Abstract
The effects of enhanced late INa, a persistent component of the Na+ channel current, on the intracellular ion dynamics and the automaticity of the pulmonary vein cardiomyocytes were studied with fluorescent microscopy. Anemonia viridis toxin II (ATX- II), an enhancer of late INa, caused increases in the basal Na+ and Ca2+ concentrations, increases in the number of Ca2+ sparks and Ca2+ waves, and the generation of repetitive Ca2+ transients. These phenomena were inhibited by eleclazine, a blocker of the late INa; SEA0400, an inhibitor of the Na+/Ca2+ exchanger (NCX); H89, a protein kinase A (PKA) inhibitor; and KN-93, a Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor. These results suggest that enhancement of late INa in the pulmonary vein cardiomyocytes causes disturbance of the intracellular ion environment through activation of the NCX and Ca2+-dependent enzymes. Such mechanisms are probably involved in the ectopic electrical activity of the pulmonary vein myocardium.
1. Introduction
The present study was undertaken to clarify the intracellular mechanisms for the induction of automaticity in the pulmonary vein myocardium by the persistent component of the voltage-dependent Na+ channel current. The pulmonary vein is the blood vessel for the blood flow from the lung to the heart. Spontaneous pulsation of the pulmonary vein independent of the cardiac pacemaker has been repeatedly observed in various animal species [1,2,3]. This is due to a myocardial layer in the pulmonary vein wall connected to the left atrial myocardium capable of generating spontaneous or triggered action potentials. Since the clinical report that the automatic activity of the pulmonary vein myocardium plays a central role in the generation and maintenance of atrial fibrillation [4,5], the automaticity of the pulmonary vein myocardium has received attention as a therapeutic target and has been studied in various animal species [1,2,6,7]. The cardiomyocytes of the pulmonary vein myocardium have a smaller inward-rectifying K+ channel current density and less repolarizing power than the atrial myocardium [8,9,10], which allows otherwise masked depolarizing currents to induce a gradual depolarization of the membrane. The depolarizing membrane currents contributing to the diastolic depolarization of the pulmonary vein myocardium include the Na+/Ca2+ exchanger current [11,12], hyperpolarization-activated current [7,10], and the voltage-dependent Na+ channel current [13,14], which is the main subject of the present study.
The voltage-dependent Na+ channel current (INa) is composed of two components. The transient component, which is referred to as peak INa, is activated by rapid depolarizations from the resting membrane potential range and rapidly inactivates within a few milliseconds. Peak INa has a large current density and is responsible for the rapid upstroke and conduction of the action potential in the working myocardium [15]. After the inactivation of the peak INa, a small fraction of the INa flows persistently [16]; such current is referred to as late INa, persistent INa, or sustained INa [17]. Such a persistent Na+ current is also observed during a slow depolarization in the negative voltage range and has been suggested to contribute to abnormal automaticity underlying cardiac arrhythmia including atrial fibrillation [18,19]. Although the amplitude of late INa is small in the normal myocardium, it may be enhanced under various acquired and congenital conditions related to myocardial automaticity such as heart failure, ischemia, and arrhythmia including atrial fibrillation [20,21,22]. In the pulmonary vein myocardium, whose resting or maximum diastolic potential is about −70 mV, less negative than the −80 to −90 mV of the working myocardium [8,9], the late INa can flow depending on the situation. It was previously reported in the pulmonary vein myocardium that anemonia viridis toxin II (ATX-II), an enhancer of late INa, increased the pacemaker depolarization slope, as well as the automatic firing frequency [13,23]. Agents with inhibitory effects on late INa, such as GS-458967, reduced the pacemaker depolarization slope and automatic activity in the pulmonary vein myocardium [13]. Thus, blockade of late INa appears to be an effective pharmacological strategy for the treatment of atrial fibrillation.
While late INa can directly contribute to depolarization as a depolarizing current, it may also cause disturbances in intracellular ion homeostasis and consequently lead to arrhythmic activity. An increase in intracellular Na+ concentration through inhibition of the Na+/K+-ATPase by ouabain induced automatic electrical activity in the pulmonary vein myocardium, which was accompanied by an increase in the number of Ca2+ sparks and Ca2+ waves; these were both inhibited by pretreatment with a selective NCX inhibitor, SEA0400 [11]. Thus, a probable explanation for the induction of electrical activity by an increase in intracellular Na+ concentration in the pulmonary vein myocardium is the induction of Ca2+ overload through activation of Ca2+ influx through NCX [11]. Ca2+ overload is reported to cause hyperactivation of Ca2+-dependent enzymes such as Ca2+/calmodulin -dependent protein kinase II (CaMKII) and protein kinase A (PKA), and leads to arrhythmias in ventricular muscle [17,24,25,26]. If an enhancement of the late INa can induce automatic activity in the pulmonary vein myocardium through similar mechanisms, their inhibitors would be promising as drugs for the treatment of atrial fibrillation caused by pulmonary vein automaticity.
In the present study, fluorescent ion measurements and pharmacological studies were conducted with isolated pulmonary vein cardiomyocytes to clarify the following points. Firstly, does enhancement of the late INa in isolated pulmonary vein myocytes induce automatic electrical activity? Secondly, does the enhancement of the late INa cause intracellular Ca2+ overload through NCX function? Finally, are Ca2+-dependent enzymes involved in the induction of automatic activity? Given the positive results obtained, their implication for drug selection and development was discussed.
2. Results
2.1. The Spontaneous and Evoked Ca2+ Transients of Isolated Pulmonary Vein Cardiomyocytes
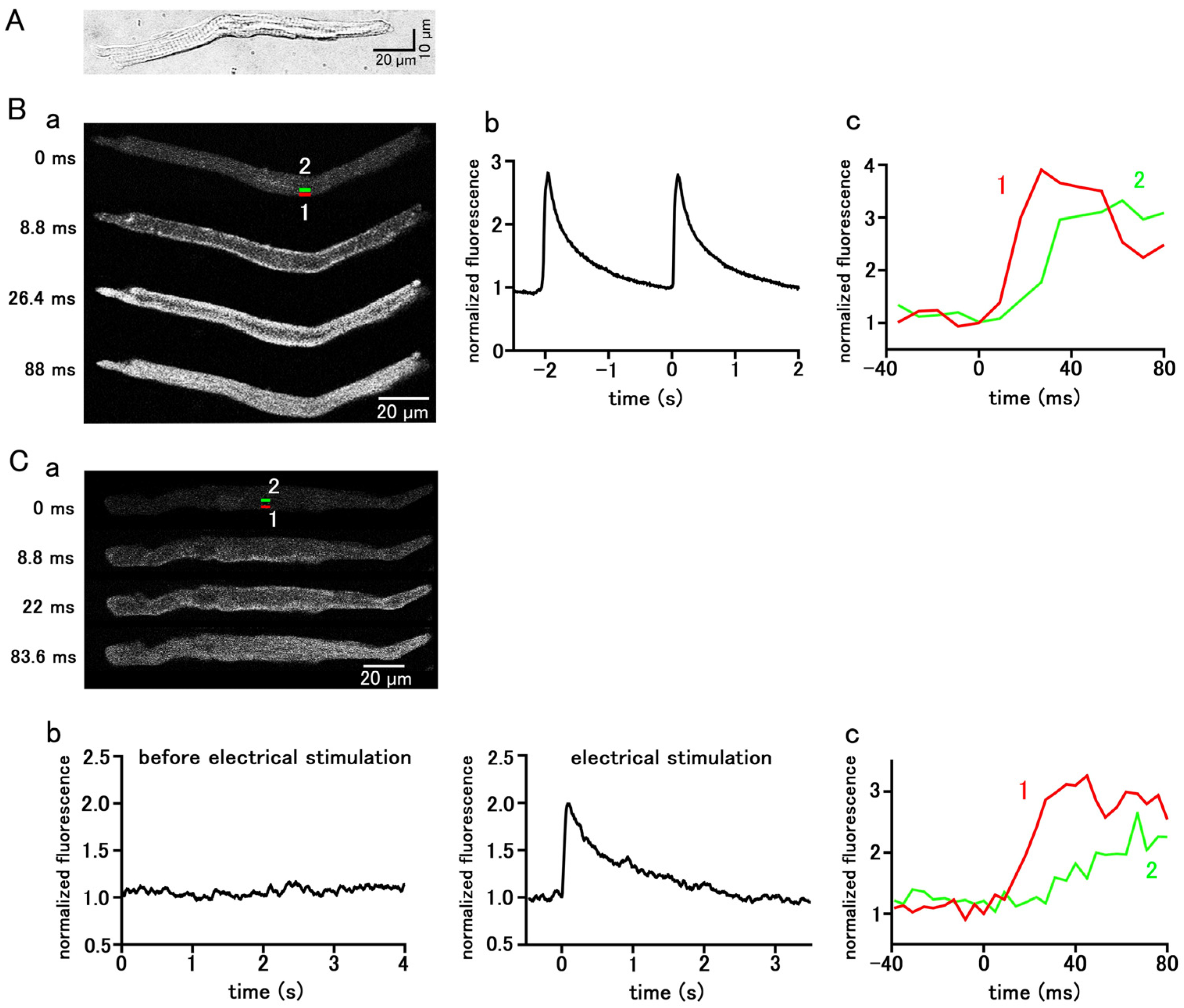
Pulmonary vein cardiomyocytes isolated from guinea pigs were spindle-shaped and had transverse striations. (Figure 1A). They were loaded with the Ca2+-sensitive fluorescent probe, Fluo-4, and observed with a confocal microscope. Among the 787 isolated guinea pig pulmonary vein cardiomyocytes examined, 220 cells showed spontaneous Ca2+ transients, which is a rapid rise in the Ca2+ concentration in the entire subsarcolemmal cytoplasm (Figure 1B); the average firing frequency of the Ca2+ transient was 1.85 ± 0.13 times in 5 s. During the early phase of the Ca2+ transient, fluorescence intensity increased from the subsarcolemmal region and then spread to the cell interior (Figure 1B). In the quiescent pulmonary vein cardiomyocytes, action potentials were elicited by field electrical stimulation, and the resulting Ca2+ transients were observed; the rise in Ca2+ concentration started from the subsarcolemmal region and spread to the central region of the cell (Figure 1C).
