
Modulation of Ceramide-Induced Apoptosis in Enteric Neurons by Aryl Hydrocarbon Receptor Signaling: Unveiling a New Pathway beyond ER Stress

 , ,
, , 
Abstract
1. Introduction
2. Results
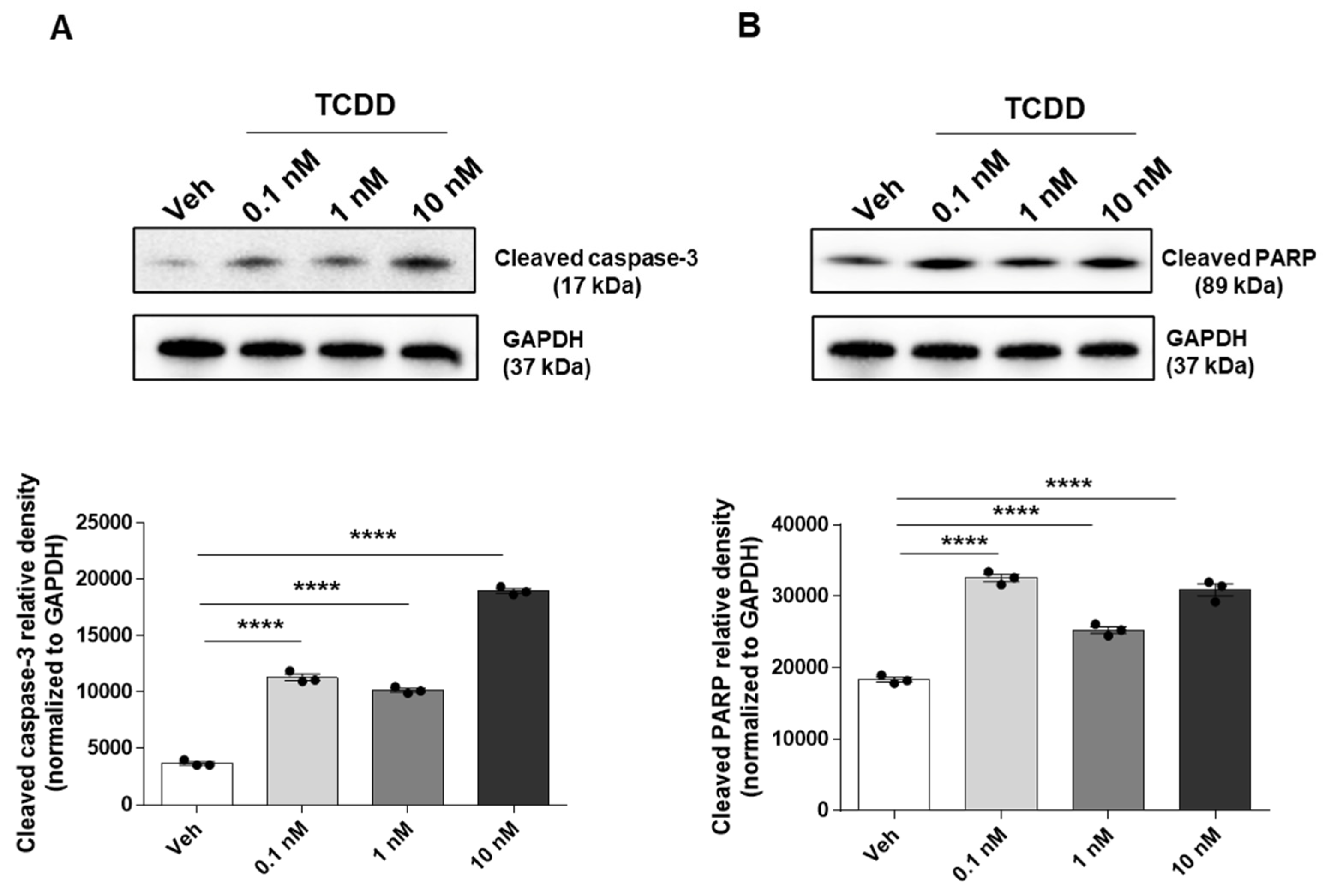
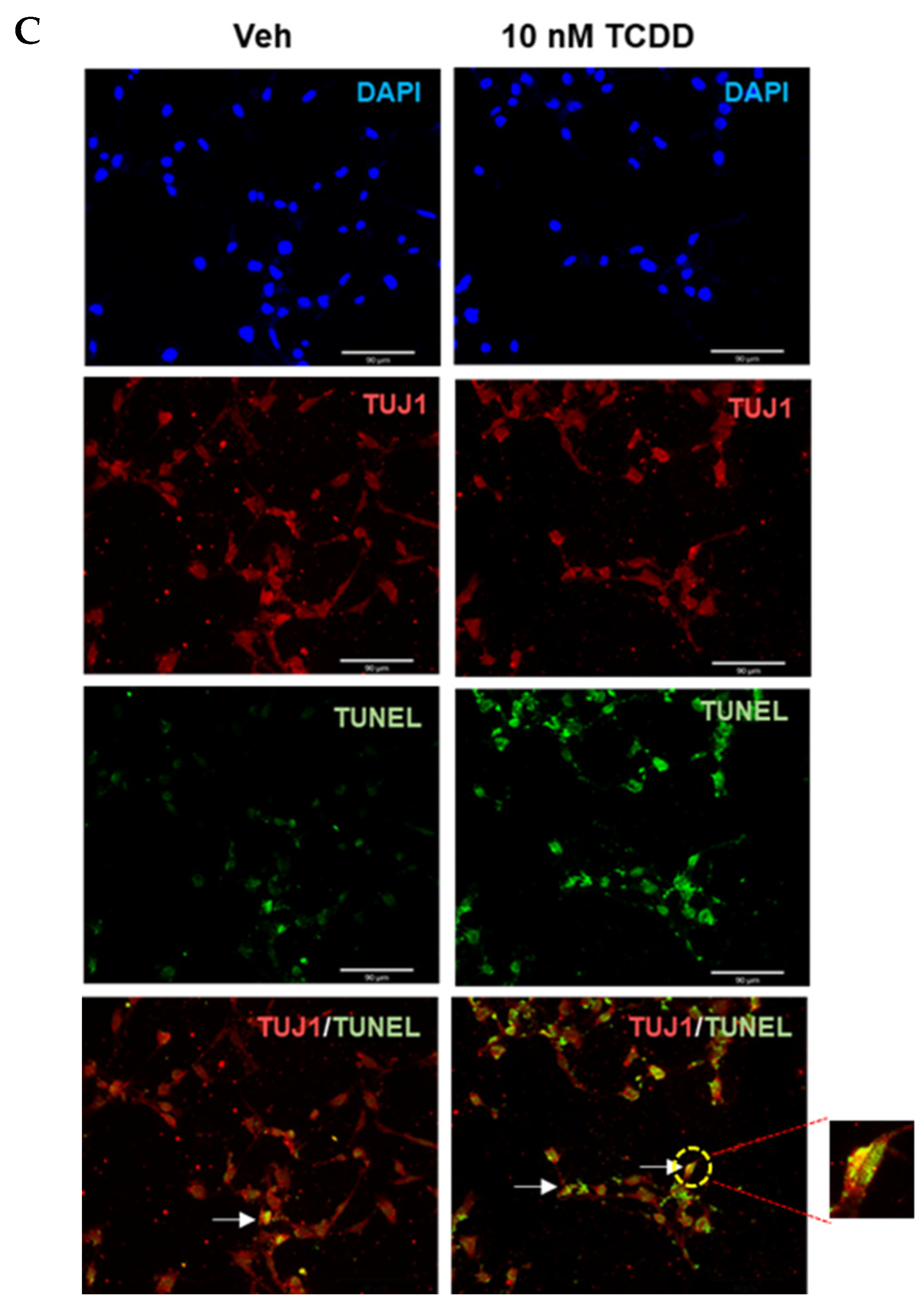
2.1. TCDD-Induced Apoptosis in IM-FEN Cells
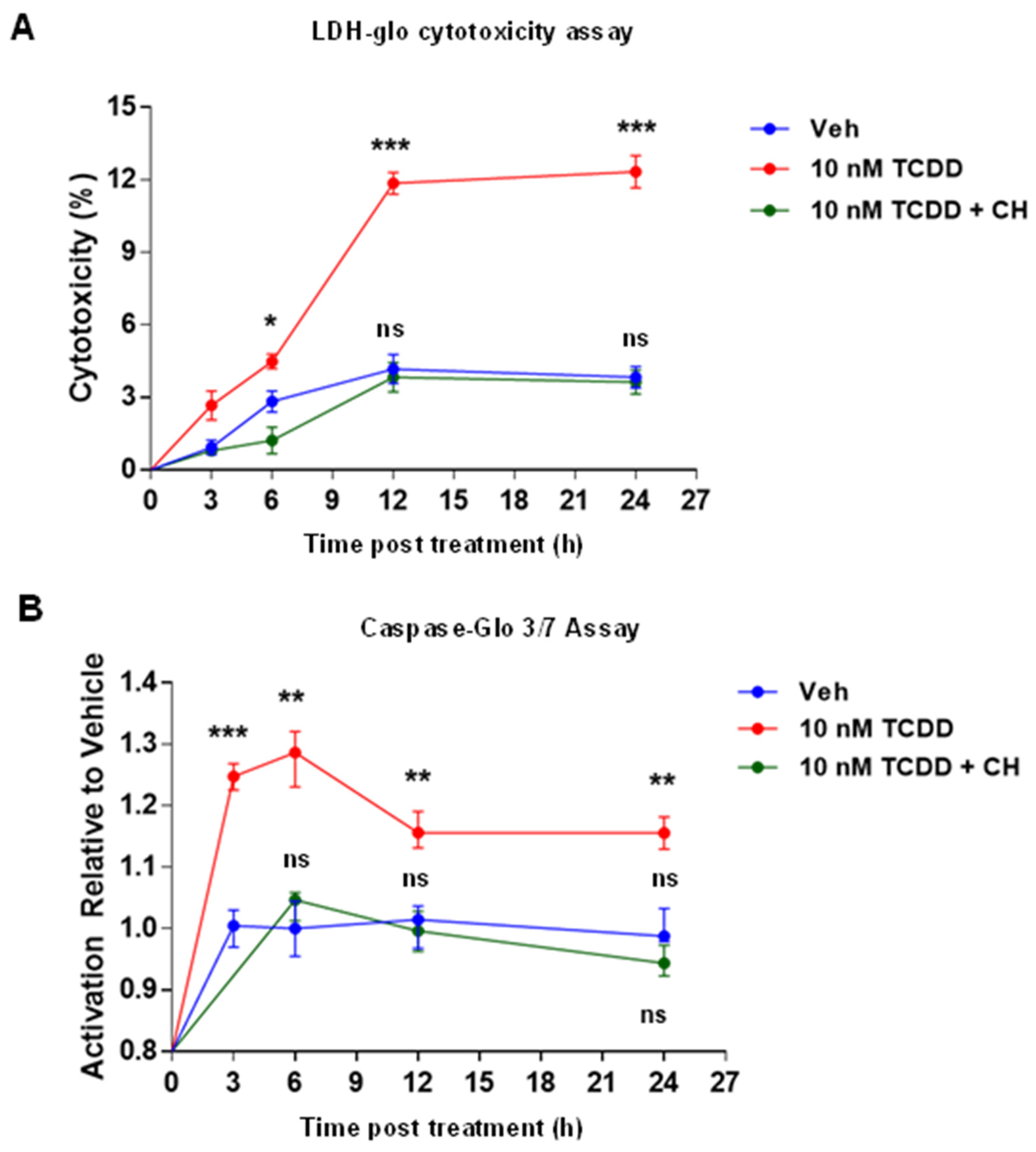
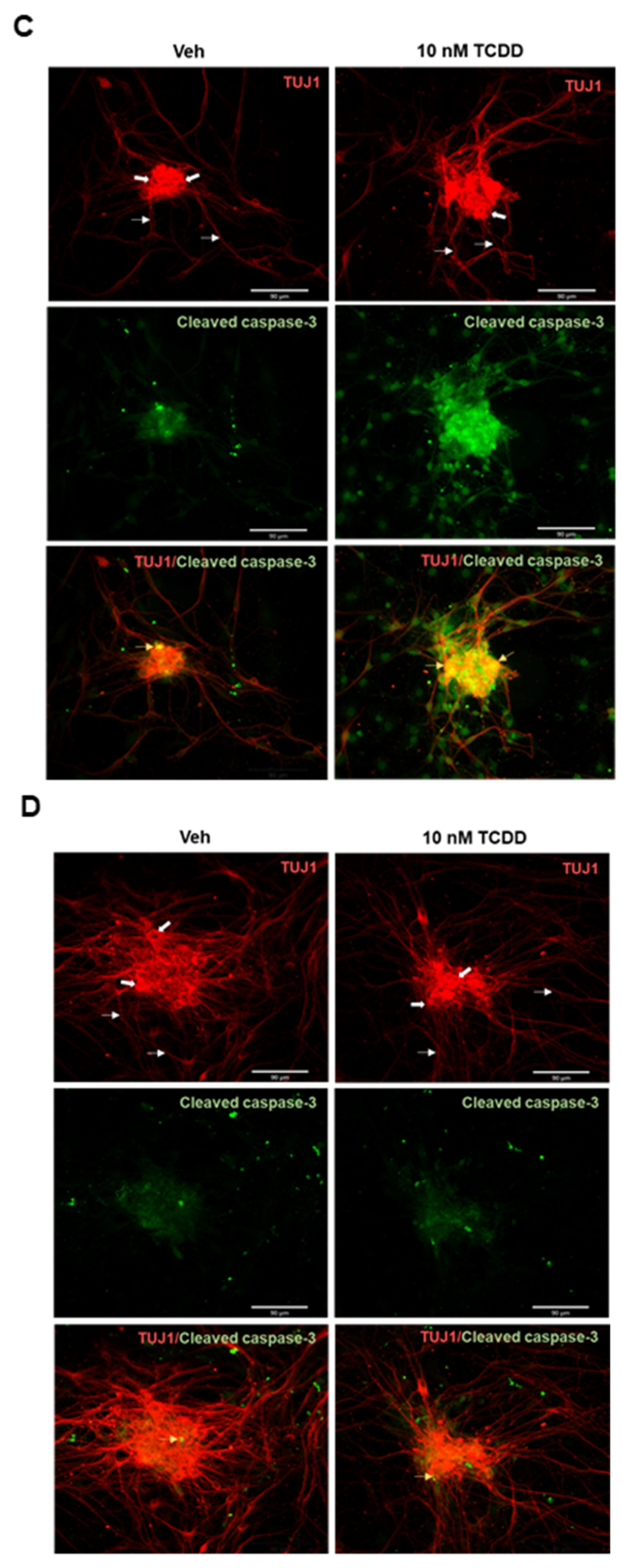
2.2. AHR-Dependent TCDD-Induced Cytotoxicity and Apoptosis in IM-FEN Cells
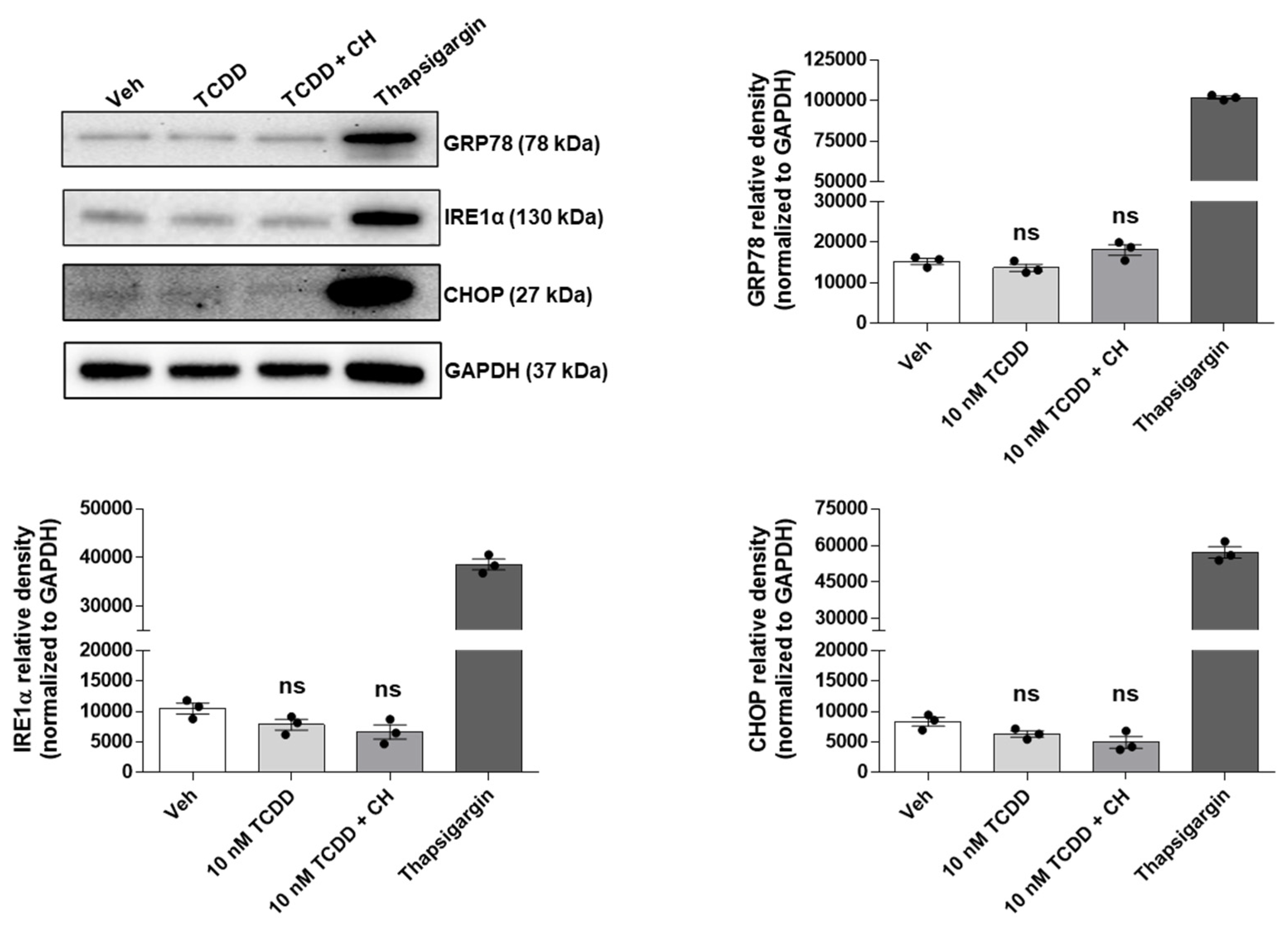
2.3. TCDD-Induced Apoptosis in IM-FEN Cells Proceeds via an ER Stress-Independent Pathway
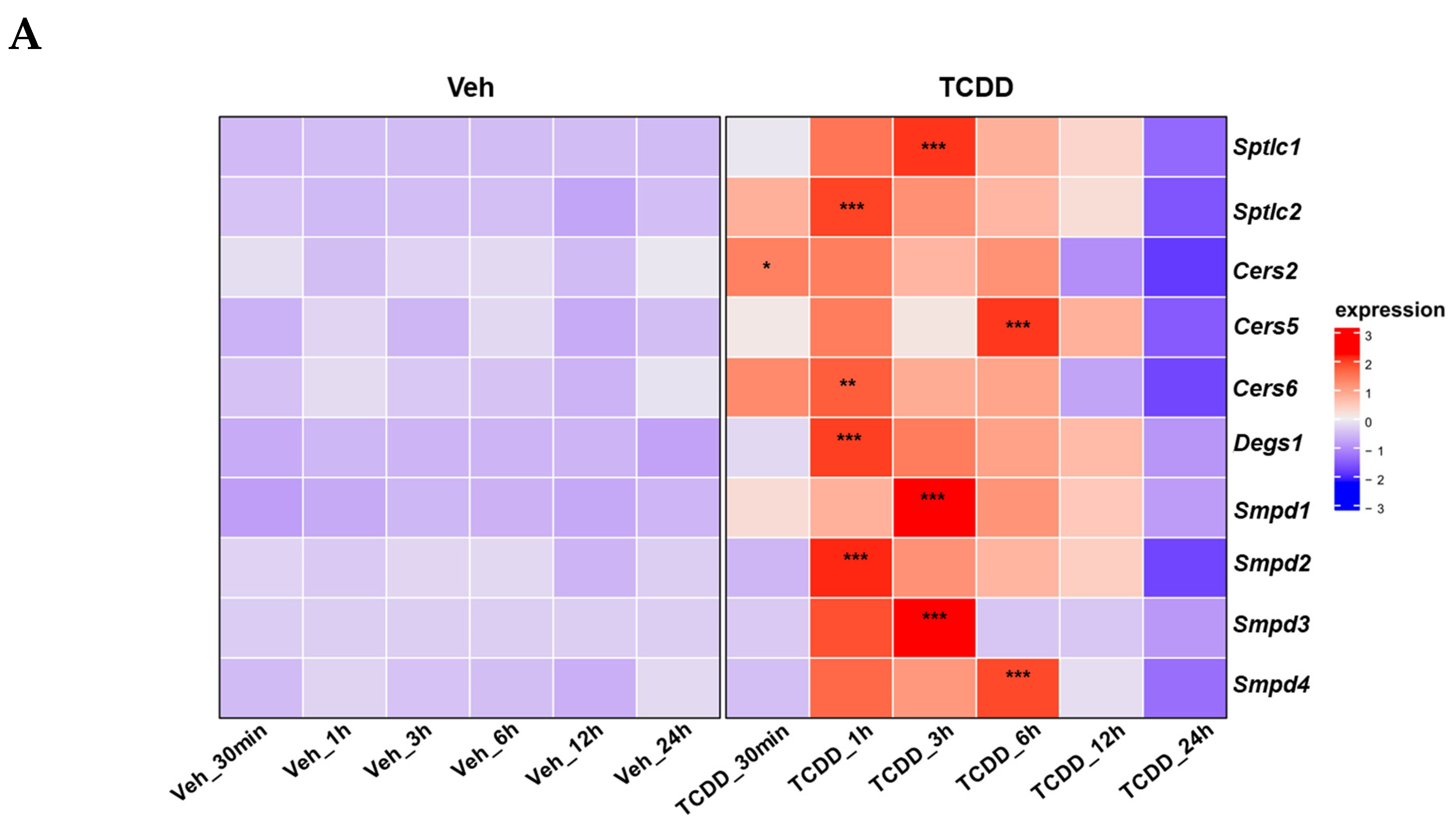
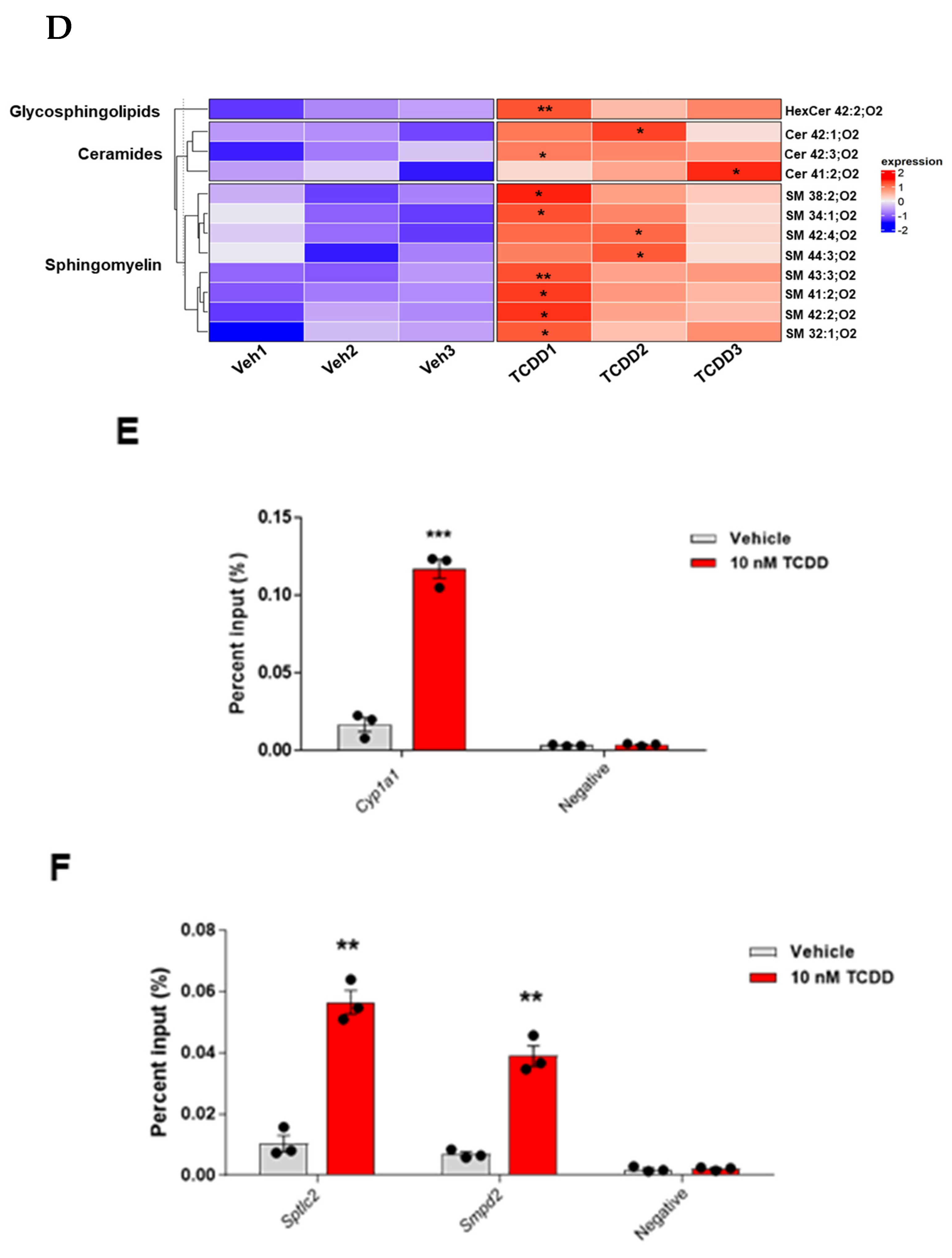
2.4. TCDD Regulates Ceramide Metabolism: Insights from Gene Expression, Enzymatic Activity, and Lipidomic Analyses
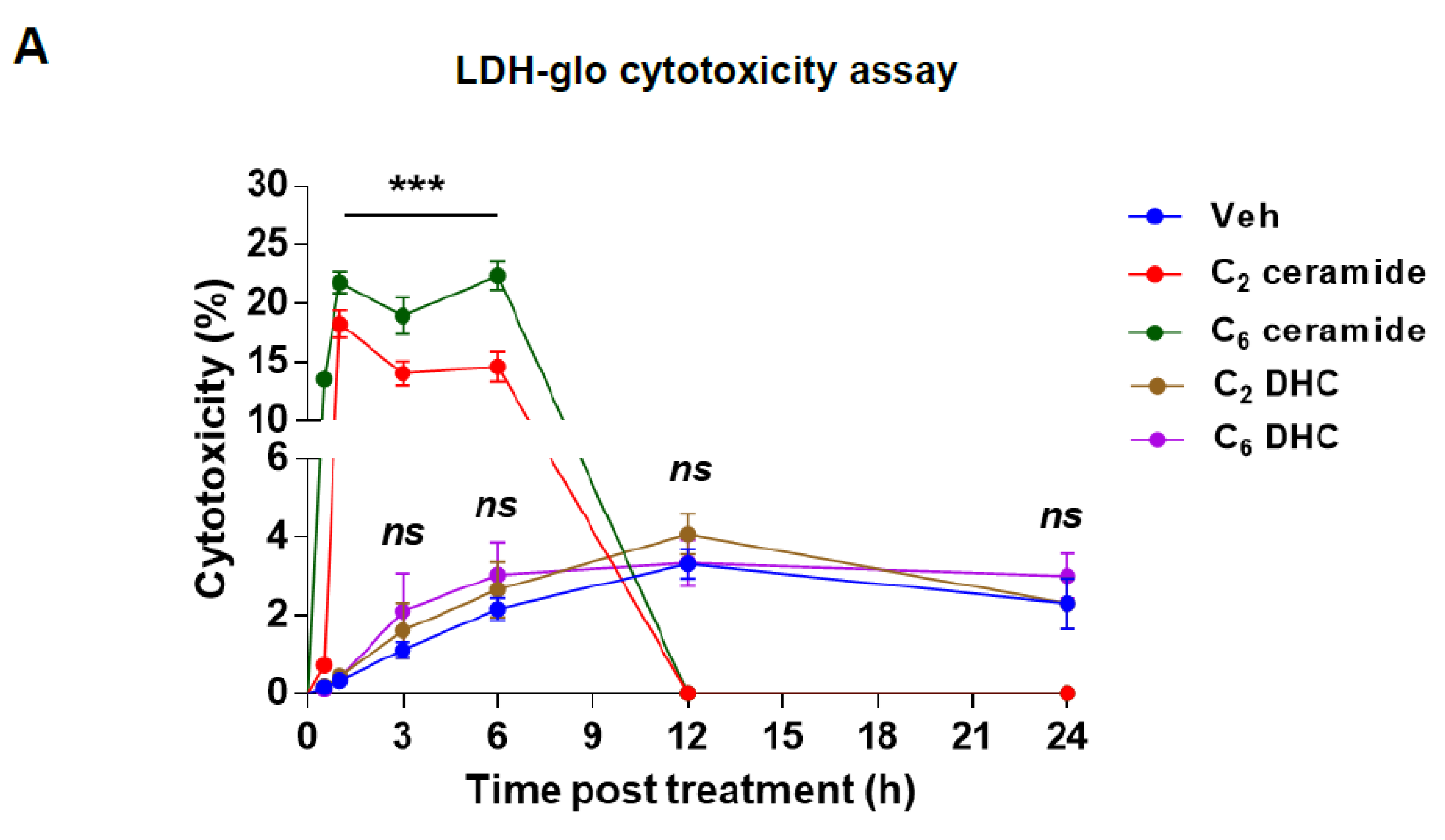
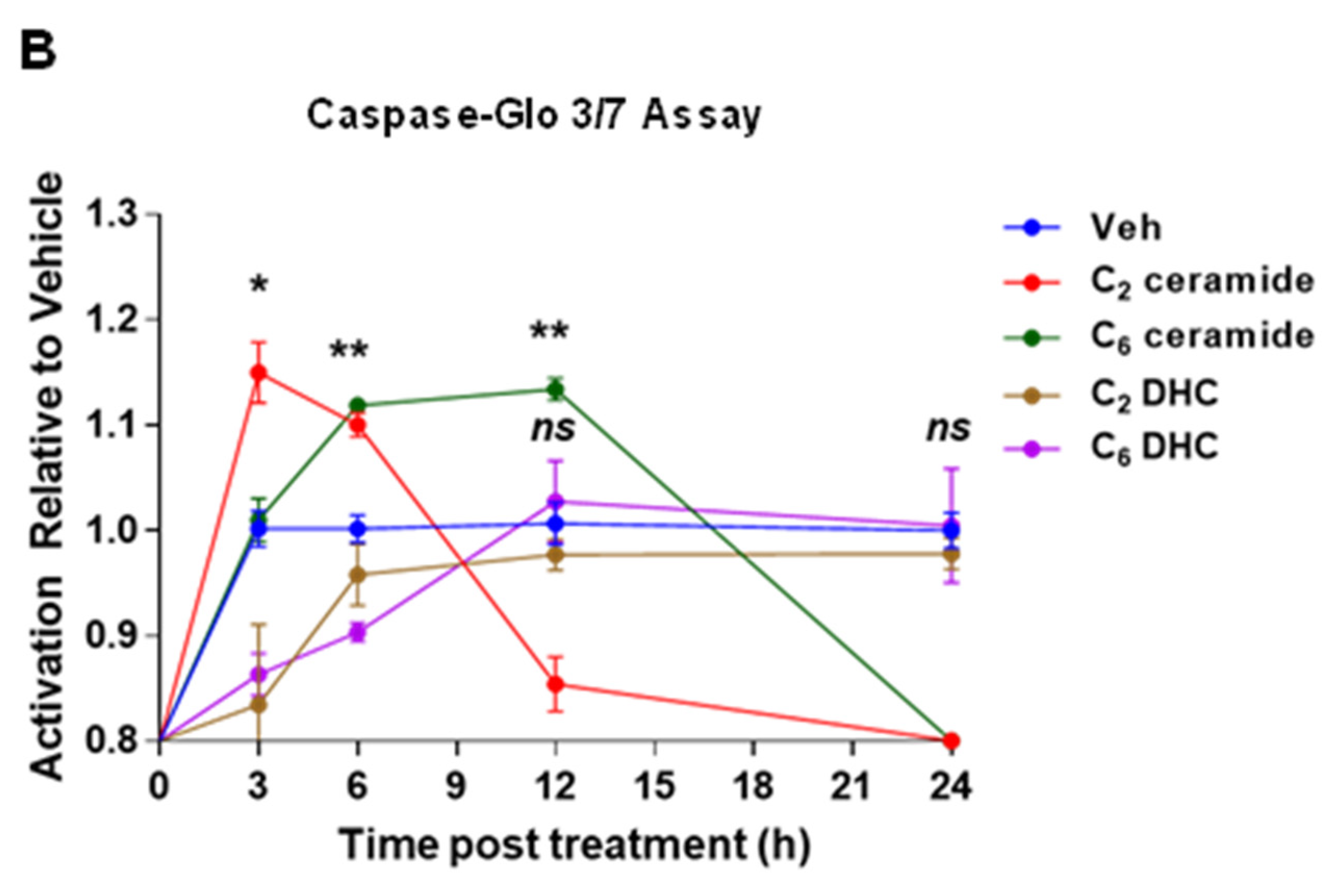
2.5. Induction of Cytotoxicity and Apoptosis by Short-Chain Ceramides in IM-FEN Cells
2.6. TCDD-Induced Cytotoxicity in IM-FEN Cells: The Mediating Role of Ceramides
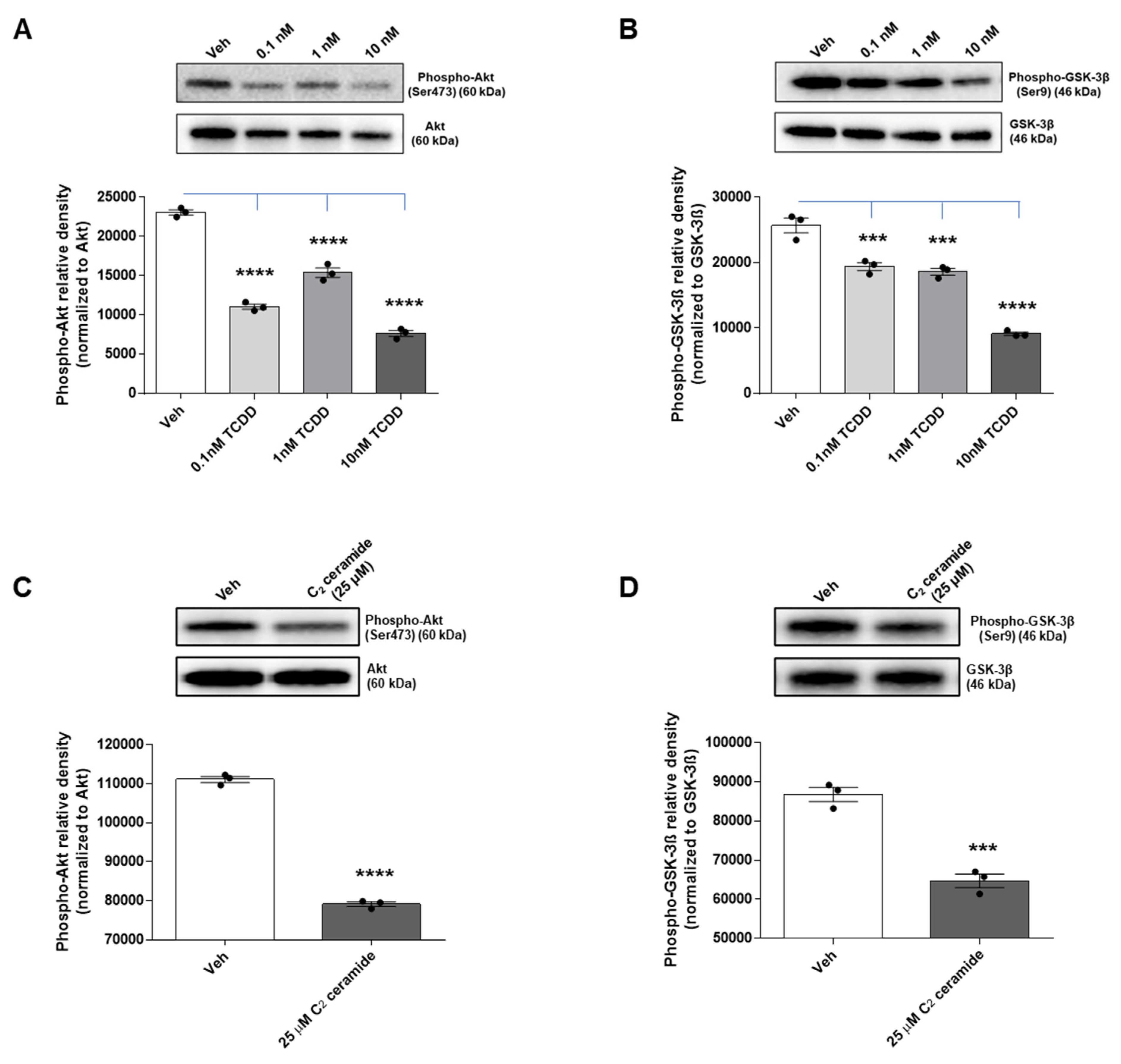
2.7. TCDD and Ceramide Impair Neuronal Survival Pathway by Modulating Akt and GSK-3β Signaling
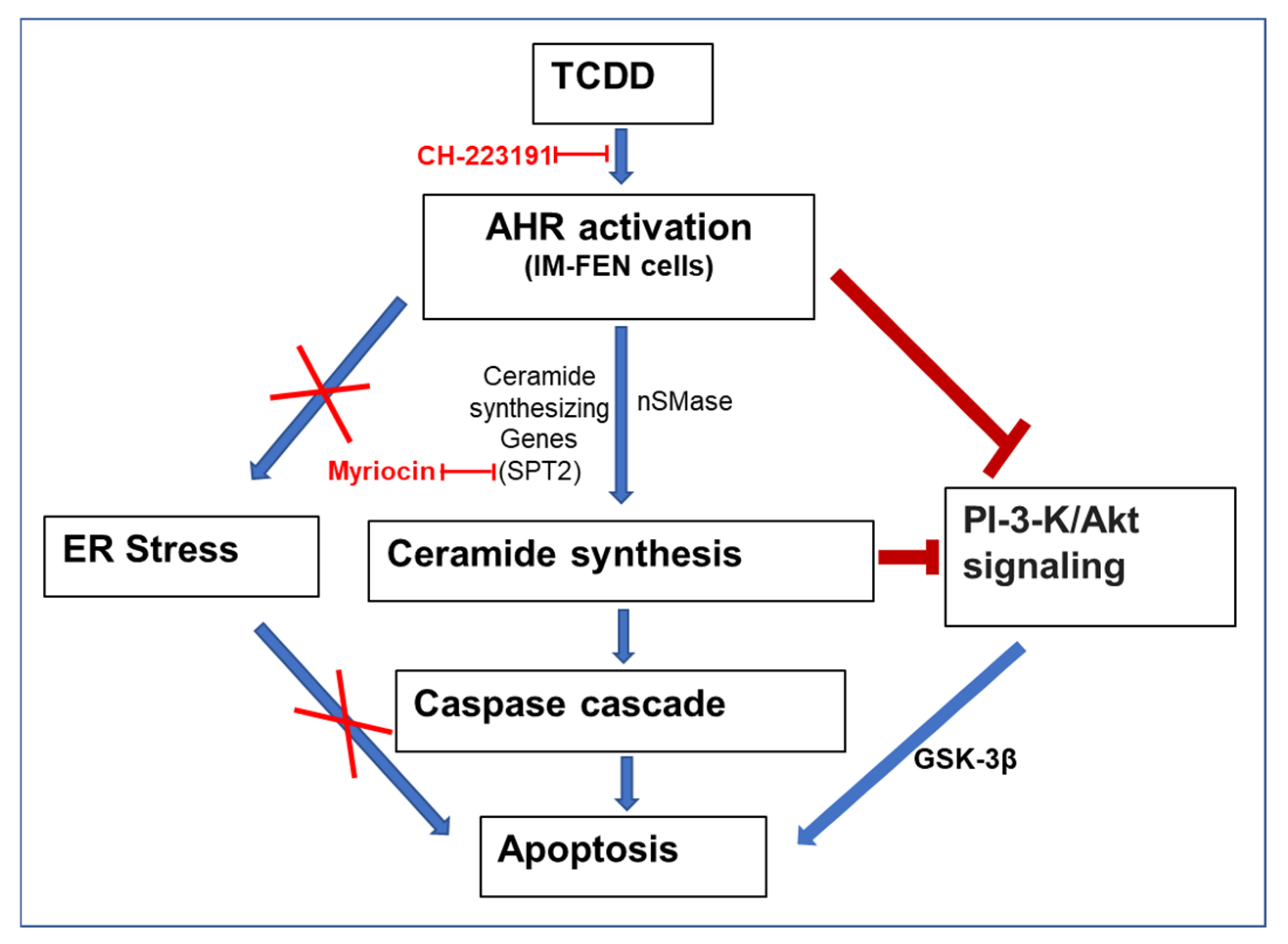
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Culture
4.3. Primary Culture Preparation
4.4. LDH-Glo Cytotoxicity Assay
4.5. Caspase-Glo 3/7 Assay
4.6. Real-Time PCR
4.7. ChIP-PCR/qPCR Assay
4.8. Sphingomyelinase Assay
4.9. Orbitrap-MS Analysis of Lipids
4.10. Western Blotting
4.11. TUNEL Staining
4.12. Immunofluorescence Staining of Primary Culture Cells
4.13. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Van Den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Håkansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization Reevaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-Like Compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.V.D.; De Jongh, J.; Poiger, H.; Olson, J.R. The Toxicokinetics and Metabolism of Polychlorinated Dibenzo-p-Dioxins (PCDDs) and Dibenzofurans (PCDFs) and Their Relevance for Toxicity. Crit. Rev. Toxicol. 1994, 24, 1–74. [Google Scholar] [CrossRef] [PubMed]
- Knerr, S.; Schrenk, D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol. Nutr. Food Res. 2006, 50, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Hogaboam, J.P.; Moore, A.J.; Lawrence, B.P. The Aryl Hydrocarbon Receptor Affects Distinct Tissue Compartments during Ontogeny of the Immune System. Toxicol. Sci. 2007, 102, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.B.; Kerkvliet, N.I. Dioxin and immune regulation. Ann. N. Y. Acad. Sci. 2010, 1183, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, X.; Ren, X.; Zhang, X.; Wu, J.; Wang, S.; Wang, Z. Animal Toxicology Studies on the Male Reproductive Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin: Data Analysis and Health Effects Evaluation. Front. Endocrinol. 2021, 12, 696106. