
Caveolin and NOS in the Development of Muscular Dystrophy


{kind=link}
{kind=link}
{kind=link}
Abstract
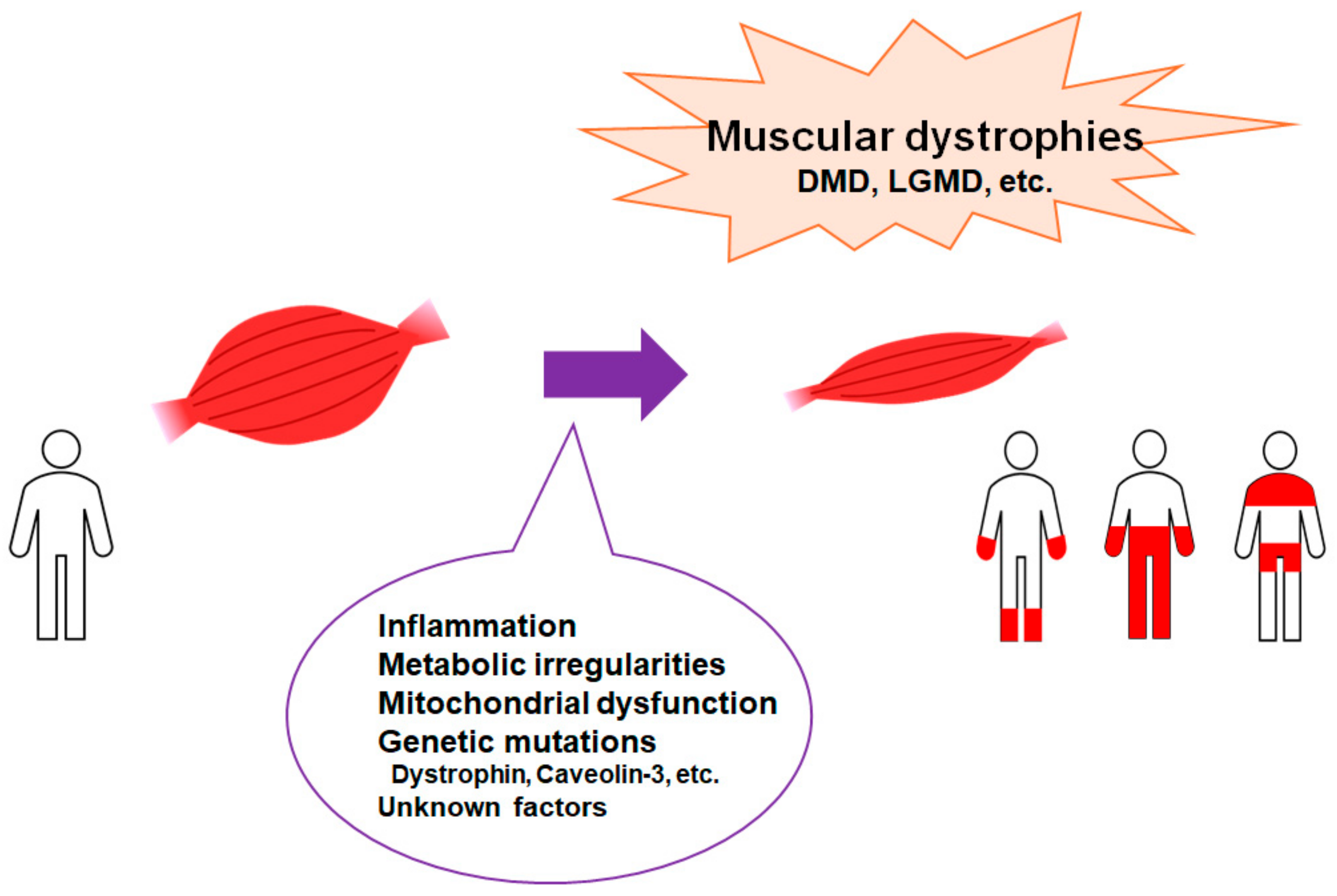
:1. Introduction
2. Inflammation and Redox System May Be Involved in the Regulation of Muscular Dystrophy
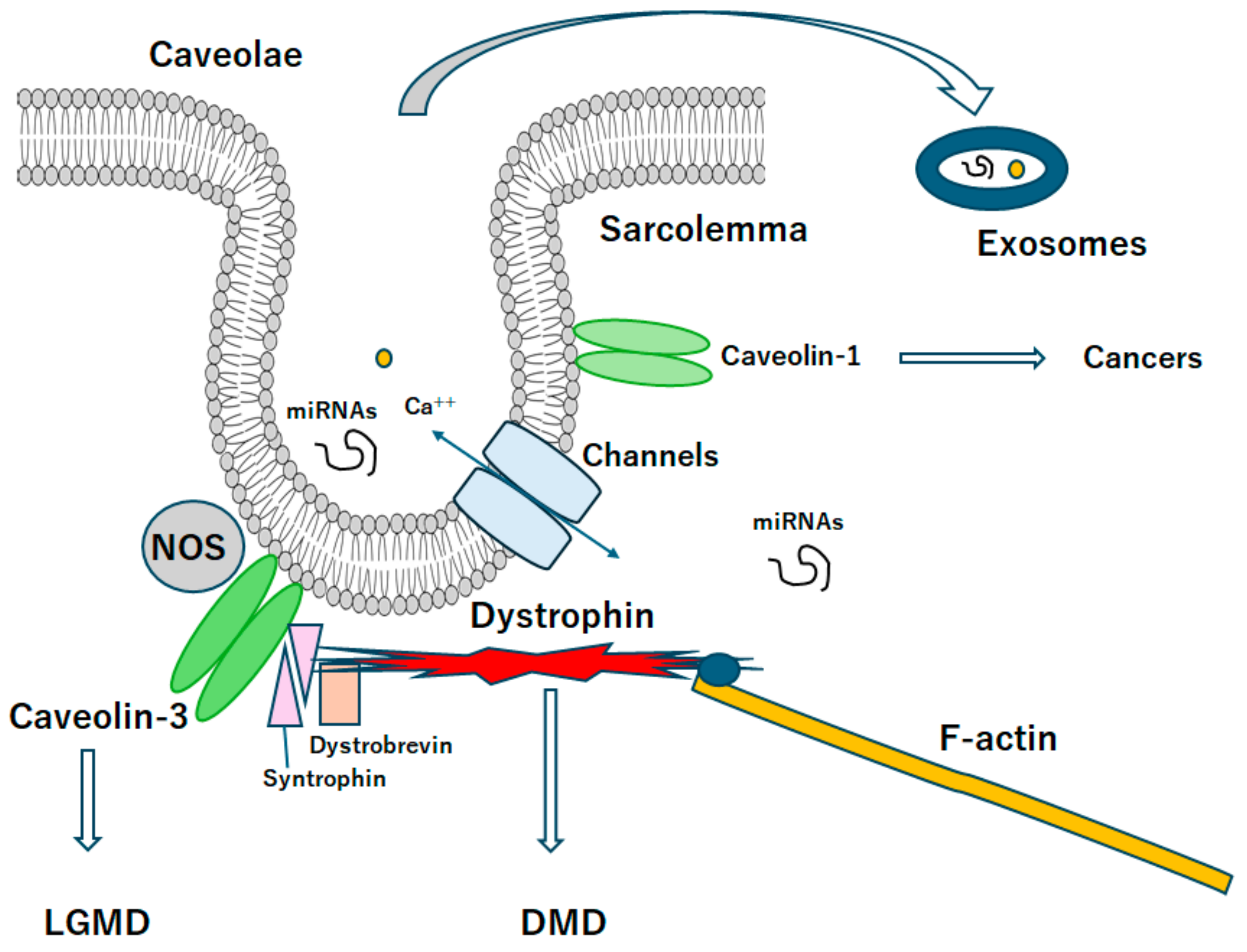
3. Relationship between Caveolin and Muscular Dystrophy
4. MicroRNAs Might Be Involved in the Pathogenesis of Muscular Dystrophies
5. Possible Treatment Tactics with the Alteration of Gut Microbiota against Muscular Dystrophies
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAV1 | caveolin-1 |
| CAV3 | caveolin-3 |
| CNS | central nervous system |
| DMD | Duchenne muscular dystrophy |
| FMT | fecal microbiota transplantation |
| iNOS | inducible nitric oxide synthase |
| LGMD | Limb–girdle muscular dystrophy |
| mRNA | messenger RNA |
| mdx mice | genetic model of Duchenne muscular dystrophy |
| miRNA | microRNA |
| miR | miRNA |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| QOL | quality of life |
| ROS | reactive oxygen species |
| SCFAs | short-chain fatty acids |
| siRNA | short interference RNA |
| UTR | untranslated region |
References
- Cossu, G.; Sampaolesi, M. New therapies for Duchenne muscular dystrophy: Challenges, prospects and clinical trials. Trends Mol. Med. 2007, 13, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Cho, D.S.; Doles, J.D. Metabolomic analyses reveal extensive progenitor cell deficiencies in a mouse model of duchenne muscular dystrophy. Metabolites 2018, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Starosta, A.; Konieczny, P. Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell Mol. Life Sci. 2021, 78, 4867–4891. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Razini, P.; Erratico, S.; Torrente, Y.; Meregalli, M. Cell based therapy for Duchenne muscular dystrophy. J. Cell Physiol. 2009, 221, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Scripture-Adams, D.D.; Chesmore, K.N.; Barthélémy, F.; Wang, R.T.; Nieves-Rodriguez, S.; Wang, D.W.; Mokhonova, E.I.; Douine, E.D.; Wan, J.; Little, I.; et al. Single nuclei transcriptomics of muscle reveals intra-muscular cell dynamics linked to dystrophin loss and rescue. Commun. Biol. 2022, 5, 989. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and nutriceutical approaches to address energy dysregulation and skeletal muscle wasting in Duchenne muscular dystrophy. Nutrients 2015, 7, 9734–9767. [Google Scholar] [CrossRef]
- Xu, H.; Cai, X.; Xu, K.; Wu, Q.; Xu, B. The metabolomic plasma profile of patients with Duchenne muscular dystrophy: Providing new evidence for its pathogenesis. Orphanet J. Rare Dis. 2023, 18, 273. [Google Scholar] [CrossRef]