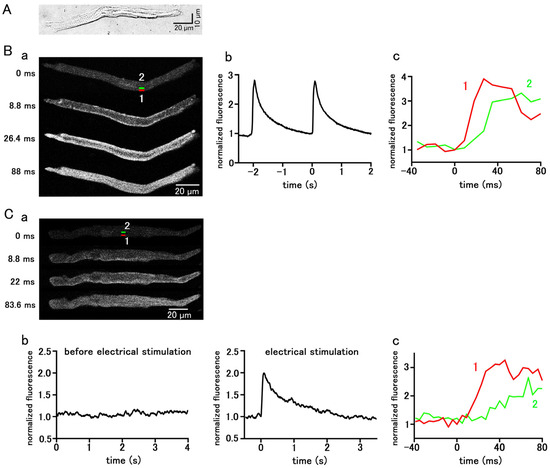
Figure 1.
Spontaneous and stimulation-evoked Ca2+ transients in pulmonary vein myocytes. (A) A typical differential interference contrast (DIC) image. (B,C) Spontaneous (B) and electrical-stimulation-evoked (C) Ca2+ transients. Typical x−y images of a cell loaded with Fluo−4/AM (B(a),C(a)). Time course of fluorescence intensity of the whole cell (B(b),C(b)) and quantified in rectangular (1 × 4 µm) regions located at 0–1 µm (1 red) and 4–5 µm (2 green) from the sarcolemma (B(c),C(c)), as shown in the top panel in (B(a),C(a)).
2.2. Effect of Enhanced Late INa on Automatic Activity and Intracellular Ca2+ Dynamics
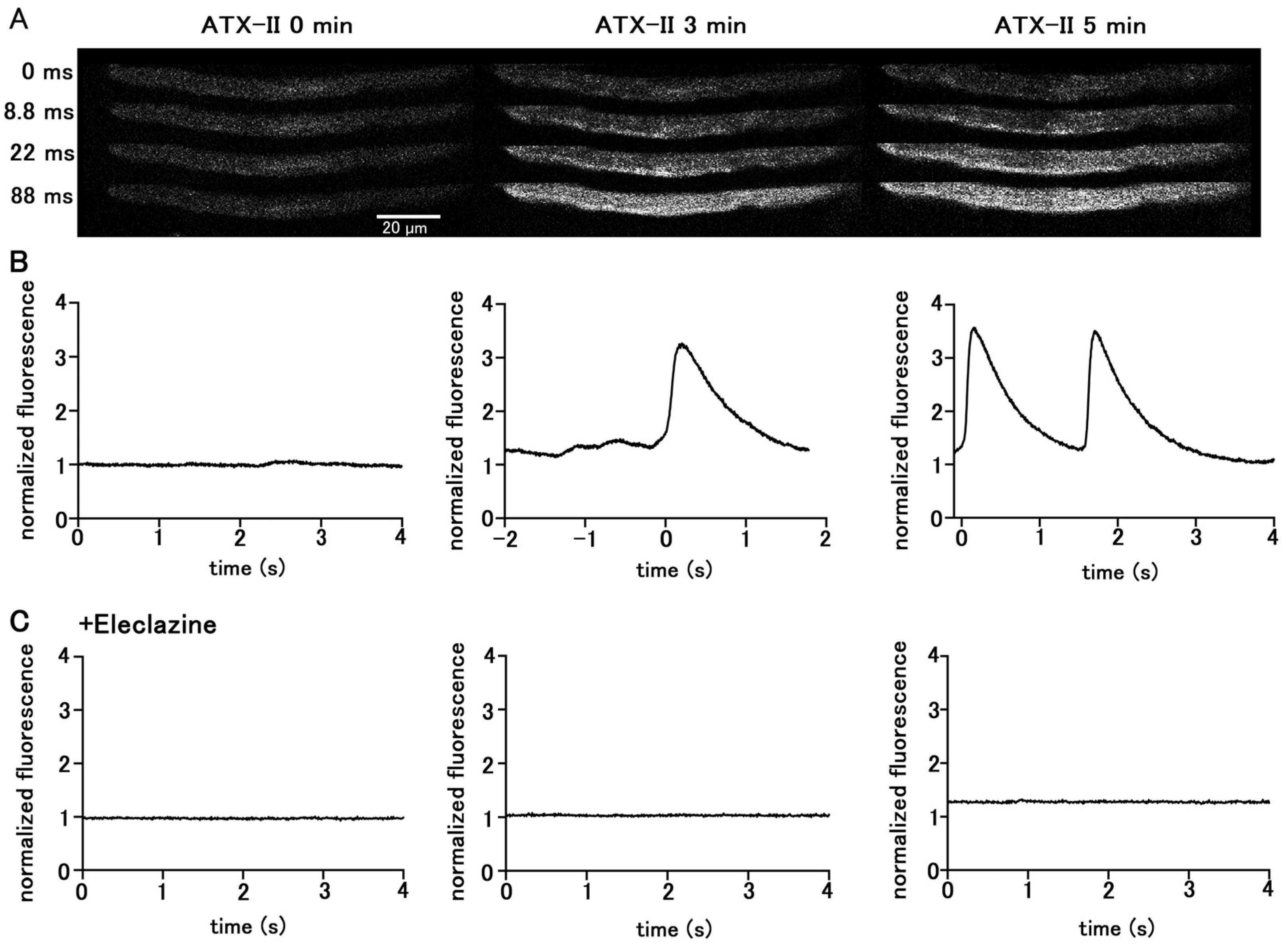
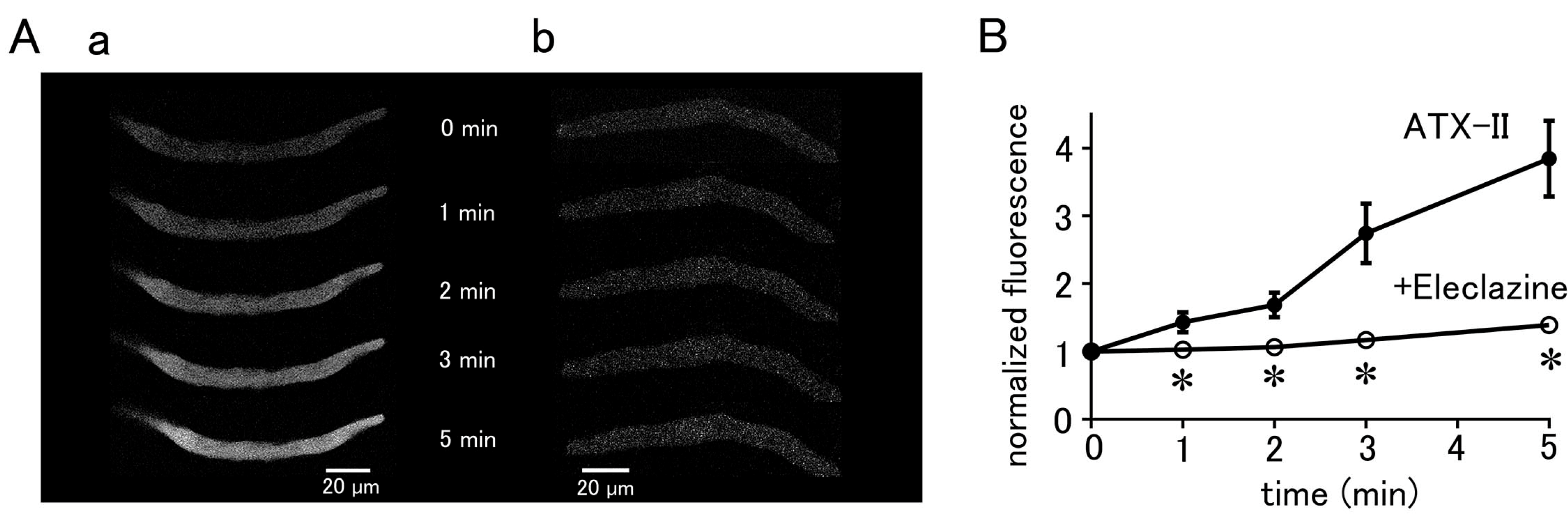
Treatment of quiescent pulmonary vein myocytes with 100 nM ATX-II induced automatic Ca2+ transients. Among the 40 isolated pulmonary vein cardiomyocytes examined, automatic Ca2+ transients were induced in 18 cells. The firing frequency of Ca2+ transients increased in a time-dependent manner (Figure 2); the average frequency was 1.25 ± 0.25 times in 5 s at 3 min and 2.25 ± 0.25 times in 5 s at 5 min. Regarding the automatic Ca2+ transient induced by ATX-II, the rise in the Ca2+ concentration first occurred at the subsarcolemmal region and spread to the central region of the cell, similarly to electrical-stimulation-evoked Ca2+ transients. Inhibition of late INa by pretreatment with 10 µM eleclazine significantly reduced the incidence of Ca2+ transients induced by ATX-II (Figure 2). Such induction of automatic activity by ATX-II was less frequently observed in atrial cardiomyocytes. Among the 56 isolated atrial cardiomyocytes examined, automatic Ca2+ transients were induced only in seven cells.