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, M.C.; Beggiato, S.; Ferraro, L.; Tanganelli, S.; Marani, L.; Lorenzini, L.; Antonelli, T. Prenatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin produces alterations in cortical neuron development and a long-term dysfunction of glutamate transmission in rat cerebral cortex. Neurochem. Int. 2012, 61, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Urban, P.; Pelclová, D.; Lukáš, E.; Kupka, K.; Preiss, J.; Fenclová, Z.; Šmerhovský, Z. Neurological and neurophysiological examinations on workers with chronic poisoning by 2,3,7,8-TCDD: Follow-up 35 years after exposure. Eur. J. Neurol. 2007, 14, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Vijay, A.; Boyle, N.R.; Kumar, S.M.; Perdew, G.H.; Srinivasan, S.; Patterson, A.D. Aryl hydrocarbon receptor activation affects nitrergic neuronal survival and delays intestinal motility in mice. Toxicol. Sci. 2023, 192, 117–128. [Google Scholar] [CrossRef]
- Zhou, X.; Chakraborty, D.; Murray, I.A.; Coslo, D.; Kehs, Z.; Vijay, A.; Ton, C.; Desai, D.; Amin, S.G.; Patterson, A.D.; et al. Aryl Hydrocarbon Receptor Activation Coordinates Mouse Small Intestinal Epithelial Cell Programming. Mod. Pathol. 2023, 103, 100012. [Google Scholar] [CrossRef]
- Nebert, D.W. Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic-helix/loop/helix per-Arnt-sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog. Lipid Res. 2017, 67, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Raggi, F.; Russo, D.; Urbani, C.; Sardella, C.; Manetti, L.; Cappellani, D.; Lupi, I.; Tomisti, L.; Martino, E.; Marcocci, C.; et al. Divergent Effects of Dioxin- or Non-Dioxin-Like Polychlorinated Biphenyls on the Apoptosis of Primary Cell Culture from the Mouse Pituitary Gland. PLoS ONE 2016, 11, e0146729. [Google Scholar] [CrossRef] [PubMed]
- Tomblin, J.K.; Salisbury, T.B. Insulin like growth factor 2 regulation of aryl hydrocarbon receptor in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 443, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.-J.; Stokes, J.V.; Kummari, E.; Eells, J.; Kaplan, B.L. Immunomodulation by Subchronic Low Dose 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Experimental Autoimmune Encephalomyelitis in the Absence of Pertussis Toxin. Toxicol. Sci. 2016, 151, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.K.; Jin, U.-H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, J.; Liang, H.; Hu, K.; Jiang, S.; Yang, L.; Mei, S.; Zhu, X.; Yu, J.; Kijlstra, A.; et al. Aryl Hydrocarbon Receptor Regulates Apoptosis and Inflammation in a Murine Model of Experimental Autoimmune Uveitis. Front. Immunol. 2018, 9, 1713. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Wójtowicz, A.; Maćkowiak, M.; Lasoń, W. Aryl hydrocarbon receptor-mediated apoptosis of neuronal cells: A possible interaction with estrogen receptor signaling. Neuroscience 2009, 158, 811–822. [Google Scholar] [CrossRef]