- Lindsay, A.; McCourt, P.M.; Karachunski, P.; Lowe, D.A.; Ervasti, J.M. Xanthine oxidase is hyper-active in Duchenne muscular dystrophy. Free Radic. Biol. Med. 2018, 129, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Dabaj, I.; Ferey, J.; Marguet, F.; Gilard, V.; Basset, C.; Bahri, Y.; Brehin, A.C.; Vanhulle, C.; Leturcq, F.; Marret, S.; et al. Muscle metabolic remodelling patterns in Duchenne muscular dystrophy revealed by ultra-high-resolution mass spectrometry imaging. Sci. Rep. 2021, 11, 1906. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Cozzoli, A.; Capogrosso, R.F.; Sblendorio, V.T.; Dinardo, M.M.; Jagerschmidt, C.; Namour, F.; Camerino, G.M.; De Luca, A. GLPG0492, a novel selective androgen receptor modulator, improves muscle performance in the exercised-mdx mouse model of muscular dystrophy. Pharmacol. Res. 2013, 72, 9–24. [Google Scholar] [CrossRef]
- Yao, S.; Chen, Z.; Yu, Y.; Zhang, N.; Jiang, H.; Zhang, G.; Zhang, Z.; Zhang, B. Current pharmacological strategies for Duchenne muscular dystrophy. Front. Cell Dev. Biol. 2021, 9, 689533. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, D.; Wu, L.; Li, M.; Song, L.; Wei, C.; Lu, C.; Zhang, X.; Hong, S.; Ma, M.; et al. Genotype characterization and delayed loss of ambulation by glucocorticoids in a large cohort of patients with Duchenne muscular dystrophy. Orphanet J. Rare Dis. 2021, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Klein, A.; Rubino, D.; Gocheva, V.; Schmidt, S.; Schroeder, J.; Bernert, G.; Laugel, V.; Steinlin, M.; et al. Effect of combination l-Citrulline and metformin treatment on motor function in patients with Duchenne muscular dystrophy: A Randomized Clinical Trial. JAMA Netw. Open. 2019, 2, e1914171. [Google Scholar] [CrossRef]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Montecino-Rodriguez, E.; Dorshkind, K.; Tidball, J.G. Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 2001, 98, 235–243. [Google Scholar] [CrossRef]
- Denis, M.C.; Desjardins, Y.; Furtos, A.; Marcil, V.; Dudonne, S.; Montoudis, A.; Garofalo, C.; Delvin, E.; Marette, A.; Levy, E. Prevention of oxidative stress, inflammation and mitochondrial dysfunction in the intestine by different cranberry phenolic fractions. Clin. Sci. 2015, 128, 197–212. [Google Scholar] [CrossRef]
- Vo, A.H.; McNally, E.M. Modifier genes and their effect on Duchenne muscular dystrophy. Curr. Opin. Neurol. 2015, 28, 528–534. [Google Scholar] [CrossRef]
- Uryash, A.; Mijares, A.; Esteve, E.; Adams, J.A.; Lopez, J.R. Cardioprotective Effect of Whole Body Periodic Acceleration in Dystrophic Phenotype mdx Rodent. Front. Physiol. 2021, 12, 658042. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.R.; McNally, E.M. Non-Glycanated Biglycan and LTBP4: Leveraging the extracellular matrix for Duchenne Muscular Dystrophy therapeutics. Matrix Biol. 2018, 68–69, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef]
- Yang, R.; Beqiri, D.; Shen, J.B.; Redden, J.M.; Dodge-Kafka, K.; Jacobson, K.A.; Liang, B.T. P2X4 receptor-eNOS signaling pathway in cardiac myocytes as a novel protective mechanism in heart failure. Comput. Struct. Biotechnol. J. 2015, 13, 1–7. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar]
