
Steroidomics in Men with Schizophrenia

 ,
, 
Abstract
1. Introduction
2. Results
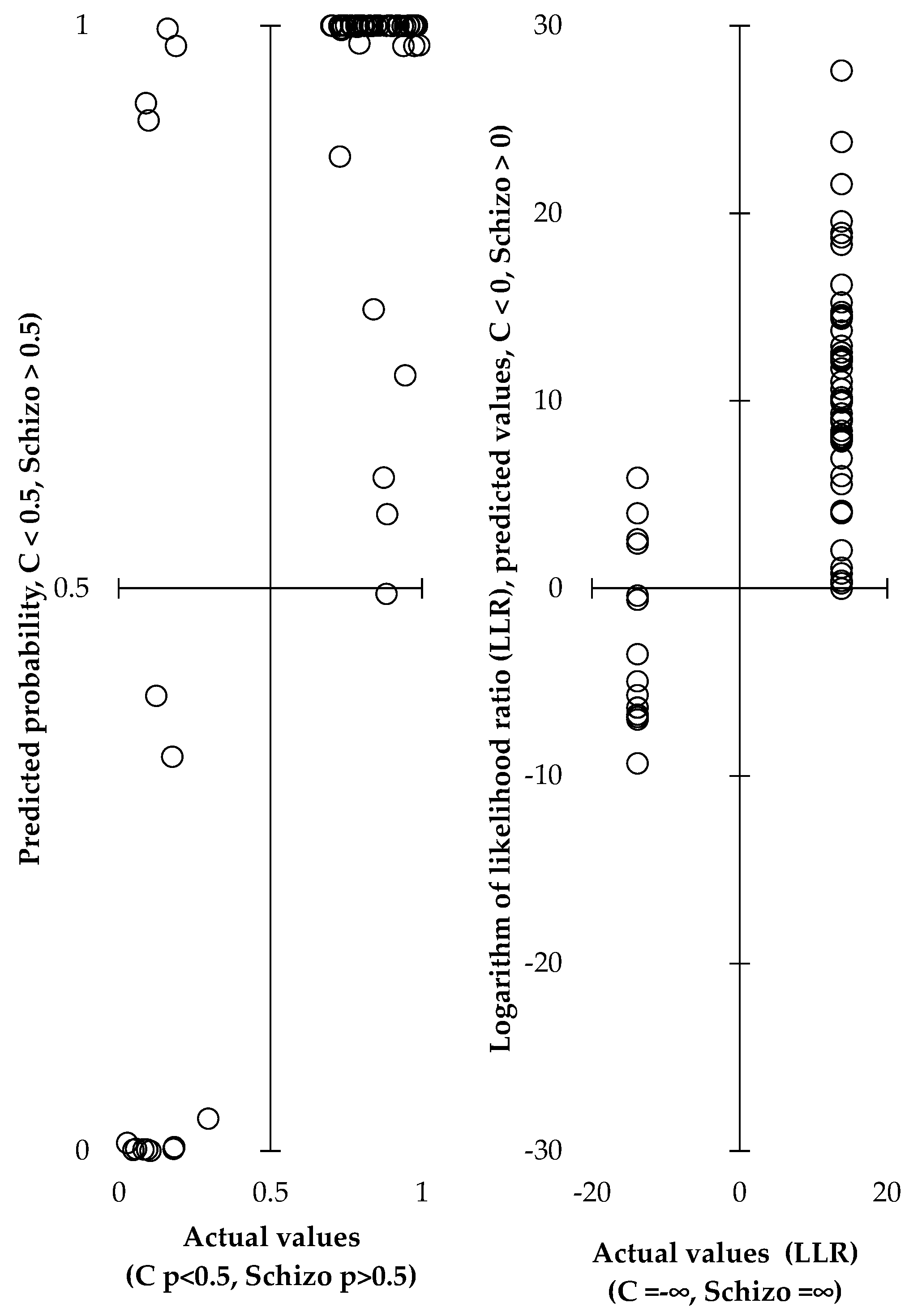
2.1. Discriminating Schizophrenics from Controls Based on Circulating Steroids
2.2. Altered Steroid Levels in Schizophrenic Men
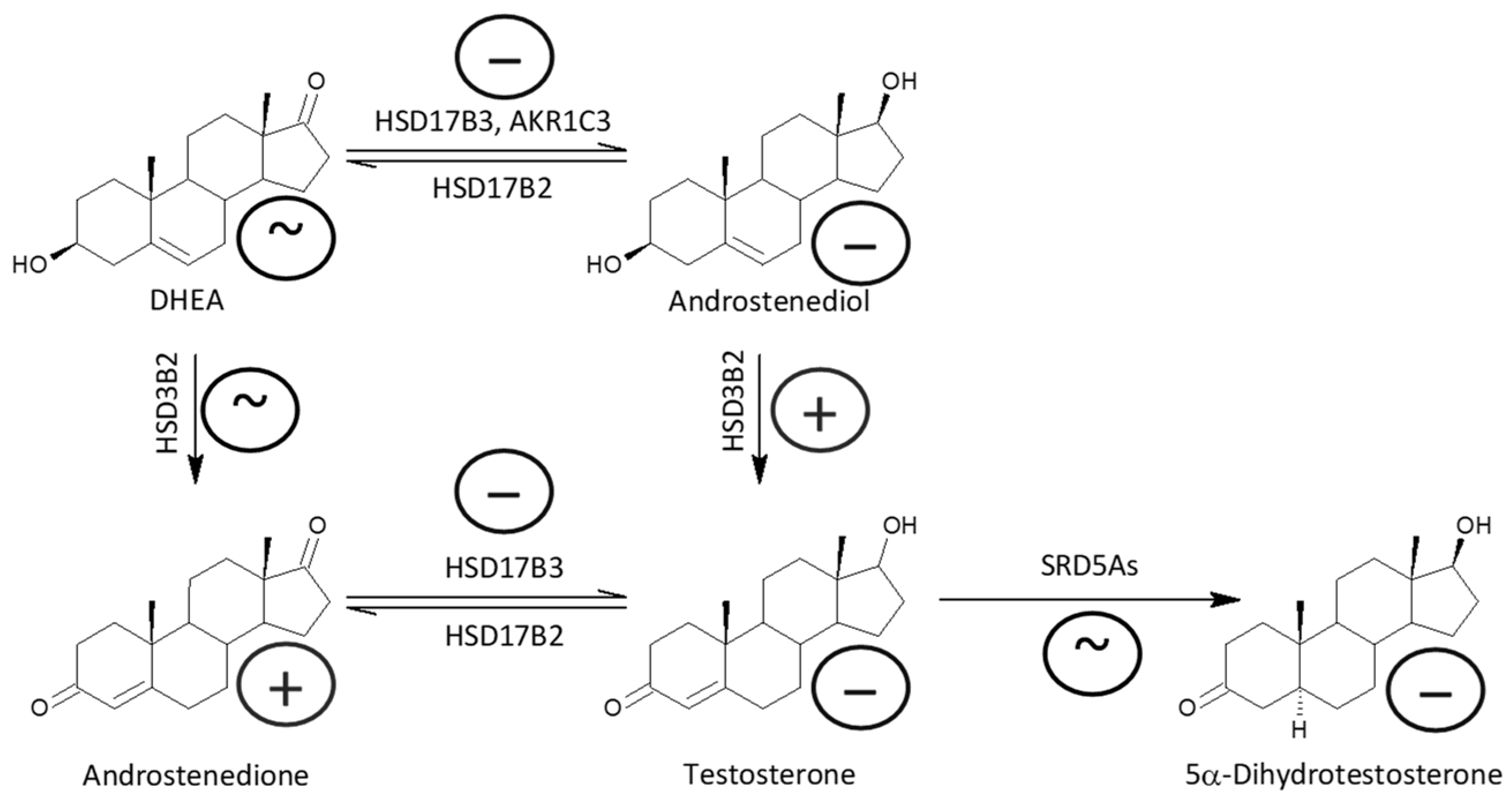
2.2.1. Δ5 and Δ4 Steroids
2.2.2. Glucocorticoids (C21 Δ4 Steroids) and 11β-Hydroxy-androgens (C19 Δ4 and 5α/β Steroids)
2.2.3. Progestogens
2.2.4. 5α-Reduced Steroids
2.2.5. 5β-Reduced Steroids
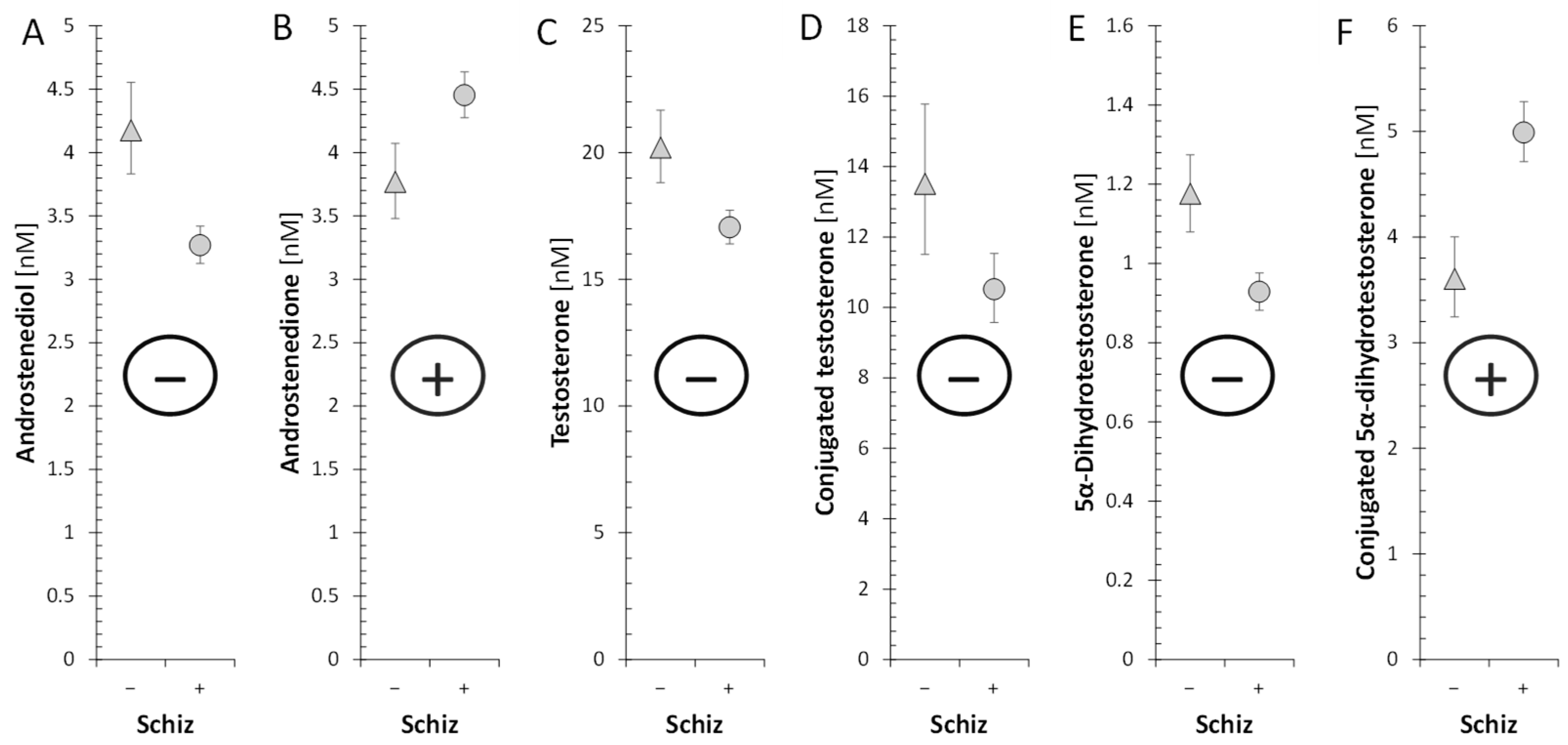
2.2.6. Androstenedione and Active Androgens
2.3. Changes in the Steroid Molar Ratios
2.3.1. The Overall Conversion of 17-Deoxy-pregnanes to Corresponding Androstanes
2.3.2. The Conversion of 17-Deoxypregnanes to Corresponding 17-Hydroxy-pregnanes
2.3.3. The Conversion of 17-Hydroxy-pregnanes to Corresponding Androstanes
2.3.4. The Conversion of Δ5 Steroids to Their Δ4 Counterparts
2.3.5. The Balances between Sulfated and Unconjugated Steroids
2.3.6. 11β-Hydroxylation of Androstane Steroids
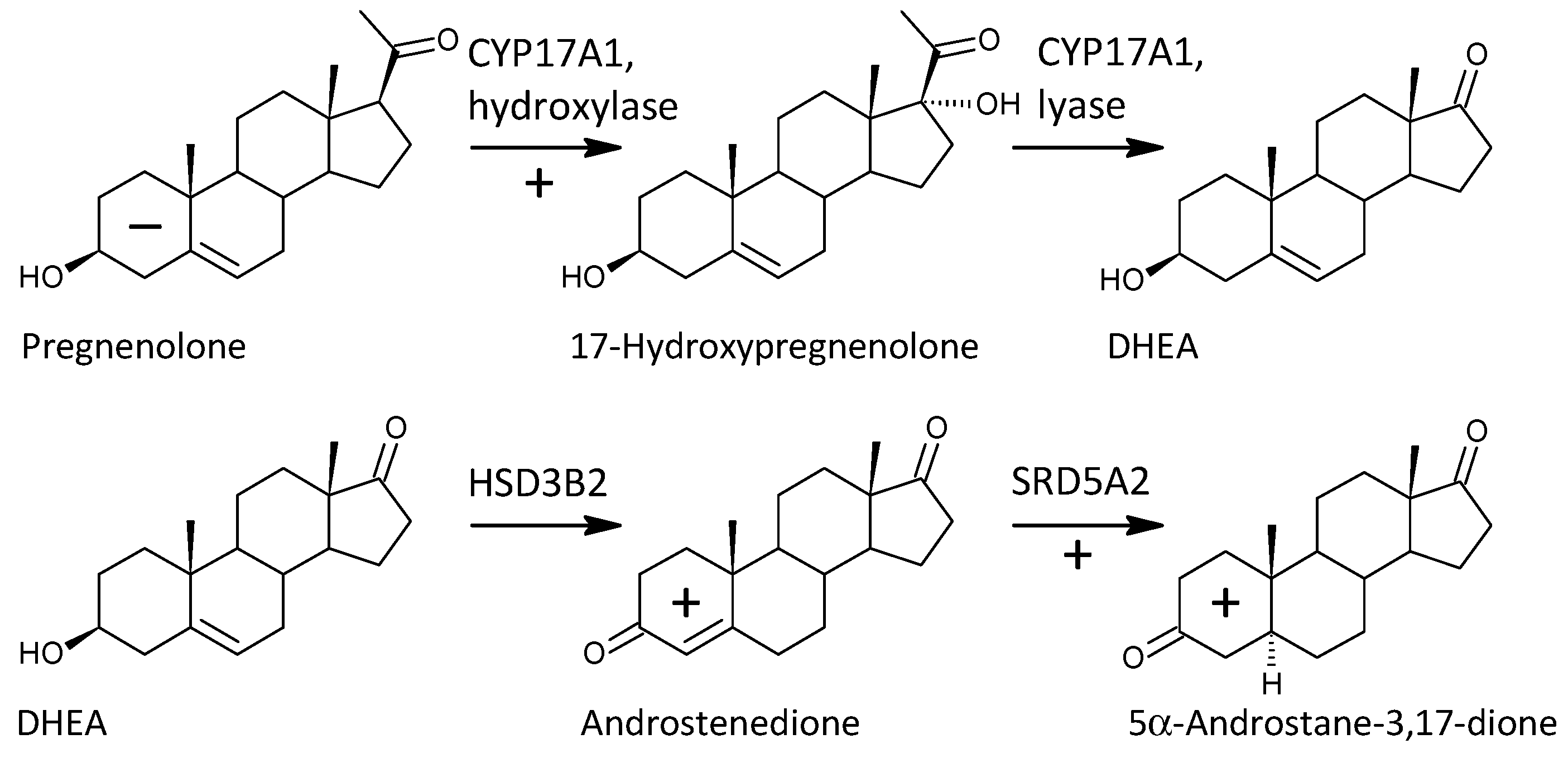
2.3.7. 7α-, 7β-, and 16α-Hydroxylation of Δ5 Androstanes
2.3.8. Steroid Molar Ratios Probably Related to HSD11B1
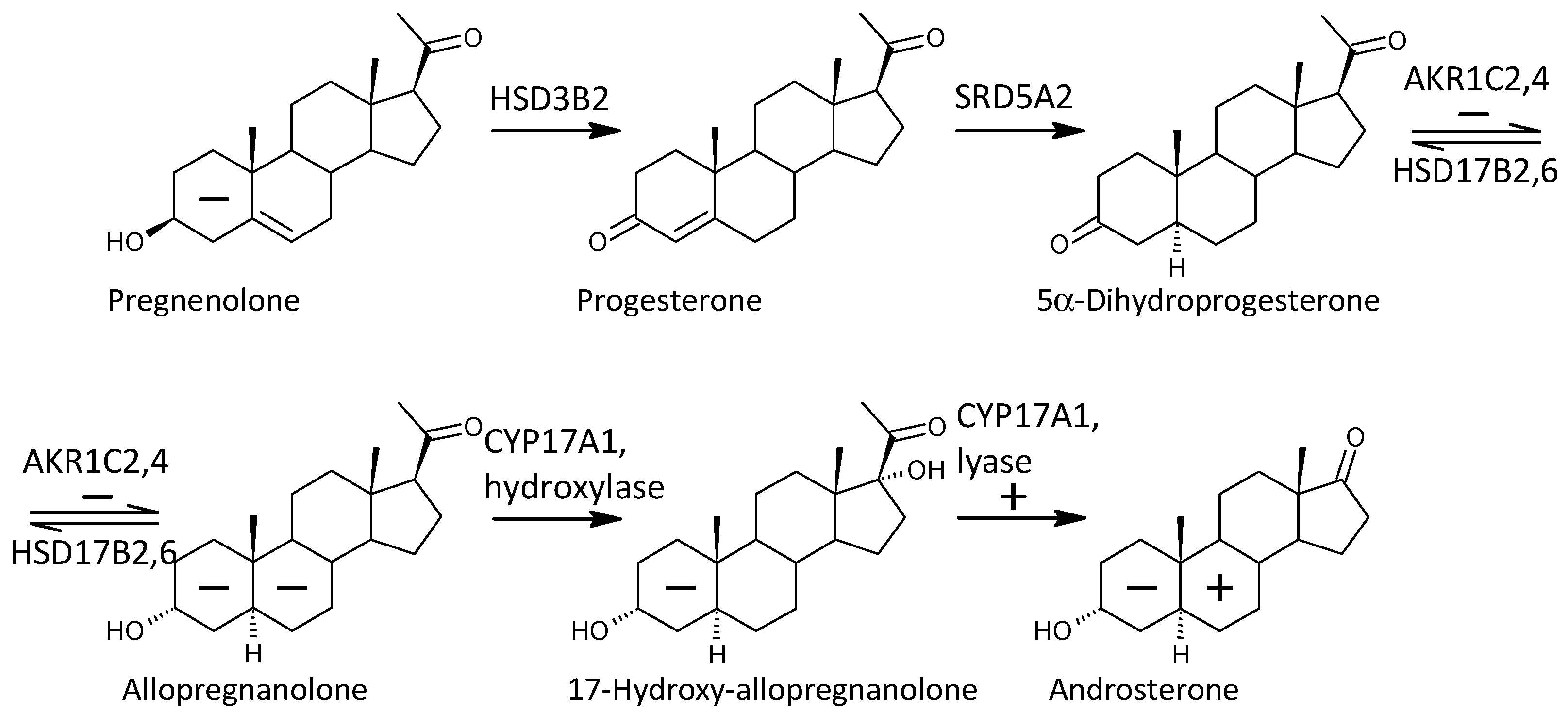
2.3.9. Steroid Molar Ratios Probably Related to 5α-Reductases (SRD5A1 and SRD5A2)
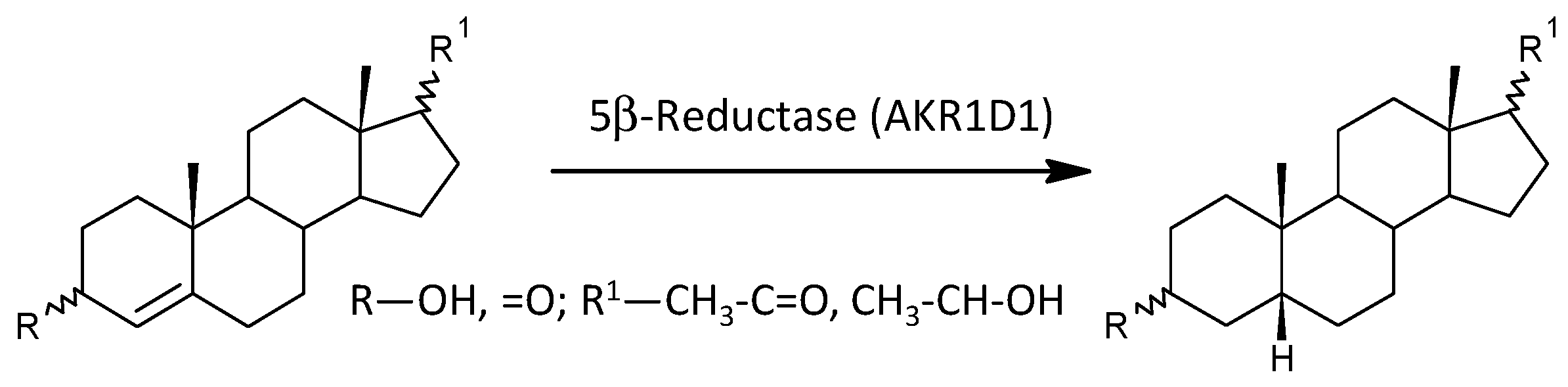
2.3.10. Steroid Molar Ratios Probably Related to AKR1D1
2.3.11. Steroid Molar Ratios Probably Related to a Balance between AKR1C1 and HSD17B2
2.3.12. Steroid Molar Ratios Probably Related to a Balance between AKR1C2 on One Side and HSD17B2 and HSD17B2 on the Other
2.3.13. Steroid Molar Ratios Probably Related to Balance between AKR1C3 and HSD17B2
2.4. Effect of Antipsychotic Treatment
3. Discussion
4. Limitations of this Study
5. Materials and Methods
5.1. Subjects
5.2. Steroid Analysis
5.3. Statistical Analysis
- Transformation of the original data to obtain the values with symmetric distribution and constant variance;
- Checking the data homogeneity in predictors using Hotelling’s statistics and the eventual elimination of non-homogeneities;
- Testing the relevance of predictors using variable importance statistics and the elimination of irrelevant predictors;
- Calculating component loadings for individual variables to evaluate their correlations with the predictive component;
- Calculating regression coefficients for the multiple regression model to evaluate the mutual independence of predictors after comparison with the corresponding component loadings from the OPLS model;
- Calculating predicted values of the logarithm of the ratio of the probability of pathology presence to the probability of pathology absence (LLR);
- Calculating the probability of the pathology presence for individual subjects;
- Calculating the sensitivity and specificity of the prediction.
6. Conclusions
- (1)
- Demonstrated the ability to effectively differentiate men with schizophrenia from controls, which helped to clarify the role of steroids in the pathophysiology of schizophrenia and suggested possibilities for their therapeutic use.
- (2)
- Showed substantially altered adrenal and testicular steroidogenesis in the patients compared with controls.
- (3)
- Showed an altered metabolic pathway from PREG/S to cortisol with several metabolic bottlenecks such as increased PREG sulfation and/or suppressed PREGS desulfation, lower pregnenolone levels, impaired conversion of 17-OH-PREG to 17-OH-P, and suppressed CYP11B1 function; however, two counterregulatory steps, increased conversion of PREG/S to 17-OH-PREG/S and decreased conversion of cortisol to cortisone, are likely to maintain unchanged basal cortisol levels but may not guarantee a sufficient cortisol response to stress.
- (4)
- Indicated a trend towards higher 7α-, 7β- and 16α-hydroxylation whereby enzymes catalyzing these conversions may counteract the autoimmune complications and pro-inflammatory processes accompanying schizophrenia.
- (5)
- Showed lower T levels at higher A levels, suggesting suppression of HSD17B3 functioning.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5α-DHA | 5α-Androstane-3,17-dione |
| 5α-DHT | 5α-Dihydrotestosterone |
| 3α,5α-THA | Androsterone |
| 3β,5α-THA | Epiandrosterone |
| 3α,5β-THA | Etiocholanolone |
| 3α,5β-THA | Epietiocholanolone |
| 5-HT3 | Type-3 5-Hydroxytryptamine receptor (serotonin receptor) |
| 8-iso-PGF2α | 8-iso-Prostaglandin F2α |
| σ1R | σ1-Receptor |
| A | Androstenedione |
| AD | Androstanediol |
| ADIOL | Androstenediol |
| AKR1C1 | Aldo-keto reductase family 1 member C1 |
| AKR1C2 | Aldo-keto reductase family 1 member C2 |
| AKR1C3 | Aldo-keto reductase family 1 member C3 |
| AKR1D1 | 5β-Reductase |
| ALLO | 3α,5α-Tetrahydroprogesterone, allopregnanolone |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPAR | AMPA receptor |
| ANOVA | Analysis of variance |
| AT | Androstenetriol (5-androstene) |
| CI | Confidence interval |
| C | Conjugated steroid |
| C+U | Conjugated + unconjugated steroids |
| CLCN3 | Chloride voltage-gated channel 3 |
| CNS | Central nervous system |
| CYP11A1 | Cholesterol desmolase |
| CYP11B1 | 11β-Hydroxylase |
| CYP17A1 | C17-hydroxylase-C17,20-lyase |
| CYP19A1 | Aromatase |
| CYP21A2 | 21-Hydroxylase |
| CYP3A4 | Cytochrome P450 3A4 |
| CYP3A7 | Cytochrome P450 3A7 |
| CYP7B1 | 7α-hydroxylase |
| HSD3B1 | Type 1 3β-Hydroxysteroid dehydrogenase |
| HSD3B2 | Type 2 3β-Hydroxysteroid dehydrogenase |
| HSD3Bs | Type 1 and 2 3β-Hydroxysteroid dehydrogenases |
| HSD11B1 | Type 1 11β-Hydroxysteroid dehydrogenase |
| HSD11B2 | Type 2 11β-Hydroxysteroid dehydrogenase |
| HSD17B2 | Type 2 17β-Hydroxysteroid dehydrogenase |
| HSD17B3 | Type 3 17β-Hydroxysteroid dehydrogenase |
| HSD17B6 | Type 6 17β-Hydroxysteroid dehydrogenase (3α/β epimerase) |
| DHEA | Dehydroepiandrosterone |
| DHEAS | Dehydroepiandrosterone sulfate |
| DHP | Dihydroprogesterone |
| FEP | First-episode patient |
| GABA | γ-Aminobutyric acid |
| GABAAR | Type A γ-aminobutyric acid receptor |
| GC-MS/MS | Gas chromatography tandem mass spectrometry |
| HC | Healthy control |
| HPA | Hypothalamic-pituitary adrenal |
| IL-6 | Interleukin 6 |
| IL-17 | Interleukin 17 |
| L-type VGCC | L-type voltage gated calcium channels |
| LLR | Logarithm of likelihood ratio |
| LSD | Least significant difference |
| MC | LSD multiple comparisons |
| mRNA | Messenger ribonucleic acid |
| NAS | Neuroactive steroid |
| NMDAR | N-methyl-D-aspartate receptor |
| OPLS | Orthogonal predictions to latent structure |
| P | Progesterone |
| PD | Pregnanediol |
| PGF2α | Prostaglandin F2α |
| PNS | Peripheral nervous system |
| PREG | Pregnenolone |
| PREGS | Pregnenolone sulfate |
| PT | Pregnanetriol |
| ROS | Reactive oxygen species |
| SRD5A1 | Type 1 5α-reductase |
| SRD5A2 | Type 2 5α-reductase |
| SULT2A1 | Steroid sulfotransferase type 2A1 |
| STS | Steroid sulfatase |
| T | Testosterone |
| THP | Tetrahydroprogesterone |
| TNFα | Tumor necrosis factor α |
| TRPC5 | Short transient receptor potential channels 5 |
| TRPV1 | Vanilloid receptor |
| TRPM3 | Melastatin receptor |
References
- Mikulska, J.; Juszczyk, G.; Gawronska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef] [PubMed]