- Bredesen, D.E. Apoptosis: Overview and Signal Transduction Pathways. J. Neurotrauma 2000, 17, 801–810. [Google Scholar] [CrossRef]
- Eldadah, B.A.; Faden, A.I. Caspase Pathways, Neuronal Apoptosis, and CNS Injury. J. Neurotrauma 2000, 17, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Rong, F.; Gao, X.; Liu, K.; Wu, J. Methotrexate remediates spinal cord injury in vivo and in vitro via suppression of endoplasmic reticulum stress-induced apoptosis. Exp. Ther. Med. 2018, 15, 4191–4198. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Yuan, M.; Guo, Y.-S.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress Is Regulated by ATF4–CHOP-Mediated Induction of the Bcl-2 Homology 3-Only Member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.-R.; Jeong, Y.J.; Hwang, C.Y.; Yoon, K.-S.; Choe, W.; Ha, J.; Kim, S.S.; Pak, Y.K.; Yeo, E.-J.; Kang, I. Alpha-naphthoflavone induces apoptosis through endoplasmic reticulum stress via c-Src-, ROS-, MAPKs-, and arylhydrocarbon receptor-dependent pathways in HT22 hippocampal neuronal cells. NeuroToxicology 2018, 71, 39–51. [Google Scholar] [CrossRef]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in Cell Growth Regulation by C18:0-Ceramide and Longevity Assurance Gene 1 in Human Head and Neck Squamous Cell Carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef] [PubMed]
- Kagan, T.; Stoyanova, G.; Lockshin, R.A.; Zakeri, Z. Ceramide from sphingomyelin hydrolysis induces neuronal differentiation, whereas de novo ceramide synthesis and sphingomyelin hydrolysis initiate apoptosis after NGF withdrawal in PC12 Cells. Cell Commun. Signal 2022, 20, 15. [Google Scholar] [CrossRef]
- Czubowicz, K.; Strosznajder, R. Ceramide in the Molecular Mechanisms of Neuronal Cell Death. The Role of Sphingosine-1-Phosphate. Mol. Neurobiol. 2014, 50, 26–37. [Google Scholar] [CrossRef]
- Plotegher, N.; Bubacco, L.; Greggio, E.; Civiero, L. Ceramides in Parkinson’s Disease: From Recent Evidence to New Hypotheses. Front. Neurosci. 2019, 13, 330. [Google Scholar] [CrossRef]
- Mandon, E.; Ehses, I.; Rother, J.; van Echten, G.; Sandhoff, K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Shimeno, H.; Soeda, S.; Sakamoto, M.; Kouchi, T.; Kowakame, T.; Kihara, T. Partial purification and characterization of sphingosine N-acyltransferase (ceramide synthase) from bovine liver mitochondrion-rich fraction. Lipids 1998, 33, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, W.D.; Kolesnick, R.N.; A Fornari, F.; Traylor, R.S.; A Gewirtz, D.; Grant, S. Induction of apoptotic DNA damage and cell death by activation of the sphingomyelin pathway. Proc. Natl. Acad. Sci. USA 1994, 91, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Gillard, B.K.; Clement, R.G.; Marcus, D.M. Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology 1998, 8, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Dbaibo, G.S.; El-Assaad, W.; Krikorian, A.; Liu, B.; Diab, K.; Idriss, N.Z.; El-Sabban, M.; A Driscoll, T.; Perry, D.K.; A Hannun, Y. Ceramide generation by two distinct pathways in tumor necrosis factor α-induced cell death. FEBS Lett. 2001, 503, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Haimovitz-Friedman, A.; Kan, C.C.; Ehleiter, D.; Persaud, R.S.; McLoughlin, M.; Fuks, Z.; Kolesnick, R.N. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J. Exp. Med. 1994, 180, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Dbaibo, G.S.; Pushkareva, M.Y.; A Rachid, R.; Alter, N.; Smyth, M.J.; Obeid, L.M.; A Hannun, Y. p53-dependent ceramide response to genotoxic stress. J. Clin. Investig. 1998, 102, 329–339. [Google Scholar] [CrossRef]
- Rotolo, J.A.; Zhang, J.; Donepudi, M.; Lee, H.; Fuks, Z.; Kolesnick, R. Caspase-dependent and -independent Activation of Acid Sphingomyelinase Signaling. J. Biol. Chem. 2005, 280, 26425–26434. [Google Scholar] [CrossRef]
- Fillet, M.; Bentires-Alj, M.; Deregowski, V.; Greimers, R.; Gielen, J.; Piette, J.; Bours, V.; Merville, M.-P. Mechanisms involved in exogenous C2- and C6-ceramide-induced cancer cell toxicity. Biochem. Pharmacol. 2003, 65, 1633–1642. [Google Scholar] [CrossRef]
- Ghidoni, R.; Sala, G.; Giuliani, A. Use of sphingolipid analogs: Benefits and risks. Biochim. Biophys. Acta. 1999, 1439, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed Cell Death Induced by Ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- A Stoica, B.; A Movsesyan, V.; Iv, P.M.L.; I Faden, A. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3β, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol. Cell Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Majumder, S.; Kono, M.; Lee, Y.T.; Byrnes, C.; Li, C.; Tuymetova, G.; Proia, R.L. A genome-wide CRISPR/Cas9 screen reveals that the aryl hydrocarbon receptor stimulates sphingolipid levels. J. Biol. Chem. 2020, 295, 4341–4349. [Google Scholar] [CrossRef] [PubMed]
- Slováčková, J.; Slavík, J.; Kulich, P.; Večeřa, J.; Kováč, O.; Paculová, H.; Straková, N.; Fedr, R.; Silva, J.P.; Carvalho, F.; et al. Polychlorinated environmental toxicants affect sphingolipid metabolism during neurogenesis in vitro. Toxicology 2021, 463, 152986. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Allman, E.L.; Hubbard, T.D.; Murray, I.A.; Hao, F.; Tian, Y.; Gui, W.; Nichols, R.G.; Smith, P.B.; et al. The aryl hydrocarbon receptor activates ceramide biosynthesis in mice contributing to hepatic lipogenesis. Toxicology 2021, 458, 152831. [Google Scholar] [CrossRef]
- Anitha, M.; Joseph, I.; Ding, X.; Torre, E.R.; Sawchuk, M.A.; Mwangi, S.; Hochman, S.; Sitaraman, S.V.; Anania, F.; Srinivasan, S. Characterization of Fetal and Postnatal Enteric Neuronal Cell Lines with Improvement in Intestinal Neural Function. Gastroenterology 2008, 134, 1424–1435. [Google Scholar] [CrossRef]
- Pavuk, M.; Schecter, A.J.; Akhtar, F.Z.; E Michalek, J. Serum 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) Levels and Thyroid Function in Air Force Veterans of the Vietnam War. Ann. Epidemiol. 2003, 13, 335–343. [Google Scholar] [CrossRef]
