
Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Metabolic Demands of RGCs
2.1. High Metabolic Demands of RGCs
2.2. Role of Mitochondria in Meeting the Metabolic and Physiological Demands of RGCs
2.3. Metabolic Interdependence between RGCs and Astrocytes
3. Mitochondrial Dysfunction in RGCs
3.1. Factors Contributing to Mitochondrial Dysfunction in RGCs
3.2. Susceptibility of RGCs to Mitochondrial Dysfunction
3.3. RGC Degenerative Diseases Associated with Mitochondrial Dysfunction
3.3.1. Hereditary Optic Neuropathies: Pathophysiology
- Fission
- Cellular Respiratory Function
- Mitochondrial DNA Replication
- Lipid Metabolism
3.3.2. Hereditary Optic Neuropathy: LHON
3.3.3. Hereditary Optic Neuropathy: OPA1
3.3.4. Hereditary Optic Neuropathy: OPA13
3.3.5. Glaucoma
3.3.6. Divergent Patterns of Vision Loss in LHON and Glaucoma
4. Oxidative Stress in RGC Health
4.1. Definition of Oxidative Stress
4.2. ROS Metabolism and Antioxidant Defense Pathway in RGC Degeneration
4.2.1. Conversion of Various ROS
4.2.2. Cofactors Responsible for Replenishing Redox Enzymes
4.2.3. Components and Mechanisms Involved in Antioxidant Defense Pathways
4.2.4. Biomarkers for Oxidative Stress
4.3. Impact of Oxidative Stress on RGC Degenerative Diseases
5. Mitochondrial-Based Therapeutic Approaches
5.1. Antioxidative Therapeutic Target in Mitochondrial Dysfunction
5.2. Potential Therapeutics Involving Mitophagy
5.3. Potential Therapeutics Involving Mitobiogenesis
5.4. Potential Therapeutics Involving Mitochondrial Transplantation
5.5. Clinical Trials of Pharmacological and Genetic Therapeutics in LHON Patients
5.6. Efficacies, Limitations, and Future Perspectives in Mitochondrial-Based and Gene Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Provencio, I.; Rodriguez, I.R.; Jiang, G.; Hayes, W.P.; Moreira, E.F.; Rollag, M.D. A novel human opsin in the inner retina. J. Neurosci. 2000, 20, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, T.; Habib, S. ON and OFF Signaling Pathways in the Retina and the Visual System. Front. Ophthalmol. 2022, 2, 989002. [Google Scholar] [CrossRef]
- Yang, T.H.; Kang, E.Y.; Lin, P.H.; Wu, P.L.; Sachs, J.A.; Wang, N.K. The Value of Electroretinography in Identifying Candidate Genes for Inherited Retinal Dystrophies: A Diagnostic Guide. Diagnostics 2023, 13, 3041. [Google Scholar] [CrossRef] [PubMed]
- Dacey, D.M.; Liao, H.W.; Peterson, B.B.; Robinson, F.R.; Smith, V.C.; Pokorny, J.; Yau, K.W.; Gamlin, P.D. Melanopsin-expressing ganglion cells in primate retina signal colour and irradiance and project to the LGN. Nature 2005, 433, 749–754. [Google Scholar] [CrossRef]
- Do, M.T.; Kang, S.H.; Xue, T.; Zhong, H.; Liao, H.W.; Bergles, D.E.; Yau, K.W. Photon capture and signalling by melanopsin retinal ganglion cells. Nature 2009, 457, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.J.; Hattar, S.; Takao, M.; Berson, D.M.; Foster, R.G.; Yau, K.W. Diminished pupillary light reflex at high irradiances in melanopsin-knockout mice. Science 2003, 299, 245–247. [Google Scholar] [CrossRef]
- Markwell, E.L.; Feigl, B.; Zele, A.J. Intrinsically photosensitive melanopsin retinal ganglion cell contributions to the pupillary light reflex and circadian rhythm. Clin. Exp. Optom. 2010, 93, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Spitschan, M. Melanopsin contributions to non-visual and visual function. Curr. Opin. Behav. Sci. 2019, 30, 67–72. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.-N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef]
- Milosavljevic, N.; Storchi, R.; Eleftheriou, C.G.; Colins, A.; Petersen, R.S.; Lucas, R.J. Photoreceptive retinal ganglion cells control the information rate of the optic nerve. Proc. Natl. Acad. Sci. USA 2018, 115, E11817–E11826. [Google Scholar] [CrossRef]
- Kang, E.Y.-C.; Liu, P.-K.; Wen, Y.-T.; Quinn, P.M.; Levi, S.R.; Wang, N.-K.; Tsai, R.-K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants 2021, 10, 1948. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Carelli, V. Mitochondrial Retinopathies. Int. J. Mol. Sci. 2021, 23, 210. [Google Scholar] [CrossRef]
- Han, G. Aspects of Retinal Energy Metabolism. Ph.D. Thesis, University of Adelaide, Adelaide, Australia, 2014. [Google Scholar]
- Strachan, E.L.; Mac White-Begg, D.; Crean, J.; Reynolds, A.L.; Kennedy, B.N.; O’Sullivan, N.C. The Role of Mitochondria in Optic Atrophy With Autosomal Inheritance. Front. Neurosci. 2021, 15, 784987. [Google Scholar] [CrossRef] [PubMed]
- Shimba, K.; Asahina, T.; Sakai, K.; Kotani, K.; Jimbo, Y. Recording Saltatory Conduction Along Sensory Axons Using a High-Density Microelectrode Array. Front. Neurosci. 2022, 16, 854637. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H. Neurotransmitters in the Retina. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 15, 139–165. [Google Scholar] [CrossRef]
- Coussa, R.G.; Merat, P.; Levin, L.A. Propagation and Selectivity of Axonal Loss in Leber Hereditary Optic Neuropathy. Sci. Rep. 2019, 9, 6720. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2021, 81, 100881. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment strategies for inherited optic neuropathies: Past, present and future. Eye 2014, 28, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.; Guymer, C.; Wood, J.; Daskalaki, E.; Chidlow, G.; Cardozo, B.H.; Foxworth, N.; Cochran, K.E.; Ouellette, T.B.; et al. Oral pyruvate prevents glaucomatous neurodegeneration. bioRxiv 2020. [Google Scholar] [CrossRef]
- Magistretti, P.J. Role of glutamate in neuron-glia metabolic coupling. Am. J. Clin. Nutr. 2009, 90, 875s–880s. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.-M.; Magistretti, P.J. Regulation of neuron-astrocyte metabolic coupling across the sleep-wake cycle. Neuroscience 2016, 323, 135–356. [Google Scholar] [CrossRef]
- Laughton, J.D.; Bittar, P.; Charnay, Y.; Pellerin, L.; Kovari, E.; Magistretti, P.J.; Bouras, C. Metabolic compartmentalization in the human cortex and hippocampus: Evidence for a cell- and region-specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci. 2007, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Naka, M.; Kanamori, A.; Negi, A.; Nakamura, M. Reduced expression of aquaporin-9 in rat optic nerve head and retina following elevated intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4618–4626. [Google Scholar] [CrossRef]
- Miki, A.; Kanamori, A.; Negi, A.; Naka, M.; Nakamura, M. Loss of aquaporin 9 expression adversely affects the survival of retinal ganglion cells. Am. J. Pathol. 2013, 182, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.L.; Bek, T.; la Cour, M.; Nielsen, S.; Prause, J.U.; Hamann, S.; Heegaard, S. Altered aquaporin expression in glaucoma eyes. Apmis 2014, 122, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Akashi, A.; Miki, A.; Kanamori, A.; Nakamura, M. Aquaporin 9 expression is required for l-lactate to maintain retinal neuronal survival. Neurosci. Lett. 2015, 589, 185–190. [Google Scholar] [CrossRef]
- Philp, N.J.; Ochrietor, J.D.; Rudoy, C.; Muramatsu, T.; Linser, P.J. Loss of MCT1, MCT3, and MCT4 expression in the retinal pigment epithelium and neural retina of the 5A11/basigin-null mouse. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1305–1311. [Google Scholar] [CrossRef]
- Chidlow, G.; Wood, J.P.; Graham, M.; Osborne, N.N. Expression of monocarboxylate transporters in rat ocular tissues. Am. J. Physiol. Cell Physiol. 2005, 288, C416–C428. [Google Scholar] [CrossRef] [PubMed]
- Badr, G.A.; Tang, J.; Ismail-Beigi, F.; Kern, T.S. Diabetes downregulates GLUT1 expression in the retina and its microvessels but not in the cerebral cortex or its microvessels. Diabetes 2000, 49, 1016–1021. [Google Scholar] [CrossRef]
- Mori, S.; Kurimoto, T.; Miki, A.; Maeda, H.; Kusuhara, S.; Nakamura, M. Aqp9 Gene Deletion Enhances Retinal Ganglion Cell (RGC) Death and Dysfunction Induced by Optic Nerve Crush: Evidence that Aquaporin 9 Acts as an Astrocyte-to-Neuron Lactate Shuttle in Concert with Monocarboxylate Transporters To Support RGC Function and Survival. Mol. Neurobiol. 2020, 57, 4530–4548. [Google Scholar] [CrossRef] [PubMed]
- Murai, Y.; Mori, S.; Okuda, M.; Kusuhara, S.; Kurimoto, T.; Nakamura, M. Effects of Elevated Intraocular Pressure on Retinal Ganglion Cell Density and Expression and Interaction of Retinal Aquaporin 9 and Monocarboxylate Transporters. Ophthalmic Res. 2023, 66, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.M.; Guymer, C.; Wood, J.P.M.; Daskalaki, E.; Chidlow, G.; Zhang, C.; Balasubramanian, R.; Cardozo, B.H.; Foxworth, N.E.; Deering, K.E.; et al. Disturbed glucose and pyruvate metabolism in glaucoma with neuroprotection by pyruvate or rapamycin. Proc. Natl. Acad. Sci. USA 2020, 117, 33619–33627. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.; Nordgaard, C.; Olsen, T.; Ferrington, D. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C.C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Shu, D.Y.; Chaudhary, S.; Cho, K.S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in ce ll fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Gustafson, M.A.; Sullivan, E.D.; Copeland, W.C. Consequences of compromised mitochondrial genome integrity. DNA Repair 2020, 93, 102916. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Mercieca, K.; Prokosch, V. Mitochondrial Markers in Aging and Primary Open-Angle Glaucoma. J. Glaucoma 2020, 29, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, E.G.; Rosenberg, P.A.; Benowitz, L.I. Non-Cell-Autonomous Regulation of Optic Nerve Regeneration by Amacrine Cells. Front. Cell. Neurosci. 2021, 15, 666798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.Y.; Aguzzi, E.A.; Johnson, T.V. Retinal Ganglion Cell Transplantation: Approaches for Overcoming Challenges to Functional Integration. Cells 2021, 10, 1426. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef]
- Hagins, W.A.; Penn, R.D.; Yoshikami, S. Dark current and photocurrent in retinal rods. Biophys. J. 1970, 10, 380–412. [Google Scholar] [CrossRef] [PubMed]
- Narayan, D.S.; Chidlow, G.; Wood, J.P.; Casson, R.J. Glucose metabolism in mammalian photoreceptor inner and outer segments. Clin. Exp. Ophthalmol. 2017, 45, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog. Retin. Eye Res. 2018, 64, 131–156. [Google Scholar] [CrossRef]
- Mahabadi, N.; Al Khalili, Y. Neuroanatomy, Retina. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2024. [Google Scholar]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef]
- Cohen, L.H.; Noell, W.K. Glucose catabolism of rabbit retina before and after development of visual function. J. Neurochem. 1960, 5, 253–276. [Google Scholar] [CrossRef]
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689. [Google Scholar] [CrossRef]
- Hurley, J.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. Optic neuropathy induced by reductions in mitochondrial superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Kang, E.Y.-C.; Liu, P.-K.; Levi, S.R.; Wang, H.-H.; Tseng, Y.-J.; Seo, G.H.; Lee, H.; Yeh, L.-K.; Chen, K.-J.; et al. Photoreceptor Manifestations of Primary Mitochondrial Optic Nerve Disorders. Investig. Ophthalmol. Vis. Sci. 2022, 63, 5. [Google Scholar] [CrossRef]
- Karanjia, R.; Yu-Wai-Man, P.; Newman, N.J. Hereditary Optic Neuropathies. In Albert and Jakobiec’s Principles and Practice of Ophthalmology; Albert, D.M., Miller, J.W., Azar, D.T., Young, L.H., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 4575–4607. [Google Scholar]
- McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. Online Mendelian Inheritance in Man, OMIM®. Available online: https://omim.org/ (accessed on 15 May 2024).