- Kim, D.Y.; Lim, S.G.; Suk, K.; Lee, W.H. Mitochondrial dysfunction regulates the JAK-STAT pathway via LKB1-mediated AMPK activation ER-stress-independent manner. Biochem. Cell Biol. 2020, 98, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Duong, M.N.; Turner, R.; Le Guiner, C.; Boyatzis, A.; Kettle, A.J.; Grounds, M.D.; Arthur, P.G. Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy. Redox Biol. 2016, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Pinniger, G.J.; Graves, J.A.; Grounds, M.D.; Arthur, P.G. Increasing taurine intake and taurine synthesis improves skeletal muscle function in the mdx mouse model for Duchenne muscular dystrophy. J. Physiol. 2016, 594, 3095–3110. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Oviedo, P.J.; Sobrino, A.; Laguna-Fernandez, A.; Novella, S.; Tarín, J.J.; García-Pérez, M.A.; Sanchís, J.; Cano, A.; Hermenegildo, C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol. Cell Endocrinol. 2011, 335, 96–103. [Google Scholar] [CrossRef]
- D’Andrea, L.; Del Gatto, A.; De Rosa, L.; Romanelli, A.; Pedone, C. Peptides targeting angiogenesis related growth factor receptors. Curr. Pharm. Des. 2009, 15, 2414–2429. [Google Scholar] [CrossRef]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Iwakiri, Y. S-nitrosylation of proteins: A new insight into endothelial cell function regulated by eNOS-derived NO. Nitric Oxide 2011, 25, 95–101. [Google Scholar] [CrossRef]
- Schwencke, C.; Braun-Dullaeus, R.C.; Wunderlich, C.; Strasser, R.H. Caveolae and caveolin in transmembrane signaling: Implications for human disease. Cardiovasc. Res. 2006, 70, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. Regulation of eNOS in caveolae. Adv. Exp. Med. Biol. 2012, 729, 51–62. [Google Scholar] [PubMed]
- Ariotti, N.; Parton, R.G. SnapShot: Caveolae, caveolins, and cavins. Cell 2013, 154, 704–704.e1. [Google Scholar] [CrossRef]
- Razani, B.; Lisanti, M.P. Caveolin-deficient mice: Insights into caveolar function human disease. J. Clin. Investig. 2001, 108, 1553–1561. [Google Scholar] [CrossRef]
- Han, B.; Copeland, C.A.; Tiwari, A.; Kenworthy, A.K. Assembly and turnover of caveolae: What do we really know? Front. Cell Dev. Biol. 2016, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.; Bai, J.; Gupta-Rossi, N.; Jimenez, A.J.; Delevoye, C.; Lamaze, C.; Echard, A. Caveolae promote successful abscission by controlling intercellular bridge tension during cytokinesis. Sci. Adv. 2022, 8, eabm5095. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R. Critical role of caveolin-1 loss/dysfunction in pulmonary hypertension. Med. Sci. 2021, 9, 58. [Google Scholar] [CrossRef]
- Kirkham, M.; Nixon, S.J.; Howes, M.T.; Abi-Rached, L.; Wakeham, D.E.; Hanzal-Bayer, M.; Ferguson, C.; Hill, M.M.; Fernandez-Rojo, M.; Brown, D.A.; et al. Evolutionary analysis and molecular dissection of caveola biogenesis. J. Cell Sci. 2008, 121, 2075–2086. [Google Scholar] [CrossRef]