- Marder, S.R.; Cannon, T.D. Schizophrenia. N. Engl. J. Med. 2019, 381, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Sbisa, A.M.; Sun, J.; Gibbons, A.; Udawela, M.; Dean, B. A Role for Estrogen in Schizophrenia: Clinical and Preclinical Findings. Int. J. Endocrinol. 2015, 2015, 615356. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.; Riordan, A.; Thornton, J. Estrogens and the cognitive symptoms of schizophrenia: Possible neuroprotective mechanisms. Front. Neuroendocr. 2017, 47, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Matuszewska, A.; Kowalski, K.; Jawien, P.; Tomkalski, T.; Gawel-Dabrowska, D.; Merwid-Lad, A.; Szelag, E.; Blaszczak, K.; Wiatrak, B.; Danielewski, M.; et al. The Hypothalamic-Pituitary-Gonadal Axis in Men with Schizophrenia. Int. J. Mol. Sci. 2023, 24, 6492. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Cao, T.; Zhou, X.; Yao, J.K. Neurosteroids in Schizophrenia: Pathogenic and Therapeutic Implications. Front. Psychiatry 2018, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.M.; Odontiadis, J.; Le Melledo, J.M.; Prior, T.I.; Baker, G.B. The relevance of neuroactive steroids in schizophrenia, depression, and anxiety disorders. Cell Mol. Neurobiol. 2007, 27, 541–574. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.L.; Kolsch, H. Effects of estrogen on brain development and neuroprotection—Implications for negative symptoms in schizophrenia. Psychoneuroendocrinology 2003, 28 (Suppl. 2), S83–S96. [Google Scholar] [CrossRef] [PubMed]
- Dogan Bulut, S.; Bulut, S.; Guriz, O. The relationship between sex hormone profiles and symptoms of schizophrenia in men. Compr. Psychiatry 2016, 69, 186–192. [Google Scholar] [CrossRef]
- Kamin, H.S.; Kertes, D.A. Cortisol and DHEA in development and psychopathology. Horm. Behav. 2017, 89, 69–85. [Google Scholar] [CrossRef]
- da Silva, T.L.; Ravindran, A.V. Contribution of sex hormones to gender differences in schizophrenia: A review. Asian J. Psychiatr. 2015, 18, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.J.; Dekker, C.F.; van Lunenburg, M.; Sommer, I.E. Estrogen augmentation in schizophrenia: A quantitative review of current evidence. Schizophr. Res. 2012, 141, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.S. The clinical and therapeutic potentials of dehydroepiandrosterone and pregnenolone in schizophrenia. Neuroscience 2011, 191, 91–100. [Google Scholar] [CrossRef]
- Qaiser, M.Z.; Dolman, D.E.M.; Begley, D.J.; Abbott, N.J.; Cazacu-Davidescu, M.; Corol, D.I.; Fry, J.P. Uptake and metabolism of sulphated steroids by the blood-brain barrier in the adult male rat. J. Neurochem. 2017, 142, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Bicikova, M.; Hill, M.; Ripova, D.; Mohr, P.; Hampl, R. Determination of steroid metabolome as a possible tool for laboratory diagnosis of schizophrenia. J. Steroid Biochem. Mol. Biol. 2013, 133, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.E.; Keefe, R.S.; Buchanan, R.W.; Hamer, R.M.; Kilts, J.D.; Bradford, D.W.; Strauss, J.L.; Naylor, J.C.; Payne, V.M.; Lieberman, J.A.; et al. Proof-of-concept trial with the neurosteroid pregnenolone targeting cognitive and negative symptoms in schizophrenia. Neuropsychopharmacology 2009, 34, 1885–1903. [Google Scholar] [CrossRef]
- Powrie, Y.S.L.; Smith, C. Central intracrine DHEA synthesis in ageing-related neuroinflammation and neurodegeneration: Therapeutic potential? J. Neuroinflamm. 2018, 15, 289. [Google Scholar] [CrossRef] [PubMed]
- Honcu, P.; Hill, M.; Bicikova, M.; Jandova, D.; Velikova, M.; Kajzar, J.; Kolatorova, L.; Bestak, J.; Macova, L.; Kancheva, R.; et al. Activation of Adrenal Steroidogenesis and an Improvement of Mood Balance in Postmenopausal Females after Spa Treatment Based on Physical Activity. Int. J. Mol. Sci. 2019, 20, 3687. [Google Scholar] [CrossRef] [PubMed]
- Misiak, B.; Frydecka, D.; Loska, O.; Moustafa, A.A.; Samochowiec, J.; Kasznia, J.; Stanczykiewicz, B. Testosterone, DHEA and DHEA-S in patients with schizophrenia: A systematic review and meta-analysis. Psychoneuroendocrinology 2018, 89, 92–102. [Google Scholar] [CrossRef]
- Cherian, K.; Schatzberg, A.F.; Keller, J. HPA axis in psychotic major depression and schizophrenia spectrum disorders: Cortisol, clinical symptomatology, and cognition. Schizophr. Res. 2019, 213, 72–79. [Google Scholar] [CrossRef]
- Misiak, B.; Piotrowski, P.; Chec, M.; Samochowiec, J. Cortisol and dehydroepiandrosterone sulfate in patients with schizophrenia spectrum disorders with respect to cognitive performance. Compr. Psychoneuroendocrinol. 2021, 6, 100041. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.; Maayan, R.; Gibel, A.; Strous, R.D.; Modai, I.; Weizman, A. Elevation of the cortisol/dehydroepiandrosterone ratio in schizophrenia patients. Eur. Neuropsychopharmacol. 2004, 14, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.; Gibel, A.; Maayan, R.; Ratner, Y.; Ram, E.; Modai, I.; Weizman, A. State and trait related predictors of serum cortisol to DHEA(S) molar ratios and hormone concentrations in schizophrenia patients. Eur. Neuropsychopharmacol. 2007, 17, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Oettel, M.; Mukhopadhyay, A.K. Progesterone: The forgotten hormone in men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef] [PubMed]
- Belvederi Murri, M.; Fanelli, F.; Pagotto, U.; Bonora, E.; Triolo, F.; Chiri, L.; Allegri, F.; Mezzullo, M.; Menchetti, M.; Mondelli, V.; et al. Neuroactive Steroids in First-Episode Psychosis: A Role for Progesterone? Schizophr. Res. Treat. 2016, 2016, 1942828. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Jung, S.W.; Joe, S.H.; Lee, C.H.; Jung, H.G.; Jung, I.K.; Kim, S.H.; Lee, M.S. Association between serum testosterone levels and the severity of negative symptoms in male patients with chronic schizophrenia. Psychoneuroendocrinology 2007, 32, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Shirayama, Y.; Hashimoto, K.; Suzuki, Y.; Higuchi, T. Correlation of plasma neurosteroid levels to the severity of negative symptoms in male patients with schizophrenia. Schizophr. Res. 2002, 58, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Sisek-Sprem, M.; Krizaj, A.; Jukic, V.; Milosevic, M.; Petrovic, Z.; Herceg, M. Testosterone levels and clinical features of schizophrenia with emphasis on negative symptoms and aggression. Nord J. Psychiatry 2015, 69, 102–109. [Google Scholar] [CrossRef]
- Bicikova, M.; Hill, M.; Ripova, D.; Mohr, P. Altered levels of circulating GABAergic 5alfa/beta-reduced pregnane and androstane steroids in schizophrenic men. Horm Mol. Biol. Clin. Investig. 2011, 6, 227–230. [Google Scholar] [CrossRef]
- Cai, H.; Zhou, X.; Dougherty, G.G.; Reddy, R.D.; Haas, G.L.; Montrose, D.M.; Keshavan, M.; Yao, J.K. Pregnenolone-progesterone-allopregnanolone pathway as a potential therapeutic target in first-episode antipsychotic-naive patients with schizophrenia. Psychoneuroendocrinology 2018, 90, 43–51. [Google Scholar] [CrossRef]
- Ritsner, M.; Maayan, R.; Gibel, A.; Weizman, A. Differences in blood pregnenolone and dehydroepiandrosterone levels between schizophrenia patients and healthy subjects. Eur. Neuropsychopharmacol. 2007, 17, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Ji, E.; Weickert, C.S.; Purves-Tyson, T.; White, C.; Handelsman, D.J.; Desai, R.; O’Donnell, M.; Liu, D.; Galletly, C.; Lenroot, R.; et al. Cortisol-dehydroepiandrosterone ratios are inversely associated with hippocampal and prefrontal brain volume in schizophrenia. Psychoneuroendocrinology 2021, 123, 104916. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Li, Y. Association among serum cortisol, dehydroepiandrosterone-sulfate levels and psychiatric symptoms in men with chronic schizophrenia. Compr. Psychiatry 2017, 76, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Brzezinski-Sinai, N.A.; Brzezinski, A. Schizophrenia and Sex Hormones: What Is the Link? Front. Psychiatry 2020, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Girshkin, L.; Matheson, S.L.; Shepherd, A.M.; Green, M.J. Morning cortisol levels in schizophrenia and bipolar disorder: A meta-analysis. Psychoneuroendocrinology 2014, 49, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Arlt, W.; Storbeck, K.H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef] [PubMed]