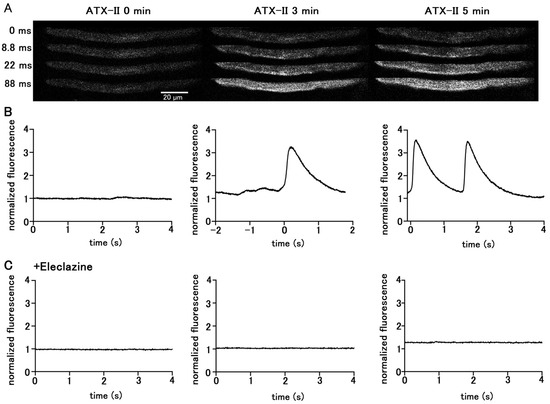
Figure 2.
Induction of Ca2+ transients by ATX-II in quiescent pulmonary vein cardiomyocytes. Typical x−y images (A) and time course (B) of a cell loaded with Fluo−4 before ATX-II treatment and at 3 and 5 min after treatment with 100 nM ATX-II. Pretreatment of eleclazine (10 µM) inhibited the induction of automatic activity (C).
The basal Ca2+ fluorescence intensity of the cells was increased by ATX-II in a time-dependent manner, regardless of the induction of automatic Ca2+ transients (Figure 3). Pretreatment of the cells with 10 µM eleclazine for 5 min significantly suppressed the ATX-II-induced increase in basal Ca2+ concentration (Figure 3).

Figure 3.
ATX-II-induced changes in intracellular Ca2+ concentration. (A) Typical x−y images of pulmonary vein cardiomyocytes loaded with Fluo−4 after treatment with 100 nM ATX-II alone (a) and in the presence of 10 μM eleclazine (b). (B) Time course of the changes in basal Ca2+ concentration induced by 100 nM ATX-II alone (closed circles) and in the presence of 10 μM eleclazine (open circles). Each point with vertical bars represents the mean ± S.E.M of 30–40 cells. Asterisks indicate significant differences (p < 0.05) from the corresponding values in ATX-II-treated cells.
2.3. Effect of Enhanced Late INa on Intracellular Na+ Concentration
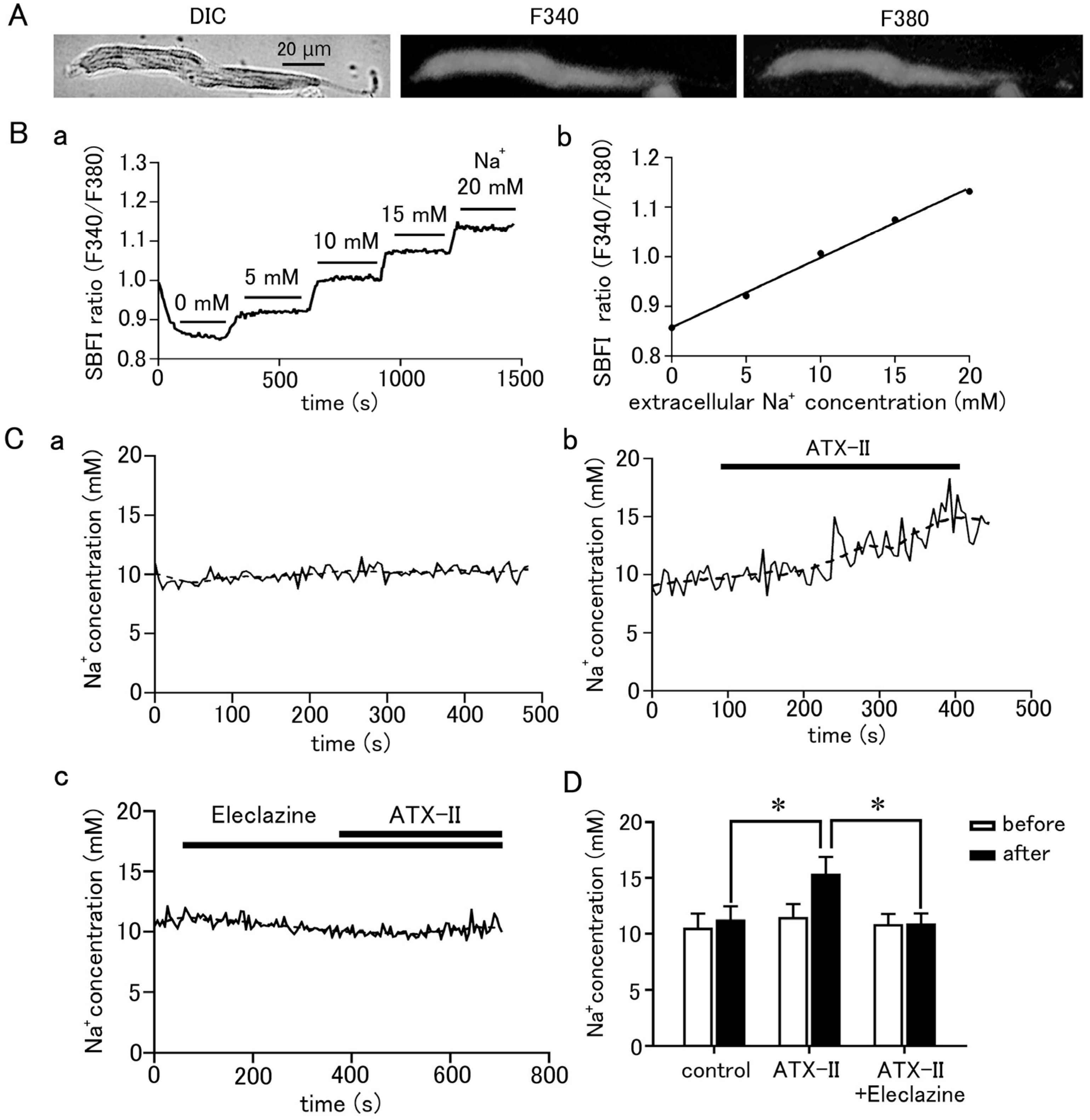
To measure the intracellular Na+ concentration, the pulmonary vein cardiomyocytes were loaded with SBFI/AM and observed by epifluorescence microscopy. SBFI was excited at 340 nm when bound to Na+ and at 380 nm when unbound, and the ratio of their fluorescence (F340/F380) can be taken to quantitatively measure Na+ concentration. Fluorescence excited at either wavelength was found to be homogeneously distributed in the cell (Figure 4A). The intracellular Na+ concentration could be quantified using a calibration curve based on a staircase graph of F340/F380 obtained by adjusting the Na+ concentrations in the extracellular fluid to 0, 5, 10, 15, and 20 mM (Figure 4B). The intracellular Na+ concentration in quiescent pulmonary vein cardiomyocytes was 11.19 ± 0.16 mM (n = 23). Intracellular Na+ concentration was increased by 3.91 ± 0.7 mM in 5 min by treatment with 100 nM ATX-II (n = 7; Figure 4C,D). This was significantly higher than the 0.73 ± 0.1 mM increase in Na+ concentration seen in the time control (n = 7). Pretreatment with 10 µM eleclazine significantly suppressed the ATX-II-induced increase in Na+ concentration; the ATX-II-induced increase in Na+ concentration was 0.06 ± 0.2 mM (n = 7).
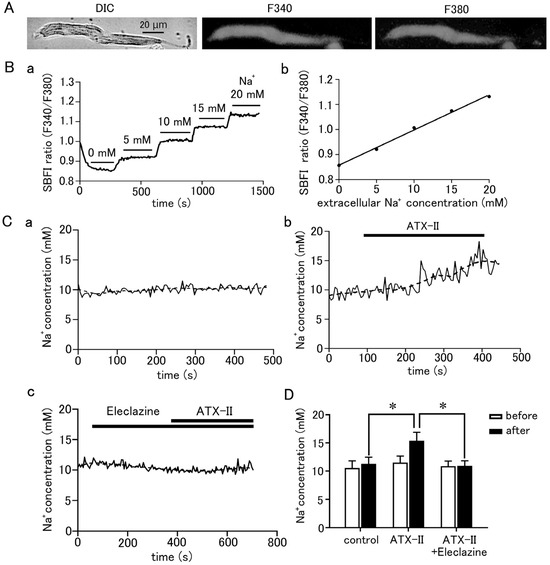
Figure 4.
