tallino, S.; Bartholomew, S.K.; Bilal, A.; Sandler, J.; et al. Dietary Choline Intake Is Necessary to Prevent Systems-Wide Organ Pathology and Reduce Alzheimer’s Disease Hallmarks. Aging Cell 2023, 22, e13775. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Derbyshire, E.; Schön, C. Association between Maternal Choline, Fetal Brain Development, and Child Neurocognition: Systematic Review and Meta-Analysis of Human Studies. Adv. Nutr. 2022, 13, 2445–2457. [Google Scholar] [CrossRef]
- Velazquez, R.; Ferreira, E.; Winslow, W.; Dave, N.; Piras, I.S.; Naymik, M.; Huentelman, M.J.; Tran, A.; Caccamo, A.; Oddo, S. Maternal Choline Supplementation Ameliorates Alzheimer’s Disease Pathology by Reducing Brain Homocysteine Levels across Multiple Generations. Mol. Psychiatry 2020, 25, 2620–2629. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong Choline Supplementation Ameliorates Alzheimer’s Disease Pathology and Associated Cognitive Deficits by Attenuating Microglia Activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [PubMed]
- Moradian, H.; Gabriel, T.; Barrau, M.; Roblin, X.; Paul, S. New Methods to Unveil Host-Microbe Interaction Mechanisms along the Microbiota-Gut-Brain-Axis. Gut Microbes 2024, 16, 2351520. [Google Scholar] [CrossRef]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota-Gut-Brain Axis and Its Therapeutic Applications in Neurodegenerative Diseases. Signal. Transduct. Target. Ther. 2024, 9, 37. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Liu, L.; Huh, J.R.; Shah, K. Microbiota and the Gut-Brain-Axis: Implications for New Therapeutic Design in the CNS. EBioMedicine 2022, 77, 103908. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Su, J.; Gao, X.; Yang, H.; Weng, R.; Ni, W.; Gu, Y. The Microbiota-Gut-Brain Axis Participates in Chronic Cerebral Hypoperfusion by Disrupting the Metabolism of Short-Chain Fatty Acids. Microbiome 2022, 10, 62. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Pimentel, M. Is Hepatic Encephalopathy the First True Disorder of Gut-Brain Interaction? Am. J. Gastroenterol. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Teratani, T.; Mikami, Y.; Nakamoto, N.; Suzuki, T.; Harada, Y.; Okabayashi, K.; Hagihara, Y.; Taniki, N.; Kohno, K.; Shibata, S.; et al. The Liver–Brain–Gut Neural Arc Maintains the Treg Cell Niche in the Gut. Nature 2020, 585, 591–596. [Google Scholar] [CrossRef]
- Yan, M.; Man, S.; Sun, B.; Ma, L.; Guo, L.; Huang, L.; Gao, W. Gut Liver Brain Axis in Diseases: The Implications for Therapeutic Interventions. Sig. Transduct. Target Ther. 2023, 8, 1–26. [Google Scholar] [CrossRef]
- Vitturi, B.K.; Sanvito, W.L. Claude Bernard (1813–1878). J. Neurol. 2021, 268, 2301–2303. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.M.; Oderberg, I.M.; Goessling, W. Hepatic Nervous System in Development, Regeneration, and Disease. Hepatology 2021, 74, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Kong, S.-H.; Park, J.-H.; Choi, J.-H.; Park, S.-H.; Zhu, C.-C.; Alzahrani, F.; Alzahrani, K.; Suh, Y.-S.; Park, D.-J.; et al. Preservation of Hepatic Branch of the Vagus Nerve Reduces the Risk of Gallstone Formation after Gastrectomy. Gastric. Cancer 2021, 24, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Dudás, B. Anatomy and Cytoarchitectonics of the Human Hypothalamus. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 179, pp. 45–66. [Google Scholar]