- Rege, J.; Nakamura, Y.; Wang, T.; Merchen, T.D.; Sasano, H.; Rainey, W.E. Transcriptome profiling reveals differentially expressed transcripts between the human adrenal zona fasciculata and zona reticularis. J. Clin. Endocrinol. Metab. 2014, 99, E518–E527. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.J.; Purohit, A.; Woo, L.W.; Newman, S.P.; Potter, B.V. Steroid sulfatase: Molecular biology, regulation, and inhibition. Endocr. Rev. 2005, 26, 171–202. [Google Scholar] [CrossRef]
- Tuckey, R.C. Side-chain cleavage of cholesterol sulfate by ovarian mitochondria. J. Steroid Biochem. Mol. Biol. 1990, 37, 121–127. [Google Scholar] [CrossRef]
- Neunzig, J.; Sanchez-Guijo, A.; Mosa, A.; Hartmann, M.F.; Geyer, J.; Wudy, S.A.; Bernhardt, R. A steroidogenic pathway for sulfonated steroids: The metabolism of pregnenolone sulfate. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt B, 324–333. [Google Scholar] [CrossRef]
- Mondelli, V. From stress to psychosis: Whom, how, when and why? Epidemiol. Psychiatr. Sci. 2014, 23, 215–218. [Google Scholar] [CrossRef]
- Plumbly, W.; Brandon, N.; Deeb, T.Z.; Hall, J.; Harwood, A.J. L-type voltage-gated calcium channel regulation of in vitro human cortical neuronal networks. Sci. Rep. 2019, 9, 13810. [Google Scholar] [CrossRef] [PubMed]
- van Rooyen, D.; Yadav, R.; Scott, E.E.; Swart, A.C. CYP17A1 exhibits 17alphahydroxylase/17,20-lyase activity towards 11beta-hydroxyprogesterone and 11-ketoprogesterone metabolites in the C11-oxy backdoor pathway. J. Steroid Biochem. Mol. Biol. 2020, 199, 105614. [Google Scholar] [CrossRef] [PubMed]
- Barnard, L.; du Toit, T.; Swart, A.C. Back where it belongs: 11beta-hydroxyandrostenedione compels the re-assessment of C11-oxy androgens in steroidogenesis. Mol. Cell Endocrinol. 2021, 525, 111189. [Google Scholar] [CrossRef] [PubMed]
- Barnard, L.; Gent, R.; van Rooyen, D.; Swart, A.C. Adrenal C11-oxy C(21) steroids contribute to the C11-oxy C(19) steroid pool via the backdoor pathway in the biosynthesis and metabolism of 21-deoxycortisol and 21-deoxycortisone. J. Steroid Biochem. Mol. Biol. 2017, 174, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Guryev, O.L.; Auchus, R.J. 5alpha-reduced C21 steroids are substrates for human cytochrome P450c17. Arch. Biochem. Biophys. 2003, 418, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Bottasso, O.; Bay, M.L.; Besedovsky, H.; del Rey, A. The immuno-endocrine component in the pathogenesis of tuberculosis. Scand. J. Immunol. 2007, 66, 166–175. [Google Scholar] [CrossRef]
- Du, C.; Khalil, M.W.; Sriram, S. Administration of dehydroepiandrosterone suppresses experimental allergic encephalomyelitis in SJL/J mice. J. Immunol. 2001, 167, 7094–7101. [Google Scholar] [CrossRef] [PubMed]
- Rontzsch, A.; Thoss, K.; Petrow, P.K.; Henzgen, S.; Brauer, R. Amelioration of murine antigen-induced arthritis by dehydroepiandrosterone (DHEA). Inflamm. Res. 2004, 53, 189–198. [Google Scholar]
- Tan, X.D.; Dou, Y.C.; Shi, C.W.; Duan, R.S.; Sun, R.P. Administration of dehydroepiandrosterone ameliorates experimental autoimmune neuritis in Lewis rats. J. Neuroimmunol. 2009, 207, 39–44. [Google Scholar] [CrossRef]
- Choi, I.S.; Cui, Y.; Koh, Y.A.; Lee, H.C.; Cho, Y.B.; Won, Y.H. Effects of dehydroepiandrosterone on Th2 cytokine production in peripheral blood mononuclear cells from asthmatics. Korean J. Intern. Med. 2008, 23, 176–181. [Google Scholar] [CrossRef]
- Sudo, N.; Yu, X.N.; Kubo, C. Dehydroepiandrosterone attenuates the spontaneous elevation of serum IgE level in NC/Nga mice. Immunol. Lett. 2001, 79, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Dehydroepiandrosterone and dehydroepiandrosterone sulphate in atopic allergy and chronic urticaria. Inflammation 2008, 31, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Sterzl, I.; Hampl, R.; Sterzl, J.; Votruba, J.; Starka, L. 7Beta-OH-DHEA counteracts dexamethasone induced suppression of primary immune response in murine spleenocytes. J. Steroid Biochem. Mol. Biol. 1999, 71, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S.; Kapsenberg, M.; Radbruch, A.; Adorini, L. Th1 and Th2 cells. Res. Immunol. 1998, 149, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Pratschke, S.; von Dossow-Hanfstingl, V.; Dietz, J.; Schneider, C.P.; Tufman, A.; Albertsmeier, M.; Winter, H.; Angele, M.K. Dehydroepiandrosterone modulates T-cell response after major abdominal surgery. J. Surg. Res. 2014, 189, 117–125. [Google Scholar] [CrossRef]
- Reading, C.L.; Frincke, J.M.; White, S.K. Molecular targets for 17alpha-ethynyl-5-androstene-3beta,7beta,17beta-triol, an anti-inflammatory agent derived from the human metabolome. PLoS ONE 2012, 7, e32147. [Google Scholar] [CrossRef]
- Pettersson, H.; Lundqvist, J.; Norlin, M. Effects of CYP7B1-mediated catalysis on estrogen receptor activation. Biochim. Biophys. Acta 2010, 1801, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Eggertsen, G.; Chiang, J.Y.; Norlin, M. Estrogen-mediated regulation of CYP7B1: A possible role for controlling DHEA levels in human tissues. J. Steroid Biochem. Mol. Biol. 2006, 100, 42–51. [Google Scholar] [CrossRef]
- Ahlem, C.N.; Page, T.M.; Auci, D.L.; Kennedy, M.R.; Mangano, K.; Nicoletti, F.; Ge, Y.; Huang, Y.; White, S.K.; Villegas, S.; et al. Novel components of the human metabolome: The identification, characterization and anti-inflammatory activity of two 5-androstene tetrols. Steroids 2011, 76, 145–155. [Google Scholar] [CrossRef]
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-adrenal glucocorticoid biosynthesis: Implications for autoimmune and inflammatory disorders. Genes Immun. 2020, 21, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. 11beta-hydroxysteroid dehydrogenases: A growing multi-tasking family. Mol. Cell Endocrinol. 2021, 526, 111210. [Google Scholar] [CrossRef] [PubMed]
- Hennebert, O.; Chalbot, S.; Alran, S.; Morfin, R. Dehydroepiandrosterone 7alpha-hydroxylation in human tissues: Possible interference with type 1 11beta-hydroxysteroid dehydrogenase-mediated processes. J. Steroid Biochem. Mol. Biol. 2007, 104, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Le Mee, S.; Hennebert, O.; Ferrec, C.; Wulfert, E.; Morfin, R. 7beta-Hydroxy-epiandrosterone-mediated regulation of the prostaglandin synthesis pathway in human peripheral blood monocytes. Steroids 2008, 73, 1148–1159. [Google Scholar] [CrossRef]
- Tchernof, A.; Mansour, M.F.; Pelletier, M.; Boulet, M.M.; Nadeau, M.; Luu-The, V. Updated survey of the steroid-converting enzymes in human adipose tissues. J. Steroid Biochem. Mol. Biol. 2015, 147, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hornsby, P.J.; Casson, P.; Morimoto, R.; Satoh, F.; Xing, Y.; Kennedy, M.R.; Sasano, H.; Rainey, W.E. Type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. J. Clin. Endocrinol. Metab. 2009, 94, 2192–2198. [Google Scholar] [CrossRef]
- Ostinelli, G.; Vijay, J.; Vohl, M.C.; Grundberg, E.; Tchernof, A. AKR1C2 and AKR1C3 expression in adipose tissue: Association with body fat distribution and regulatory variants. Mol. Cell Endocrinol. 2021, 527, 111220. [Google Scholar] [CrossRef]
- Rizner, T.L.; Penning, T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef]