Quantitative measurement of changes in intracellular Na+ concentration in pulmonary vein myocardium induced by ATX-II. (A) Differential interference contrast (DIC) images and fluorescence images excited at 340 nm and 380 nm in pulmonary vein cardiomyocytes loaded with SBFI. (B) Determination of intracellular Na+ concentration in pulmonary vein cardiomyocytes. After measuring the Na+ fluorescence ratio, calibration of the fluorescence ratio to Na+ concentration was performed as described in the methods section (B(a)); the calibration curve was prepared by plotting the fluorescence intensity ratio of SBFI obtained in (B(a)) for each extracellular Na+ concentration (B(b)). (C) Effect of 100 nM ATX-II on intracellular Na+ concentration. Time course of Na+ concentration for time control (C(a)), ATX-II (C(b)), and ATX-II in the presence of 10 μM eleclazine (C(c)). The solid line and the dashed line represent the Na+ concentration obtained every 5 s and its 15-point moving average, respectively. (D) Intracellular Na+ concentration at 5 min after ATX-II treatment. Each column with vertical bars represents the mean ± S.E.M. of 7 cells. Asterisks indicate significant differences (p < 0.05) from the corresponding values.
2.4. Contribution of NCX to Automatic Activity
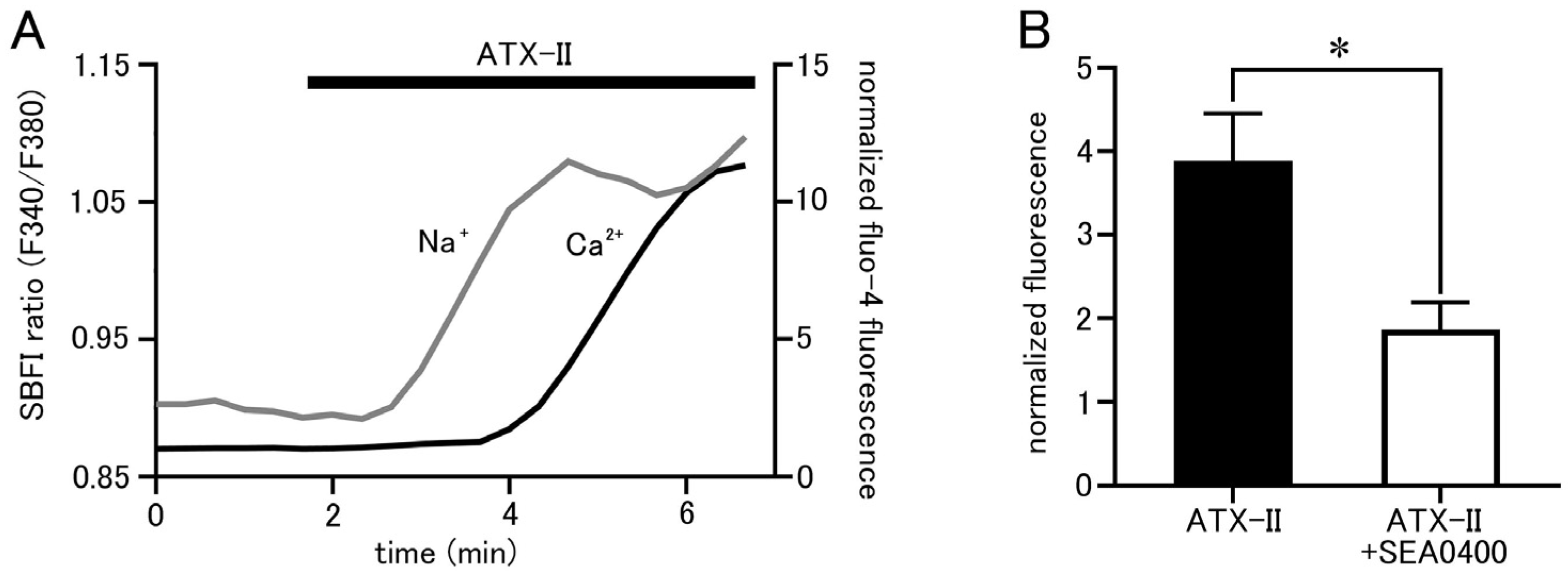
Isolated pulmonary vein cardiomyocytes were loaded with SBFI and Fluo-4, and intracellular Na+ and Ca2+ concentrations were simultaneously measured by epifluorescence microscopy. ATX-II (100 nM) caused increases in both Na+ and Ca2+ concentrations, but it took 1.23 ± 0.2 min for the Na+ concentration to reach half of its peak value and 3.46 ± 0.2 min for the Ca2+ concentration (n = 5; Figure 5A). Thus, ATX-II induced increases in intracellular Na+ and Ca2+ concentrations sequentially in this order. Furthermore, pretreatment with 1 µM of SEA0400, a selective NCX inhibitor, significantly reduced the ATX-II-induced increase in basal Ca2+ concentration (Figure 5B).
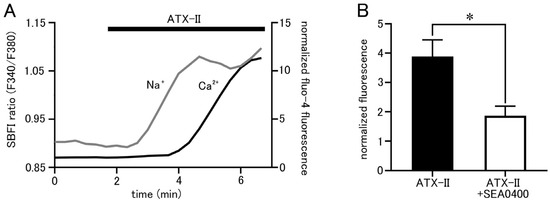
Figure 5.
ATX-II-induced changes in intracellular ion concentrations and the involvement of NCX. (A) Time course of respective fluorescence intensity in cells loaded with SBFI and Fluo−4. The gray line shows the fluorescence intensity ratio of SBFI, and the black line shows the fluorescence intensity of Fluo-4. Note that ATX-II (100 nM) induced an increase in the fluorescence intensity ratio of SBFI, followed by an increase in Fluo-4 fluorescence intensity. (B) Effect of SEA0400 (1 μM) on the elevation of basal Ca2+ concentration induced by ATX-II. Each column with vertical bars represents the mean ± S.E.M. of 30–40 cells. Asterisks indicate significant differences (p < 0.05) from the corresponding values in ATX-II-treated cells.
2.5. ATX-II-Induced Ca2+ Oscillation and Involvement of Ca2+-Dependent Enzymes
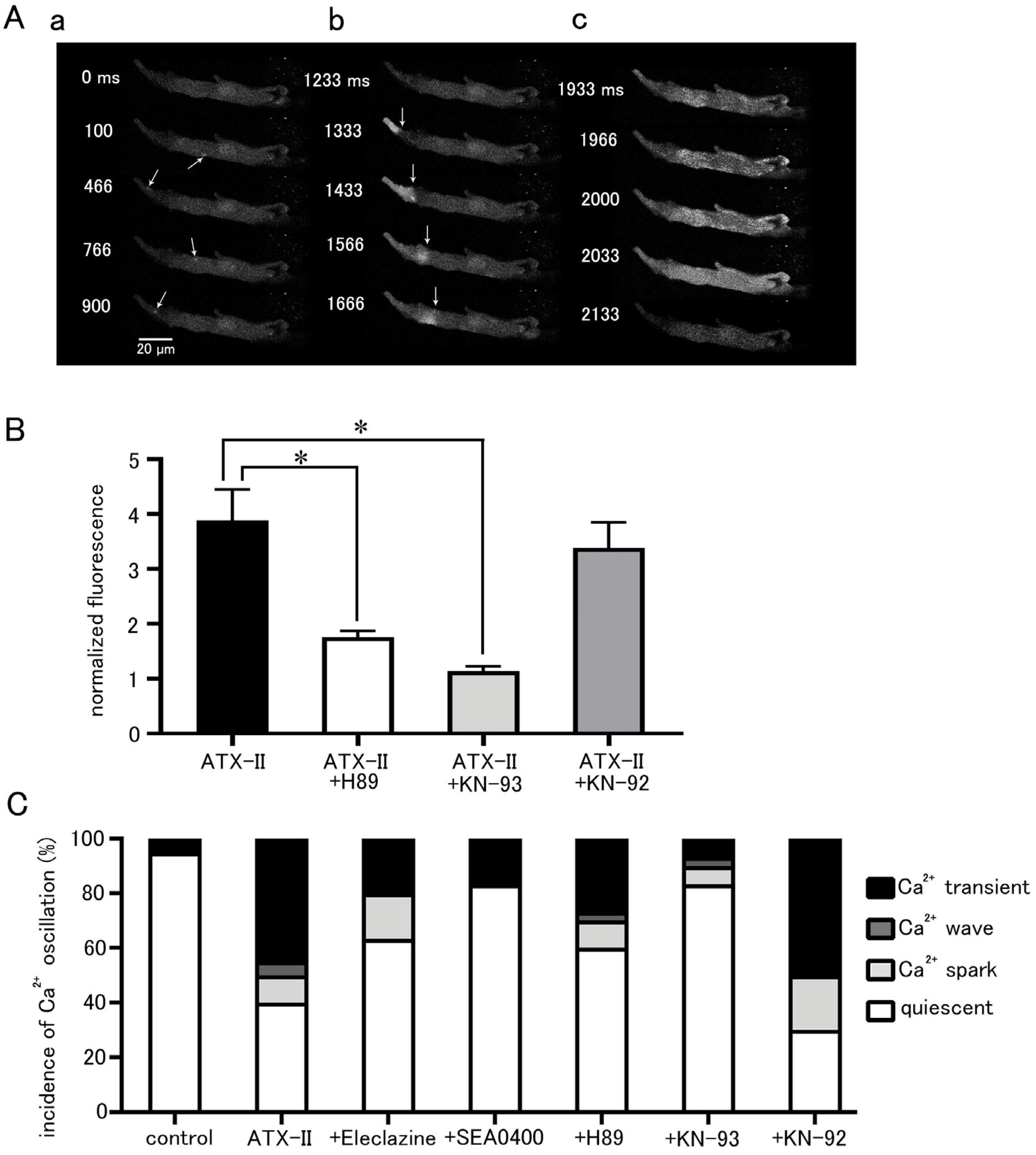
The effect of ATX-II on intracellular Ca2+ dynamics appeared in the form of an increase in basal Ca2+ concentration, Ca2+ sparks, and Ca2+ waves as well as Ca2+ transients (Figure 6A). These Ca2+ oscillations often occurred after 3 min of ATX-II treatment. The increase in basal Ca2+ concentration by 100 nM ATX-II occurred in all cells examined; it was suppressed by pretreatment with 5 µM H89 (n = 30), a PKA inhibitor, and 3 µM KN-93 (n = 30), a CaMKII inhibitor (Figure 6B). Such suppression was not observed with 3 µM KN-92 (n = 20), an inert analog of KN-93. The percentage of cells in which these Ca2+ oscillations were induced by ATX-II was significantly higher than the time control, and Ca2+ transients were observed in approximately half of the cells (Figure 6C). Pretreatment with eleclazine, SEA0400, H89, and KN-93 reduced the incidence of Ca2+ oscillations induced by ATX-II (Figure 6C).