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Baderna, V.; Schultz, J.; Kearns, L.S.; Fahey, M.; Thompson, B.A.; Ruddle, J.B.; Huq, A.; Maltecca, F. A novel AFG3L2 mutation close to AAA domain leads to aberrant OMA1 and OPA1 processing in a family with optic atrophy. Acta Neuropathol. Commun. 2020, 8, 93. [Google Scholar] [CrossRef]
- Charif, M.; Chevrollier, A.; Gueguen, N.; Bris, C.; Goudenège, D.; Desquiret-Dumas, V.; Leruez, S.; Colin, E.; Meunier, A.; Vignal, C.; et al. Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy. Neurol. Genet. 2020, 6, e428. [Google Scholar] [CrossRef]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef]
- Da Cruz, S.; Xenarios, I.; Langridge, J.; Vilbois, F.; Parone, P.A.; Martinou, J.C. Proteomic analysis of the mouse liver mitochondrial inner membrane. J. Biol. Chem. 2003, 278, 41566–41571. [Google Scholar] [CrossRef]
- Ryu, S.W.; Jeong, H.J.; Choi, M.; Karbowski, M.; Choi, C. Optic atrophy 3 as a protein of the mitochondrial outer membrane induces mitochondrial fragmentation. Cell. Mol. Life Sci. 2010, 67, 2839–2850. [Google Scholar] [CrossRef] [PubMed]
- Reynier, P.; Amati-Bonneau, P.; Verny, C.; Olichon, A.; Simard, G.; Guichet, A.; Bonnemains, C.; Malecaze, F.; Malinge, M.C.; Pelletier, J.B.; et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J. Med. Genet. 2004, 41, e110. [Google Scholar] [CrossRef]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.Y.; Gingras, A.C.; et al. SLC25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife 2015, 4, e07739. [Google Scholar] [CrossRef]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef]
- Wallace, D.; Singh, G.; Lott, M.; Hodge, J.A.; Schurr, T.; Lezza, A.; Elsas, L.; Nikoskelainen, E. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Michaelides, M.; Tee, L.J.; Robson, A.G.; Rahman, F.; Pasha, S.; Luxon, L.M.; Moore, A.T.; Maher, E.R. Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy. Mol. Vis. 2010, 16, 650–664. [Google Scholar] [PubMed]
- Formosa, L.E.; Reljic, B.; Sharpe, A.J.; Hock, D.H.; Muellner-Wong, L.; Stroud, D.A.; Ryan, M.T. Optic atrophy-associated TMEM126A is an assembly factor for the ND4-module of mitochondrial complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2019665118. [Google Scholar] [CrossRef]
- D’Angelo, L.; Astro, E.; De Luise, M.; Kurelac, I.; Umesh-Ganesh, N.; Ding, S.; Fearnley, I.M.; Gasparre, G.; Zeviani, M.; Porcelli, A.M.; et al. NDUFS3 depletion permits complex I maturation and reveals TMEM126A/OPA7 as an assembly factor binding the ND4-module intermediate. Cell Rep. 2021, 35, 109002. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.H.; Hausmann, O.N.; Yan, M.S.; Walters, W.M.; Wong, P.K.; Bethea, J.R. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP). J. Neurochem. 2002, 81, 36–45. [Google Scholar] [CrossRef] [PubMed]
- D’Gama, A.M.; England, E.; Madden, J.A.; Shi, J.; Chao, K.R.; Wojcik, M.H.; Torres, A.R.; Tan, W.H.; Berry, G.T.; Prabhu, S.P.; et al. Exome sequencing identifies novel missense and deletion variants in RTN4IP1 associated with optic atrophy, global developmental delay, epilepsy, ataxia, and choreoathetosis. Am. J. Med. Genet. A 2021, 185, 203–207. [Google Scholar] [CrossRef]
- Mao, H.; Chen, W.; Chen, L.; Li, L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharmacol. 2022, 199, 115011. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.D.; Fischer, T.T.; Abreu, D.; Arroyo, A.; Urano, F.; Ehrlich, B.E. Calpain inhibitor and ibudilast rescue β cell functions in a cellular model of Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 17389–17398. [Google Scholar] [CrossRef] [PubMed]
- La Morgia, C.; Maresca, A.; Amore, G.; Gramegna, L.L.; Carbonelli, M.; Scimonelli, E.; Danese, A.; Patergnani, S.; Caporali, L.; Tagliavini, F.; et al. Calcium mishandling in absence of primary mitochondrial dysfunction drives cellular pathology in Wolfram Syndrome. Sci. Rep. 2020, 10, 4785. [Google Scholar] [CrossRef]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Cell Death Dis. 2018, 9, 364. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Hofmann, S.; Hamasaki, D.I.; Yamamoto, H.; Kreczmanski, P.; Schmitz, C.; Parel, J.M.; Schmidt-Kastner, R. Wolfram syndrome 1 (WFS1) protein expression in retinal ganglion cells and optic nerve glia of the cynomolgus monkey. Exp. Eye Res. 2006, 83, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Charif, M.; Gueguen, N.; Ferré, M.; Elkarhat, Z.; Khiati, S.; LeMao, M.; Chevrollier, A.; Desquiret-Dumas, V.; Goudenège, D.; Bris, C.; et al. Dominant ACO2 mutations are a frequent cause of isolated optic atrophy. Brain Commun. 2021, 3, fcab063. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, G.L.; Bermek, O.; Rosado-Ruiz, F.A.; Hovde, S.L.; Neitzke, O.J.; Griffith, J.D.; Kaguni, L.S. Mitochondrial Single-stranded DNA-binding Proteins Stimulate the Activity of DNA Polymerase γ by Organization of the Template DNA. J. Biol. Chem. 2015, 290, 28697–28707. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.T.; Kaguni, L.S. Reduced stimulation of recombinant DNA polymerase γ and mitochondrial DNA (mtDNA) helicase by variants of mitochondrial single-stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J. Biol. Chem. 2011, 286, 40649–40658. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Orssaud, C.; Kaplan, J.; Johansson, C.; Rozet, J.M. MCAT Mutations Cause Nuclear LHON-like Optic Neuropathy. Genes 2021, 12, 521. [Google Scholar] [CrossRef]
- Fiorini, C.; Degiorgi, A.; Cascavilla, M.L.; Tropeano, C.V.; La Morgia, C.; Battista, M.; Ormanbekova, D.; Palombo, F.; Carbonelli, M.; Bandello, F.; et al. Recessive MECR pathogenic variants cause an LHON-like optic neuropathy. J. Med. Genet. 2023, 61, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Joshi, A.K.; Smith, S. Cloning, expression, characterization, and interaction of two components of a human mitochondrial fatty acid synthase. Malonyltransferase and acyl carrier protein. J. Biol. Chem. 2003, 278, 40067–40074. [Google Scholar] [CrossRef] [PubMed]
- Heimer, G.; Kerätär, J.M.; Riley, L.G.; Balasubramaniam, S.; Eyal, E.; Pietikäinen, L.P.; Hiltunen, J.K.; Marek-Yagel, D.; Hamada, J.; Gregory, A.; et al. MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder. Am. J. Hum. Genet. 2016, 99, 1229–1244. [Google Scholar] [CrossRef]
- Tucci, S.; Alatibi, K.I.; Wehbe, Z. Altered Metabolic Flexibility in Inherited Metabolic Diseases of Mitochondrial Fatty Acid Metabolism. Int. J. Mol. Sci. 2021, 22, 3799. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Yu-Wai-Man, P. Leber hereditary optic neuropathy: Bridging the translational gap. Curr. Opin. Ophthalmol. 2017, 28, 403–409. [Google Scholar] [CrossRef]
- Giordano, C.; Montopoli, M.; Perli, E.; Orlandi, M.; Fantin, M.; Ross-Cisneros, F.N.; Caparrotta, L.; Martinuzzi, A.; Ragazzi, E.; Ghelli, A.; et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain 2011, 134, 220–234. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Maresca, A.; Peron, C.; Raimondi, A.; Caporali, L.; Marchi, S.; La Morgia, C.; Del Dotto, V.; Zanna, C.; et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. Cell Rep. 2022, 40, 111124. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene-environment interactions in Leber hereditary optic neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Ross-Cisneros, F.N.; Caporali, L.; Maresca, A.; Loguercio Polosa, P.; Barboni, P.; De Negri, A.; et al. Parsing the differences in affected with LHON: Genetic versus environmental triggers of disease conversion. Brain 2016, 139, e17. [Google Scholar] [CrossRef]
- Carelli, V.; Giordano, C.; d’Amati, G. Pathogenic expression of homoplasmic mtDNA mutations needs a complex nuclear-mitochondrial interaction. Trends Genet. 2003, 19, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Jin, X.; Peng, Y.; Wang, M.; Liu, H.; Liu, X.; Zhang, Z.; Ji, Y.; Zhang, J.; Liang, M.; et al. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 2016, 25, 584–596. [Google Scholar] [CrossRef]