- Luu-The, V. Assessment of steroidogenesis and steroidogenic enzyme functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 176–182. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Starka, L.; Duskova, M.; Hill, M. Dehydroepiandrosterone: A neuroactive steroid. J. Steroid Biochem. Mol. Biol. 2015, 145, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, W.; You, X.; Leimert, K.; Popowycz, K.; Fang, X.; Wood, S.L.; Slater, D.M.; Sun, Q.; Gu, H.; et al. PGF2alpha modulates the output of chemokines and pro-inflammatory cytokines in myometrial cells from term pregnant women through divergent signaling pathways. Mol. Hum. Reprod. 2015, 21, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Fei, J.; Feng, C.M.; Xu, Z.; Fu, L.; Zhao, H. Serum 8-iso-PGF2alpha Predicts the Severity and Prognosis in Patients With Community-Acquired Pneumonia: A Retrospective Cohort Study. Front. Med. 2021, 8, 633442. [Google Scholar] [CrossRef] [PubMed]
- Sharma, I.; Dhaliwal, L.K.; Saha, S.C.; Sangwan, S.; Dhawan, V. Role of 8-iso-prostaglandin F2alpha and 25-hydroxycholesterol in the pathophysiology of endometriosis. Fertil. Steril. 2010, 94, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Vallee, A. Neuroinflammation in Schizophrenia: The Key Role of the WNT/beta-Catenin Pathway. Int. J. Mol. Sci. 2022, 23, 2810. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.; Hana, V., Jr.; Velikova, M.; Parizek, A.; Kolatorova, L.; Vitku, J.; Skodova, T.; Simkova, M.; Simjak, P.; Kancheva, R.; et al. A method for determination of one hundred endogenous steroids in human serum by gas chromatography-tandem mass spectrometry. Physiol. Res. 2019, 68, 179–207. [Google Scholar] [CrossRef]
- Dehennin, L.; Peres, G. Plasma and urinary markers of oral testosterone misuse by healthy men in presence of masking epitestosterone administration. Int. J. Sports Med. 1996, 17, 315–319. [Google Scholar] [CrossRef]

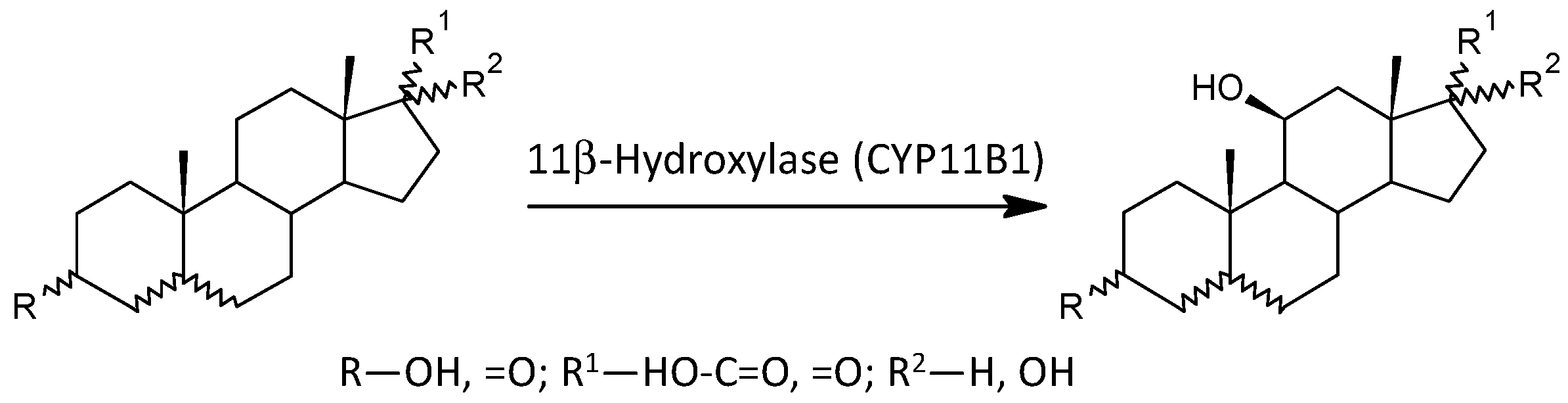


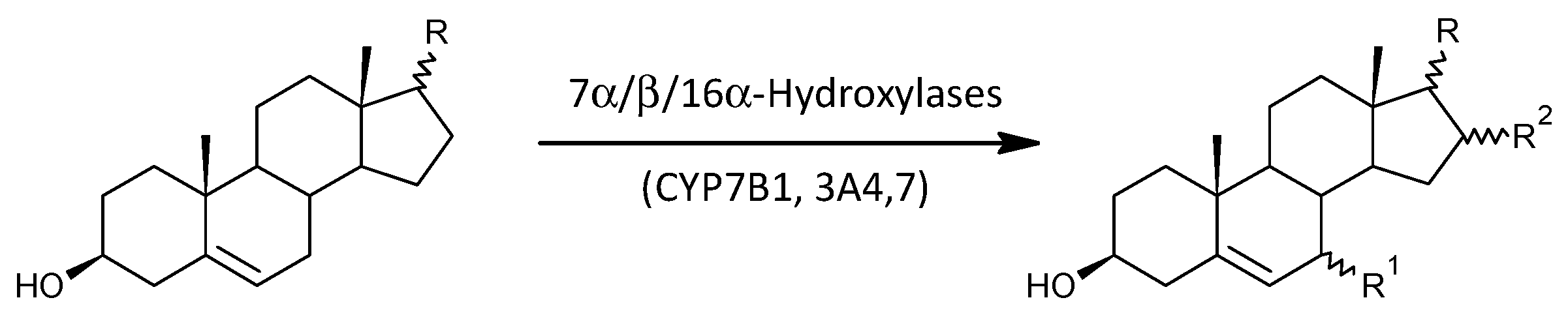







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OPLS, Predictive Component | MR | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Variable importance (VIP) | t-statistics | Component loading | t-statistics | R a | Regression coefficient | t-statistics | ||||||
| Explaining Variables | Steroids | PREG | 1.164 | 5.97 | ** | −0.169 | −5.11 | −0.576 | ** | −0.036 | −4.51 | ** | |
| 20α-Dihydro-PREG | 1.012 | 4.05 | ** | −0.169 | −6.75 | −0.575 | ** | −0.032 | −4.51 | ** | |||
| 17-OH-P | 0.907 | 5.85 | ** | −0.173 | −5.24 | −0.588 | ** | −0.028 | −4.40 | ** | |||
| 17-OH−20α-DHP | 1.413 | 4.80 | ** | −0.185 | −5.18 | −0.629 | ** | −0.044 | −3.45 | ** | |||
| T | 1.011 | 3.58 | ** | −0.157 | −4.30 | −0.535 | ** | −0.032 | −3.85 | ** | |||
| 5α-DHT | 0.745 | 3.13 | ** | −0.152 | −4.59 | −0.518 | ** | −0.023 | −3.26 | ** | |||
| 3β,5α-THP | 0.736 | 3.32 | ** | −0.135 | −3.72 | −0.460 | ** | −0.023 | −3.23 | ** | |||
| 17-OH-ALLO | 0.666 | 3.22 | ** | −0.150 | −5.35 | −0.509 | ** | −0.021 | −3.05 | ** | |||
| 3β,5α,20α-PD | 0.833 | 2.57 | * | −0.138 | −3.48 | −0.470 | ** | −0.026 | −2.72 | * | |||
| 3α,5β,17,20α-PT | 1.117 | 2.69 | * | −0.153 | −3.79 | −0.521 | ** | −0.035 | −2.16 | * | |||
| 3α,5α-THA | 0.895 | 2.23 | * | −0.053 | −1.13 | −0.182 | −0.028 | −1.89 | |||||
| 3β,5α,17β-AD | 1.036 | 2.71 | * | −0.105 | −2.12 | −0.358 | * | −0.032 | −2.26 | * | |||
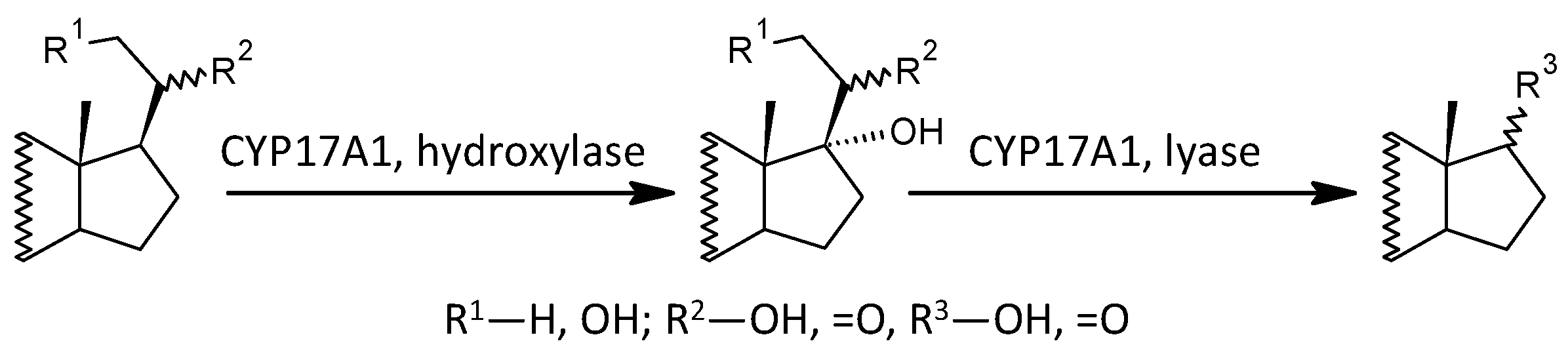
| CYP17A1 | hydroxylase + lyase | DHEA/PREG | 1.51 | 29.63 | ** | 0.239 | 15.17 | 0.816 | ** | 0.047 | 9.96 | ** | |
| DHEA/PREGS | 0.727 | 2.80 | * | 0.107 | 2.25 | 0.366 | * | 0.023 | 2.90 | * | |||
| DHEA/20α-Dihydro-PREG | 1.137 | 5.55 | ** | 0.206 | 10.40 | 0.702 | ** | 0.035 | 8.17 | ** | |||
| A/20α-DHP | 0.777 | 2.80 | * | 0.147 | 3.76 | 0.500 | ** | 0.024 | 2.72 | * | |||
| 5α-DHA/5α,20α-THP | 0.797 | 2.45 | * | 0.130 | 3.58 | 0.442 | ** | 0.025 | 2.67 | * | |||
| 3α,5α-THA, C/3α,5α,20α-PD, C | 1.077 | 4.02 | ** | 0.168 | 8.86 | 0.576 | ** | 0.034 | 5.25 | ** | |||
| 3β,5α-THA/3β,5α-THP | 0.925 | 3.44 | ** | 0.164 | 5.77 | 0.542 | ** | 0.029 | 3.69 | ** | |||
| 3β,5α-THA/3β,5α,20α-PD | 0.895 | 2.74 | * | 0.154 | 5.52 | 0.511 | ** | 0.028 | 3.23 | ** | |||
| hydroxylase | 17-OH-PREG/PREG | 1.364 | 8.04 | ** | 0.185 | 6.02 | 0.630 | ** | 0.043 | 6.05 | ** | ||
| 17-OH-PREGS/PREGS | 1.016 | 3.69 | ** | 0.114 | 3.29 | 0.393 | ** | 0.032 | 3.71 | ** | |||
| 16α-OH-PREG/PREG | 1.073 | 6.83 | ** | 0.165 | 4.29 | 0.562 | ** | 0.033 | 4.92 | ** | |||
| lyase | A/17-OH-P | 1.287 | 4.85 | ** | 0.218 | 10.14 | 0.742 | ** | 0.040 | 3.77 | ** | ||
| A/17-OH−20α-DHP | 1.233 | 3.60 | ** | 0.167 | 3.50 | 0.569 | ** | 0.038 | 2.88 | * | |||
| 3α,5α-THA, C/3α,5α,17,20α-PT, C | 0.906 | 3.04 | ** | 0.180 | 6.07 | 0.617 | ** | 0.028 | 3.35 | ** | |||
| 3α,5β-THA, C/3α,5β,17,20α-PT, C | 0.696 | 2.10 | * | 0.136 | 2.93 | 0.471 | * | 0.022 | 2.13 | * | |||
| HSD3Bs (HSD3B1,2) | 17-OH-P/17-OH-PREG | 1.214 | 4.74 | ** | −0.190 | −4.98 | −0.648 | ** | −0.038 | −4.22 | ** | ||
| 17-OH-P/17-OH-PREGS | 1.201 | 4.28 | ** | −0.171 | −4.46 | −0.587 | ** | −0.037 | −4.31 | ** | |||
| SULT2A1/STS | PREG, C/U | 0.864 | 3.46 | ** | 0.140 | 3.94 | 0.482 | ** | 0.027 | 3.54 | ** | ||
| 5α-DHT, C/U | 0.959 | 3.88 | ** | 0.138 | 4.25 | 0.473 | ** | 0.030 | 2.86 | * | |||
| 3α,5α-THA, C/U | 1.016 | 3.07 | ** | 0.157 | 4.06 | 0.536 | ** | 0.032 | 3.36 | ** | |||
| 3β,5α,17β-AD, C/U | 0.826 | 2.81 | * | 0.159 | 4.62 | 0.547 | ** | 0.026 | 2.48 | * | |||
| CYP11B1 | 11β-OH−3α,5α-THA, C/3α,5α-THA, C | 0.832 | 2.87 | * | −0.096 | −2.85 | −0.330 | * | −0.026 | −3.15 | ** | ||
| 11β-OH−3β,5α-THA/3β,5α-THA | 1.109 | 3.88 | ** | −0.111 | −2.69 | −0.368 | * | −0.035 | −3.51 | ** | |||
| 11β-OH−3β,5α-THA, C/3β,5α-THA, C | 0.94 | 3.49 | ** | −0.142 | −5.84 | −0.492 | ** | −0.029 | −3.33 | ** | |||
| SRD5As | (5α-DHT + 3α,5α,17β-AD + 3β,5α,17β-AD)/T, U + C | 0.879 | 3.50 | ** | 0.124 | 2.57 | 0.426 | * | 0.027 | 3.38 | ** | ||
| AKR1C2/HSD17B2,6 | 3α,5β-THP, C/3β,5β-THP, C | 0.741 | 2.80 | * | −0.088 | −2.95 | −0.303 | * | −0.023 | −2.36 | * | ||
| 3α,5α-THA, C/3β,5α-THA, C | 0.646 | 2.75 | * | 0.100 | 4.99 | 0.343 | ** | 0.020 | 3.01 | ** | |||
| AKR1C3/HSD17B2 | ADIOL/DHEA | 0.963 | 3.12 | ** | −0.155 | −4.42 | −0.527 | ** | −0.030 | −3.04 | ** | ||
| T/A | 0.964 | 5.63 | ** | −0.155 | −6.36 | −0.526 | ** | −0.030 | −5.78 | ** | |||
| 5α-DHT/5α-DHA | 1.105 | 4.66 | ** | −0.173 | −7.15 | −0.587 | ** | −0.034 | −4.67 | ** | |||
| 3β,5α,17β-AD/3β,5α-THA | 0.907 | 3.36 | ** | −0.175 | −6.49 | −0.581 | ** | −0.028 | −3.39 | ** | |||
| Explained Variable | Male schizophrenics, LLR b (vs. controls) | 1.000 | 17.71 | 0.695 | ** | ||||||||
| Explained variability = 48.3% (43.4% after cross-validation), sensitivity = 0.923 (0.851–0.996), specificity = 0.917 (0.76–1), shown as means with 95% CI. | |||||||||||||
| Steroids | Schizophrenia | Controls | ANOVA | MC, Stage 1 | OPLS, Stage 1 | |
|---|---|---|---|---|---|---|
| Δ5 Pregnanes (C21 steroids) | ||||||
| Pregnenolone [nM] | 1.31 (1.25, 1.39) | 2.12 (1.92, 2.34) | ↓ | *** | ↓ | ↓ |
| Pregnenolone sulfate [nM] | 397 (378, 417) | 430 (394, 469) | ||||
| 17-Hydroxypregnenolone [nM] | 13.1 (12.3, 14) | 12 (10.6, 13.6) | ||||
| 17-Hydroxypregnenolone sulfate [nM] | 32.3 (31, 33.7) | 32 (29.6, 34.5) | ↑ | |||
| 16α-Hydroxypregnenolone [nM] | 0.652 (0.629, 0.676) | 0.723 (0.676, 0.772) | ||||
| 20α-Dihydropregnenolone [nM] | 2.69 (2.6, 2.78) | 3.71 (3.49, 3.94) | ↓ | *** | ↓ | ↓ |
| 20α-Dihydropregnenolone sulfate [μM] | 1.58 (1.5, 1.66) | 1.92 (1.74, 2.12) | ↓ | * | ||
| Δ5 Androstanes (C19 steroids) | ||||||
| Dehydroepiandrosterone (DHEA) [nM] | 11.9 (11.4, 12.4) | 11.6 (10.7, 12.6) | ||||
| DHEA sulfate [μM] | 7.51 (7.19, 7.84) | 7.2 (6.67, 7.77) | ||||
| 7α-Hydroxy-DHEA [nM] | 1.33 (1.28, 1.39) | 1.42 (1.31, 1.53) | ||||