- Saper, C.B.; Lowell, B.B. The Hypothalamus. Curr. Biol. 2014, 24, R1111–R1116. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.; Pinto, S.; Segal, J.; Pérez, C.A.; Viale, A.; DeFalco, J.; Cai, X.; Heisler, L.K.; Friedman, J.M. Identification of Neuronal Subpopulations That Project from Hypothalamus to Both Liver and Adipose Tissue Polysynaptically. Proc. Natl. Acad. Sci. USA 2010, 107, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sheng, S.; Guo, C.; Qiao, W.; Jin, Y.; Tan, L.; Gao, Y.; Zhang, L.; Dong, X.; Zhang, J.; et al. Forkhead Box I2 Transcription Factor Regulates Systemic Energy Metabolism Via Neuropeptide AgRP. Diabetes 2022, 71, 2106–2122. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Rubinstein, M.; Low, M.J. Developmental Single-Cell Transcriptomics of Hypothalamic POMC Neurons Reveal the Genetic Trajectories of Multiple Neuropeptidergic Phenotypes. eLife 2022, 11, e72883. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Choudhury, A.I.; Glegola, J.A.; Viskaitis, P.; Irvine, E.E.; de Campos Silva, P.C.C.; Khadayate, S.; Zeilhofer, H.U.; Withers, D.J. Extrahypothalamic GABAergic Nociceptin-Expressing Neurons Regulate AgRP Neuron Activity to Control Feeding Behavior. J. Clin. Investig. 2020, 130, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Michael, N.J.; Lee-Young, R.S.; Mangiafico, S.P.; Pryor, J.T.; Munder, A.C.; Simonds, S.E.; Brüning, J.C.; Zhang, Z.-Y.; Cowley, M.A.; et al. Insulin Regulates POMC Neuronal Plasticity to Control Glucose Metabolism. Elife 2018, 7, e38704. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef]
- López-Gambero, A.J.; Martínez, F.; Salazar, K.; Cifuentes, M.; Nualart, F. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Mol. Neurobiol. 2019, 56, 769–796. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, J.D.; Zsombok, A. Brain-Liver Connections: Role of the Preautonomic PVN Neurons. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E183–E189. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Molinas, A.J.R.; Miyata, K.; Qiao, X.; Zsombok, A. Overactivity of Liver-Related Neurons in the Paraventricular Nucleus of the Hypothalamus: Electrophysiological Findings in Db/Db Mice. J. Neurosci. 2017, 37, 11140–11150. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-H.; Fujikawa, T.; Lee, J.; Reuter, A.; Kim, K.W. Revisiting the Ventral Medial Nucleus of the Hypothalamus: The Roles of SF-1 Neurons in Energy Homeostasis. Front. Neurosci. 2013, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.-C.; et al. Activation of SF1 Neurons in the Ventromedial Hypothalamus by DREADD Technology Increases Insulin Sensitivity in Peripheral Tissues. Diabetes 2017, 66, 2372–2386. [Google Scholar] [CrossRef] [PubMed]
- Felsted, J.A.; Chien, C.-H.; Wang, D.; Panessiti, M.; Ameroso, D.; Greenberg, A.; Feng, G.; Kong, D.; Rios, M. Alpha2delta-1 in SF1+ Neurons of the Ventromedial Hypothalamus Is an Essential Regulator of Glucose and Lipid Homeostasis. Cell Rep. 2017, 21, 2737–2747. [Google Scholar] [CrossRef]
- Meek, T.H.; Nelson, J.T.; Matsen, M.E.; Dorfman, M.D.; Guyenet, S.J.; Damian, V.; Allison, M.B.; Scarlett, J.M.; Nguyen, H.T.; Thaler, J.P.; et al. Functional Identification of a Neurocircuit Regulating Blood Glucose. Proc. Natl. Acad. Sci. USA 2016, 113, E2073–E2082. [Google Scholar] [CrossRef]