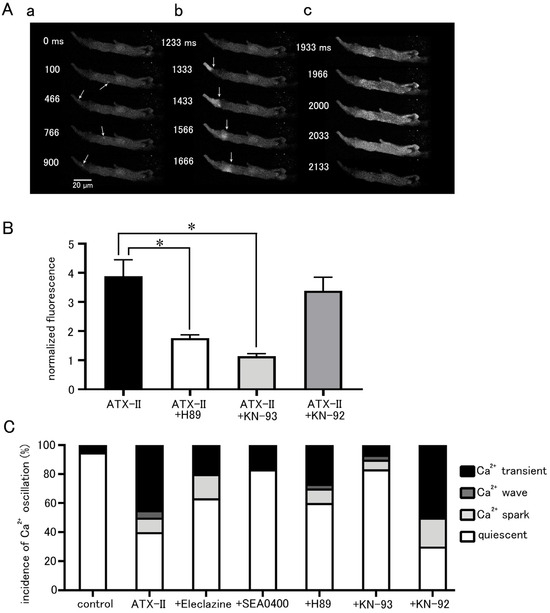
Figure 6.
Induction of intracellular Ca2+ oscillation by ATX-II and the effects of pharmacological agents. (A) Typical x−y images of a Fluo-4-loaded pulmonary vein cardiomyocyte after ATX-II treatment (100 nM) showing Ca2+ sparks (arrows; (a)), a Ca2+ wave (arrows indicate wavefront of Ca2+ wave; (b)), and a Ca2+ transient (c). The images in ((a–c)) were obtained in this order in the same cell. (B) The effects of H89 (5 μM), KN-93 (3 μM), and KN-92 (3 μM) on the ATX-II-induced increase in basal Ca2+ concentration. Asterisks indicate significant differences (p < 0.05) from the corresponding control values. (C) The effects of eleclazine (10 μM), H89 (5 μM), KN-93 (3 μM), and KN-92 (3 μM) on the incidence of ATX-II-induced Ca2+ oscillations. Each column with vertical bars in (B,C) represents the mean ± S.E.M of 20–40 cells.
3. Discussion
3.1. ATX-II-Induced Changes in Intracellular Ca2+ Concentration
The present study was undertaken to examine whether a pharmacological enhancement of the late INa can induce automatic activity in the pulmonary vein cardiomyocytes and to clarify the intracellular mechanisms involved. We first confirmed the relationship between action potential generation and the Ca2+ transient, a temporal increase in Ca2+ concentration throughout the cell. Both in the spontaneous Ca2+ transients and action-potential evoked Ca2+ transients, the rise in Ca2+ concentration occurred initially in the subsarcolemmal region and then spread to the cell center (Figure 1). This is a characteristic feature of Ca2+ transients in cardiomyocytes lacking T-tubules, including the atrial and pulmonary vein cardiomyocytes [27]. In cells lacking T-tubules, the trans-sarcolemmal Ca2+ influx elicited by an action potential first triggers Ca2+ release from the Ca2+ release channel (ryanodine receptor) in the sarcoplasmic reticulum (SR) in the subsarcolemmal region [27,28,29]. Next, in the central region lacking T-tubules, a propagated Ca2+-induced Ca2+ release mechanism in the SR activates the Ca2+ release from SR [28,29]. This pattern of rise in Ca2+ indicates the firing of action potentials. The same pattern was observed in ATX-II-induced Ca2+ transients (Figure 2), indicating that they were accompanied by action potentials. A characteristic feature of ATX-II-induced Ca2+ transients is that they were preceded by a gradual rise in basal Ca2+ concentration (Figure 3). This suggests that the rise in Ca2+ concentration is a cause of action potential generation. The rise in basal Ca2+ concentration, as well as the Ca2+ transient, was inhibited by eleclazine, confirming that both phenomena were caused by enhancement of late INa (Figure 2 and Figure 3).
3.2. NCX Is Involved in ATX-II-Induced Increases in Intracellular Na+ and Ca2+ Concentrations
ATX-II caused an increase in intracellular Na+ concentration in quiescent pulmonary vein cardiomyocytes, which was inhibited by eleclazine (Figure 4). These results also indicated that late INa could be activated at negative membrane potentials near the resting potential. This is consistent with our previous results with voltage-clamped pulmonary vein cardiomyocytes that the late INa can flow at membrane potentials near the resting potential of about −70 mV [13,30]. On the other hand, in the ventricular cardiomyocytes, ATX-II increased the Na+ concentration in electrically stimulated cardiomyocytes but had no effects on Na+ concentration in quiescent cardiomyocytes [31]. Our results also showed that ATX-II tended not to induce automatic activity in atrial cardiomyocytes. This is probably because the resting membrane potential of the atrial and ventricular myocardium of about −80 to −90 mV is more negative than that of the pulmonary vein myocardium [8,9]; the late INa is unlikely to flow near the resting membrane potential in the working myocardium. The pulmonary vein myocardium has a smaller inward rectifying K+ channel current density and less repolarizing power than the working myocardium [8,9,10]; this allows the membrane potential to stay in a less negative voltage range in which the late INa can flow.
Simultaneous measurement of intracellular Na+ and Ca2+ concentrations revealed that the ATX-II-induced increase in intracellular Na+ concentration was followed by an increase in Ca2+ concentration (Figure 5A); this order suggested the involvement of the reverse-mode NCX. The NCX has two modes of action: the forward mode, in which Na+ is taken into the cell and Ca2+ is excreted, and the reverse mode, in which Na+ is excreted from the cell and Ca2+ is taken into the cell. The forward mode is dominant in normal cardiomyocytes, but under conditions with elevated intracellular Na+ concentration such as ischemia–reperfusion or cardiac glycoside treatment, the NCX can function in the reverse mode and cause an increase in intracellular Ca2+ concentration [32]. In the present study, the treatment of quiescent cardiomyocytes with ATX-II increased the Na+ concentration from 11 to 15 mM, and, as mentioned above, the resting membrane potential of the pulmonary vein myocardium is less negative than that of the working myocardium [8,9]; these are conditions which favor Ca2+ entry through the reverse-mode NCX. Some reports suggested that the reverse-mode NCX may be more dominant at intracellular Na+ concentrations above 12 mM [33,34]. The result that SEA0400, a selective inhibitor of NCX, inhibited the increase in basal Ca2+ concentration by ATX-II (Figure 5B) also confirmed the involvement of NCX. Among the three known types of NCX, the expression of the SEA0400-sensitive NCX1 was reported in the pulmonary vein myocardium of rats [12].
3.3. ATX-II Ca2+ Overload Triggers Further Spontaneous Activity
ATX-II induced not only an increase in basal Ca2+ concentration but also Ca2+ oscillations such as Ca2+ sparks and Ca2+ waves, which are local non-propagating increases in Ca2+ concentration that occur independently of action potentials [28,29]. Ca2+ sparks, which are the unit of Ca2+ release from the SR Ca2+ release channel, occur in a small area of 1 to 2 μm in diameter, while on Ca2+ waves, the Ca2+ release propagates within the cell at a velocity of about 100 μm/s [27]. Confocal imaging in pulmonary vein tissue showed that Ca2+ waves, as well as Ca2+ sparks, do not propagate between connected cells, which confirms that they occur independently of action potential firing [35]. Ca2+ waves and Ca2+ sparks are considered to reflect spontaneous Ca2+ leaks from the sarcoplasmic reticulum which are enhanced in Ca2+ overload myocytes [36,37,38]. The increase in cytoplasmic Ca2+ concentration not only stimulates the SR Ca2+ release channel from the cytoplasmic side through the Ca2+-induced Ca2+ release mechanism but also causes an increase in Ca2+ concentration in the lumen of the SR, which affects the Ca2+ release channel and increases the Ca2+ sparks and Ca2+ waves. The abnormal increase in cytoplasmic Ca2+ causes hyperactivation of voltage-dependent ion channels, Ca2+-activated channels, and Ca2+-driven transporters, which leads to spontaneous electrical activity and arrhythmias [39]. The induction of Ca2+ sparks, Ca2+ waves, and Ca2+ transients in the pulmonary vein myocardium was also observed with ouabain, which increases intracellular Na+ concentration through inhibition of the Na+/K+ ATPase [11]. We also observed that the Ca2+ waves and Ca2+ transients were suppressed by carbachol, which hyperpolarized the resting membrane potential of the pulmonary vein myocardium to the same level as the working myocardium [8]. Thus, less repolarizing power of the pulmonary vein cardiomyocytes appears to play a permissive role in the generation of Ca2+ overload and the subsequent induction of automatic activity.