- Zhao, B.; DeGroot, D.E.; Hayashi, A.; He, G.; Denison, M.S. CH223191 Is a Ligand-Selective Antagonist of the Ah (Dioxin) Receptor. Toxicol. Sci. 2010, 117, 393–403. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, X.; Zhou, Y.; Wang, H. C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2014, 15, 3336–3355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Summers, S.A.; Birnbaum, M.J.; Pittman, R.N. Inhibition of Akt Kinase by Cell-permeable Ceramide and Its Implications for Ceramide-induced Apoptosis. J. Biol. Chem. 1998, 273, 16568–16575. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Garza, L.A.; Zhou, H.; Birnbaum, M.J. Regulation of Insulin-Stimulated Glucose Transporter GLUT4 Translocation and Akt Kinase Activity by Ceramide. Mol. Cell Biol. 1998, 18, 5457–5464. [Google Scholar] [CrossRef] [PubMed]
- Mulero-Navarro, S.; Fernandez-Salguero, P.M. New Trends in Aryl Hydrocarbon Receptor Biology. Front. Cell Dev. Biol. 2016, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fernández-Salguero, P.M.; Merino, J.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces apoptosis in neural growth factor (NGF)-differentiated pheochromocytoma PC12 cells. NeuroToxicology 2010, 31, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, F.J.; Fernández-Salguero, P.M.; Merino, J.M. Aryl hydrocarbon receptor-dependent induction of apoptosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells from mouse. J. Neurochem. 2011, 118, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Howard, C.V.; Strahle, U.; Cossins, A. Neurodevelopmental Defects in Zebrafish (Danio rerio) at Environmentally Relevant Dioxin (TCDD) Concentrations. Toxicol. Sci. 2003, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Hankinson, O. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses the growth of human liver cancer HepG2 cells in vitro: Involvement of cell signaling factors. Int. J. Oncol. 2018, 53, 1657–1666. [Google Scholar] [CrossRef]
- Chopra, M.; Schrenk, D. Dioxin toxicity, aryl hydrocarbon receptor signaling, and apoptosis—Persistent pollutants affect programmed cell death. Crit. Rev. Toxicol. 2011, 41, 292–320. [Google Scholar] [CrossRef]
- Heimler, I.; Trewin, A.L.; Chaffin, C.L.; Rawlins, R.G.; Hutz, R.J. Modulation of ovarian follicle maturation and effects on apoptotic cell death in holtzman rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin(TCDD) in utero and lactationally. Reprod. Toxicol. 1998, 12, 69–73. [Google Scholar] [CrossRef]