- Yu, J.; Liang, X.; Ji, Y.; Ai, C.; Liu, J.; Zhu, L.; Nie, Z.; Jin, X.; Wang, C.; Zhang, J.; et al. PRICKLE3 linked to ATPase biogenesis manifested Leber’s hereditary optic neuropathy. J. Clin. Investig. 2020, 130, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Achilli, A.; Valentino, M.L.; Rengo, C.; Semino, O.; Pala, M.; Olivieri, A.; Mattiazzi, M.; Pallotti, F.; Carrara, F.; et al. Haplogroup effects and recombination of mitochondrial DNA: Novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am. J. Hum. Genet. 2006, 78, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Strobbe, D.; Caporali, L.; Iommarini, L.; Maresca, A.; Montopoli, M.; Martinuzzi, A.; Achilli, A.; Olivieri, A.; Torroni, A.; Carelli, V.; et al. Haplogroup J mitogenomes are the most sensitive to the pesticide rotenone: Relevance for human diseases. Neurobiol. Dis. 2018, 114, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Franceschini, F.; Venturi, S.; Barboni, P.; Savini, G.; Barbieri, G.; Pirro, E.; La Morgia, C.; Valentino, M.L.; Zanardi, F.; et al. Grand rounds: Could occupational exposure to n-hexane and other solvents precipitate visual failure in leber hereditary optic neuropathy? Environ. Health Perspect. 2007, 115, 113–115. [Google Scholar] [CrossRef]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Vidoni, S.; Mattioli, S.; Barbieri, A.; Iommarini, L.; Pala, M.; Achilli, A.; Torroni, A.; et al. The background of mitochondrial DNA haplogroup J increases the sensitivity of Leber’s hereditary optic neuropathy cells to 2,5-hexanedione toxicity. PLoS ONE 2009, 4, e7922. [Google Scholar] [CrossRef]
- Lenaers, G.; Neutzner, A.; Le Dantec, Y.; Jüschke, C.; Xiao, T.; Decembrini, S.; Swirski, S.; Kieninger, S.; Agca, C.; Kim, U.S.; et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog. Retin. Eye Res. 2021, 83, 100935. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain A J. Neurol. 2008, 131, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Leu, C.; Pogoda, H.M.; Arno, G.; Robson, A.G.; Nürnberg, G.; Altmüller, J.; Thiele, H.; Motameny, S.; Toliat, M.R.; et al. SSBP1 mutations in dominant optic atrophy with variable retinal degeneration. Ann. Neurol. 2019, 86, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A.; Caburet, S.; Birchler, J.A. Mechanisms of Mendelian dominance. Clin. Genet. 2018, 93, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Del Dotto, V.; Ullah, F.; Di Meo, I.; Magini, P.; Gusic, M.; Maresca, A.; Caporali, L.; Palombo, F.; Tagliavini, F.; Baugh, E.H.; et al. SSBP1 mutations cause mtDNA depletion underlying a complex optic atrophy disorder. J. Clin. Investig. 2020, 130, 108–125. [Google Scholar] [CrossRef]
- Meunier, I.; Bocquet, B.; Defoort-Dhellemmes, S.; Smirnov, V.; Arndt, C.; Picot, M.C.; Dollfus, H.; Charif, M.; Audo, I.; Huguet, H.; et al. Characterization of SSBP1-related optic atrophy and foveopathy. Sci. Rep. 2021, 11, 18703. [Google Scholar] [CrossRef] [PubMed]
- Dietze, J.; Blair, K.; Havens, S.J. Glaucoma. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2024. [Google Scholar]
- Tribble, J.R.; Hui, F.; Quintero, H.; Hajji, S.E.; Bell, K.; Di Polo, A.; Williams, P.A. Neuroprotection in glaucoma: Mechanisms beyond intraocular pressure lowering. Mol. Asp. Med. 2023, 92, 101193. [Google Scholar] [CrossRef]
- Ju, W.-K.; Perkins, G.A.; Kim, K.-Y.; Bastola, T.; Choi, W.-Y.; Choi, S.-H. Glaucomatous optic neuropathy: Mitochondrial dynamics, dysfunction and protection in retinal ganglion cells. Prog. Retin. Eye Res. 2022, 95, 101136. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M. Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef]
- Kondkar, A.A.; Azad, T.A.; Sultan, T.; Osman, E.A.; Almobarak, F.A.; Al-Obeidan, S.A. Elevated plasma level of 8-Hydroxy-2′-deoxyguanosine is associated with primary open-angle glaucoma. J. Ophthalmol. 2020, 2020, 6571413. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sheck, L.; Crowston, J.G.; Van Bergen, N.J.; O’Neill, E.C.; O’Hare, F.; Kong, Y.X.; Chrysostomou, V.; Vincent, A.L.; Trounce, I.A. Impaired complex-I-linked respiration and ATP synthesis in primary open-angle glaucoma patient lymphoblasts. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Leung, K.W.; Zhang, Y.H.; Duan, S.; Zhong, X.F.; Jiang, R.Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Geyman, L.S.; Suwan, Y.; Garg, R.; Field, M.G.; Krawitz, B.D.; Mo, S.; Pinhas, A.; Ritch, R.; Rosen, R.B. Noninvasive Detection of Mitochondrial Dysfunction in Ocular Hypertension and Primary Open-angle Glaucoma. J. Glaucoma 2018, 27, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dai, Y.; Zhang, R.; Shang, K.; Sun, X. Overexpression of Optic Atrophy Type 1 Protects Retinal Ganglion Cells and Upregulates Parkin Expression in Experimental Glaucoma. Front. Mol. Neurosci. 2018, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Luo, X.Y.; Zhao, Z.C.; Zheng, W.; Lv, H.Y.; Luo, W.M. Association between optic atrophy 1 polymorphisms and primary open angle glaucoma risk: Based on a meta-analysis. Eur. J. Ophthalmol. 2024, 34, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, N.J.; Crowston, J.G.; Craig, J.E.; Burdon, K.P.; Kearns, L.S.; Sharma, S.; Hewitt, A.W.; Mackey, D.A.; Trounce, I.A. Measurement of Systemic Mitochondrial Function in Advanced Primary Open-Angle Glaucoma and Leber Hereditary Optic Neuropathy. PLoS ONE 2015, 10, e0140919. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Soto, I.; Libby, R.T.; John, S.W. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp. Neurol. 2013, 246, 54–61. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Introductory Remarks. In Oxidative Stress; Sies, H., Ed.; Academic Press: London, UK, 1985; pp. 1–8. [Google Scholar]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants 2021, 10, 1538. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Vissers, M.C.; Winterbourn, C.C. Living with a killer: The effects of hypochlorous acid on mammalian cells. IUBMB Life 2000, 50, 259–266. [Google Scholar] [CrossRef]
- Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Meyerstein, D.; van Eldik, R. New mechanistic aspects of the Fenton reaction. Chemistry 2009, 15, 8303–8309. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Adhikary, A.; Dingfelder, M.; Dizdaroglu, M. Hydroxyl radical is a significant player in oxidative DNA damage in vivo. Chem. Soc. Rev. 2021, 50, 8355–8360. [Google Scholar] [CrossRef] [PubMed]
- Subramanian Vignesh, K.; Deepe, G.S., Jr. Metallothioneins: Emerging Modulators in Immunity and Infection. Int. J. Mol. Sci. 2017, 18, 2197. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.M.; Pagliarini, D.J. Coenzyme Q biochemistry and biosynthesis. Trends Biochem. Sci. 2023, 48, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Wu, L.; Sedgwick, A.C.; Sun, X.; Bull, S.D.; He, X.P.; James, T.D. Reaction-Based Fluorescent Probes for the Detection and Imaging of Reactive Oxygen, Nitrogen, and Sulfur Species. Acc. Chem. Res. 2019, 52, 2582–2597. [Google Scholar] [CrossRef]
- Corral-Domenge, C.; de la Villa, P.; Mansilla, A.; Germain, F. Tools and Biomarkers for the Study of Retinal Ganglion Cell Degeneration. Int. J. Mol. Sci. 2022, 23, 4287. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Stockwell, B.R. SnapShot: Ferroptosis. Cell 2020, 181, 1188. [Google Scholar] [CrossRef] [PubMed]
- Raederstorff, D.; Wyss, A.; Calder, P.C.; Weber, P.; Eggersdorfer, M. Vitamin E function and requirements in relation to PUFA. Br. J. Nutr. 2015, 114, 1113–1122. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Goldman, R.; Darrow, R.M.; Organisciak, D.T.; Kagan, V.E. Endogenous ascorbate regenerates vitamin E in the retina directly and in combination with exogenous dihydrolipoic acid. Curr. Eye Res. 1995, 14, 181–189. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Kozumbo, W.J. The phytoprotective agent sulforaphane prevents inflammatory degenerative diseases and age-related pathologies via Nrf2-mediated hormesis. Pharmacol. Res. 2021, 163, 105283. [Google Scholar] [CrossRef]