- Karhan, A.N.; Zammouri, J.; Auclair, M.; Capel, E.; Apaydin, F.D.; Ates, F.; Verpont, M.C.; Magré, J.; Fève, B.; Lascols, O.; et al. Biallelic CAV1 null variants induce congenital generalized lipodystrophy with achalasia. Eur. J. Endocrinol. 2021, 185, 841–854. [Google Scholar] [CrossRef]
- Lee, H.; Park, D.S.; Razani, B.; Russell, R.G.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: Caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (−/−) null mice show mammary epithelial cell hyperplasia. Am. J. Pathol. 2002, 161, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.K.; Lee, M.G.; Kim, N.H.; Lee, K.Y.; Oh, S.J.; Moon, J.R.; Kim, H.J.; Chi, S.G. Bidirectional alteration of Cav-1 expression is associated with mitogenic conversion of its function in gastric tumor progression. BMC Cancer 2017, 17, 766. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, B.S.; Proszynski, T.J. A role for caveolin-3 in the pathogenesis of muscular dystrophies. Int. J. Mol. Sci. 2020, 21, 8736. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.S.; Nisr, R.B.; Stretton, C.; Krasteva-Christ, G.; Hundal, H.S. Caveolin-3 deficiency associated with the dystrophy P104L mutation impairs skeletal muscle mitochondrial form and function. J. Cachexia Sarcopenia Muscle 2020, 11, 838–858. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E.; Sotgia, F.; Galbiati, F.; Minetti, C.; Lisanti, M.P. Caveolinopathies: Mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology 2004, 62, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, F.; Volonte, D.; Minetti, C.; Chu, J.B.; Lisanti, M.P. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J. Biol. Chem. 1999, 274, 25632–25641. [Google Scholar] [CrossRef]
- Sotgia, F.; Woodman, S.E.; Bonuccelli, G.; Capozza, F.; Minetti, C.; Scherer, P.E.; Lisanti, M.P. Phenotypic behavior of caveolin-3 R26Q, a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am. J. Physiol. Cell Physiol. 2003, 285, C1150–C1160. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Deng, Y.; Shang, L.; Yang, L.; Huang, J.; Ma, J.; Liao, X.; Zhou, H.; Xian, J.; Liang, G.; et al. Effect of type 2 diabetes mellitus caveolin-3 K15N mutation on glycometabolism. Exp. Ther. Med. 2019, 18, 2531–2539. [Google Scholar] [PubMed]
- Morales-Paytuví, F.; Ruiz-Mirapeix, C.; Fajardo, A.; Rae, J.; Bosch, M.; Enrich, C.; Collins, B.M.; Parton, R.G.; Pol, A. Proteostatic regulation of caveolins avoids premature oligomerisation and preserves ER homeostasis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Huang, H.; Bae, C.; Sachs, F.; Suchyna, T.M. Caveolae regulation of mechanosensitive channel function in myotubes. PLoS ONE 2013, 8, e72894. [Google Scholar] [CrossRef]
- Reilly, S.N.; Liu, X.; Carnicer, R.; Recalde, A.; Muszkiewicz, A.; Jayaram, R.; Carena, M.C.; Wijesurendra, R.; Stefanini, M.; Surdo, N.C.; et al. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci. Transl. Med. 2016, 8, 340ra374. [Google Scholar] [CrossRef]