- Mizuno, K.; Ueno, Y. Autonomic Nervous System and the Liver. Hepatol. Res. 2017, 47, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Aspelund, G.; Sritharan, K.C.; Wang, J.P.; Slezak, L.A.; Andersen, D.K. Isolated Hepatic Cholinergic Denervation Impairs Glucose and Glycogen Metabolism. J. Surg. Res. 2000, 90, 19–25. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The Many Faces of Insulin-like Peptide Signalling in the Brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Luo, L. How Does the Brain Regulate the Functions of Internal Organs? In Principles of Neurobiology, 1st ed.; Garland Science: New York, NY, USA, 2015; pp. 351–355. ISBN 978-0-8153-4492-6. [Google Scholar]
- Xiao, F.; Xia, T.; Lv, Z.; Zhang, Q.; Xiao, Y.; Yu, J.; Liu, H.; Deng, J.; Guo, Y.; Wang, C.; et al. Central Prolactin Receptors (PRLRs) Regulate Hepatic Insulin Sensitivity in Mice via Signal Transducer and Activator of Transcription 5 (STAT5) and the Vagus Nerve. Diabetologia 2014, 57, 2136–2144. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakamura, Y.; Kataoka, N. A Hypothalamomedullary Network for Physiological Responses to Environmental Stresses. Nat. Rev. Neurosci. 2022, 23, 35–52. [Google Scholar] [CrossRef]
- Kalsbeek, A.; Bruinstroop, E.; Yi, C.-X.; Klieverik, L.; Liu, J.; Fliers, E. Hormonal Control of Metabolism by the Hypothalamus-Autonomic Nervous System-Liver Axis. Front. Horm. Res. 2014, 42, 1–28. [Google Scholar] [CrossRef]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and Brainstem Neuronal Circuits Controlling Homeostatic Energy Balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef]
- Parent, A.D.; Perkins, E. The Hypothalamus. In Fundamental Neuroscience for Basic and Clinical Applications, 5th ed.; Haines, D.E., Mihailoff, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 442–456. ISBN 978-0-323-39632-5. [Google Scholar]
- Koike, N.; Tadokoro, T.; Ueno, Y.; Okamoto, S.; Kobayashi, T.; Murata, S.; Taniguchi, H. Development of the Nervous System in Mouse Liver. World J. Hepatol. 2022, 14, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Yang, L.; Wang, G.; Liu, J.; Zhao, X.; Wang, Y.; Li, J.; Yang, J. Metabolic Stress Drives Sympathetic Neuropathy within the Liver. Cell Metab. 2021, 33, 666–675.e4. [Google Scholar] [CrossRef]
- Jha, S.K.; Kannan, S. Serum Prolactin in Patients with Liver Disease in Comparison with Healthy Adults: A Preliminary Cross-Sectional Study. Int. J. Appl. Basic Med. Res. 2016, 6, 8–10. [Google Scholar] [CrossRef]
- Alukal, J.J.; John, S.; Thuluvath, P.J. Hyponatremia in Cirrhosis: An Update. Am. J. Gastroenterol. 2020, 115, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- TeSlaa, T.; Bartman, C.R.; Jankowski, C.S.R.; Zhang, Z.; Xu, X.; Xing, X.; Wang, L.; Lu, W.; Hui, S.; Rabinowitz, J.D. The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab. 2021, 33, 367–378.e5. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose Feeds the TCA Cycle via Circulating Lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Radenkovic, S.; Vuckovic, I.; Lanza, I.R. Metabolic Flux Analysis: Moving beyond Static Metabolomics. Trends Biochem. Sci. 2020, 45, 545–546. [Google Scholar] [CrossRef] [PubMed]