- Ren, F.; Ji, C.; Huang, Y.; Aniagu, S.; Jiang, Y.; Chen, T. AHR-mediated ROS production contributes to the cardiac developmental toxicity of PM2.5 in zebrafish embryos. Sci. Total Environ. 2020, 719, 135097. [Google Scholar] [CrossRef]
- Aranda, M.L.; González Fleitas, M.F.; De Laurentiis, A.; Keller Sarmiento, M.I.; Chianelli, M.; Sande, P.H.; Dorfman, D.; Rosenstein, R.E. Neuroprotective effect of melatonin in experimental optic neuritis in rats. J. Pineal Res. 2016, 60, 360–372. [Google Scholar] [CrossRef]
- VanderWall, K.B.; Vij, R.; Ohlemacher, S.K.; Sridhar, A.; Fligor, C.M.; Feder, E.M.; Edler, M.C.; Baucum, A.J., 2nd; Cummins, T.R.; Meyer, J.S. Astrocytes Regulate the Development and Maturation of Retinal Ganglion Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 12, 201–212. [Google Scholar] [CrossRef]
- Rodriguez-Muela, N.; Germain, F.; Mariño, G.; Fitze, P.S.; Boya, P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012, 19, 162–169. [Google Scholar] [CrossRef]
- Lee, D.; Shim, M.; Kim, K.-Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.; Ju, W. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, G.; Konani, A.M.; Miraftabi, A. Variability of Antioxidant Defense Enzymes in Primary Angle Closure Glaucoma Patients in Comparison with Healthy Subjects. Arak Med. Univ. J. 2015, 18, 41–49. [Google Scholar]
- Yabana, T.; Sato, K.; Shiga, Y.; Himori, N.; Omodaka, K.; Nakazawa, T. The relationship between glutathione levels in leukocytes and ocular clinical parameters in glaucoma. PLoS ONE 2019, 14, e0227078. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Lee, Y.; Kim, M.; Bhanvadia, S.; Kim, K.Y.; Ju, W.K. Effect of Ubiquinol on Glaucomatous Neurodegeneration and Oxidative Stress: Studies for Retinal Ganglion Cell Survival and/or Visual Function. Antioxidants 2020, 9, 952. [Google Scholar] [CrossRef] [PubMed]
- Farkas, R.H.; Chowers, I.; Hackam, A.S.; Kageyama, M.; Nickells, R.W.; Otteson, D.C.; Duh, E.J.; Wang, C.; Valenta, D.F.; Gunatilaka, T.L.; et al. Increased expression of iron-regulating genes in monkey and human glaucoma. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Wang, S.Y.; Yoo, C.; Singh, K.; Lin, S.C. Association between serum ferritin and glaucoma in the South Korean population. JAMA Ophthalmol. 2014, 132, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Zhu, Y.; Shi, Y.; Meng, X.; Dong, X.; Zhang, H.; Wang, X.; Du, M.; Yan, H. Inhibition of ferroptosis promotes retina ganglion cell survival in experimental optic neuropathies. Redox Biol. 2022, 58, 102541. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Ueno, M.; Ikeda, Y.; Oya, T.; Mori, K.; Kinoshita, S. Lipid Peroxidation in Aqueous Humor of Primary Open Angle Glaucoma and Pseudoexfoliative Glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3842. [Google Scholar]
- Nucci, C.; Di Pierro, D.; Varesi, C.; Ciuffoletti, E.; Russo, R.; Gentile, R.; Cedrone, C.; Pinazo Duran, M.D.; Coletta, M.; Mancino, R. Increased malondialdehyde concentration and reduced total antioxidant capacity in aqueous humor and blood samples from patients with glaucoma. Mol. Vis. 2013, 19, 1841–1846. [Google Scholar]
- Zanon-Moreno, V.; Marco-Ventura, P.; Lleo-Perez, A.; Pons-Vazquez, S.; Garcia-Medina, J.J.; Vinuesa-Silva, I.; Moreno-Nadal, M.A.; Pinazo-Duran, M.D. Oxidative stress in primary open-angle glaucoma. J. Glaucoma 2008, 17, 263–268. [Google Scholar] [CrossRef]
- Yuki, K.; Tsubota, K. Increased urinary 8-hydroxy-2′-deoxyguanosine (8-OHdG)/creatinine level is associated with the progression of normal-tension glaucoma. Curr. Eye Res. 2013, 38, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Han, J.S.; Park, C.K. Neuroprotective Effects of Nicotinamide (Vitamin B(3)) on Neurodegeneration in Diabetic Rat Retinas. Nutrients 2022, 14, 1162. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.l.n.; Yang, X.; Cai, J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Saccà, S.C.; Cutolo, C.A.; Rossi, T. Visual Defects and Ageing. Subcell. Biochem. 2019, 91, 393–434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxid. Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal 2015, 22, 686–729. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Kline, A.E.; Amoscato, A.; Samhan-Arias, A.K.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 2012, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.H.; Kim, K.Y.; Shim, M.S.; Choi, S.H.; Choi, S.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A.; Ju, W.K. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013, 4, e820. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Chen, T.; Xu, Z.; Huo, F.; Wei, Y.; Yang, X. Crocin protects retinal ganglion cells against H2O2-induced damage through the mitochondrial pathway and activation of NF-κB. Int. J. Mol. Med. 2016, 37, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hondur, G.; Tezel, G. Antioxidant Treatment Limits Neuroinflammation in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2344–2354. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Shim, M.S.; Kim, K.Y.; Bu, J.H.; Park, T.L.; Ahn, S.; Weinreb, R.N. Ubiquinol promotes retinal ganglion cell survival and blocks the apoptotic pathway in ischemic retinal degeneration. Biochem. Biophys. Res. Commun. 2018, 503, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.D.; Duan, J.; Samuelson, A.T.; Gaffrey, M.J.; Merrihew, G.E.; Egertson, J.D.; Wang, L.; Bammler, T.K.; Moore, R.J.; White, C.C.; et al. Improving mitochondrial function with SS-31 reverses age-related redox stress and improves exercise tolerance in aged mice. Free Radic. Biol. Med. 2019, 134, 268–281. [Google Scholar] [CrossRef]
- Genrikhs, E.E.; Stelmashook, E.V.; Alexandrova, O.P.; Novikova, S.V.; Voronkov, D.N.; Glibka, Y.A.; Skulachev, V.P.; Isaev, N.K. The single intravenous administration of mitochondria-targeted antioxidant SkQR1 after traumatic brain injury attenuates neurological deficit in rats. Brain Res. Bull. 2019, 148, 100–108. [Google Scholar] [CrossRef]
- Kang, L.; Liu, S.; Li, J.; Tian, Y.; Xue, Y.; Liu, X. The mitochondria-targeted anti-oxidant MitoQ protects against intervertebral disc degeneration by ameliorating mitochondrial dysfunction and redox imbalance. Cell Prolif. 2020, 53, e12779. [Google Scholar] [CrossRef]
- Amato, R.; Canovai, A.; Melecchi, A.; Maci, S.; Quintela, F.; Fonseca, B.A.; Cammalleri, M.; Dal Monte, M. Efficacy of a Spearmint (Mentha spicata L.) Extract as Nutritional Support in a Rat Model of Hypertensive Glaucoma. Transl. Vis. Sci. Technol. 2023, 12, 6. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Zhang, X.; Feng, Y.; Yuan, Y. Neuroprotective effects of idebenone on hydrogen peroxide-induced oxidative damage in retinal ganglion cells-5. Int. Ophthalmol. 2023, 43, 3831–3839. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Ma, X.H.; Zhong, Z.; Zhao, Y.; Chen, X.H.; Sun, X.F. Low-Dose Trans-Resveratrol Ameliorates Diabetes-Induced Retinal Ganglion Cell Degeneration via TyrRS/c-Jun Pathway. Investig. Ophthalmol. Vis. Sci. 2023, 64, 2. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.L.; Chen, S.L.; Xu, Y.; Yao, Y.; Liang, J.J.; Zhuang, X.; Hald, E.S.; Ng, T.K. Green tea extract enhances retinal ganglion cell survival and axonal regeneration in rats with optic nerve injury. J. Nutr. Biochem. 2023, 117, 109333. [Google Scholar] [CrossRef] [PubMed]
- García-Ayuso, D.; Di Pierdomenico, J.; Martínez-Vacas, A.; Vidal-Sanz, M.; Picaud, S.; Villegas-Pérez, M.P. Taurine: A promising nutraceutic in the prevention of retinal degeneration. Neural Regen. Res. 2024, 19, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Loygorri, J.I.; Benítez-Fernández, R.; Viedma-Poyatos, Á.; Zapata-Muñoz, J.; Villarejo-Zori, B.; Gómez-Sintes, R.; Boya, P. Mitophagy in the retina: Viewing mitochondrial homeostasis through a new lens. Prog. Retin. Eye Res. 2023, 96, 101205. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef] [PubMed]