- Groh, W.J.; Bhakta, D.; Tomaselli, G.F.; Aleong, R.G.; Teixeira, R.A.; Amato, A.; Asirvatham, S.J.; Cha, Y.M.; Corrado, D.; Duboc, D.; et al. 2022 HRS expert consensus statement on evaluation and management of arrhythmic risk in neuromuscular disorders. Heart Rhythm 2022, 19, e61–e120. [Google Scholar] [CrossRef]
- Llano-Diez, M.; Ortez, C.I.; Gay, J.A.; Alvarez-Cabado, L.; Jou, C.; Medina, J.; Nascimento, A.; Jimenez-Mallebrera, C. Digital PCR quantification of miR-30c and miR-181a as serum biomarkers for Duchenne muscular dystrophy. Neuromuscul. Disord. 2017, 27, 15–23. [Google Scholar] [CrossRef]
- Mizuno, H.; Nakamura, A.; Aoki, Y.; Ito, N.; Kishi, S.; Yamamoto, K.; Sekiguchi, M.; Takeda, S.; Hashido, K. Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: Promising novel blood-based markers for muscular dystrophy. PLoS ONE 2011, 6, e18388. [Google Scholar] [CrossRef]
- Nguyen, M.N.; Hooper, C.; Stefanini, M.; Vrellaku, B.; Carnicer, R.; Wood, M.J.; Simon, J.N.; Casadei, B. Why is early-onset atrial fibrillation uncommon in patients with Duchenne muscular dystrophy? Insights from the mdx mouse. Cardiovasc. Res. 2024, 120, 519–530. [Google Scholar] [CrossRef]
- Jelinkova, S.; Fojtik, P.; Kohutova, A.; Vilotic, A.; Marková, L.; Pesl, M.; Jurakova, T.; Kruta, M.; Vrbsky, J.; Gaillyova, R.; et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells. 2019, 8, 53. [Google Scholar] [CrossRef]
- Bender, A.T.; Demady, D.R.; Osawa, Y. Ubiquitination of neuronal nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 2000, 275, 17407–17411. [Google Scholar] [CrossRef]
- Paula, S.M.; Fernandes, T.; Couto, G.K.; Jordão, M.T.; Oliveira, E.M.; Michelini, L.C.; Rossoni, L.V. Molecular Pathways Involved in Aerobic Exercise Training Enhance Vascular Relaxation. Med. Sci. Sports Exerc. 2020, 52, 2117–2126. [Google Scholar] [CrossRef]
- Huang-Doran, I.; Zhang, C.Y.; Vidal-Puig, A. Extracellular vesicles: Novel mediators of cell communication in metabolic disease. Trends Endocrinol. Metab. 2017, 28, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Silva-Palacios, A.; Arroyo-Campuzano, M.; Flores-García, M.; Patlán, M.; Hernández-Díazcouder, A.; Alcántara, D.; Ramírez-Camacho, I.; Arana-Hidalgo, D.; Soria-Castro, E.; Sánchez, F.; et al. Citicoline Modifies the Expression of Specific miRNAs Related to Cardioprotection in Patients with ST-Segment Elevation Myocardial Infarction Subjected to Coronary Angioplasty. Pharmaceuticals 2022, 15, 925. [Google Scholar] [CrossRef]
- Benzoni, P.; Gazzerro, E.; Fiorillo, C.; Baratto, S.; Bartolucci, C.; Severi, S.; Milanesi, R.; Lippi, M.; Langione, M.; Murano, C.; et al. Caveolin-3 and Caveolin-1 Interaction Decreases Channel Dysfunction Due to Caveolin-3 Mutations. Int. J. Mol. Sci. 2024, 25, 980. [Google Scholar] [CrossRef]