| 7-oxo-DHEA [nM] | 0.381 (0.336, 0.432) | 0.359 (0.279, 0.459) | ||||
| 7β-Hydroxy-DHEA [nM] | 0.693 (0.675, 0.712) | 0.682 (0.647, 0.717) | ||||
| Androstenediol [nM] | 3.27 (3.12, 3.42) | 4.18 (3.83, 4.55) | ↓ | *** | ↓ | |
| Androstenediol sulfate [μM] | 2.21 (2.06, 2.37) | 2.2 (1.93, 2.51) | ||||
| 5-Androstene-3β,7α,17β-triol [nM] | 0.698 (0.667, 0.73) | 0.784 (0.718, 0.854) | ||||
| 5-Androstene-3β,7β,17β-triol [nM] | 0.349 (0.331, 0.368) | 0.379 (0.342, 0.42) | ||||
| 5-Androstene-3β,16α,17β-triol [pM] | 187 (173, 201) | 143 (123, 166) | ↑ | * | ||
| 5-Androstene-3β,16α,17β-triol sulfate [nM] | 148 (137, 160) | 131 (114, 150) | ||||
| Δ4 Pregnanes (C21 steroids) | ||||||
| Progesterone [pM] | 101 (83.5, 122) | 152 (105, 228) | ||||
| 17-Hydroxyprogesterone [nM] | 3.25 (3.1, 3.41) | 4.43 (4.03, 4.88) | ↓ | *** | ↓ | ↓ |
| 17,20α-Dihydroxy-4-pregnen-3-one [nM] | 1.55 (1.47, 1.63) | 2.52 (2.27, 2.79) | ↓ | *** | ↓ | ↓ |
| Conjugated 17,20α-dihydroxy-4-pregnen-3-one [nM] | 9.68 (9.2, 10.2) | 11.9 (10.8, 13.1) | ↓ | * | ||
| 16α-Hydroxypogesterone [nM] | 0.733 (0.526, 0.679) | 0.824 (0.403, 0.594) | ||||
| 20α-Dihydroprogesterone [pM] | 162 (151, 174) | 213 (188, 241) | ↓ | * | ↓ | ↓ |
| Conjugated 20α-dihydroprogesterone [nM] | 1.44 (1.35, 1.54) | 1.28 (1.13, 1.44) | ||||
| Δ4 Androstanes (C19 steroids) | ||||||
| Androstenedione [nM] | 4.45 (4.28, 4.64) | 3.77 (3.48, 4.07) | ↑ | ** | ||
| Testosterone [nM] | 17 (16.4, 17.7) | 20.2 (18.8, 21.7) | ↓ | ** | ↓ | ↓ |
| Conjugated testosterone [nM] | 10.5 (9.57, 11.5) | 13.5 (11.5, 15.8) | ↓ | |||
| 5α Pregnanes (C21 steroids) | ||||||
| 5α-Dihydroprogesterone [pM] | 33.6 (29.9, 37.7) | 37.4 (30, 46.5) | ||||
| Allopregnanolone (3α,5α-THP) [pM] | 107 (98.3, 117) | 160 (137, 186) | ↓ | ** | ↓ | |
| Allopregnanolone (3α,5α-THP) sulfate [nM] | 6.46 (6.12, 6.82) | 7.74 (7, 8.54) | ↓ | * | ||
| Isopregnanolone (3β,5α-THP) [pM] | 76.9 (76.6, 102) | 128 (135, 212) | ↓ | ** | ↓ | ↓ |
| Isopregnanolone (3β,5α-THP) sulfate [nM] | 18.4 (17.7, 19.2) | 20 (18.5, 21.6) | ||||
| 17-Hydroxyallopregnanolone [pM] | 23.4 (21.1, 25.9) | 39.9 (33, 47.9) | ↓ | ** | ↓ | ↓ |
| 17-Hydroxyallopregnanolone sulfate [nM] | 4.54 (4.31, 4.77) | 5.21 (4.75, 5.68) | ||||
| 5α,20α-Tetrahydroprogesterone [pM] | 91.9 (86.1, 97.9) | 109 (97.2, 122) | ↓ | |||
| Conjugated 5α,20α-tetrahydroprogesterone [pM] | 122 (109, 136) | 78.9 (67.5, 93.2) | ↑ | ** | ||
| 5α-Pregnane-3α,20α-diol [pM] | 214 (196, 234) | 217 (183, 256) | ||||
| Conjugated 5α-pregnane-3α,20α-diol [nM] | 28.8 (27.3, 30.4) | 38.8 (35.3, 42.6) | ↓ | *** | ||
| 5α-Pregnane-3β,20α-diol [nM] | 1.21 (1.03, 1.38) | 1.94 (1.53, 2.28) | ↓ | * | ↓ | ↓ |
| Conjugated 5α-pregnane-3β,20α-diol [μM] | 2.66 (2.52, 2.81) | 3.58 (3.26, 3.92) | ↓ | *** | ↓ | ↓ |
| 5α-Pregnane-3α,17,20α-triol [pM] | 277 (259, 295) | 409 (367, 454) | ↓ | *** | ↓ | ↓ |
| Conjugated 5α-pregnane-3α,17,20α-triol [nM] | 57.2 (52.1, 62.7) | 52.5 (44, 62.3) | ||||
| 5α-Pregnane-3β,17,20α-triol [pM] | 255 (232, 281) | 361 (300, 433) | ↓ | * | ||
| Conjugated 5α-pregnane-3β,17,20α-triol [nM] | 5.12 (4.5, 5.83) | 7.19 (5.71, 9.09) | ||||
| 5β Pregnanes (C21 steroids) | ||||||
| Pregnanolone (THP-3α,5;) [pM] | 10.5 (9.21, 12.1) | 13.6 (10.5, 17.5) | ||||
| Pregnanolone (3α,5β-THP) sulfate [nM] | 27.8 (26.6, 29.1) | 32.4 (29.8, 35.3) | ↓ | * | ||
| Epipregnanolone (3β,5β-THP) sulfate [nM] | 3.58 (2.5, 2.89) | 3.75 (2.38, 2.97) | ||||
| 17-Hydroxypregnanolone [pM] | 40.8 (36.1, 45.9) | 65.7 (53.2, 80.8) | ↓ | ** | ↓ | |
| Conjugated 17-hydroxypregnanolone [nM] | 16.5 (15.7, 17.3) | 19.4 (17.9, 21.1) | ↓ | * | ↓ | |
| Conjugated 5β,20α-tetrahydroprogesterone [pM] | 178 (167, 189) | 249 (220, 281) | ↓ | *** | ↓ | |
| 5β-Pregnane-3α,20α-diol [pM] | 97.1 (88.5, 106) | 96 (79.8, 115) | ||||
| Conjugated 5β-pregnane-3α,20α-diol [nM] | 13.5 (12.7, 14.3) | 15.7 (14, 17.5) | ||||
| 5β-Pregnane-3β,20α-diol [pM] | 88.1 (74.6, 104) | 68.1 (53.3, 87.5) | ||||
| Conjugated 5β-pregnane-3β,20α-diol [nM] | 10.6 (14.2, 16.9) | 10.9 (15.9, 21.1) | ||||
| 5β-Pregnane-3α,17,20α-triol [nM] | 1.8 (1.7, 1.91) | 2.69 (2.45, 2.94) | ↓ | *** | ↓ | |
| Conjugated 5β-pregnane-3α,17,20α-triol [nM] | 128 (121, 136) | 183 (163, 205) | ↓ | *** | ↓ | |
| 5α Androstanes (C19 steroids) | ||||||
| 5α-Androstane-3,17-dione [pM] | 301 (286, 316) | 249 (226, 275) | ↑ | * | ↑ | |
| Androsterone (THA-3α,5α) [nM] | 1 (0.962, 1.04) | 1.18 (1.09, 1.27) | ↓ | ** | ↓ | |
| Androsterone (THA-3α,5α) sulfate [μM] | 4.15 (3.94, 4.36) | 3.24 (2.93, 3.58) | ↑ | ** | ↑ | ↑ |
| Epiandrosterone (THA-3β,5α) [pM] | 419 (400, 440) | 387 (351, 426) | ||||
| Epiandrosterone (THA-3β,5α) sulfate [μM] | 0.85 (0.807, 0.894) | 0.73 (0.663, 0.802) | ↑ | * | ↑ | |
| 5α-Dihydrotestosterone [nM] | 0.929 (0.882, 0.976) | 1.18 (1.08, 1.27) | ↓ | ** | ↓ | ↓ |
| Conjugated 5α-dihydrotestosterone [nM] | 4.99 (4.71, 5.28) | 3.6 (3.24, 4) | ↑ | *** | ↑ | |
| 5α-Androstane-3α,17β-diol [pM] | 388 (367, 410) | 373 (331, 416) | ||||
| Conjugated 5α-androstane-3α,17β-diol [nM] | 199 (191, 208) | 195 (180, 212) | ||||
| 5α-Androstane-3β,17β-diol [pM] | 85.7 (79.5, 92.3) | 128 (111, 147) | ↓ | *** | ↓ | ↓ |
| Conjugated 5α-androstane-3β,17β-diol [nM] | 190 (179, 202) | 170 (153, 190) | ||||
| 5β Androstanes (C19 steroids) | ||||||
| Etiocholanolone (THA-3α,5β) [pM] | 166 (159, 174) | 157 (144, 171) | ||||
| Etiocholanolone (THA-3α,5β) sulfate [nM] | 121 (115, 126) | 105 (96, 114) | ↑ | * | ||
| Epietiocholanolone (THA-3α,5β) sulfate [nM] | 35.3 (32.7, 38.2) | 36.1 (31.2, 41.7) | ||||
| 5β-Androstane-3α,17β-diol [pM] | 5.85 (5.18, 6.61) | 4.91 (3.91, 6.18) | ||||
| Conjugated 5β-androstane-3α,17β-diol [nM] | 15.9 (15.2, 16.7) | 18.7 (17.2, 20.4) | ↓ | * | ||
| Conjugated 5β-androstane-3β,17β-diol [nM] | 0.61 (0.416, 0.517) | 0.642 (0.433, 0.599) | ||||
| Glucocorticoids (Δ4 C21 steroids) and 11β-hydroxy-androstanes (Δ4, 5α/β- C19 steroids) | ||||||
| Cortisol [nM] | 378 (367, 389) | 388 (368, 408) | ||||
| Cortisone [nM] | 108 (103, 112) | 130 (121, 140) | ↓ | ** | ↓ | |
| Corticosterone [nM] | 18.4 (17.1, 19.7) | 17.2 (14.9, 19.6) | ↑ | |||
| 21-Deoxycortisol [pM] | 39.1 (33.9, 45.3) | 61.4 (46.6, 81.7) | ↓ | * | ||
| 11β-Hydroxyandrostenedione [nM] | 39.6 (37.4, 41.8) | 44 (39.7, 48.6) | ||||
| 11β-Hydroxyandrosterone [nM] | 2.21 (2.05, 2.37) | 3.48 (3.06, 3.95) | ↓ | *** | ↓ | ↓ |
| 11β-Hydroxyandrosterone sulfate [nM] | 44.5 (42.6, 46.5) | 52.1 (48, 56.4) | ↓ | * | ||
| 11β-Hydroxyepiandrosterone [pM] | 95.1 (87.7, 103) | 142 (124, 162) | ↓ | *** | ↓ | ↓ |
| 11β-Hydroxyepiandrosterone sulfate [nM] | 1.01 (0.939, 1.09) | 1.57 (1.37, 1.81) | ↓ | *** | ↓ | |
| 11β-Hydroxyetiocholanolone [nM] | 1.33 (1.24, 1.41) | 1.56 (1.39, 1.75) | ↓ | |||
| 11β-Hydroxyetiocholanolone sulfate [nM] | 8.37 (7.8, 8.98) | 12.4 (11, 14) | ↓ | *** | ↓ | |
| Molar Ratios | Schizophrenia | Controls | ANOVA | MC, Stage 1 | OPLS, Stage 1 | |
|---|---|---|---|---|---|---|
| C17-Hydroxylase, C17,20-lyase (CYP17A1), hydroxylase + lyase | ||||||
| DHEA/PREG | 5.49 (5.12, 5.88) | 8.79 (8.49, 9.09) | ↑ | *** | ↑ | ↑ |
| DHEA/PREG, C | 16.2 (15.3, 17.2) | 19.1 (18.4, 19.7) | ↑ | ** | ↑ | ↑ |
| DHEA/20α-dihydro-PREG | 3.01 (2.75, 3.29) | 4.36 (4.14, 4.59) | ↑ | *** | ↑ | ↑ |
| DHEA/20α-dihydro-PREG, C | 3.23 (3.02, 3.46) | 4.46 (4.29, 4.65) | ↑ | *** | ↑ | ↑ |
| A/P | 25.7 (18.2, 35.8) | 40.1 (33.7, 47.5) | ||||
| A/20α-DHP | 18.6 (17, 20.3) | 27.9 (26.3, 29.5) | ↑ | *** | ↑ | |
| 5α-DHA/5α-DHP | 7.38 (5.91, 9.23) | 8.66 (7.69, 9.76) | ||||
| 5α-DHA/5α,20α-THP | 2.44 (2.21, 2.71) | 3.48 (3.27, 3.71) | ↑ | *** | ↑ | ↑ |
| 3α,5α-THA/ALLO | 6.81 (6.14, 7.58) | 9.87 (9.31, 10.5) | ↑ | *** | ↑ | |