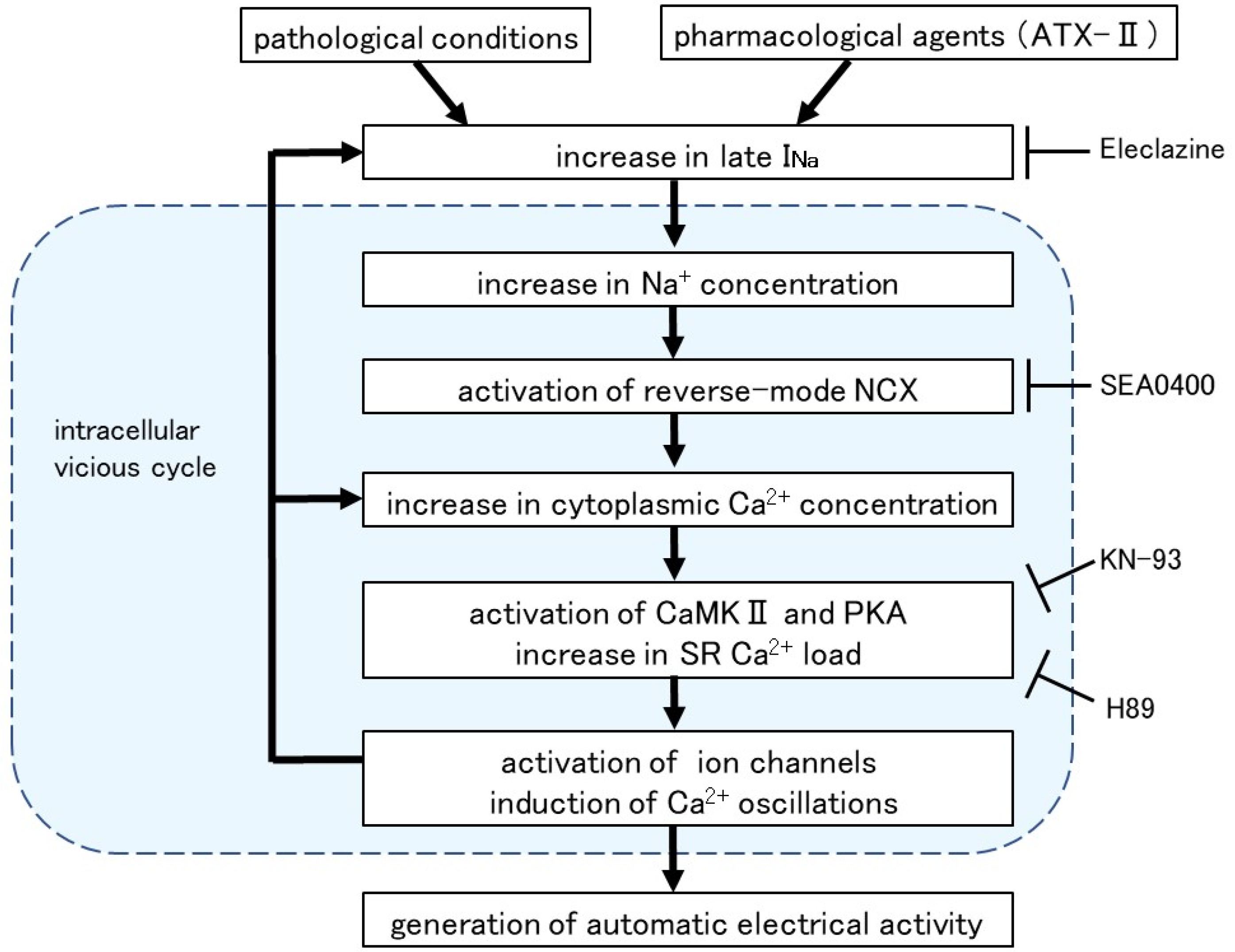
Intracellular Ca2+ overload in cardiomyocytes not only causes alterations in ion channels and transporters but also causes activation of various Ca2+-dependent enzymes [40]. The ATX-II-induced increase in basal Ca2+ concentration and generation of Ca2+ oscillation was suppressed in the presence of H89, a PKA inhibitor, and KN-93, a CaMKII inhibitor (Figure 6). A similar suppression of automatic activity after inhibition of CaMKII and PKA has been reported in rabbit pulmonary vein myocardium [41]. The activation of adenylate cyclase (AC) and PKA was reported in cardiomyocytes from the rat pulmonary vein [42] and guinea pig atria [43]. CaMKII, a multifunctional serine/threonine protein kinase is activated by the Ca2+/calmodulin complex when intracellular Ca2+ levels are increased [44,45]. CaMKII could phosphorylate the Na+ channels and Ca2+ channels [46], which could cause membrane depolarization. The hyperphosphorylation of the respective target sites on ryanodine receptors and phospholamban by CaMKII and PKA triggered a significant increase in Ca2+ leaks from SR [43]. Thus, enhancement of late INa and the resulting intracellular Ca2+ overload causes hyperactivation of various Ca2+-dependent ion channels, transporters, and enzymes, which in turn causes further accumulation of intracellular Ca2+ (Figure 7).
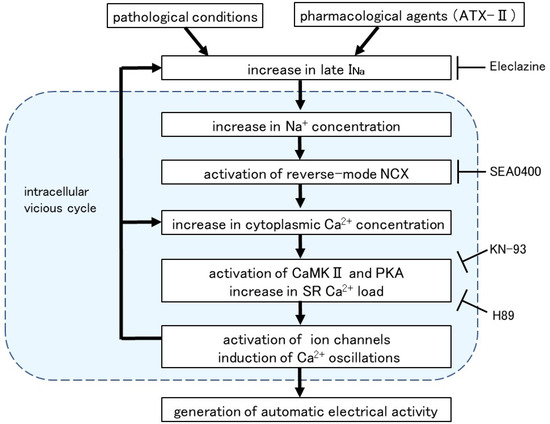
Figure 7.
Intracellular mechanism for the induction of automatic activity by enhanced late INa. Arrows denote stimulation and T-shaped lines indicate inhibition.
3.4. Clinical Implications of This Study
The present study showed that induction of pulmonary vein automaticity by enhancement of the late INa is partly mediated by mechanisms including the NCX and enzymes such as CaMKII and PKA. This implies that inhibitors of these mechanisms may be promising as drugs for the treatment of atrial fibrillation caused by pulmonary vein automaticity. Ranolazine, which selectively blocks the late INa, was shown to be anti-arrhythmic in human patients in some situations such as post-operative atrial fibrillation [47]; the mechanisms of action still remain to be clarified because the drug also has effects on many other ion channels and the α-adrenoceptor. Agents with higher selectivity towards late INa such as GS-458967 [13] and NCC-3902 [30] have been shown to inhibit pulmonary vein automaticity and to be effective against animal models of atrial fibrillation but have not been applied to human patients at present. Some of the class I antiarrhythmic drugs were shown to have inhibitory effects on the late INa and reduce the diastolic depolarization slope of the isolated guinea pig pulmonary vein myocardium [48]; whether such effects contribute to the antiarrhythmic effect of these drugs in human patients is unknown at present. A novel inhibitor of the NCX, SAR296968, was reported to reduce the frequency of Ca2+ sparks in isolated human atrial cardiomyocytes [49]; whether such effects can lead to antiarrhythmic effects in vivo awaits further investigation because the effect of NCX inhibitors appears to largely vary depending on the experimental condition [50,51]. Some of the class I antiarrhythmic drugs were reported to have inhibitory effects on the NCX, but only at concentrations higher than the therapeutic blood concentration [52]. A CaMKII inhibitor, AS105, which is more potent and selective than KN-93, was reported to inhibit SR Ca2+ leaks in human atrial cardiomyocytes [53,54]. Whether the mechanisms clarified in this study are involved in the pulmonary vein automaticity of human patients and whether these inhibitors are effective in clinical practice await further investigation.
3.5. Conclusions
The present results suggested that enhancement of late INa in pulmonary vein cardiomyocytes causes Ca2+ overload through the reverse-mode NCX and induces automatic activity. Such mechanisms are particularly likely to occur in the pulmonary vein myocardium, whose repolarizing membrane current density is lower than the working myocardium.
4. Materials and Methods
4.1. General
All experiments were performed in compliance with the Guiding Principles for the Care and Use of Laboratory Animals of the Japanese Pharmacological Society and were approved by the Toho University Animal Care and User Committee (21-41-507, 7 May 2022).
4.2. Isolation of Pulmonary Vein Cardiomyocytes
Pulmonary vein cardiomyocytes were obtained according to the methods described in our previous study [13,27,30]. Male Hartley guinea pigs (weight, 300–450 g) were anesthetized with isoflurane, and the hearts with lungs were isolated and perfused via the aorta with a Tyrode’s solution of the following composition (mM concentration): NaCl 143, KCl 4, MgCl2 0.5, CaCl2 1.8, NaH2PO4 0.33, glucose 5.5, and HEPES 5 (pH 7.4, gassed with 100% O2, and warmed to 36 °C). The heart was further perfused successively with nominally Ca2+-free Tyrode’s solution and the same solution containing 0.1 mg/mL protease (type XIV; Sigma-Aldrich, St. Louis, MO, USA) and 0.5 mg/mL collagenase (YK-102; Yakult, Tokyo, Japan) for about 20 min. After the washout of the enzymes, the cardiomyocytes were isolated by gentle disruption of the pulmonary vein.