- Murfitt, L.; Whiteley, G.; Iqbal, M.M.; Kitmitto, A. Targeting caveolin-3 for the treatment of diabetic cardiomyopathy. Pharmacol. Ther. 2015, 151, 50–71. [Google Scholar] [CrossRef]
- O’Rourke, J.R.; Georges, S.A.; Seay, H.R.; Tapscott, S.J.; McManus, M.T.; Goldhamer, D.J.; Swanson, M.S.; Harfe, B.D. Essential role for Dicer during skeletal muscle development. Dev. Biol. 2007, 311, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef]
- McCarthy, J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport. Sci. Rev. 2011, 39, 150–154. [Google Scholar] [CrossRef]
- Bagheri, A.; Khorram Khorshid, H.R.; Mowla, S.J.; Mowla, S.J.; Mohebbi, H.A.; Mohammadian, A.; Yaseri, M.; Solaymani-Dodaran, M.; Sherafatian, M.; Tavallaie, M. Altered miR-223 Expression in Sputum for Diagnosis of Non-Small Cell Lung Cancer. Avicenna J. Med. Biotechnol. 2017, 9, 189–195. [Google Scholar]
- Zanotti, S.; Gibertini, S.; Blasevich, F.; Bragato, C.; Ruggieri, A.; Saredi, S.; Fabbri, M.; Bernasconi, P.; Maggi, L.; Mantegazza, R.; et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018, 74, 77–100. [Google Scholar] [CrossRef]
- Wan, R.; Liu, S.; Feng, X.; Luo, W.; Zhang, H.; Wu, Y.; Chen, S.; Shang, X. The Revolution of exosomes: From biological functions to therapeutic applications in skeletal muscle diseases. J. Orthop. Trans. 2024, 45, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yedigaryan, L.; Sampaolesi, M. Extracellular vesicles and Duchenne muscular dystrophy pathology: Modulators of disease progression. Front. Physiol. 2023, 14, 1130063. [Google Scholar] [CrossRef]
- Su, X.; Shen, Y.; Kim, I.M.; Weintraub, N.L.; Hamrick, M.; Tang, Y. Extracellular Vesicles for Muscle Atrophy Treatment. Adv. Exp. Med. Biol. 2023, 1418, 119–126. [Google Scholar]
- Shapira, M. Gut microbiotas and host evolution: Scaling up symbiosis. Trends Ecol. Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef]
- Jollet, M.; Mariadassou, M.; Rué, O.; Pessemesse, L.; Ollendorff, V.; Ramdani, S.; Vernus, B.; Bonnieu, A.; Bertrand-Gaday, C.; Goustard, B.; et al. Insight into the Role of Gut Microbiota in Duchenne Muscular Dystrophy: An Age-Related Study in Mdx Mice. Am. J. Pathol. 2024, 194, 264–279. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Kosiewicz, M.M.; Zirnheld, A.L.; Alard, P. Gut microbiota, immunity, and disease: A complex relationship. Front. Microbiol. 2011, 2, 180. [Google Scholar] [CrossRef]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Donovan, S.M. Introduction to the special focus issue on the impact of diet on gut microbiota composition and function and future opportunities for nutritional modulation of the gut microbiome to improve human health. Gut Microbes 2017, 8, 75–81. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Heiskanen, M.A.; Aatsinki, A.; Hakonen, P.; Kartiosuo, N.; Munukka, E.; Lahti, L.; Keskitalo, A.; Huovinen, P.; Niinikoski, H.; Viikari, J.; et al. Association of Long-Term Habitual Dietary Fiber Intake since Infancy with Gut Microbiota Composition in Young Adulthood. J. Nutr. 2024, 154, 744–754. [Google Scholar] [CrossRef]