- Moiz, B.; Sriram, G.; Clyne, A.M. Interpreting Metabolic Complexity via Isotope-Assisted Metabolic Flux Analysis. Trends Biochem. Sci. 2023, 48, 553–567. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, Y.; Lin, C.-W.; Alvarez, S.; Oh, D.; Yang, P.; Groves, J.T. Nanopore-Mediated Protein Delivery Enabling Three-Color Single-Molecule Tracking in Living Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2012229118. [Google Scholar] [CrossRef]
- Pang, Z.; Schafroth, M.A.; Ogasawara, D.; Wang, Y.; Nudell, V.; Lal, N.K.; Yang, D.; Wang, K.; Herbst, D.M.; Ha, J.; et al. In Situ Identification of Cellular Drug Targets in Mammalian Tissue. Cell 2022, 185, 1793–1805.e17. [Google Scholar] [CrossRef]
- Chau, C.; Actis, P.; Hewitt, E. Methods for Protein Delivery into Cells: From Current Approaches to Future Perspectives. Biochem. Soc. Trans. 2020, 48, 357–365. [Google Scholar] [CrossRef]
- Luo, L. Architectures of Neuronal Circuits. Science 2021, 373, eabg7285. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Xiang, B.; Xiong, J.; He, Z.; Xiang, H. Use of Viruses for Interrogating Viscera-Specific Projections in Central Nervous System. J. Neurosci. Methods 2020, 341, 108757. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Callaway, E.M.; Svoboda, K. Genetic Dissection of Neural Circuits: A Decade of Progress. Neuron 2018, 98, 256–281. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the Mechanism of Tissue-Specific mRNA Delivery by Selective Organ Targeting Nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Sun, Y.; Yu, X.; Lee, S.M.; Cheng, Q.; Wei, T.; Gong, J.; Robinson, J.; Zhang, D.; et al. Preparation of Selective Organ-Targeting (SORT) Lipid Nanoparticles (LNPs) Using Multiple Technical Methods for Tissue-Specific mRNA Delivery. Nat. Protoc. 2023, 18, 265–291. [Google Scholar] [CrossRef]
- Brown, C.R.; Gupta, S.; Qin, J.; Racie, T.; He, G.; Lentini, S.; Malone, R.; Yu, M.; Matsuda, S.; Shulga-Morskaya, S.; et al. Investigating the Pharmacodynamic Durability of GalNAc-siRNA Conjugates. Nucleic. Acids Res. 2020, 48, 11827–11844. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, X.-Y.; Lu, A.; Wang, X.-Y.; Jiang, L.-X.; Wang, J.-C. Non-Viral Vectors for RNA Delivery. J. Control. Release 2022, 342, 241–279. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering Exosomes as Refined Biological Nanoplatforms for Drug Delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef]
- Gabisonia, K.; Khan, M.; Recchia, F.A. Extracellular Vesicle-Mediated Bidirectional Communication between Heart and Other Organs. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H769–H784. [Google Scholar] [CrossRef]
- Ji, Y.; Luo, Z.; Gao, H.; Dos Reis, F.C.G.; Bandyopadhyay, G.; Jin, Z.; Manda, K.A.; Isaac, R.; Yang, M.; Fu, W.; et al. Hepatocyte-Derived Exosomes from Early Onset Obese Mice Promote Insulin Sensitivity through miR-3075. Nat. Metab. 2021, 3, 1163–1174. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.-F.; Jiang, S.; Wu, J.; Liu, J.; Yuan, X.-W.; Shen, D.; Zhang, J.-Z.; Zhou, N.; He, J.; et al. Liver Governs Adipose Remodelling via Extracellular Vesicles in Response to Lipid Overload. Nat. Commun. 2020, 11, 719. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Zhang, Z.; Zhang, X.; Zhu, Y.; Zhang, C.; Bi, Y. Extracellular Vesicles Mediate the Communication of Adipose Tissue with Brain and Promote Cognitive Impairment Associated with Insulin Resistance. Cell Metab. 2022, 34, 1264–1279.e8. [Google Scholar] [CrossRef]