- Jin, M.; Lou, J.; Yu, H.; Miao, M.; Wang, G.; Ai, H.; Huang, Y.; Han, S.; Han, D.; Yu, G. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin promotes inflammation in mouse testes: The critical role of Klotho in Sertoli cells. Toxicol. Lett. 2018, 295, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-S.; Xu, H.; He, X.-J.; Li, Y.; He, B.; Zhao, D. Endoplasmic reticulum stress mediates cortical neuron apoptosis after experimental subarachnoid hemorrhage in rats. Int. J. Clin. Exp. Pathol. 2020, 13, 1569–1577. [Google Scholar] [PubMed]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Ellerby, H.M.; E Bredesen, D. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhao, J.; Fan, X.; Tang, C.; Liang, L.; Nie, X.; Liu, J.; Wu, Q.; Xu, G. The PERK-eIF2α signaling pathway is involved in TCDD-induced ER stress in PC12 cells. NeuroToxicology 2014, 44, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull. 1997, 53, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Futerman, A.H. Distinct Roles for Ceramide and Glucosylceramide at Different Stages of Neuronal Growth. J. Neurosci. 1997, 17, 2929–2938. [Google Scholar] [CrossRef] [PubMed]
- Brann, A.B.; Scott, R.; Neuberger, Y.; Abulafia, D.; Boldin, S.; Fainzilber, M.; Futerman, A.H. Ceramide Signaling Downstream of the p75 Neurotrophin Receptor Mediates the Effects of Nerve Growth Factor on Outgrowth of Cultured Hippocampal Neurons. J. Neurosci. 1999, 19, 8199–8206. [Google Scholar] [CrossRef] [PubMed]
- Siskind, L.J. Mitochondrial Ceramide and the Induction of Apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef]
- Ravid, T.; Tsaba, A.; Gee, P.; Rasooly, R.; Medina, E.A.; Goldkorn, T. Ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Am. J. Physiol. Cell Mol. Physiol. 2003, 284, L1082–L1092. [Google Scholar] [CrossRef]
- Lièvremont, J.-P.; Sciorati, C.; Morandi, E.; Paolucci, C.; Bunone, G.; Della Valle, G.; Meldolesi, J.; Clementi, E. The p75NTR-induced Apoptotic Program Develops through a Ceramide-Caspase Pathway Negatively Regulated by Nitric Oxide. J. Biol. Chem. 1999, 274, 15466–15472. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Kolesnick, R. Stress signals for apoptosis: Ceramide and c-Jun kinase. Oncogene 1998, 17, 3277–3285. [Google Scholar] [CrossRef]
- Hartfield, P.J.; Bilney, A.J.; Murray, A.W. Neurotrophic Factors Prevent Ceramide-Induced Apoptosis Downstream of c-Jun N-Terminal Kinase Activation in PC12 Cells. J. Neurochem. 1998, 71, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Break-down. In Sphingolipids as Signaling and Regulatory Molecules; Chalfant, C., Poeta, M.D., Eds.; Advances in Experi-mental Medicine and Biology; Springer New York: New York, NY, USA, 2010; Volume 688, pp. 1–23. ISBN 978-1-4419-6740-4. [Google Scholar]
- Jaffrezou, J.-P.; Maestre, N.; de Mas-Mansat, V.; Bezombes, C.; Levade, T.; Laurent, G. Positive feedback control of neutral sphingomyelinase activity by ceramide. FASEB J. 1998, 12, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- France-Lanord, V.; Brugg, B.; Michel, P.P.; Agid, Y.; Ruberg, M. Mitochondrial Free Radical Signal in Ceramide-Dependent Apoptosis: A Putative Mechanism for Neuronal Death in Parkinson’s Disease. J. Neurochem. 