- Lewis Luján, L.M.; McCarty, M.F.; Di Nicolantonio, J.J.; Gálvez Ruiz, J.C.; Rosas-Burgos, E.C.; Plascencia-Jatomea, M.; Iloki Assanga, S.B. Nutraceuticals/Drugs Promoting Mitophagy and Mitochondrial Biogenesis May Combat the Mitochondrial Dysfunction Driving Progression of Dry Age-Related Macular Degeneration. Nutrients 2022, 14, 1985. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Liu, P.F.; Chang, C.H.; Lin, Y.C.; Chen, Y.J.; Shu, C.W. The interplay of autophagy and oxidative stress in the pathogenesis and therapy of retinal degenerative diseases. Cell Biosci. 2022, 12, 1. [Google Scholar] [CrossRef]
- Um, J.H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, W.K.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Metformin Restores Parkin-Mediated Mitophagy, Suppressed by Cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Hass, D.T.; Barnstable, C.J. Cell Autonomous Neuroprotection by the Mitochondrial Uncoupling Protein 2 in a Mouse Model of Glaucoma. Front. Neurosci. 2019, 13, 201. [Google Scholar] [CrossRef]
- Sharma, L.K.; Tiwari, M.; Rai, N.K.; Bai, Y. Mitophagy activation repairs Leber’s hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum. Mol. Genet. 2019, 28, 422–433. [Google Scholar] [CrossRef]
- Liu, H.L.; Hu, F.Y.; Xu, P.; Wu, J.H. Regulation of mitophagy by metformin improves the structure and function of retinal ganglion cells following excitotoxicity-induced retinal injury. Exp. Eye Res. 2022, 217, 108979. [Google Scholar] [CrossRef]
- Zhuang, D.; Zhang, R.; Liu, H.; Dai, Y. A Small Natural Molecule S3 Protects Retinal Ganglion Cells and Promotes Parkin-Mediated Mitophagy against Excitotoxicity. Molecules 2022, 27, 4957. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Hu, X.; Zhang, J.; Wu, Y.; Zhao, N.; Ma, H.; He, H.; Lu, Q. Fucoxanthin protects retinal ganglion cells and promotes parkin-mediated mitophagy against glutamate excitotoxicity. Neuroreport 2023, 34, 385–394. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Gong, G.; Song, M.; Csordás, G.; Kelly, D.; Matkovich, S.; Dorn, G. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef]
- Patoli, D.; Mignotte, F.; Deckert, V.; Dusuel, A.; Dumont, A.; Rieu, A.; Jalil, A.; Van Dongen, K.; Bourgeois, T.; Gautier, T.; et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J. Clin. Investig. 2020, 130, 5858–5874. [Google Scholar] [CrossRef]
- Zaninello, M.; Palikaras, K.; Naon, D.; Iwata, K.; Herkenne, S.; Quintana-Cabrera, R.; Semenzato, M.; Grespi, F.; Ross-Cisneros, F.N.; Carelli, V.; et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 2020, 11, 4029. [Google Scholar] [CrossRef] [PubMed]
- Fauzi, Y.R.; Nakahata, S.; Chilmi, S.; Ichikawa, T.; Nueangphuet, P.; Yamaguchi, R.; Nakamura, T.; Shimoda, K.; Morishita, K. Antitumor effects of chloroquine/hydroxychloroquine mediated by inhibition of the NF-κB signaling pathway through abrogation of autophagic p47 degradation in adult T-cell leukemia/lymphoma cells. PLoS ONE 2021, 16, e0256320. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Satyanarayana, G.; Liu, T.; Wang, L.; Wang, J.; Cheng, J.; Itoh, K.; Sharma, A.; Bhutto, I.; et al. Mitophagy initiates retrograde mitochondrial-nuclear signaling to guide retinal pigment cell heterogeneity. Autophagy 2023, 19, 966–983. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.B.; Beeson, C.C.; Schnellmann, R.G. Development of Therapeutics That Induce Mitochondrial Biogenesis for the Treatment of Acute and Chronic Degenerative Diseases. J. Med. Chem. 2016, 59, 10411–10434. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.; Anbarasu, K.; Dutta, S.; Olivera Perez, L.J.; Huang, K.C.; Meyer, J.S.; Das, A. Enhanced mitochondrial biogenesis promotes neuroprotection in human pluripotent stem cell derived retinal ganglion cells. Commun. Biol. 2023, 6, 218. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Kumar, S.; Vijayan, M.; Bhatti, G.K.; Reddy, P.H. Therapeutic Strategies for Mitochondrial Dysfunction and Oxidative Stress in Age-Related Metabolic Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 146, 13–46. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.H.; Kreymerman, A.; Belle, K.; Ghiam, B.K.; Muscat, S.R.; Mahajan, V.B.; Enns, G.M.; Mercola, M.; Wood, E.H. The Present and Future of Mitochondrial-Based Therapeutics for Eye Disease. Transl. Vis. Sci. Technol. 2021, 10, 4. [Google Scholar] [CrossRef]
- Wilson, L.; Yang, Q.; Szustakowski, J.D.; Gullicksen, P.S.; Halse, R. Pyruvate induces mitochondrial biogenesis by a PGC-1 alpha-independent mechanism. Am. J. Physiol. Cell Physiol. 2007, 292, C1599–C1605. [Google Scholar] [CrossRef]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J. Alzheimer’s Dis. 2007, 11, 45–51. [Google Scholar] [CrossRef]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal 2013, 18, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Taub, P.R.; Ramirez-Sanchez, I.; Ciaraldi, T.P.; Perkins, G.; Murphy, A.N.; Naviaux, R.; Hogan, M.; Maisel, A.S.; Henry, R.R.; Ceballos, G.; et al. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: Effects of epicatechin rich cocoa. Clin. Transl. Sci. 2012, 5, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef] [PubMed]
- Indrieri, A.; Carrella, S.; Romano, A.; Spaziano, A.; Marrocco, E.; Fernandez-Vizarra, E.; Barbato, S.; Pizzo, M.; Ezhova, Y.; Golia, F.M.; et al. miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol. Med. 2019, 11, e8734. [Google Scholar] [CrossRef] [PubMed]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Zubricky, R.; Coughlin, L.; Jassim, A.H.; Inman, D.M. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol. Dis. 2020, 141, 104944. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.; Park, S.H.; Lee, D.; Kim, G.H.; Noh, J.E.; Lee, K.J.; Kim, G.J. Overexpression of pigment epithelium-derived factor in placenta-derived mesenchymal stem cells promotes mitochondrial biogenesis in retinal cells. Lab. Investig. 2021, 101, 51–69. [Google Scholar] [CrossRef]
- Ross, A.G.; McDougald, D.S.; Khan, R.S.; Duong, T.T.; Dine, K.E.; Aravand, P.; Bennett, J.; Chavali, V.R.M.; Shindler, K.S. Rescue of retinal ganglion cells in optic nerve injury using cell-selective AAV mediated delivery of SIRT1. Gene Ther. 2021, 28, 256–264. [Google Scholar] [CrossRef]
- Kuang, G.; Halimitabrizi, M.; Edziah, A.A.; Salowe, R.; O’Brien, J.M. The potential for mitochondrial therapeutics in the treatment of primary open-angle glaucoma: A review. Front. Physiol. 2023, 14, 1184060. [Google Scholar] [CrossRef]
- Subramaniam, M.D.; Iyer, M.; Nair, A.P.; Venkatesan, D.; Mathavan, S.; Eruppakotte, N.; Kizhakkillach, S.; Chandran, M.K.; Roy, A.; Gopalakrishnan, A.V.; et al. Oxidative stress and mitochondrial transfer: A new dimension towards ocular diseases. Genes Dis. 2022, 9, 610–637. [Google Scholar] [CrossRef] [PubMed]
- Muench, N.A.; Patel, S.J.; Maes, M.E.; Donahue, R.J.; Ikeda, A.; Nickells, R. The Influence of Mitochondrial Dynamics and Function on Retinal Ganglion Cell Susceptibility in Optic Nerve Disease. Cells 2021, 10, 1593. [Google Scholar] [CrossRef] [PubMed]
- Aharoni-Simon, M.; Ben-Yaakov, K.; Sharvit-Bader, M.; Raz, D.; Haim, Y.; Ghannam, W.; Porat, N.; Leiba, H.; Marcovich, A.; Eisenberg-Lerner, A.; et al. Oxidative stress facilitates exogenous mitochondria internalization and survival in retinal ganglion precursor-like cells. Sci. Rep. 2022, 12, 5122. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Tanabe, T.; Dezawa, M.; Ishikawa, H.; Yoshimura, N. Effects of bone marrow stromal cell injection in an experimental glaucoma model. Biochem. Biophys. Res. Commun. 2006, 344, 1071–1079. [Google Scholar] [CrossRef]