| 3α,5α-THA/3α,5α,20α-PD | 5 (4.28, 5.88) | 4.65 (4.27, 5.07) | ||||
| 3α,5α-THA/ALLO, C | 396 (368, 427) | 633 (608, 660) | ↑ | *** | ↑ | |
| 3α,5α-THA/3α,5α,20α-PD, C | 77.4 (71.8, 83.4) | 130 (125, 136) | ↑ | *** | ↑ | ↑ |
| 3β,5α-THA/3β,5α-THP | 3.27 (2.98, 3.58) | 5.94 (5.62, 6.3) | ↑ | *** | ↑ | ↑ |
| 3β,5α-THA/3β,5α,20α-PD | 0.236 (0.203, 0.275) | 0.349 (0.32, 0.38) | ↑ | ** | ↑ | ↑ |
| 3β,5α-THA/3β,5α-THP, C | 35.7 (33, 38.5) | 44.7 (42.9, 46.5) | ↑ | *** | ↑ | |
| 3β,5α-THA/3β,5α,20α-PD, C | 0.22 (0.2, 0.242) | 0.326 (0.311, 0.342) | ↑ | *** | ↑ | ↑ |
| 3α,5β-THA/3α,5β-THP | 11.7 (9.24, 14.9) | 15.2 (13.3, 17.3) | ||||
| 3α,5β-THA/3α,5β,20α-PD | 1.66 (1.47, 1.89) | 1.8 (1.68, 1.92) | ||||
| 3α,5β-THA/3α,5β-THP, C | 3.21 (3.02, 3.41) | 4.26 (4.12, 4.41) | ↑ | *** | ↑ | |
| 3α,5β-THA/3α,5β,20α-PD, C | 6.31 (5.83, 6.83) | 8.33 (7.97, 8.71) | ↑ | *** | ↑ | |
| 3β,5β-THA/3β,5β-THP, C | 9.82 (8.77, 11) | 10.2 (9.56, 10.8) | ||||
| 3β,5β-THA/3α,5β,20α-PD, C | 3.56 (3.18, 3.98) | 3.62 (3.4, 3.85) | ||||
| 11β-OH-A/corticosterone | 2.56 (2.3, 2.85) | 2.32 (2.19, 2.46) | ↓ | |||
| C17-Hydroxylase, C17,20-lyase (CYP17A1), hydroxylase | ||||||
| 103·17-OH-PREG/PREG | 66 (60.5, 71.8) | 88.1 (84.5, 91.7) | ↑ | *** | ↑ | ↑ |
| 103·17-OH-PREG/PREGS | 5.96 (5.5, 6.46) | 9.26 (8.85, 9.69) | ↑ | *** | ↑ | ↑ |
| 17-OH-P/P | 31.2 (22.1, 43.4) | 32.9 (27.5, 39.3) | ||||
| 17-OH-20α-DHP/20α-DHP | 31.2 (22.1, 43.4) | 32.9 (27.5, 39.3) | ||||
| 17-OH-20α-DHP/20α-DHP, C | 10.1 (8.91, 11.4) | 6.31 (5.94, 6.72) | ↓ | *** | ↓ | ↓ |
| 17-OH-ALLO/ALLO | 0.27 (0.222, 0.331) | 0.22 (0.199, 0.243) | ||||
| 17-OH-ALLO/ALLO, C | 0.735 (0.683, 0.79) | 0.728 (0.7, 0.757) | ||||
| 17-OH-3α,5β-THP/3α,5β-THP | 3.57 (2.68, 4.75) | 3.45 (2.96, 4.03) | ||||
| 17-OH-3α,5β-THP/3α,5β-THP, C | 0.56 (0.519, 0.603) | 0.595 (0.572, 0.619) | ||||
| 3α,5α,17,20α-PT/3α,5α,20α-PD | 2.11 (1.75, 2.53) | 1.58 (1.42, 1.76) | ↓ | |||
| 3α,5α,17,20α-PT/3α,5α,20α-PD, C | 1.2 (1.03, 1.41) | 1.78 (1.63, 1.94) | ↑ | ** | ↑ | |
| 103·3β,5α,17,20α-PT/3β,5α,20α-PD | 192 (154, 238) | 230 (205, 258) | ||||
| 103·3β,5α,17,20α-PT/3β,5α,20α-PD, C | 2.32 (1.78, 3.05) | 2.11 (1.82, 2.45) | ||||
| 3α,5β,17,20α-PT/3α,5β,20α-PD | 25.7 (22, 30) | 18.9 (17.5, 20.4) | ↓ | * | ↓ | ↓ |
| 3α,5β,17,20α-PT/3α,5β,20α-PD, C | 10.1 (8.74, 11.7) | 8.97 (8.29, 9.7) | ||||
| Cortisol/corticosterone | 23 (21, 25.2) | 20.3 (19.3, 21.3) | ↓ | ↓ | ||
| C17-Hydroxylase, C17,20-lyase (CYP17A1), lyase | ||||||
| DHEA/17-OH-PREG | 0.933 (0.864, 1.01) | 0.927 (0.888, 0.967) | ||||
| DHEA/17-OH-PREGS | 227 (205, 252) | 224 (212, 237) | ||||
| A/17-OH-P | 0.856 (0.811, 0.904) | 1.26 (1.22, 1.3) | ↑ | *** | ↑ | ↑ |
| A/17-OH-20α-DHP | 1.59 (1.43, 1.75) | 2.8 (2.66, 2.93) | ↑ | *** | ↑ | ↑ |
| 3α,5α-THA/17-OH-ALLO | 27.1 (23.1, 32.2) | 41.4 (37.6, 45.6) | ↑ | ** | ||
| 10−3·3α,5α-THA/17-OH-ALLO, C | 0.532 (0.483, 0.586) | 0.909 (0.862, 0.96) | ↑ | *** | ↑ | ↑ |
| 3α,5β-THA/17-OH-3α,5β-THP | 3.13 (2.54, 3.89) | 4.28 (3.8, 4.83) | ↑ | |||
| 3α,5β-THA/17-OH-3α,5β-THP, C | 5.24 (4.89, 5.62) | 7.01 (6.74, 7.29) | ↑ | *** | ↑ | ↑ |
| 3α,5α-THA/3α,5α,17,20α-PT | 2.66 (2.42, 2.94) | 3.54 (3.33, 3.76) | ↑ | ** | ↑ | |
| 3α,5α-THA/3α,5α,17,20α-PT, C | 61.8 (51.8, 73.4) | 78.2 (71.1, 85.9) | ↑ | |||
| 3β,5α-THA/3β,5α,17,20α-PT | 1.06 (0.908, 1.24) | 1.55 (1.42, 1.69) | ↑ | ** | ↑ | ↑ |
| 3β,5α-THA/3β,5α,17,20α-PT, C | 102 (80.8, 130) | 151 (132, 172) | ↑ | * | ↑ | |
| 103·3α,5β-THA/3α,5β,17,20α-PT | 62.9 (57.3, 69) | 96.6 (92.2, 101) | ↑ | *** | ↑ | ↑ |
| 3α,5β-THA/3α,5β,17,20α-PT, C | 0.563 (0.503, 0.629) | 0.938 (0.884, 0.996) | ↑ | *** | ↑ | |
| 103·11β-OH-A/cortisol | 111 (103, 120) | 109 (104, 113) | ||||
| 10−3·11β-OH-A/21-deoxycortisol | 0.687 (0.528, 0.892) | 0.969 (0.843, 1.11) | ||||
| 3β-Hydroxysteroid dehydrogenases (type 1 and 2) (HSD3B1,2) | ||||||
| 103·P/PREG | 82.7 (60.5, 116) | 72.9 (61.9, 86.4) | ||||
| 103·P/PREGS | 0.352 (0.252, 0.506) | 0.261 (0.22, 0.311) | ||||
| 103·20α-DHP/20α-dihydro-PREG | 57 (50.9, 63.5) | 58 (54.6, 61.4) | ||||
| 103·20α-DHP/20α-dihydro-PREGS | 100 (85.7, 118) | 83.4 (76.9, 90.4) | ||||
| 103·20α-DHP/20α-dihydro-PREGS | 0.699 (0.624, 0.782) | 0.916 (0.865, 0.971) | ↑ | ** | ||
| 17-OH-P/17-OH-PREG | 0.374 (0.346, 0.404) | 0.263 (0.253, 0.274) | ↓ | *** | ↓ | ↓ |
| 103·17-OH-P/17-OH-PREGS | 155 (144, 167) | 106 (101, 111) | ↓ | *** | ↓ | ↓ |
| 16α-OH-P/16α-OH-PREG | 1.24 (1.18, 1.31) | 1.14 (1.1, 1.17) | ||||
| A/DHEA | 0.376 (0.348, 0.407) | 0.361 (0.346, 0.376) | ||||
| 103·A/DHEA sulfate | 0.54 (0.479, 0.609) | 0.544 (0.509, 0.58) | ||||
| T/ADIOL | 4.66 (4.43, 4.91) | 5.08 (4.93, 5.23) | ↑ | * | ||
| 103·T, C/ADIOLS | 7.15 (5.95, 8.59) | 4.15 (3.75, 4.58) | ↓ | *** | ↓ | ↓ |
| Conjugated/unconjugated steroid ratio—Steroid sulfotransferase 2A1 (SULT2A1) vs. steroid sulfatase (STS) | ||||||
| PREG, C/U | 215 (196, 236) | 288 (275, 301) | ↑ | *** | ↑ | ↑ |
| 20α-Dihydro-PREG, C/U | 595 (529, 671) | 636 (599, 676) | ||||
| 17-OH-PREG, C/U | 2.61 (2.38, 2.86) | 2.61 (2.48, 2.74) | ||||
| DHEA, C/U | 609 (551, 676) | 642 (609, 678) | ||||
| ADIOL, C/U | 598 (525, 680) | 656 (613, 703) | ||||
| 10−3·3β,16α,17β-AT, C/U | 1 (0.854, 1.18) | 0.841 (0.774, 0.915) | ||||
| 20α-DHP, C/U | 6.36 (5.42, 7.5) | 9.65 (8.82, 10.6) | ↑ | ** | ↑ | ↑ |
| 17-OH-20α-DHP, C/U | 4.76 (4.17, 5.47) | 5.9 (5.47, 6.38) | ↑ | ↑ | ||
| T, C/U | 0.619 (0.505, 0.759) | 0.581 (0.522, 0.647) | ||||
| 5α-DHT, C/U | 3.59 (3.1, 4.15) | 6.43 (5.99, 6.9) | ↑ | *** | ↑ | ↑ |
| ALLO, C/U | 46.7 (41.2, 53.3) | 64.5 (60, 69.6) | ↑ | ** | ↑ | |
| 3β,5α-THP, C/U | 185 (163, 210) | 252 (235, 270) | ↑ | ** | ↑ | |
| 10−3·3α,5β-THP, C/U | 2.63 (1.97, 3.51) | 2.79 (2.4, 3.23) | ||||
| 17-OH-ALLO, C/U | 114 (95.5, 136) | 187 (169, 207) | ↑ | ** | ||
| 17-OH-3α,5β-THP, C/U | 317 (260, 390) | 413 (369, 464) | ||||
| 5α,20α-THP, C/U | 0.811 (0.665, 1) | 1.43 (1.26, 1.62) | ↑ | ** | ↑ | |
| 3α,5α,20α-PD, C/U | 213 (179, 253) | 174 (158, 192) | ||||
| 10−3·3β,5α,20α-PD, C/U | 1.86 (1.6, 2.19) | 2.4 (2.19, 2.63) | ↑ | |||
| 3α,5β,20α-PD, C/U | 180 (150, 215) | 174 (159, 191) | ||||
| 3β,5β,20α-PD, C/U | 149 (111, 197) | 134 (110, 162) | ↑ | |||
| 3α,5α,17,20α-PT, C/U | 97.8 (81.9, 117) | 173 (156, 192) | ↑ | *** | ↑ | |
| 3β,5α,17,20α-PT, C/U | 20 (17.1, 23.4) | 20.8 (19.1, 22.6) | ||||
| 10−3·3α,5β,17,20α-PT, C/U | 2.66 (2.33, 3.03) | 4.29 (4.03, 4.57) | ↑ | *** | ||
| 10−3·3α,5α-THA, C/U | 2.66 (2.33, 3.03) | 4.29 (4.03, 4.57) | ↑ | *** | ↑ | ↑ |
| 10−3·3β,5α-THA, C/U | 1.9 (1.65, 2.18) | 1.93 (1.81, 2.06) | ||||
| 10−3·3α,5β-THA, C/U | 0.68 (0.621, 0.744) | 0.782 (0.746, 0.819) | ↑ | |||
| 10−3·3α,5α,17β-AD, C/U | 0.5 (0.452, 0.555) | 0.512 (0.486, 0.539) | ||||
| 10−3·3β,5α,17β-AD, C/U | 1.53 (1.25, 1.89) | 2.35 (2.12, 2.61) | ↑ | * | ↑ | |
| 10−3·3α,5β,17β-AD, C/U | 2.39 (1.86, 3.08) | 2.66 (2.34, 3.03) | ||||
| 11β-OH-3α,5α-THA, C/U | 15 (13.5, 16.7) | 20.4 (19.2, 21.6) | ↑ | ** | ↑ | |
| 11β-OH-3β,5α-THA, C/U | 10.1 (8.65, 11.8) | 10.5 (9.67, 11.5) | ||||
| 11β-OH-3β,5α-THA, C/U | 8.2 (7.08, 9.44) | 6.7 (6.16, 7.28) | ||||
| 11β-Hydroxylase (CYP11B1) | ||||||
| 11β-OH-A/A | 11.1 (10.2, 12.1) | 8.96 (8.56, 9.38) | ↓ | ** | ↓ | |
| 11β-OH-3α,5α-THA/3α,5α-THA | 3.03 (2.68, 3.4) | 2.36 (2.2, 2.53) | ↓ | * | ↓ | ↓ |
| 103·11β-OH-3α,5α-THA/3α,5α-THA, C | 16.4 (14.7, 18.3) | 10.9 (10.3, 11.5) | ↓ | *** | ↓ | ↓ |
| 11β-OH-3β,5α-THA/3β,5α-THA | 0.348 (0.303, 0.398) | 0.234 (0.216, 0.254) | ↓ | ** | ↓ | ↓ |
| 103·11β-OH-3β,5α-THA/3β,5α-THA, C | 1.9 (1.67, 2.17) | 1.17 (1.1, 1.25) | ↓ | *** | ↓ | ↓ |
| 11β-OH-3β,5α-THA/3α,5β-THA | 10.1 (9.25, 11.1) | 8.53 (8.11, 8.97) | ↓ | * | ↓ | |