- Machado, M.G.; Patente, T.A.; Rouillé, Y.; Heumel, S.; Melo, E.M.; Deruyter, L.; Pourcet, B.; Sencio, V.; Teixeira, M.M.; Trottein, F. Acetate Improves the Killing of Streptococcus pneumoniae by Alveolar Macrophages via NLRP3 Inflammasome and Glycolysis-HIF-1α Axis. Front. Immunol. 2022, 13, 773261. [Google Scholar] [CrossRef]
- Noris, M.; Todeschini, M.; Casiraghi, F.; Roccatello, D.; Martina, G.; Minetti, L.; Imberti, B.; Gaspari, F.; Atti, M.; Remuzzi, G. Effect of Acetate, Bicarbonate Dialysis, and Acetate-Free Biofiltration on Nitric Oxide Synthesis: Implications for Dialysis Hypotension. Am. J. Kidney Dis. 1998, 32, 115–124. [Google Scholar] [CrossRef]
- Cerdá, B.; Pérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; González-Soltero, R.; Larrosa, M. Gut microbiota modification: Another piece in the puzzle of the benefits of physical exercise in health? Front. Physiol. 2016, 7, 51. [Google Scholar] [CrossRef]
- Baccari, M.C.; Nistri, S.; Vannucchi, M.G.; Calamai, F.; Bani, D. Reversal by relaxin of altered ileal spontaneous contractions in dystrophic (mdx) mice through a nitric oxide-mediated mechanism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R662–R668. [Google Scholar] [CrossRef]
- Mulè, F.; Amato, A.; Serio, R. Gastric emptying, small intestinal transit and fecal output in dystrophic (mdx) mice. J. Physiol. Sci. 2010, 60, 75–79. [Google Scholar] [CrossRef]
- Swiderski, K.; Bindon, R.; Trieu, J.; Naim, T.; Schokman, S.; Swaminathan, M.; Leembruggen, A.J.L.; Hill-Yardin, E.L.; Koopman, R.; Bornstein, J.C.; et al. Spatiotemporal mapping reveals regional gastrointestinal dysfunction in mdx dystrophic mice ameliorated by oral L-arginine supplementation. J. Neurogastroenterol. Motil. 2020, 26, 133–146. [Google Scholar] [CrossRef]
- Mancinelli, R.; Tonali, P.; Servidei, S.; Azzena, G.B. Analysis of peristaltic reflex in young mdx dystrophic mice. Neurosci. Lett. 1995, 192, 57–60. [Google Scholar] [CrossRef]
- Farini, A.; Tripodi, L.; Villa, C.; Strati, F.; Facoetti, A.; Baselli, G.; Troisi, J.; Landolfi, A.; Lonati, C.; Molinaro, D.; et al. Microbiota dysbiosis influences immune system and muscle pathophysiology of dystrophin-deficient mice. EMBO Mol. Med. 2023, 15, e16244. [Google Scholar] [CrossRef]
- Kalkan, H.; Pagano, E.; Paris, D.; Panza, E.; Cuozzo, M.; Moriello, C.; Piscitelli, F.; Abolghasemi, A.; Gazzerro, E.; Silvestri, C.; et al. Targeting gut dysbiosis against inflammation and impaired autophagy in Duchenne muscular dystrophy. EMBO Mol. Med. 2023, 15, e16225. [Google Scholar] [CrossRef]
- Liu, D.; Ji, Y.; Cheng, Q.; Zhu, Y.; Zhang, H.; Guo, Y.; Cao, X.; Wang, H. Dietary astaxanthin-rich extract ameliorates atherosclerosis/retinopathy and restructures gut microbiome in apolipoprotein E-deficient mice fed on a high-fat diet. Food Funct. 2022, 13, 10461–10475. [Google Scholar] [CrossRef]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef]