- Ma, J.; Guo, C.; Guo, C.; Sun, Y.; Liao, T.; Beattie, U.; López, F.J.; Chen, D.F.; Lashkari, K. Transplantation of Human Neural Progenitor Cells Expressing IGF-1 Enhances Retinal Ganglion Cell Survival. PLoS ONE 2015, 10, e0125695. [Google Scholar] [CrossRef]
- Jiang, D.; Xiong, G.; Feng, H.; Zhang, Z.; Chen, P.; Yan, B.; Chen, L.; Gandhervin, K.; Ma, C.; Li, C.; et al. Donation of mitochondria by iPSC-derived mesenchymal stem cells protects retinal ganglion cells against mitochondrial complex I defect-induced degeneration. Theranostics 2019, 9, 2395–2410. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Lee, J.Y.; Sanberg, P.R.; Napoli, E.; Borlongan, C.V. Eye Opener in Stroke. Stroke 2019, 50, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Nascimento-Dos-Santos, G.; de-Souza-Ferreira, E.; Lani, R.; Faria, C.C.; Araújo, V.G.; Teixeira-Pinheiro, L.C.; Vasconcelos, T.; Gonçalo, T.; Santiago, M.F.; Linden, R.; et al. Neuroprotection from optic nerve injury and modulation of oxidative metabolism by transplantation of active mitochondria to the retina. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165686. [Google Scholar] [CrossRef]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front. Neurol. 2021, 12, 651639. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef] [PubMed]
- Karanjia, R.; Sadun, A.A. Elamipretide Topical Ophthalmic Solution for the Treatment of Subjects with Leber Hereditary Optic Neuropathy: A Randomized Trial. Ophthalmology 2024, 131, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Subramanian, P.S.; Moster, M.L.; Wang, A.G.; Donahue, S.P.; Leroy, B.P.; Carelli, V.; Biousse, V.; Vignal-Clermont, C.; et al. Randomized trial of bilateral gene therapy injection for m.11778G>A MT-ND4 Leber optic neuropathy. Brain 2023, 146, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients With the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuroophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136, e230. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Carelli, V.; Newman, N.J.; Silva, M.J.; Linden, A.; Van Stavern, G.; Szaflik, J.P.; Banik, R.; Lubiński, W.; Pemp, B.; et al. Therapeutic benefit of idebenone in patients with Leber hereditary optic neuropathy: The LEROS nonrandomized controlled trial. Cell Rep. Med. 2024, 5, 101437. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Biousse, V.; Moster, M.L.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Girmens, J.F.; et al. Intravitreal Gene Therapy vs. Natural History in Patients With Leber Hereditary Optic Neuropathy Carrying the m.11778G>A ND4 Mutation: Systematic Review and Indirect Comparison. Front. Neurol. 2021, 12, 662838. [Google Scholar] [CrossRef]
- Vignal-Clermont, C.; Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Subramanian, P.S.; Wang, A.G.; Donahue, S.P.; Leroy, B.P.; et al. Safety of Lenadogene Nolparvovec Gene Therapy Over 5 Years in 189 Patients With Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2023, 249, 108–125. [Google Scholar] [CrossRef]
- VanderWall, K.B.; Lu, B.; Alfaro, J.S.; Allsop, A.R.; Carr, A.S.; Wang, S.; Meyer, J.S. Differential susceptibility of retinal ganglion cell subtypes in acute and chronic models of injury and disease. Sci. Rep. 2020, 10, 17359. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef] [PubMed]
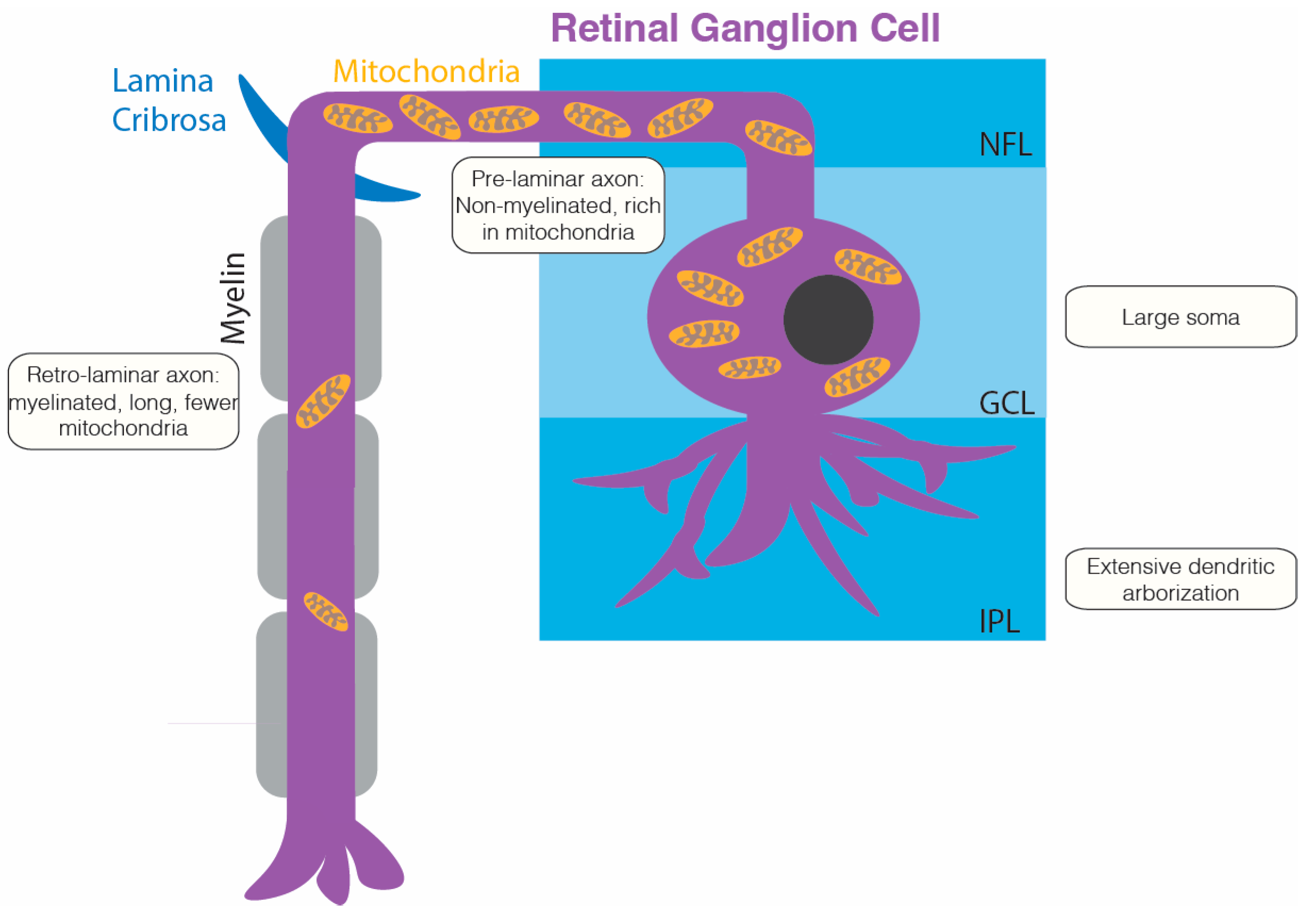
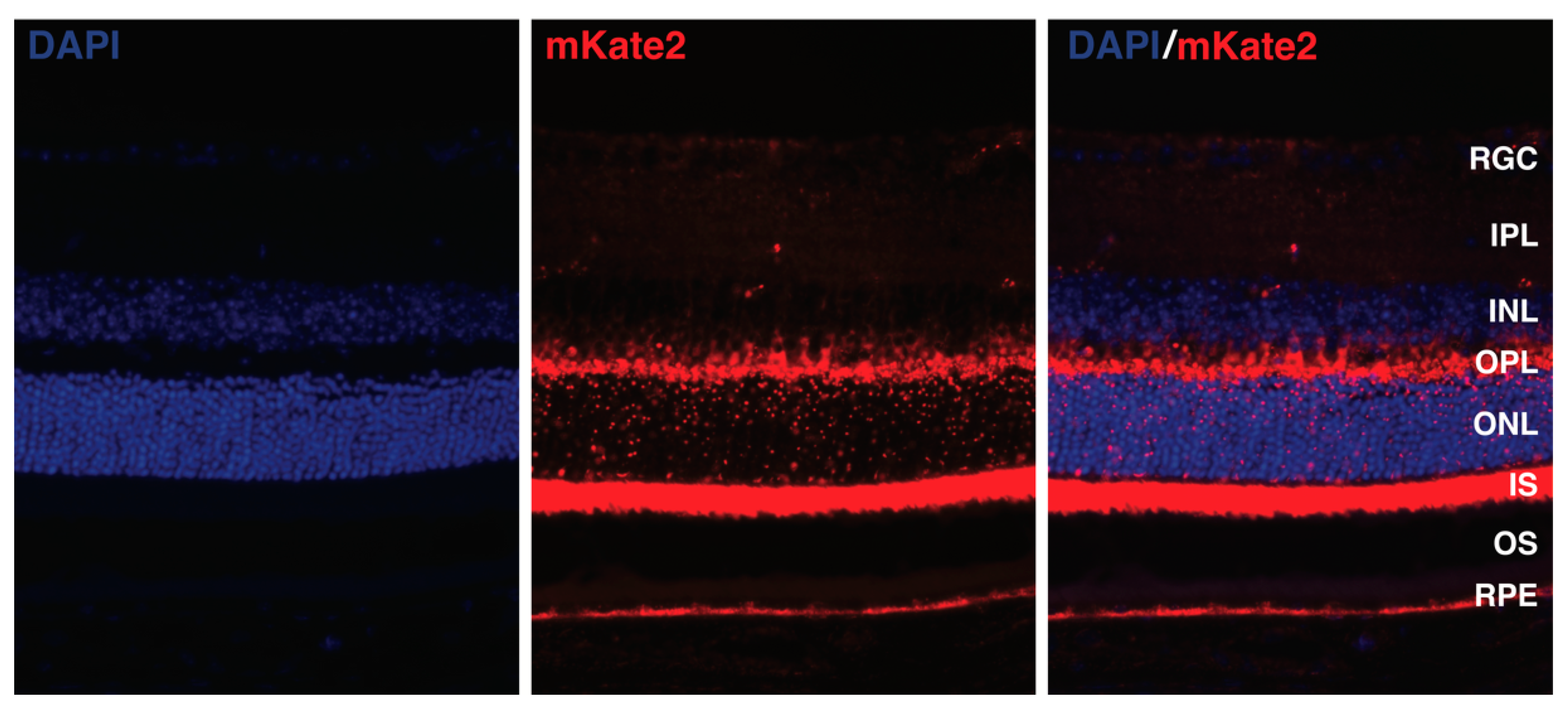
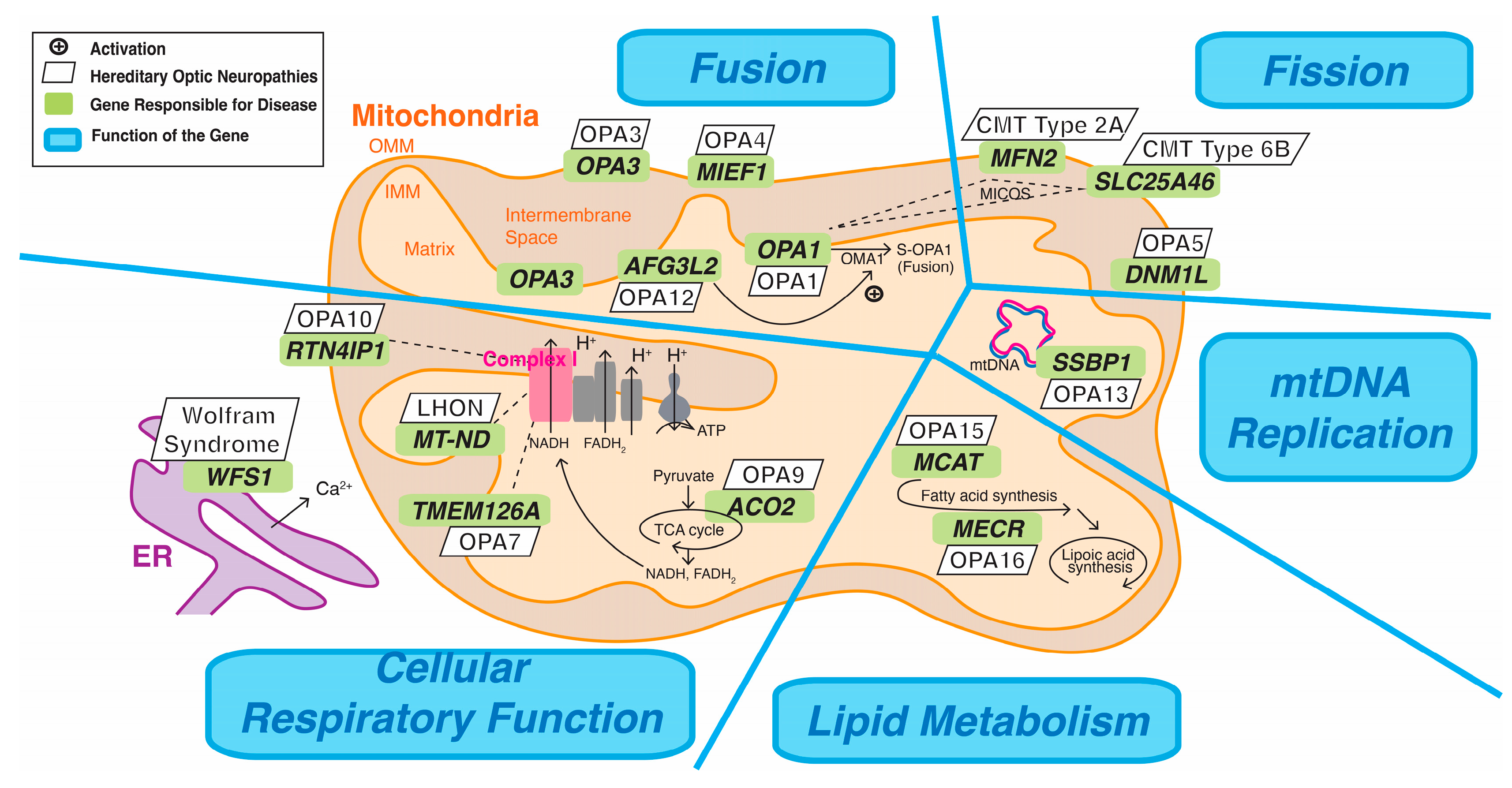
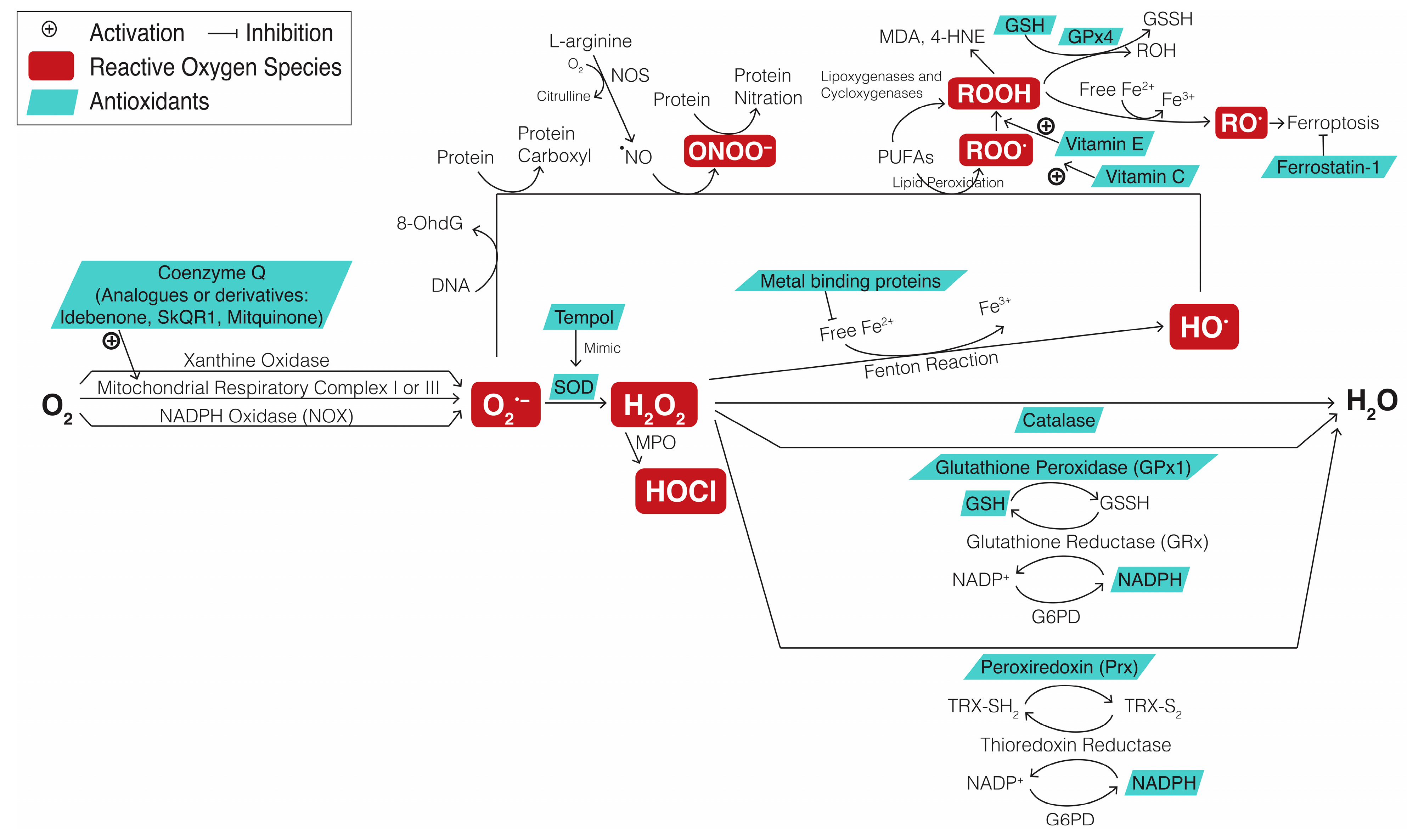

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Function | Gene; Location; MIM | Function and Location of the Encoded Protein | Hereditary Optic Neuropathies and Their Classifications: (1) Nonsyndromic, (2) Syndromic with Optic Neuropathy as a Necessary Manifestation, (3) Syndromic with Optic Neuropathy as a Possible Manifestation, (4) Typically Does Not Manifest Optic Neuropathy |
|---|---|---|---|
| Fusion or Fission | OPA1 (Mitochondrial dynamin-like GTPase); 3q29; MIM #605290 | Fusion; in the IMM | (1) Optic atrophy 1 (OPA1), AD (also known as Kjer’s type optic atrophy) (MIM #165500) (2) Dominant Optic atrophy plus syndrome (DOA-plus), AD (MIM #125250) (2)Behr syndrome, AR (MIM #210000) |
| OPA3 (Outer mitochondrial membrane lipid metabolism regulator); 19q13.32; MIM #606580 | Fusion; conflicting evidence regarding the whether OPA3 is embedded in the IMM or OMM | (2) Optic atrophy 3 (OPA3) with cataract, AD (MIM #165300) (2) 3-methylglutaconic aciduria, type III (Costeff syndrome), AR (MIM #258501) | |
| AFG3L2 (AFG3-like matrix AAA peptidase, subunit 2); 18p11.21; MIM #604581 | Fusion; in the IMM | (1) Optic atrophy 12 (OPA12), AD (MIM #618977) (4) Spastic ataxia 5, AR (MIM #614487) (4) Spinocerebellar ataxia 28 (SCA28), AD (MIM #610246) | |
| MIEF1 (Mitochondrial elongation factor 1); 22q13.1; MIM #615497 | Fusion; in the OMM | (1) Optic atrophy 14 (OPA14), AD (MIM #620550) | |
| DNM1L (Dynamin 1-like); 12p11.21; MIM #603850 | Encodes DRP1; Fission; in the OMM | (1) Optic atrophy 5 (OPA5), AD (MIM #610708) (3) Encephalopathy, lethal, due to defective mitochondrial peroxisomal fission 1, AD or AR (MIM #614388) | |
| MFN2 (Mitofusin 2); 1p36.22; MIM #608507 | Fission; in the OMM | (3) Charcot-Marie-Tooth Disease (CMT) type 2A2A, AD (MIM #609260) (3) Charcot-Marie-Tooth Disease (CMT) type 2A2B, AR (MIM #617087) (3) Hereditary motor and sensory neuropathy, type VIA, AD (MIM #601152) (4) Lipomatosis, multiple symmetric, with or without peripheral neuropathy, AR (MIM #151800) | |
| SLC25A46 (Solute carrier family 25, member 46); 5q22.1; MIM #610826 | Cristae maintenance and fission; in the OMM | (3) Charcot-Marie-Tooth Disease (CMT) type 6B, AR (MIM #616505) (3) Pontocerebellar hypoplasia, type 1E (MIM #619303) | |
| Cellular Respiratory Function | MT-ND (Mitochondrial NADH dehydrogenase subunit); MT-ND1 3460G>A, MT-ND4 11778G>A, and MT-ND6 14484T>C accouts for approximately 90% of LHON cases. | Subunits of the NADH dehydrogenase, or known as complex I of the ETC; in the IMM | (1) Leber’s hereditary optic neuropathy (LHON) (MIM #535000) |
| TMEM126A (Transmembrane protein 126A); 11q14.1; MIM #612988 | Assembly of complex I of the ETC; in the IMM | (2) Optic atrophy 7 (OPA7), AR (MIM #612989) | |
| ACO2 (Aconitase 2); 22q13.2; MIM #100850 | Catalyzes citrate to isocitrate in the citric acid cycle; in the mitochondrial matrix | (2) Optic atrophy 9 (OPA9), AR (MIM #616289) (4) Infantile cerebellar-retinal degeneration, AR (MIM #614559) | |
| RTN4IP1 (Reticulon 4-interacting protein 1); 6q21; MIM #610502 | Contains an oxidoreductase domain act on complex I of the ETC; in the OMM | (2) Optic atrophy 10 (OPA10) with or without ataxia, impaired intellectual development and seizures, AR (MIM #616732) | |
| WFS1 (Wolframin ER transmembrane glycoprotein); 4p16.1; MIM #606201 | Comprising mitochondria-associated endoplasmic reticulum membranes (MAM); in the endoplasmic reticulum membrane | (2) Wolfram syndrome type 1, or DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness), AR (MIM #222300) (2) Wolfram-like syndrome, AD (MIM #614296) | |
| Mitochondrial DNA Replication | SSBP1 (Single-stranded DNA-binding protein 1); 7q34; MIM #600439 | Mitochondrial single-strand binding protein, essential for mtDNA replication; in the mitochondrial matrix | (1) Optic atrophy 13 (OPA13) with retinal and foveal abnormalities, AD (MIM #165510) |
| Lipid Metabolism | MCAT (Malonyl CoA:ACP acyltransferase); 22q13.2; MIM #614479 | Catalyze the initial step of mtFAS; in the mitochondrial matrix | (1) Optic atrophy 15 (OPA15), AR (MIM #620583) |
| MECR (Mitochondrial trans-2-enoyl-CoA reductase); 1p35.3; MIM #608205 | Catalyze the last step of mtFAS; in the mitochondrial matrix | (1) Optic atrophy 16 (OPA16), AR (MIM #620629) (2) Dystonia, childhood-onset, with optic atrophy and basal ganglia abnormalities, AR: optic atrophy, involuntary movements, mainly dystonia (MIM #617282) |