| 103·11β-OH-3β,5α-THA/3α,5β-THA, C | 94.8 (83.4, 108) | 67.6 (63, 72.5) | ↓ | ** | ↓ | ↓ |
| 7α/β-,16α-Hydroxylases (CYP7B1, CYP3A4,7) | ||||||
| 103·7α-OH-DHEA/DHEA | 118 (110, 126) | 114 (110, 118) | ||||
| 103·3β,7α,17β-AT/ADIOL | 180 (168, 193) | 210 (203, 218) | ↑ | ** | ↑ | |
| 103·7β-OH-DHEA/DHEA | 52 (47.8, 56.6) | 58.4 (55.9, 61.1) | ||||
| 103·3β,7β,17β-AT/ADIOL | 88.5 (82.1, 95.4) | 110 (105, 114) | ↑ | *** | ↑ | |
| 103·16α-OH-PREG/PREG | 347 (322, 373) | 448 (430, 467) | ↑ | *** | ↑ | ↑ |
| 103·3β,16α,17β-AT/ADIOL | 32.1 (27.8, 36.8) | 54.5 (50.8, 58.4) | ↑ | *** | ↑ | ↑ |
| 103·3β,16α,17β-AT/ADIOLS | 54.8 (49.1, 61.4) | 62.8 (58.9, 67) | ||||
| 16α-OH-P/P | 5.82 (4.27, 7.8) | 7.73 (6.62, 9) | ||||
| 11β-Hydroxysteroid dehydrogenase, type 1 (HSD11B1) | ||||||
| 7-oxo-DHEA/7α-OH-DHEA | 0.285 (0.225, 0.355) | 0.323 (0.287, 0.362) | ||||
| 7β-OH-DHEA/7α-OH-DHEA | 0.475 (0.448, 0.503) | 0.533 (0.516, 0.549) | ↑ | * | ||
| 3β,7β,17β-AT/3β,7α,17β-AT | 0.493 (0.47, 0.518) | 0.523 (0.51, 0.538) | ||||
| Cortisol/cortisone | 3.04 (2.83, 3.26) | 3.32 (3.19, 3.44) | ||||
| 5α-Reductases, type 1 and 2 (SRD5A1,2) | ||||||
| (5α-DHP+3α/β,5α-THP)/P | 1.76 (1.26, 2.43) | 1.98 (1.66, 2.35) | ||||
| (3α/β,5α-THP, C)/P | 169 (115, 243) | 252 (209, 303) | ||||
| (5α-DHP+3α/β,5α-THP+3α/β,5α-THP, all C)/P | 169 (115, 243) | 252 (209, 303) | ||||
| (5α,20α-THP+3α/β,5α,20α-PD)/20α-DHP | 10.1 (8.78, 11.7) | 9.67 (8.95, 10.5) | ||||
| 10−3·(5α,20α-THP+3α/β,5α,20α-PD)/20α-DHP, all C | 2.44 (2.19, 2.71) | 1.79 (1.7, 1.9) | ↓ | *** | ↓ | |
| 10−3·(5α,20α-THP+3α/β,5α,20α-PD)/20α-DHP, all C+U | 1.94 (1.73, 2.16) | 1.56 (1.47, 1.65) | ↓ | * | ||
| 103·3α,5α,17-PD/17-OH-P | 9.87 (8.22, 11.8) | 6.64 (6.03, 7.31) | ↓ | ** | ||
| 3α,5α,17-PD, C/17-OH-P | 1.08 (0.973, 1.21) | 1.31 (1.24, 1.39) | ↑ | * | ↑ | |
| 3α,5α,17-PD, C+U/17-OH-P | 1.07 (0.936, 1.21) | 1.34 (1.25, 1.43) | ↑ | * | ↑ | |
| 3α,5α,17,20α-PT/17-OH-20α-DHP | 0.281 (0.244, 0.324) | 0.322 (0.299, 0.346) | ||||
| 3α,5α,17,20α-PT/17-OH-20α-DHP, all C | 4.66 (3.95, 5.51) | 6.53 (5.94, 7.19) | ↑ | * | ||
| 3α,5α,17,20α-PT/17-OH-20α-DHP, all C+U | 3.82 (3.25, 4.49) | 5.54 (5.06, 6.07) | ↑ | ** | ||
| (5α-DHA+3α/β,5α-THA)/A | 0.437 (0.406, 0.47) | 0.384 (0.37, 0.398) | ↓ | * | ↓ | |
| 10−3·3α/β,5α-THA, C/A | 0.971 (0.846, 1.11) | 1.16 (1.08, 1.24) | ↑ | ↑ | ||
| 10−3·(5α-DHA+3α/β,5α-THA, all C+U)/A | 0.992 (0.863, 1.13) | 1.15 (1.08, 1.23) | ↑ | |||
| 10−3·(5α-DHT+3α/β,5α-AD)/T | 84 (78.4, 90.1) | 78.7 (75.8, 81.6) | ||||
| (5α-DHT+3α/β,5α-AD)/T, all C | 31.1 (25.2, 38.4) | 44.3 (39.5, 49.6) | ↑ | * | ↑ | |
| (5α-DHT+3α/β,5α-AD)/T, all C+U | 11.8 (10.3, 13.3) | 14.6 (13.7, 15.6) | ↑ | * | ↑ | ↑ |
| 103·11β-OH-3α/β,5α-THA/11β-OH-A | 82.7 (74.7, 91.1) | 62.2 (58.5, 66) | ↓ | ** | ↓ | |
| 11β-OH-3α/β,5α-THA, C/11β-OH-A | 1.17 (1.06, 1.3) | 1.18 (1.12, 1.25) | ||||
| 11β-OH-3α/β,5α-THA, C+U/11β-OH-A | 1.27 (1.15, 1.4) | 1.25 (1.19, 1.32) | ||||
| 5β-Reductase (AKR1D1) | ||||||
| 103·3α,5β-THP/P | 76 (49.5, 115) | 92.1 (73.7, 115) | ||||
| 3α/β,5β-THP, C/P | 230 (156, 333) | 323 (267, 390) | ||||
| 3α/β,5β-THP, C+U/P | 230 (156, 333) | 323 (267, 390) | ||||
| (3α/β,5β,20α-PD)/20α-DHP | 0.834 (0.699, 1) | 1.09 (0.963, 1.25) | ||||
| (5β,20α-THP+3α/β,5β,20α-PD)/20α-DHP, all C | 19.8 (17.7, 22.2) | 16.4 (15.5, 17.4) | ↓ | * | ||
| (5β,20α-THP+3α/β,5β,20α-PD)/20α-DHP, all C+U | 16.2 (14.5, 18.1) | 16.4 (15.2, 17.6) | ↑ | |||
| 103·3α,5β,17-PD/17-OH-P | 11.9 (8.96, 15.6) | 11.6 (9.93, 13.4) | ||||
| 3α,5β,17-PD, C/17-OH-P | 4.02 (3.65, 4.43) | 4.94 (4.69, 5.2) | ↑ | * | ||
| 3α,5β,17-PD, C+U/17-OH-P | 4.03 (3.66, 4.44) | 4.96 (4.7, 5.22) | ↑ | * | ||
| 3α,5β,17,20α-PT/17-OH-20α-DHP | 0.985 (0.89, 1.09) | 1.05 (0.992, 1.1) | ||||
| 3α,5β,17,20α-PT/17-OH-20α-DHP, all C | 12.2 (10.8, 13.7) | 12.8 (12.1, 13.6) | ||||
| 3α,5β,17,20α-PT/17-OH-20α-DHP, all C+U | 9.98 (8.91, 11.2) | 11 (10.4, 11.7) | ||||
| 103·3α,5β-THA/A | 40.5 (36.6, 44.8) | 36.9 (34.9, 39) | ||||
| 3α/β,5β-THA, C/A | 37.3 (32.7, 42.6) | 37.2 (34.7, 39.9) | ||||
| 3α/β,5β-THA, C+U/A | 37.3 (32.7, 42.6) | 37.2 (34.7, 39.9) | ||||
| 3α,5β,17β-AD/T | 1.42 (1.2, 1.68) | 1.66 (1.51, 1.83) | ||||
| 3α/β,5β,17β-AD/T, C | 1.42 (1.2, 1.68) | 1.66 (1.51, 1.83) | ||||
| 3α/β,5β,17β-AD/T, C+U | 0.554 (0.499, 0.615) | 0.561 (0.531, 0.592) | ||||
| 103·11β-OH-3α,5β-THA/11β-OH-A | 38.2 (35.4, 41.1) | 33.1 (31.7, 34.6) | ↓ | * | ↓ | |
| 11β-OH-3α,5β-THA, C/11β-OH-A | 0.269 (0.228, 0.317) | 0.215 (0.196, 0.236) | ||||
| 11β-OH-3α,5β-THA, C+U/11β-OH-A | 0.314 (0.273, 0.363) | 0.251 (0.233, 0.272) | ||||
| Aldoketoreductase 1C1 (AKR1C1) vs. 17β-hydroxysteroid dehydrogenase, type 2 (HSD17B2) | ||||||
| 20α-Dihydro-PREG/PREG | 1.78 (1.62, 1.95) | 1.88 (1.79, 1.98) | ||||
| 20α-Dihydro-PREGS/PREGS | 4.78 (4.59, 4.98) | 4.07 (3.98, 4.15) | ↓ | *** | ↓ | |
| 20α-DHP/P | 1.39 (1.01, 1.89) | 1.5 (1.27, 1.76) | ||||
| 20α-DHP, C/P | 7.89 (5.41, 11.4) | 13.5 (11.1, 16.4) | ↑ | |||
| 17-OH-20α-DHP/17-OH-P | 0.523 (0.479, 0.574) | 0.456 (0.437, 0.477) | ||||
| 17-OH-20α-DHP, C/17-OH-P | 2.69 (2.41, 3.01) | 2.95 (2.77, 3.13) | ↑ | |||
| 5α,20α-THP/5α-DHP | 2.65 (2.11, 3.33) | 2.35 (2.07, 2.66) | ||||
| 5α,20α-THP, C/5α-DHP | 2.1 (1.61, 2.79) | 3.33 (2.85, 3.91) | ↑ | * | ||
| 3α,5α,20α-PD/ALLO | 1.16 (1.01, 1.33) | 2.03 (1.87, 2.2) | ↑ | *** | ↑ | |
| 3α,5α,20α-PD, C/ALLO, C | 5.27 (4.95, 5.61) | 4.57 (4.43, 4.72) | ↓ | ** | ↓ | |
| 3β,5α,20α-PD/3β,5α-THP | 13.6 (11.6, 16.1) | 15.1 (13.9, 16.6) | ||||
| 3β,5α,20α-PD, C/3β,5α-THP, C | 173 (165, 182) | 141 (137, 145) | ↓ | *** | ↓ | |
| 3α,5β,20α-PD/3α,5β-THP | 7.42 (5.79, 9.57) | 7.58 (6.64, 8.67) | ||||
| 3α,5β,20α-PD, C/3α,5β-THP, C | 0.488 (0.448, 0.532) | 0.498 (0.475, 0.522) | ||||
| 3β,5β,20α-PD, C/3α,5β-THP, C | 2.84 (2.65, 3.05) | 3 (2.89, 3.11) | ||||
| 3α,5α,17,20α-PT/17-OH-ALLO | 8.37 (7.26, 9.7) | 11.3 (10.5, 12.3) | ↑ | * | ||
| 3α,5α,17,20α-PT, C/17-OH-ALLO, C | 9.33 (8.01, 10.9) | 11.3 (10.4, 12.3) | ||||
| 3α,5β,17,20α-PT/17-OH-3α,5β-THP | 42.5 (35.9, 50.6) | 45.1 (41, 49.6) | ||||
| 3α,5β,17,20α-PT, C/17-OH-3α,5β-THP, C | 7.69 (6.95, 8.51) | 7.68 (7.28, 8.11) | ||||
| Aldoketoreductase 1C2 (AKR1C2) vs. 17β-hydroxysteroid dehydrogenases, type 2 and 6 (HSD17B2,6) | ||||||
| ALLO/3β,5α-THP | 1.43 (1.25, 1.63) | 1.63 (1.52, 1.74) | ||||
| ALLO, C/3β,5α-THP, C | 0.383 (0.353, 0.415) | 0.338 (0.323, 0.352) | ||||
| 3α,5β-THP, C/3β,5β-THP, C | 8.69 (8.19, 9.22) | 7.83 (7.59, 8.09) | ↓ | * | ↓ | |
| 1000·3α,5α,20α-PD/3β,5α,20α-PD | 118 (99.5, 141) | 162 (148, 179) | ↑ | * | ||
| 1000·3α,5α,20α-PD, C/3β,5α,20α-PD, C | 11.2 (10.4, 12.1) | 11 (10.5, 11.4) | ||||
| 3α,5β,20α-PD, C/3β,5β,20α-PD, C | 1.49 (1.39, 1.6) | 1.36 (1.31, 1.41) | ||||
| 3α,5α,17,20α-PT/3β,5α,17,20α-PT | 1.05 (0.943, 1.17) | 1.1 (1.04, 1.17) | ||||
| 3α,5α,17,20α-PT, C/3β,5α,17,20α-PT, C | 6.63 (5.22, 8.48) | 9.39 (8.2, 10.8) | ||||
| 3α,5α-THA/3β,5α-THA | 6.63 (5.22, 8.48) | 9.39 (8.2, 10.8) | ||||
| 3α,5α-THA, C/3β,5α-THA, C | 4.31 (4.05, 4.57) | 4.76 (4.61, 4.92) | ↑ | * | ↑ | ↑ |
| 3α,5β-THA/3β,5β-THA, C | 3.1 (2.79, 3.42) | 3.6 (3.41, 3.79) | ||||
| 3α,5α,17β-AD, C/3β,5α,17β-AD, C | 2.79 (2.39, 3.23) | 4.62 (4.29, 4.98) | ↑ | *** | ↑ | |
| 3α,5α,17β-AD, C/3β,5α,17β-AD, C | 1.27 (1.19, 1.36) | 1.06 (1.02, 1.1) | ↓ | ** | ||
| 3α,5β,17β-AD, C/3β,5β,17β-AD, C | 32 (28.6, 35.9) | 26.4 (24.9, 28) | ↓ | * | ↓ | |
| 11β-OH-3α,5α-THA/11β-OH-3β,5α-THA | 32 (28.6, 35.9) | 26.4 (24.9, 28) | ↓ | * | ↓ | |
| 11β-OH-3α,5α-THA, C11β-OH-3β,5α-THA, C | 35.4 (31.5, 39.9) | 41.9 (39.1, 44.8) | ||||
| ALLO/5α-DHP | 3.9 (3.15, 4.81) | 2.86 (2.54, 3.21) | ↓ | |||
| ALLO, C/5α-DHP | 191 (152, 241) | 184 (163, 207) | ||||
| ALLO, C+U/5α-DHP | 196 (156, 247) | 188 (168, 212) | ||||