4.3. Measurement of Intracellular Ca2+ Dynamics
For the measurement of intracellular Ca2+ dynamics, the isolated pulmonary vein cardiomyocytes were loaded with 5 µM Fluo-4/AM (Dojindo, Kumamoto, Japan) for about 30 min and superfused with the Tyrode’s solution described above at room temperature. The cardiomyocytes were observed with a rapid-scanning laser confocal microscope (A1R; Nikon Corporation, Tokyo, Japan) so that the Ca2+ sparks and Ca2+ waves could be detected. The objective lens was Apochromat ×40, 1.15 numerical aperture (water immersion). The excitation wavelength was 488 nm, and the emission in the wavelength range of 500 to 550 nm was detected. The data were analyzed with computer software, NIS elements (Nikon Corporation). The fluorescence intensity of Fluo-4 at each time point was normalized against the basal intensity. The scanning was performed every 4.4 ms at 1024 × 64 pixels or every 8.8 ms at 1024 × 128 pixels. For the triggering of action potentials, field electrical stimulation by a rectangle voltage pulse of 3 ms duration was applied to cells through a platinum wire electrode paired with an electric stimulator (SEN-3303; Nihon Kohden Corporation, Tokyo, Japan).
4.4. Measurement of Intracellular Na+ Concentration
Isolated cardiomyocytes were loaded with SBFI (5 μM SBFI/AM) and 0.05% pluronic F-127 at 36 °C (Invitrogen, Carlsbad, CA, USA), and the total Na+ fluorescence from single cardiomyocytes was measured with epifluorescence microscopy. The cells were excited at 340 and 380 nm from a xenon lamp, and the emission (>500 nm) was separated with a dichroic mirror, detected by a cooled CCD camera (C6790, Hamamatsu Photonics, Shizuoka, Japan) at a time resolution of 5 s, and ratioed after correction of background fluorescence (Aquacosmos software, version 2.52, Hamamatsu Photonics, Shizuoka, Japan). A cooled CCD camera was used because a rapid-scanning laser confocal microscope is not suitable for excitation using two wavelengths. In situ calibration of SBFI was performed by exposing the myocytes to various extracellular Na+ solutions in the presence of 10 μM ouabain, 2 µM gramicidin, and 40 μM monensin. The Na+ concentration was increased from 0 mM to 20 mM in 5 mM steps.
4.5. Simultaneous Measurement of Intracellular Ca2+ and Na+ Concentrations
For the simultaneous measurement of intracellular Ca2+ and Na+ concentration, the isolated pulmonary vein cardiomyocytes were loaded with Fluo-4/AM and SBFI/AM (see above). The cells were excited at 480 nm for Fluo-4 from an LED lamp (COLIBRI, Carl Zeiss, Oberkochen, Germany), and 340 and 380 nm for SBFI from a mercury lamp (HXP120V, Zeiss, Oberkochen, Germany), and the emission (>500 nm) was separated with a dichroic mirror, detected by a digital camera (AxioCam MRm, Carl Zeiss, Oberkochen, Germany) at a time resolution of 10 s, and ratioed after correction of background fluorescence (Zen Pro, Carl Zeiss).
4.6. Chemicals
ATX-II and H89 were purchased from Alomone Labs (Jerusalem, Israel); gramicidin, monensin, ouabain, and SEA0400 were purchased from Sigma-Aldrich (St. Louis, USA); eleclazine was purchased from Cosmo Bio (Tokyo, Japan); and KN-93 and KN-92 were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan). ATX-II was dissolved in distilled water, and monensin was dissolved in ethanol. All other chemicals were dissolved in dimethyl sulfoxide.
4.7. Data Analytics and Statistics
All data were expressed as the mean ± standard error of the mean (S.E.M.). Statistical significance between means was evaluated by Welch’s t-test, or one-way repeated-measures ANOVA followed by Tukey’s or Dunnett’s multiple comparisons. A p-value less than 0.05 was considered statistically significant.
Author Contributions
Conceptualization, I.N. and H.T.; methodology, I.N. and H.T.; software, S.H.; validation, T.S., M.S. and A.O.; formal analysis, T.S. and M.S.; investigation, T.S., M.S. and A.O.; resources, I.N. and H.T.; data curation, T.S.; writing—original draft preparation, T.S.; writing—review and editing, I.N. and H.T.; visualization, T.S.; supervision, H.T.; project administration, I.N. and H.T.; funding acquisition, I.N., S.H. and H.T. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by JSPS KAKENHI, grant numbers JP20K16013, JP20K07299, and JP20K07091. T.S. received the Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (N-231501).
Institutional Review Board Statement
The study was approved by the Ethics Committee of Toho University (21-55-362; 29 March 2021) and was conducted in accordance with the “Guiding Principles for the Care and Use of Laboratory Animals” approved by the Japanese Pharmacological Society.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nattel, S. Basic electrophysiology of the pulmonary veins and their role in atrial fibrillation: Precipitators, perpetuators, and perplexers. J. Cardiovasc. Electrophysiol. 2003, 14, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Namekata, I.; Tsuneoka, Y.; Tanaka, H. Electrophysiological and pharmacological properties of the pulmonary vein myocardium. Biol. Pharm. Bull. 2013, 36, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, S.; Chen, Y.; Yeh, H.; Chang, M.; Lin, C. Electrophysiology of single cardiomyocytes isolated from rabbit pulmonary veins: Implication in initiation of focal atrial fibrillation. Basic. Res. Cardiol. 2002, 97, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Haissaguerre, M.; Jais, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Metayer, P.; Clementy, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998, 339, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Hsieh, M.H.; Tai, C.T.; Tsai, C.F.; Prakash, V.S.; Yu, W.C.; Hsu, T.L.; Ding, Y.A.; Chang, M.S. Initiation of atrial fibrillation by ectopic beats originating from the pulmonary veins: Electrophysiological characteristics, pharmacological responses, and effects of radiofrequency ablation. Circulation 1999, 100, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- de Bakker, J.M.T.; Ho, S.Y.; Hocini, M. Basic and clinical electrophysiology of pulmonary vein ectopy. Cardiovasc. Res. 2002, 54, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Takagi, D.; Okamoto, Y.; Ohba, T.; Yamamoto, H.; Ono, K. Comparative study of hyperpolarization-activated currents in pulmonary vein cardiomyocytes isolated from rat, guinea pig, and rabbit. J. Physiol. Sci. 2020, 70, 6. [Google Scholar] [CrossRef] [PubMed]
- Tsuneoka, Y.; Irie, M.; Tanaka, Y.; Sugimoto, T.; Kobayashi, Y.; Kusakabe, T.; Kato, K.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Permissive role of reduced inwardly-rectifying potassium current density in the automaticity of the guinea pig pulmonary vein myocardium. J. Pharmacol. Sci. 2017, 133, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.R.; Cha, T.; Zhang, L.; Chartier, D.; Melnyk, P.; Hohnloser, S.H.; Nattel, S. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: Action potential and ionic current properties. J. Physiol. 2003, 551, 801–813. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawamura, K.; Nakamura, Y.; Ono, K. Pathological impact of hyperpolarization-activated chloride current peculiar to rat pulmonary vein cardiomyocytes. J. Mol. Cell Cardiol. 2014, 66, 53–62. [Google Scholar] [CrossRef]
- Namekata, I.; Tsuneoka, Y.; Takahara, A.; Shimada, H.; Sugimoto, T.; Takeda, K.; Nagaharu, M.; Shigenobu, K.; Kawanishi, T.; Tanaka, H. Involvement of the Na+/Ca2+ exchanger in the automaticity of guinea-pig pulmonary vein myocardium as revealed by SEA0400. J. Pharmacol. Sci. 2009, 110, 111–116. [Google Scholar] [CrossRef]
- Okamoto, Y.; Takano, M.; Ohba, T.; Ono, K. Arrhythmogenic coupling between the Na+-Ca2+ exchanger and inositol 1,4,5-triphosphate receptor in rat pulmonary vein cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Irie, M.; Hiiro, H.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Involvement of the persistent Na+ current in the diastolic depolarization and automaticity of the guinea pig pulmonary vein myocardium. J. Pharmacol. Sci. 2019, 141, 9–16. [Google Scholar] [CrossRef]
- Malecot, C.O.; Bredeloux, P.; Findlay, I.; Maupoil, V. A TTX-sensitive resting Na+ permeability contributes to the catecholaminergic automatic activity in rat pulmonary vein. J. Cardiovasc. Electrophysiol. 2015, 26, 311–319. [Google Scholar] [CrossRef]
- Cohen, C.J.; Bean, B.P.; Tsien, R.W. Maximal upstroke velocity as an index of available sodium conductance. Comparison of maximal upstroke velocity and voltage clamp measurements of sodium current in rabbit Purkinje fibers. Circ. Res. 1984, 54, 636–651. [Google Scholar] [CrossRef]