1997, 69, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A. Functions of Ceramide in Coordinating Cellular Responses to Stress. Science 1996, 274, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. Ceramide signaling in cancer and stem cells. Futur. Lipidol. 2008, 3, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, E.E.; Theibert, A.B. Functions of PI 3-kinase in development of the nervous system. Int. J. Dev. Neurosci. 2002, 20, 187–197. [Google Scholar] [CrossRef]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, 930710. [Google Scholar] [CrossRef]
- Romorini, L.; Garate, X.; Neiman, G.; Luzzani, C.; Furmento, V.A.; Guberman, A.S.; Sevlever, G.E.; Scassa, M.E.; Miriuka, S.G. AKT/GSK3β signaling pathway is critically involved in human pluripotent stem cell survival. Sci. Rep. 2016, 6, 35660. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Pollak, M.; Topisirovic, I. The role of GSK3 in metabolic pathway perturbations in cancer. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 119059. [Google Scholar] [CrossRef] [PubMed]
- Zundel, W.; Giaccia, A. Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev. 1998, 12, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Heuckeroth, R.O.; Lampe, P.A.; Johnson, E.M.; Milbrandt, J. Neurturin and GDNF Promote Proliferation and Survival of Enteric Neuron and Glial Progenitorsin Vitro. Dev. Biol. 1998, 200, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.H.; Ngwainmbi, J.; Grider, J.R.; Dewey, W.L.; Akbarali, H.I. An In-vitro Preparation of Isolated Enteric Neurons and Glia from the Myenteric Plexus of the Adult Mouse. J. Vis. Exp. 2013, e50688. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, W. Mouse Enteric Neuronal Cell Culture. In Neuronal Cell Culture; Amini, S., White, M.K., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 55–63. ISBN 978-1-62703-639-9. [Google Scholar]
- Tian, Y.; Gui, W.; Rimal, B.; Koo, I.; Smith, P.B.; Nichols, R.G.; Cai, J.; Liu, Q.; Patterson, A.D. Metabolic impact of persistent organic pollutants on gut microbiota. Gut Microbes 2020, 12, 1848209. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| B2M | CATGGCTCGCTCGGTGAC | CAGTTCAGTATGTTCGGCTTCC |
| Sptlc1 | CGAGGGTTCTATGGCACATT | GGTGGAGAAGCCATACGAGT |
| Sptlc2 | TCACCTCCATGAAGTGCATC | CAGGCGTCTCCTGAAATACC |
| Cers2 | AAGTGGGAAACGGAGTAGCG | ACAGGCAGCCATAGTCGTTC |
| Cers5 | CTTCTCCGTGAGGATGCTGT | GTGTCATTGGGTTCCACCTT |
| Cers6 | AAGCCAATGGACCACAAACT | TGCTTGGAGAGCCCTTCTAAT |
| Degs1 | AATGGGTCTACACGGACCAG | TGGTCAGGTTTCATCAAGGAC |
| Smpd1 | GTTACCAGCTGATGCCCTTC | AGCAGGATCTGTGGAGTTG |
| Smpd2 | GACATCCCCTACCTGAGCAA | CCAGGAGAGCCAGATCAAAG |
| Smpd3 | CCTGACCAGTGCCATTCTTT | AGAAACCCGGTCCTCGTACT |
| Smpd4 | ACCTGGCCCTCAATCCATTTG | ATAGGCACAGTCCGAAGTACG |
| Gene | Abbreviation | Sequence |
|---|---|---|
| Cytochrome P450, family 1, subfamily A, polypeptide 1 | Cyp1a1 | CAGAGGATGGAGCAGGCTTA AGGATCCACGCGAGACAG |
|
Serine palmitoyltransferase, long chain base subunit 2 | Sptlc2 | ACCTCTCCGAAACCGGAAAT GCGAGCCCCGTCTTCTC |