| 3α,5α,20α-PD/5α,20α-THP | 1.82 (1.54, 2.15) | 2.15 (1.96, 2.37) | ||||
| 3α,5α,20α-PD, C/5α,20α-THP, C | 387 (325, 464) | 221 (203, 241) | ↓ | *** | ↓ | |
| 3α,5α,20α-PD, C+U/5α,20α-THP, C+U | 407 (353, 471) | 361 (335, 389) | ||||
| 3α,5β,20α-PD, C/5β,20α-THP, C | 70 (62.9, 77.9) | 76.4 (72, 81) | ↑ | |||
| 3α,5α-THA/5α-DHA | 4.2 (3.78, 4.68) | 3.26 (3.08, 3.45) | ↓ | ** | ↓ | |
| 10−3·3α,5α-THA, C/5α-DHA | 12.7 (10.9, 14.6) | 14.3 (13.3, 15.3) | ↑ | |||
| 10−3·3α,5α-THA, C+U/5α-DHA | 12.7 (10.9, 14.6) | 14.3 (13.3, 15.3) | ↑ | |||
| 17β-Hydroxysteroid dehydrogenase, type 3 (HSD17B3) + aldoketoreductase 1C3 (AKR1C3) vs. 17β-hydroxysteroid dehydrogenase, type 2 (HSD17B2) | ||||||
| ADIOL/DHEA | 0.381 (0.346, 0.419) | 0.28 (0.267, 0.294) | ↓ | *** | ↓ | ↓ |
| ADIOLS/DHEAS | 0.306 (0.275, 0.342) | 0.257 (0.244, 0.272) | ↓ | * | ||
| 3β,7α,17β-AT/7α-OH-DHEA | 0.688 (0.651, 0.727) | 0.537 (0.521, 0.554) | ↓ | *** | ↓ | ↓ |
| 3β,7β,17β-AT/7β-OH-DHEA | 0.616 (0.571, 0.665) | 0.549 (0.527, 0.573) | ||||
| T/A | 5.35 (4.94, 5.78) | 4.14 (3.96, 4.33) | ↓ | *** | ↓ | ↓ |
| T, C/A | 3.31 (2.7, 4.04) | 2.28 (2.04, 2.56) | ↓ | * | ||
| T, C+U/A | 8.75 (7.78, 9.83) | 6.96 (6.53, 7.42) | ↓ | * | ||
| 5α-DHT/5α-DHA | 4.4 (3.94, 4.91) | 2.89 (2.71, 3.07) | ↓ | *** | ↓ | ↓ |
| 5α-DHT, C/5α-DHA | 14.3 (12.4, 16.6) | 16.9 (15.7, 18.3) | ||||
| 5α-DHT, C+U/5α-DHA | 19 (16.6, 21.8) | 20.4 (19, 22) | ||||
| 103·3α,5α,17β-AD/3α,5α-THA | 352 (328, 376) | 407 (394, 420) | ↑ | ** | ||
| 103·3α,5α,17β-AD, C/3α,5α-THA, C | 61.8 (58, 66) | 49.5 (47.9, 51.1) | ↓ | *** | ↓ | ↓ |
| 103·3β,5α,17β-AD/3β,5α-THA | 340 (302, 383) | 225 (211, 240) | ↓ | *** | ↓ | ↓ |
| 103·3β,5α,17β-AD/3β,5α-THA, C | 239 (219, 261) | 232 (221, 244) | ||||
| 103·3α,5β,17β-AD/3α,5β-THA, C | 174 (163, 185) | 134 (129, 139) | ↓ | *** | ↓ | ↓ |
| 103·3β,5β,17β-AD/3β,5β-THA, C | 14 (12.7, 15.4) | 15.9 (15, 16.7) | ||||
| Steroids and Steroid Molar Ratios | Controls | Schizophrenia | ANOVA, Stage main factor | MC, Controls | MC, Schizophrenia | ANOVA, Schizophrenia × Stage | |||
|---|---|---|---|---|---|---|---|---|---|
| Stage 1 | Stage 2 | Stage 1 | Stage 2 | ||||||
| 17-OH-PREGS [nM] | 27.6 (24.8, 30.7) | 37 (33.1, 41.3) | 34.8 (32.7, 37) | 30 (28.2, 31.9) | ↑ | ↓ | *** | ||
| ADIOL [nM] | 4.91 (4.35, 5.56) | 3.56 (3.15, 4.04) | 3.33 (3.12, 3.55) | 3.21 (3.01, 3.42) | ↓ | ↓ | * | ||
| 16α-OH-P [nM] | 0.74 (0.625, 0.874) | 0.915 (0.767, 1.09) | 0.841 (0.769, 0.918) | 0.637 (0.581, 0.698) | ↓ | * | |||
| 3α,5α,20α-PD [pM] | 248 (197, 308) | 190 (146, 242) | 174 (152, 198) | 262 (232, 294) | ↑ | * | |||
| 3β,5α,20α-PD [nM] | 2.35 (1.98, 2.77) | 1.58 (1.29, 1.92) | 1.1 (0.975, 1.23) | 1.34 (1.2, 1.49) | ↓ | ** | |||
| 3β,5β,20α-PD [pM] | 105 (74, 150) | 45 (31.5, 64.5) | 67.7 (53.9, 85.3) | 116 (90.8, 148) | ↓ | ↑ | ** | ||
| 3β,5β,20α-PD, C [nM] | 10.1 (8.74, 11.7) | 11.7 (10, 13.6) | 11.8 (10.8, 12.8) | 9.51 (8.75, 10.3) | ↓ | * | |||
| 3α,5α-THA sulfate [μM] | 2.88 (2.48, 3.31) | 3.63 (3.15, 4.16) | 4.36 (4.07, 4.67) | 3.94 (3.66, 4.23) | * | ||||
| Corticosterone [nM] | 15.2 (12.4, 18.4) | 19.3 (15.8, 23.2) | 20.8 (18.8, 22.9) | 16.1 (14.4, 17.9) | ↓ | * | |||
| 11β-OH-3β,5α-THA [pM] | 155 (129, 185) | 130 (105, 158) | 78.2 (69.2, 88) | 114 (103, 127) | ↑ | * | |||
| DHEA/PREG | 4.81 (4.37, 5.29) | 6.23 (5.63, 6.89) | 9.4 (8.95, 9.86) | 8.22 (7.83, 8.62) | ↑ | ↓ | *** | ||
| DHEA/20α-dihydro-PREG | 2.65 (2.34, 2.99) | 3.42 (3, 3.92) | 4.65 (4.31, 5.02) | 4.09 (3.81, 4.4) | ↑ | * | |||
| 3α,5α-THA/3α,5α,20α-PD | 4.66 (3.79, 5.82) | 5.37 (4.27, 6.87) | 5.93 (5.22, 6.78) | 3.7 (3.3, 4.16) | ↓ | * | |||
| 11β-OH-A/corticosterone | 3.08 (2.66, 3.59) | 2.13 (1.84, 2.49) | 2.17 (2, 2.35) | 2.48 (2.29, 2.7) | ↓ | ** | |||
| 17-OH-PREG/PREG | 5.17 (4.63, 5.77) | 6.88 (6.1, 7.76) | 10.8 (10.2, 11.6) | 7.94 (7.46, 8.46) | ↑ | ↓ | *** | ||
| 103·17-OH-PREG/PREG, C | 58.5 (51.1, 66.3) | 74 (65.8, 82.8) | 97.5 (92.3, 103) | 79.1 (74.4, 83.9) | ↓ | ** | |||
| Cortisol/corticosterone | 25 (22, 28.4) | 21.2 (18.6, 24.2) | 17.6 (16.4, 18.8) | 23.4 (21.8, 25.2) | ↑ | ** | |||
| DHEA/17-OH-PREG, C | 254 (220, 293) | 204 (176, 236) | 206 (190, 223) | 244 (225, 264) | ↑ | * | |||
| A/17-OH-P | 0.791 (0.734, 0.852) | 0.927 (0.855, 1.01) | 1.28 (1.22, 1.34) | 1.24 (1.18, 1.29) | ↑ | * | |||
| 10−3·3α,5α-THA/17-OH-ALLO, C | 0.486 (0.424, 0.556) | 0.583 (0.509, 0.669) | 1 (0.929, 1.08) | 0.825 (0.765, 0.889) | ↓ | * | |||
| 17-OH-P/17-OH-PREG | 0.446 (0.399, 0.5) | 0.315 (0.283, 0.352) | 0.258 (0.243, 0.273) | 0.269 (0.253, 0.286) | ↓ | * | ↓ | ** | |
| 103·17-OH-P/17-OH-PREGS | 209 (189, 231) | 114 (101, 128) | 106 (99.2, 113) | 106 (99.6, 113) | ↓ | *** | ↓ | *** | |
| 16α-OH-P/16α-OH-PREG | 1.1 (1.02, 1.19) | 1.39 (1.29, 1.5) | 1.19 (1.14, 1.24) | 1.09 (1.04, 1.14) | ↑ | ↓ | *** | ||
| 17-OH-20α-DHP, C/U | 4 (3.35, 4.8) | 5.72 (4.66, 7.12) | 6.34 (5.67, 7.11) | 5.5 (4.97, 6.12) | * | ||||
| ALLO, C/U | 38.2 (32.6, 45.2) | 57.8 (47.2, 71.6) | 70.7 (63.4, 79) | 59.1 (53.6, 65.4) | ↑ | ** | |||
| 3α,5α,20α-PD, C/U | 168 (131, 214) | 267 (207, 342) | 219 (191, 251) | 137 (119, 158) | ↓ | ** | |||
| 10−3·3β,5α,20α-PD, C/U | 1.48 (1.22, 1.81) | 2.38 (1.87, 3.09) | 2.59 (2.27, 2.97) | 2.23 (1.97, 2.53) | ↑ | * | |||
| 3β,5β,20α-PD, C/U | 93.2 (60, 139) | 229 (150, 339) | 196 (151, 252) | 88.9 (65.9, 118) | ↑ | ↓ | ** | ||
| 11β-OH-3α,5α-THA, C/U | 13.4 (11.6, 15.5) | 16.9 (14.4, 19.8) | 22 (20.2, 24) | 18.9 (17.4, 20.5) | * | ||||
| 11β-OH-3β,5α-THA/3β,5α-THA | 0.376 (0.309, 0.453) | 0.322 (0.262, 0.391) | 0.193 (0.17, 0.217) | 0.282 (0.253, 0.313) | ↑ | * | |||
| 103·16α-OH-PREG/PREG | 324 (293, 359) | 371 (333, 414) | 502 (474, 533) | 399 (377, 423) | ↓ | ** | |||
| 103·(11β-OH-3α/β,5α-THA)/11β-OH-A | 89.6 (78.3, 102) | 76 (65.1, 88) | 53.2 (48.5, 58.3) | 71.9 (66.3, 77.9) | ↑ | ** | |||
| (5β,20α-THP+3α/β,5β,20α-PD)/20α-DHP, C | 16.6 (14.2, 19.4) | 23.9 (20.2, 28.3) | 17.1 (15.7, 18.6) | 15.7 (14.5, 17.1) | ↑ | * | |||
| (5β,20α-THP+3α/β,5β,20α-PD)/20α-DHP, C+U | 13.2 (11.4, 15.3) | 19.8 (16.7, 23.5) | 17.9 (16.1, 20) | 14.9 (13.4, 16.6) | ↑ | ** | |||
| 3α,5α,20α-PT/3β,5α,20α-PT | 1.13 (0.976, 1.3) | 0.976 (0.834, 1.14) | 0.965 (0.892, 1.04) | 1.26 (1.17, 1.36) | ↑ | * | |||
| 3α,5α-THA/3β,5α-THA, C | 4.03 (3.7, 4.38) | 4.6 (4.21, 5) | 4.98 (4.75, 5.2) | 4.56 (4.36, 4.77) | * | ||||
| 3α,5β-THA/3β,5β-THA, C | 3.34 (2.91, 3.82) | 2.86 (2.45, 3.32) | 3.23 (2.99, 3.5) | 3.99 (3.71, 4.29) | ↑ | * | |||
| 3α,5β,20α-PD/5β,20α-THP, C | 56.5 (48.9, 65.4) | 86.9 (74.3, 102) | 75.4 (69.3, 82.2) | 77.4 (71.3, 84) | ↑ | * | ↑ | * | |
| 103·3α,5α,17β-AD/3α,5α-THA | 63.2 (57.9, 69) | 60.5 (55, 66.8) | 45.1 (43.1, 47.3) | 54.3 (51.8, 57) | ↑ | * | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, M.; Velíková, M.; Hovorková, T.; Bulant, J.; Janšáková, K.; Valeš, K. Steroidomics in Men with Schizophrenia. Int. J. Mol. Sci. 2024, 25, 8729. https://doi.org/10.3390/ijms25168729
Hill M, Velíková M, Hovorková T, Bulant J, Janšáková K, Valeš K. Steroidomics in Men with Schizophrenia. International Journal of Molecular Sciences. 2024; 25(16):8729. https://doi.org/10.3390/ijms25168729
Chicago/Turabian StyleHill, Martin, Marta Velíková, Tereza Hovorková, Josef Bulant, Katarína Janšáková, and Karel Valeš. 2024. "Steroidomics in Men with Schizophrenia" International Journal of Molecular Sciences 25, no. 16: 8729. https://doi.org/10.3390/ijms25168729
APA StyleHill, M., Velíková, M., Hovorková, T., Bulant, J., Janšáková, K., & Valeš, K. (2024). Steroidomics in Men with Schizophrenia. International Journal of Molecular Sciences, 25(16), 8729. https://doi.org/10.3390/ijms25168729