- Crill, W.E. Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 1996, 58, 349–362. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Nesterenko, V.; Shryock, J.C.; Rajamani, S.; Song, Y.; Belardinelli, L. The role of late INa in development of cardiac arrhythmias. Handb. Exp. Pharmacol. 2014, 221, 137–168. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shryock, J.C.; Belardinelli, L. A slowly inactivating sodium current contributes to spontaneous diastolic depolarization of atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1254–H1262. [Google Scholar] [CrossRef]
- Rota, M.; Vassalle, M. Patch-clamp analysis in canine cardiac Purkinje cells of a novel sodium component in the pacemaker range. J. Physiol. 2003, 548, 147–165. [Google Scholar] [CrossRef]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.D.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; et al. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef]
- Wimmer, N.J.; Stone, P.H. Anti-anginal and anti-ischemic effects of late sodium current inhibition. Cardiovasc. Drugs Ther. 2013, 27, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.; Bers, D.M. The late sodium current in heart failure: Pathophysiology and clinical relevance. ESC Heart Fail. 2014, 1, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cheng, C.; Chen, Y.; Chen, S.; Chen, Y. ATX-II-induced pulmonary vein arrhythmogenesis related to atrial fibrillation and long QT syndrome. Eur. J. Clin. Investig. 2012, 42, 823–831. [Google Scholar] [CrossRef]
- Hegyi, B.; Polonen, R.; Hellgren, K.T.; Ko, C.Y.; Ginsburg, K.S.; Bossuyt, J.; Mercola, M.; Bers, D.M. Cardiomyocyte Na+ and Ca2+ mishandling drives vicious cycle involving CaMKII, ROS, and ryanodine receptors. Basic Res. Cardiol. 2021, 116, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, L.; Giles, W.R.; Rajamani, S.; Karagueuzian, H.S.; Shryock, J.C. Cardiac late Na+ current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. 2015, 12, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Mallwitz, A.; Wagner, S.; Hartmann, N.; Schotola, H.; Fischer, T.H.; Ungeheuer, N.; Herting, J.; Shah, A.M.; Maier, L.S.; et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014, 76, 94–105. [Google Scholar] [CrossRef]
- Namekata, I.; Tanaka, Y.; Ohmori, T.; Tsuneoka, Y.; Hamaguchi, S.; Tanaka, H. Cell Morphology and Early-phase Ca2+ Transients of Guinea-Pig Pulmonary Vein Cardiomyocytes Compared with Atrial and Ventricular Cardiomyocytes. Bioimages 2019, 27, 1–12. [Google Scholar]
- Tanaka, H.; Nishimaru, K.; Sekine, T.; Kawanishi, T.; Nakamura, R.; Yamagaki, K.; Shigenobu, K. Two-dimensional millisecond analysis of intracellular Ca2+ sparks in cardiac myocytes by rapid scanning confocal microscopy: Increase in amplitude by isoproterenol. Biochem. Biophys. Res. Commun. 1997, 233, 413–418. [Google Scholar] [CrossRef]
- Tanaka, H.; Kawanishi, T.; Matsuda, T.; Takahashi, M.; Shigenobu, K. Intracellular free Ca2+ movements in cultured cardiac myocytes as shown by rapid scanning confocal microscopy. J. Cardiovasc. Pharmacol. 1996, 27, 761–769. [Google Scholar] [CrossRef]
- Namekata, I.; Hiiro, H.; Odaka, R.; Saito, T.; Hamaguchi, S.; Tsukamoto, T.; Ishikawa, R.; Katayama, Y.; Kondo, Y.; Tanaka, H. Inhibitory Effect of a Late Sodium Current Blocker, NCC-3902, on the Automaticity of the Guinea Pig Pulmonary Vein Myocardium. Biol. Pharm. Bull. 2022, 45, 1644–1652. [Google Scholar] [CrossRef]
- Kornyeyev, D.; El-Bizri, N.; Hirakawa, R.; Nguyen, S.; Viatchenko-Karpinski, S.; Yao, L.; Rajamani, S.; Belardinelli, L. Contribution of the late sodium current to intracellular sodium and calcium overload in rabbit ventricular myocytes treated by anemone toxin. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H426–H435. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Weber, C.R. Na/Ca exchange function in intact ventricular myocytes. Ann. N. Y. Acad. Sci. 2002, 976, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Noble, D.; Noble, P.J. Late sodium current in the pathophysiology of cardiovascular disease: Consequences of sodium-calcium overload. Heart 2006, 92 (Suppl. 4), iv1–iv5. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Namekata, I.; Tsuneoka, Y.; Akiba, A.; Nakamura, H.; Shimada, H.; Takahara, A.; Tanaka, H. Intracellular Calcium and Membrane Potential Oscillations in the Guinea Pig and Rat Pulmonary Vein Myocardium. Bioimages 2010, 18, 11–22. [Google Scholar]
- Fujiwara, K.; Tanaka, H.; Mani, H.; Nakagami, T.; Takamatsu, T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: Simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ. Res. 2008, 103, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.V.; Bovo, E.; Bers, D.M.; Blatter, L.A. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010, 588, 4743–4757. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, A.; Argenziano, M.; Aguilar-Sanchez, Y.; Mazzocchi, G.; Escobar, A.L. Ca2+ Sparks and Ca2+ waves are the subcellular events underlying Ca2+ overload during ischemia and reperfusion in perfused intact hearts. J. Mol. Cell. Cardiol. 2015, 79, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Lee, W.; Chien, Y.; Chen, T.; Yang, K. The link between abnormalities of calcium handling proteins and catecholaminergic polymorphic ventricular tachycardia. Tzu Chi Med. J. 2021, 33, 323–331. [Google Scholar] [CrossRef]
- Bers, D.M.; Guo, T. Calcium signaling in cardiac ventricular myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 86–98. [Google Scholar] [CrossRef]
- Chan, C.; Lin, F.; Chen, Y.; Lin, Y.; Higa, S.; Chen, S.; Chen, Y. Glucagon-like Peptide-1 Receptor Activation Reduces Pulmonary Vein Arrhythmogenesis and Regulates Calcium Homeostasis. Int. J. Mol. Sci. 2023, 24, 13100. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Aung, N.Y.; Tanaka, M.; Takeda, Y.; Takagi, D.; Igarashi, W.; Ishii, K.; Yamakawa, M.; Ono, K. Preferential Expression of Ca2+-Stimulable Adenylyl Cyclase III in the Supraventricular Area, including Arrhythmogenic Pulmonary Vein of the Rat Heart. Biomolecules 2022, 12, 724. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Herting, J.; Mason, F.E.; Hartmann, N.; Watanabe, S.; Nikolaev, V.O.; Sprenger, J.U.; Fan, P.; Yao, L.; Popov, A.; et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc. Res. 2015, 107, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Couchonnal, L.F.; Anderson, M.E. The role of calmodulin kinase II in myocardial physiology and disease. Physiology 2008, 23, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hudmon, A.; Schulman, H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 2002, 364, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef]
- Kaplan, A.D.; Joca, H.C.; Boyman, L.; Greiser, M. Calcium Signaling Silencing in Atrial Fibrillation: Implications for Atrial Sodium Homeostasis. Int. J. Mol. Sci. 2021, 22, 10513. [Google Scholar] [CrossRef] [PubMed]
- Irie, M.; Hiiro, H.; Kato, S.; Kuramochi, M.; Hamaguchi, S.; Namekata, I.; Tanaka, H. Differential effects of class I antiarrhythmic drugs on the guinea pig pulmonary vein myocardium: Inhibition of automatic activity correlates with blockade of a diastolic sodium current component. J. Pharmacol. Sci. 2020, 143, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Hegner, P.; Drzymalski, M.; Biedermann, A.; Memmel, B.; Durczok, M.; Wester, M.; Floerchinger, B.; Provaznik, Z.; Schmid, C.; Zausig, Y.; et al. SAR296968, a Novel Selective Na+/Ca2+ Exchanger Inhibitor, Improves Ca2+ Handling and Contractile Function in Human Atrial Cardiomyocytes. Biomedicines 2022, 10, 1932. [Google Scholar] [CrossRef] [PubMed]
- Christ, T.; Kovacs, P.P.; Acsai, K.; Knaut, M.; Eschenhagen, T.; Jost, N.; Varro, A.; Wettwer, E.; Ravens, U. Block of Na+/Ca2+ exchanger by SEA0400 in human right atrial preparations from patients in sinus rhythm and in atrial fibrillation. Eur. J. Pharmacol. 2016, 788, 286–293. [Google Scholar] [CrossRef]
- Nagy, N.; Toth, N.; Nanasi, P.P. Antiarrhythmic and Inotropic Effects of Selective Na+/Ca2+ Exchanger Inhibition: What Can We Learn from the Pharmacological Studies? Int. J. Mol. Sci. 2022, 23, 14651. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Koide, Y.; Kimura, J. Topics on the Na+/Ca2+ Exchanger: Pharmacological Characterization of Na+/Ca2+ Exchanger Inhibitors. J. Pharmacol. Sci. 2006, 102, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Dobrev, D. Non-ion channel therapeutics for heart failure and atrial fibrillation: Are CaMKII inhibitors ready for clinical use? J. Mol. Cell Cardiol. 2018, 121, 300–303. [Google Scholar] [CrossRef]
- Neef, S.; Steffens, A.; Pellicena, P.; Mustroph, J.; Lebek, S.; Ort, K.R.; Schulman, H.; Maier, L.S. Improvement of cardiomyocyte function by a novel pyrimidine-based CaMKII-inhibitor. J. Mol. Cell. Cardiol. 2018, 115, 73–81. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).