| Mechanism of Oxidative Stress Biomarker | Oxidative Stress Biomarker | Disease | Results | Ref. |
|---|---|---|---|---|
| Direct detection of specific ROS | Nitric oxide synthase 2 | Optic neuritis | Melatonin increased nitric oxide synthase 2 level and improved RGC survival and visually evoked potential loss of optic neuritis mice | [155] |
| DCFDA | Optic neuropathy | Increased H2O2 detected by DCFDA in the RGCs of an optic neuropathy animal model | [156] | |
| DHE | Optic nerve injury | Increased O2•− detected by DHE in the RGCs of an optic nerve axotomy mice model | [157] | |
| Ocular hypertension | Increased O2•− detected by DHE in the RGCs of an ocular hypertension animal model | [116] | ||
| Decreased antioxidants | SOD, OXPHOS complex IV protein expression | Glaucoma | Coenzyme Q10 increased SOD level and OXPHOS complex IV protein expression and promoted RGC survival in glaucomatous DBA/2J mice | [158] |
| SOD, catalase, glutathione peroxidase, or glutathione reductase | Glaucoma | Decreased plasma levels of SOD, catalase, glutathione peroxidase, and glutathione reductase in patients with PAOG | [159] | |
| Glutathione | Glaucoma | Decreased glutathione levels in the leukocytes of patients with glaucoma | [160] | |
| OXPHOS complex II activity | Glaucoma | Ubiquinol, the reduced form of CoQ10, enhanced TFAM expression and OXPHOS complex II activity, as well as promoting RGC survival in glaucomatous DBA/2J mice | [161] | |
| Upregulation of iron-binding proteins | Transferrin, ceruloplasmin, or ferritin | Glaucoma | Transferrin, ceruloplasmin, and ferritin were upregulated in a monkey model of glaucoma and human postmortem glaucomatous eyes | [162] |
| Glaucoma | Higher serum ferritin levels were associated with greater odds of glaucoma | [163] | ||
| Glaucoma | The ferroptosis inhibitor ferrostatin-1 (Fer-1) significantly enhanced RGC survival and preserved retinal function in mouse models of optic nerve crush and microbead-induced glaucoma | [164] | ||
| Cellular response to oxidative stress | 4-HNE | Glaucoma | Increased lipid peroxidation indicated by 4-HNE levels in the aqueous humor of patients with glaucoma | [165] |
| MDA | Glaucoma | Increased lipid peroxidation indicated by MDA levels in patients with glaucoma | [166] | |
| Glaucoma | Increased lipid peroxidation indicated by MDA-TBARS tests in the aqueous humor of patients with PAOG | [167] | ||
| 8-OhdG or 8-oxodG | Glaucoma | Increased oxidative DNA damage indicated by plasma 8-OHdG levels in patients with POAG | [117] | |
| Glaucoma | Increased oxidative DNA damage indicated by urinary 8-OHdG/creatinine associated with glaucomatous visual field progression | [168] | ||
| Diabetic retinopathy | Nicotinamide (Vitamin B3) decreased 8-OHdG level and apoptotic RGC death in a diabetic rat model | [169] | ||
| Protein carbonylation | Glaucoma | Increased protein oxidation indicated by protein carbonyl immunoreactivity in the retina in a chronic pressure–induced rat model of glaucoma | [170] | |
| Nrf2 expression | RGC death | RGC survival in Nrf2 knockout mice significantly lower than that in wild-type mice | [171] |
| Antioxidant | Study Model | Results | Ref. |
|---|---|---|---|
| MitoVit E | Rat | Protected isolated rat liver mitochondria from oxidative damage | [177] |
| Glutathione | Rat | Delayed mitochondrial depolarization and reduced ROS generation in striatal neurons | [178] |
| XJB-5-131 | Rat | Prevented cardiolipin oxidation in the brain and reduced neuronal death signals | [179] |
| CoQ10 | Rat | Increased mitochondria number/density, preserved structure, and reduced ROS generation | [180] |
| Crocin | Human RGCs | Reduced ROS production, boosted RGC viability, and protected against apoptosis | [181] |
| Melatonin | Mice | Reduced loss in RGCs and caused less oxidative damage | [155] |
| Tempol | Mice | Limited oxidative stress and reduced neuroinflammation | [182] |
| CoQ10 | Mice | Promoted RGC survival and blocked the apoptotic pathway in the ischemic mouse retina | [183] |
| SS-31 (elamipretide) | Mice | Restored redox homeostasis and improved mitochondrial quality, thereby increasing exercise tolerance | [184] |
| SkQR1 | Rat | Reduced traumatic brain damage–related disorders of limb functions and increased survivability of neurons | [185] |
| MitoQ (mitoquinone) | Human NP cells | Achieved equilibrium in mitochondrial dynamics and eliminated compromised mitochondria | [186] |
| Ferrostatin-1 (Fer-1) | Rat | Significantly promoted RGC survival and preserved retinal function in ONC and microbead-induced glaucoma mouse models | [164] |
| Spearmint | Rat | Improved RGC-related ERG responses, cell density, neurotrophins, oxidative stress, and inflammation markers | [187] |
| Idebenone | Mice | Protected against H2O2-induced oxidative damage by reducing mitochondrial damage and autophagic activity | [188] |
| Trans-Resveratrol | Mice | Relieved electrophysiological injury of the retinas and inhibited RGC apoptosis | [189] |
| Green Tea Extract | Rat | Reduced RGC death and led to significantly higher RGC numbers and regenerated axons | [190] |
| Taurine | Rat | Enhanced RGC survival and protected RGCs from NMDA excitotoxicity | [191] |
| Study Model | Intervention to Promote Mitophagy | Results | Ref. |
|---|---|---|---|
| Mice (Optic nerve injury) | Rapamycin | Increased RGC survival | [157] |
| Rat Neurons | Mitochondrial damage | Physiological mitochondrial damage in hippocampal axons recruits Parkin for mitophagy, a process absent in PINK1-mutated neurons, demonstrating the role of PINK1/Parkin pathway in local mitophagy | [197] |
| Mice (Obesity) | Metformin | Reduced ER stress and p53 levels to restore Parkin-mediated mitophagy in hepatocytes | [198] |
| Rat (Glaucoma) | Overexpression Opa1 with AAV2-OPA1 | Upregulated Parkin, leading to a greater mitochondrial surface area, better mitochondria quality, and increased fusion morphology, and increased RGC survival | [121] |
| Mice (Glaucoma) | Mitochondrial Uncoupling protein 2 (Ucp2) knock-out | Decreased ROS-mediated protein modifications and reduced RGC death | [199] |
| Cybrids of Human (LHON) | Rapamycin | Repaired mitochondrial defects and improved overall cell survival | [200] |
| Rat (NMDA-induced retinal excitotoxicity) | Metformin | Reduced RGC death and improved structure/function of RGCs | [201] |
| Rat (NMDA-induced retinal excitotoxicity) | Small molecule S3 | Promoted RGC survival and improved mitochondrial quality | [202] |
| Rat (Glutamate excitotoxicity) | Fucoxanthin | Increased mitochondrial membrane potential and reduced cytotoxicity and apoptosis | [203] |
| Study Model | Intervention to Inhibit Mitophagy | Results | Ref. |
|---|---|---|---|
| C. elegans | RNAi against PINK1 | Revealed mitochondrial biosynthetic pathway to be affected by retrograde signals through DCT-1 | [204] |
| Mice | Mfn2 mutation | Interruption of PINK1-Mfn2-Parkin mediated mitophagy in cardiomyocytes led to lethal cardiomyopathy and mitochondrial maturation arrest at the fetal stage | [205] |
| Mice | Sesn2 mutation | Interruption of SESN2-induced mitophagy in macrophages led to increased mortality in sepsis models | [206] |
| Mice (Polymicrobial infection) | Lipopolysaccharide and interferon-γ | Interruption of PINK1-dependent mitophagy promoted microphage activation and increased cellular survival | [207] |
| Mice (Opa1 mutation) | Atg7 mutation | Normalized mitochondrial content and corrected visual loss | [208] |
| Mice (Human adult T-cell leukemia/lymphoma xenograft) | Chloroquine | Suppressed tumor growth and improved overall survival rate | [209] |
| Mice | Pink1 mutation | Interruption of mitophagy and mitochondrial function in retinal pigment cells promoted death-resistant epithelial-mesenchymal transition | [210] |