| Sphingomyelin phosphodiesterase 2 | Smpd2 | AGAGAGGGTTGTGTGTGTGC GATGATGAAGTTGGCAGAGC |
| Negative control | CAGCAGCTCTTTGTGCTGAC TCCATCTGTGCAGCCTGTAG |
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| Ahr | AAGGCCACTAAAGCAATGGGATGT | AAAGTTTATGACTGGGTCACCACA |
| Wnt1Cre | CAGCGCCGCAACTATAAGAG | CATCGACCGGTAATGCAG |
| Antibody/Host | Company | Cat. No. | Dilution |
|---|---|---|---|
| Cleaved Caspase-3 (Asp175) | Cell Signaling Technology (Danvers, MA, USA) | 9661 | 1:1000 (WB) 1:200 (IF) |
| Cleaved PARP (Asp214) | Cell Signaling Technology | 94885 | 1:1000 (WB) |
| Phospho-Akt (Ser473) | Cell Signaling Technology | 9271 | 1:1000 (WB) |
| Akt | Cell Signaling Technology | 9272 | 1:1000 (WB) |
| Phospho-GSK-3β (Ser9) | Cell Signaling Technology | 9336 | 1:1000 (WB) |
| GSK-3β | Cell Signaling Technology | 12456 | 1:1000 (WB) |
| BiP (C50B12) | Cell Signaling Technology | 3177 | 1:1000 (WB) |
| IRE1α (14C10) | Cell Signaling Technology | 3294 | 1:1000 (WB) |
| CHOP (D46F1) | Cell Signaling Technology | 5554 | 1:1000 (WB) |
| GAPDH/Rabbit | Cell Signaling Technology | 2118 | 1:2500 (WB) |
| TUJ1/Mouse | abcam (Waltham, MA, USA) | ab78078 | 1:500 (IF) |
| HRP-conjugated anti-mouse | Cell Signaling Technology | 7076 | 1:2000 (WB) |
| HRP-conjugated anti-rabbit | Cell Signaling Technology | 7074 | 1:2000–5000 (WB) |
| Goat anti-Rb, Alexa Fluor 488 | Invitrogen (Waltham, MA, USA) | A11008 | 1:200 (IF) |
| Goat anti-Mouse, Alexa Fluor 594 | Invitrogen (Waltham, MA, USA) | A11005 | 1:200 (IF) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anitha, M.; Kumar, S.M.; Koo, I.; Perdew, G.H.; Srinivasan, S.; Patterson, A.D. Modulation of Ceramide-Induced Apoptosis in Enteric Neurons by Aryl Hydrocarbon Receptor Signaling: Unveiling a New Pathway beyond ER Stress. Int. J. Mol. Sci. 2024, 25, 8581. https://doi.org/10.3390/ijms25168581
Anitha M, Kumar SM, Koo I, Perdew GH, Srinivasan S, Patterson AD. Modulation of Ceramide-Induced Apoptosis in Enteric Neurons by Aryl Hydrocarbon Receptor Signaling: Unveiling a New Pathway beyond ER Stress. International Journal of Molecular Sciences. 2024; 25(16):8581. https://doi.org/10.3390/ijms25168581
Chicago/Turabian StyleAnitha, Mallappa, Supriya M. Kumar, Imhoi Koo, Gary H. Perdew, Shanthi Srinivasan, and Andrew D. Patterson. 2024. "Modulation of Ceramide-Induced Apoptosis in Enteric Neurons by Aryl Hydrocarbon Receptor Signaling: Unveiling a New Pathway beyond ER Stress" International Journal of Molecular Sciences 25, no. 16: 8581. https://doi.org/10.3390/ijms25168581
APA StyleAnitha, M., Kumar, S. M., Koo, I., Perdew, G. H., Srinivasan, S., & Patterson, A. D. (2024). Modulation of Ceramide-Induced Apoptosis in Enteric Neurons by Aryl Hydrocarbon Receptor Signaling: Unveiling a New Pathway beyond ER Stress. International Journal of Molecular Sciences, 25(16), 8581. https://doi.org/10.3390/ijms25168581