- Thietart, S.; Rautou, P.-E. Extracellular Vesicles as Biomarkers in Liver Diseases: A Clinician’s Point of View. J. Hepatol. 2020, 73, 1507–1525. [Google Scholar] [CrossRef]
- Salomon, C.; Das, S.; Erdbrügger, U.; Kalluri, R.; Kiang Lim, S.; Olefsky, J.M.; Rice, G.E.; Sahoo, S.; Andy Tao, W.; Vader, P.; et al. Extracellular Vesicles and Their Emerging Roles as Cellular Messengers in Endocrinology: An Endocrine Society Scientific Statement. Endocr. Rev. 2022, 43, 441–468. [Google Scholar] [CrossRef]
- Sancandi, M.; Uysal-Onganer, P.; Kraev, I.; Mercer, A.; Lange, S. Protein Deimination Signatures in Plasma and Plasma-EVs and Protein Deimination in the Brain Vasculature in a Rat Model of Pre-Motor Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2743. [Google Scholar] [CrossRef] [PubMed]

| From Medial to Lateral | From Anterior to Posterior | Hypothalamic Nuclei | Abbreviation | Functions on Liver Metabolism |
|---|---|---|---|---|
| Periventricular zone | Periventricular nucleus | |||
| Suprachiasmatic nucleus | SCN | SCN manipulates the circadian clock of hepatic glucose secretion. | ||
| Arcuate nucleus | ARC | ARC receives integrated information from the VMH and LH about food intake. AgRP neurons and POMC neurons control fat accumulation and hepatic glucose production in opposite ways. | ||
| Intermediate zone | Preoptic area | Periventricular nucleus | ||
| Medial preoptic nucleus | ||||
| Lateral preoptic nucleus | ||||
| Supraoptic area (anterior area) | Suprachiasmatic nucleus | SCN | / | |
| Supraoptic nucleus | ||||
| Paraventricular nucleus | PVN | PVN integrates multiple signals from different brain areas including ARC, VMH, SCN and LH. | ||
| Anterior hypothalamic nucleus | ||||
| Lateral hypothalamic nucleus | LH | LH serves as a “feeding center” and is involved in modulating feeding behavior. | ||
| Tuberal area (middle area) | Arcuate nucleus | ARC | / | |
| Dorsomedial nucleus | DMH | DMH integrates feeding behavior with circadian activity. | ||
| Ventromedial nucleus | VMH | VMH is involved in feeding behavior and is said to be a “satiety center”. | ||
| Lateral tuberal nucleus | ||||
| Mammillary area (posterior area) | Mammillary nucleus | |||
| Posterior hypothalamic nucleus | ||||
| Lateral hypothalamic nucleus | LH | / | ||
| Latera zone | Lateral preoptic nucleus | |||
| Lateral tuberal nucleus | ||||
| Lateral hypothalamic nucleus | LH | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Qiu, K.; Jiang, Y.; Huang, Y.; Zhang, Y.; Liao, Y. Metabolic Crosstalk between Liver and Brain: From Diseases to Mechanisms. Int. J. Mol. Sci. 2024, 25, 7621. https://doi.org/10.3390/ijms25147621
Yang X, Qiu K, Jiang Y, Huang Y, Zhang Y, Liao Y. Metabolic Crosstalk between Liver and Brain: From Diseases to Mechanisms. International Journal of Molecular Sciences. 2024; 25(14):7621. https://doi.org/10.3390/ijms25147621
Chicago/Turabian StyleYang, Xiaoyue, Kangli Qiu, Yaoyao Jiang, Yumei Huang, Yajuan Zhang, and Yunfei Liao. 2024. "Metabolic Crosstalk between Liver and Brain: From Diseases to Mechanisms" International Journal of Molecular Sciences 25, no. 14: 7621. https://doi.org/10.3390/ijms25147621
APA StyleYang, X., Qiu, K., Jiang, Y., Huang, Y., Zhang, Y., & Liao, Y. (2024). Metabolic Crosstalk between Liver and Brain: From Diseases to Mechanisms. International Journal of Molecular Sciences, 25(14), 7621. https://doi.org/10.3390/ijms25147621