- Srivastava, N.K.; Yadav, R.; Mukherjee, S.; Pal, L.; Sinha, N. Abnormal lipid metabolism in skeletal muscle tissue of patients with muscular dystrophy: In vitro, high-resolution NMR spectroscopy based observation in early phase of the disease. Magn. Reson. Imaging 2017, 38, 163–173. [Google Scholar] [CrossRef]
- Cardone, N.; Taglietti, V.; Baratto, S.; Kefi, K.; Periou, B.; Gitiaux, C.; Barnerias, C.; Lafuste, P.; Pharm, F.L.; Pharm, J.N.; et al. Myopathologic trajectory in Duchenne muscular dystrophy (DMD) reveals lack of regeneration due to senescence in satellite cells. Acta Neuropathol. Commun. 2023, 11, 167. [Google Scholar] [CrossRef]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2019, 20, 1205–1217. [Google Scholar] [CrossRef]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Hu, P.; Li, K.; Peng, X.; Kan, Y.; Li, H.; Zhu, Y.; Wang, Z.; Li, Z.; Liu, H.Y.; Cai, D. Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients 2023, 15, 4772. [Google Scholar] [CrossRef]
- Li, L.; Guo, W.L.; Zhang, W.; Xu, J.X.; Qian, M.; Bai, W.D.; Zhang, Y.Y.; Rao, P.F.; Ni, L.; Lv, X.C. Grifola Frondosa Polysaccharides Ameliorate Lipid Metabolic Disorders and Gut Microbiota Dysbiosis in High-Fat Diet Fed Rats. Food Funct. 2019, 10, 2560–2572. [Google Scholar] [CrossRef]
- Cao, S.; Liu, M.; Han, Y.; Li, S.; Zhu, X.; Li, D.; Shi, Y.; Liu, B. Effects of Saponins on Lipid Metabolism: The Gut-Liver Axis Plays a Key Role. Nutrients 2024, 16, 1514. [Google Scholar] [CrossRef]
- Tian, Y.; Wu, G.; Zhao, X.; Zhang, H.; Ren, M.; Song, X.; Chang, H.; Jing, Z. Probiotics combined with atorvastatin administration in the treatment of hyperlipidemia: A randomized, double-blind, placebo-controlled clinical trial. Medicine 2024, 103, e37883. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Spitali, P. Circulating Biomarkers for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, S49–S58. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakashima, M.; Suga, N.; Yoshikawa, S.; Matsuda, S. Caveolin and NOS in the Development of Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 8771. https://doi.org/10.3390/ijms25168771
Nakashima M, Suga N, Yoshikawa S, Matsuda S. Caveolin and NOS in the Development of Muscular Dystrophy. International Journal of Molecular Sciences. 2024; 25(16):8771. https://doi.org/10.3390/ijms25168771
Chicago/Turabian StyleNakashima, Moeka, Naoko Suga, Sayuri Yoshikawa, and Satoru Matsuda. 2024. "Caveolin and NOS in the Development of Muscular Dystrophy" International Journal of Molecular Sciences 25, no. 16: 8771. https://doi.org/10.3390/ijms25168771
APA StyleNakashima, M., Suga, N., Yoshikawa, S., & Matsuda, S. (2024). Caveolin and NOS in the Development of Muscular Dystrophy. International Journal of Molecular Sciences, 25(16), 8771. https://doi.org/10.3390/ijms25168771