| Study Model | Intervention | Results | Ref. |
|---|---|---|---|
| Mice | Pyruvate | Induced mitochondrial biogenesis through the pyruvate energy-sensing pathway, regulating oxidative capacity | [215] |
| Mice | Rosiglitazone (PPAR-γ agonist) | Improved glucose utilization, cellular function, and cognition | [216] |
| Human cells (Neuroblastoma) | Rosiglitazone (PPAR-γ agonist) | Increased levels of mtDNA and mitochondrial activators while maintaining constant membrane potential | [217] |
| Cybrids (LHON) | 17β-estradiol | Increased number of mtDNA and reduced levels of ROS | [95] |
| Mice | AICAR (AMPK activator) | Improved biochemical phenotype and improved motor impairment | [218] |
| Mice | Glutathione (GSH) | Protected against brain damage and reduced loss of body weight | [219] |
| Human cells (Skeletal muscle of patients with type 2 diabetes and heart failure) | Epicatechin-rich cocoa | Increased the protein content and activity of mediators of mitobiogenesis and the abundance of cristae without affecting mitochondrial volume density | [220] |
| Human cells (Down syndrome) | Epigallocatechin-3-gallate | Promoted mitobiogenesis in Down syndrome cells and increased protein and mtDNA content | [221] |
| Mice | Nicotinamide riboside (NAD+ precursor) | Improved motor performance and exercise intolerance | [222] |
| Rat | Resveratrol | Promoted mitochondrial mass and DNA copy number while improving mitochondrial homeostasis and neural function | [223] |
| Mice and fish | miR-181a/b (microRNAs) | Protected cells from mitochondrial damage and reduced RGC death | [224] |
| Mice | MCT2 overexpression by viral vector | Increased RGC density and axon number, reduced energy imbalance, and increased mitochondrial function | [225] |
| Rat | PEDF overexpression by viral vector | Improved mitochondrial activities and induced OS-damaged cell regeneration | [226] |
| Mice | SIRT1 activator (NAD+) by electroporation gene delivery system | Delayed loss of RGC function and reduced RGC death from optic nerve injury | [227] |
| Mice | BX795 (Tank binding kinase 1 inhibitor) | Restored energy homeostasis and mitigated mitochondrial swelling to reduce mitochondrial damage caused by glaucoma | [212] |
| Study Model | Results | Ref. | |
|---|---|---|---|
| In Vivo | Rat (Glaucoma) | Transplantation of bone marrow stromal cells enhanced RGC survival | [232] |
| In Vivo | Mice (Glaucoma) | Transplantation of human neuronal progenitor cells transfected with a vector that expresses IGF-1 increased RGC survival and increased neurite extension | [233] |
| In Vivo | LHON patient-derived iPSCs transplanted into immuno-deficient rats for teratoma formation assay | Cybrid technique to replace mutant mtDNA with wild-type mtDNA in iPSC-derived RGCs lowers superoxide levels and RGC apoptosis | [234] |
| In Vivo | Rat (Middle cerebral artery occlusion, modeling stroke) | Transplantation of MSCs induced mitochondrial repair and enhanced RGC survival | [235] |
| In Vivo | Rat (Optic nerve crush) | Transplantation of liver-isolated mitochondria improved respiratory capacity and increased RGC survival and regeneration capacity | [236] |
| In Vitro | RGC precursor-like cells | Exposure to moderate oxidative stress prior to MitoPlant enhanced mitochondrial uptake and prolonged survival of mitochondria by more than three-fold | [231] |
| ClinicalTrials.gov ID | Study Design | Results | Comments |
|---|---|---|---|
| NCT01793119 (RHODOS) | Randomized, double-blind. 85 patients. Intervention: idebenone (900 mg daily for 24 weeks). Control: placebo. Follow-up: 24 weeks. | Best recovery in visual acuity not statistically significant, but BCVA for patients with discordant BCVA at baseline showed significant improvement. No severe AEs reported [242]. A follow-up analysis of 70% of patients showed continued BCVA improvement even after discontinuing idebenone [243]. | Demonstrated that idebenone can benefit some patients with LHON, particularly when treatment begins early. However, the modest effect highlights the need for more effective therapies. |
| NCT02774005 (LEROS) | Open-label, Phase IV. 199 patients with vision loss of 1–5 years since onset. Intervention: idebenone (900 mg daily for 24 weeks). Control: untreated. Follow-up: 2 years. | Clinically relevant stabilization in visual acuity was maintained in treated eyes over 2 years. Confirms the long-term efficacy of idebenone in the subacute and chronic phases. No severe AEs reported [244]. | The results support the use of idebenone in slowing disease progression. However, the open-label design limits definitive conclusions. |
| NCT02693119 | Randomized followed by OLE, phase II. Twelve patients (m.11778G>A) with decreased vision of 1–10 years since onset. Interventions: Randomized 52 weeks (1% topical elamipretide in both eyes, left, or right eye), followed OLE up to 108 weeks (both eyes). Follow-up: 4 weeks. | No significant changes in BCVA in elamipretide-treated eyes compared to vehicle eyes. No severe AEs reported [239]. | Assessments of visual function beyond BCVA during the OLE and post hoc analyses merit further investigation. |
| NCT02652767 (RESCUE) | Open-label, Phase III. 39 patients (m.11778G>A) within 6 months of vision loss onset. Intervention: rAAV2/2-ND4, unilateral, single treatment. Control: sham injection. Follow-up: 96 weeks. | Sustained BCVA improvement was observed in both eyes over the 96-week follow-up period. No severe AEs reported [245]. | Early intervention with gene therapy shows promise in halting or reversing vision loss in LHON. |
| NCT02652780 (REVERSE) | Open-label, Phase III. 36 patients (m.11778G>A) with 6 months to 1 year of vision loss onset. Intervention: rAAV2/2-ND4, unilateral, single treatment. Control: sham injection. Follow-up: 96 weeks. | Sustained BCVA improvement was observed in both eyes over the 96-week follow-up period. No severe AEs reported [238]. | Bilateral improvement following unilateral injection of lenadogene nolparvovec was attributed to viral vector DNA transfer from the treated to the untreated eye, hypothesized to occur via the optic chiasm [238]. |
| NCT03406104 (RESTORE) | Follow-up study for patients from REVERSE and RESCUE. Long-term (52 months). Intervention: rAAV2/2-ND4, unilateral, single treatment. Control: sham injection. | The treatment effect at last observation remained significant after adjusting for age and follow-up duration [246]. | Long-term data solidify the efficacy and safety of gene therapy. |
| NCT03293524 (REFLECT) | Randomized, double-blind, Phase III. A total of 98 patients (m.11778G>A). Intervention: rAAV2/2-ND4, bilateral, single treatment. Control: unilateral rAAV2/2-ND4 with placebo in second eye. Follow-up: 5 years. | Bilaterally treated patients showed better improvement in BCVA and higher responder rates compared to those treated unilaterally. No severe systemic AEs; intraocular inflammation was common but mild [240,247]. | Better outcomes of bilateral treatment over unilateral treatment indicate a potential dose-response effect. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-H.; Kang, E.Y.-C.; Lin, P.-H.; Yu, B.B.-C.; Wang, J.H.-H.; Chen, V.; Wang, N.-K. Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress. Int. J. Mol. Sci. 2024, 25, 8626. https://doi.org/10.3390/ijms25168626
Yang T-H, Kang EY-C, Lin P-H, Yu BB-C, Wang JH-H, Chen V, Wang N-K. Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress. International Journal of Molecular Sciences. 2024; 25(16):8626. https://doi.org/10.3390/ijms25168626
Chicago/Turabian StyleYang, Tsai-Hsuan, Eugene Yu-Chuan Kang, Pei-Hsuan Lin, Benjamin Ben-Chi Yu, Jason Hung-Hsuan Wang, Vincent Chen, and Nan-Kai Wang. 2024. "Mitochondria in Retinal Ganglion Cells: Unraveling the Metabolic Nexus and Oxidative Stress" International Journal of Molecular Sciences 25, no. 16: 8626. https://doi.org/10.3390/ijms25168626