
Role of PRMT1 and PRMT5 in Breast Cancer
 , and
, and
Abstract
:1. Introduction
2. Protein Arginine Methyltransferases
3. PRMT1 in Breast Cancer
3.1. Regulation of Tumor Suppressor Pathways
3.2. Regulations of Oncogenic Pathways
3.3. Regulation of Alternative Splicing
3.4. Steroid Receptors Regulation
3.5. Involvement in Double-Strand Break DNA Repair
3.6. Suppression of Tumor Immune Surveillance
3.7. Chemoresistance
4. PRMT5 in Breast Cancer
4.1. Regulations of Oncogenic Pathways
4.2. Maintenance of Breast Cancer Stem Cells
4.3. Regulation of Epithelial–Mesenchymal Transition
4.4. Splicing Regulation
4.5. Regulation of Immunotherapy
4.6. Involvement in Double-Strand Break DNA Repair
4.7. Dual Role of PRMT5
5. Targeting PRMTs in Breast Cancer
5.1. PRMT1 Inhibitors
5.2. PRMT5 Inhibitors
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer Statistics, 2024. CA A Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer-Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast Cancer Intrinsic Subtype Classification, Clinical Use and Future Trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987. [Google Scholar] [CrossRef] [PubMed]
- Rosso, R.; D’Alonzo, M.; Bounous, V.E.; Actis, S.; Cipullo, I.; Salerno, E.; Biglia, N. Adherence to Adjuvant Endocrine Therapy in Breast Cancer Patients. Curr. Oncol. 2023, 30, 1461–1472. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Wang, H.; Wang, Y.; He, Q.; Xia, X.; Hu, Z.-Y.; Ouyang, Q. Clinical and Genetic Risk Factors for Fulvestrant Treatment in Post-Menopause ER-Positive Advanced Breast Cancer Patients. J. Transl. Med. 2019, 17, 27. [Google Scholar] [CrossRef]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef]
- Kinnel, B.; Singh, S.K.; Oprea-Ilies, G.; Singh, R. Targeted Therapy and Mechanisms of Drug Resistance in Breast Cancer. Cancers 2023, 15, 1320. [Google Scholar] [CrossRef]
- Poulard, C.; Corbo, L.; Le Romancer, M. Protein Arginine Methylation/Demethylation and Cancer. Oncotarget 2016, 7, 67532–67550. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein Arginine Methyltransferases: Promising Targets for Cancer Therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Kim, S. Enzymatic Methylation of Protein Fractions from Calf Thymus Nuclei. Biochem. Biophys. Res. Commun. 1967, 29, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Matsuoka, Y.; Kakimoto, Y. Isolation and Identification of N-G-Monomethyl, N-G, N-G-Dimethyl- and N-G,N’ G-Dimethylarginine from the Hydrolysate of Proteins of Bovine Brain. Biochim. Biophys. Acta 1971, 230, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Gary, J.D.; Yang, M.C.; Clarke, S.; Herschman, H.R. The Mammalian Immediate-Early TIS21 Protein and the Leukemia-Associated BTG1 Protein Interact with a Protein-Arginine N-Methyltransferase. J. Biol. Chem. 1996, 271, 15034–15044. [Google Scholar] [CrossRef]
- Bedford, M.T. Arginine Methylation at a Glance. J. Cell Sci. 2007, 120, 4243–4246. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Lopez, C.I.; Sandberg, T.; Kelly, R.; Clarke, S.G. Human Protein Arginine Methyltransferase 7 (PRMT7) Is a Type III Enzyme Forming ω-NG-Monomethylated Arginine Residues. J. Biol. Chem. 2012, 287, 7859–7870. [Google Scholar] [CrossRef]
- Lorton, B.M.; Shechter, D. Cellular Consequences of Arginine Methylation. Cell. Mol. Life Sci. 2019, 76, 2933–2956. [Google Scholar] [CrossRef]
- LITT, M.; QIU, Y.; HUANG, S. Histone Arginine Methylations: Their Roles in Chromatin Dynamics and Transcriptional Regulation. Biosci. Rep. 2009, 29, 131–141. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein Arginine Methyltransferases and Cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The Regulation, Functions and Clinical Relevance of Arginine Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Zhang, J.; Lu, Y.; Liu, X.; Geng, P.; Huang, B.; Zhang, Y.; Lu, J. The Dual Function of PRMT1 in Modulating Epithelial-Mesenchymal Transition and Cellular Senescence in Breast Cancer Cells through Regulation of ZEB1. Sci. Rep. 2016, 6, 19874. [Google Scholar] [CrossRef]
- Lin, C.-C.; Chang, T.-C.; Wang, Y.; Guo, L.; Gao, Y.; Bikorimana, E.; Lemoff, A.; Fang, Y.V.; Zhang, H.; Zhang, Y.; et al. PRMT5 Is an Actionable Therapeutic Target in CDK4/6 Inhibitor-Resistant ER+/RB-Deficient Breast Cancer. Nat. Commun. 2024, 15, 2287. [Google Scholar] [CrossRef]
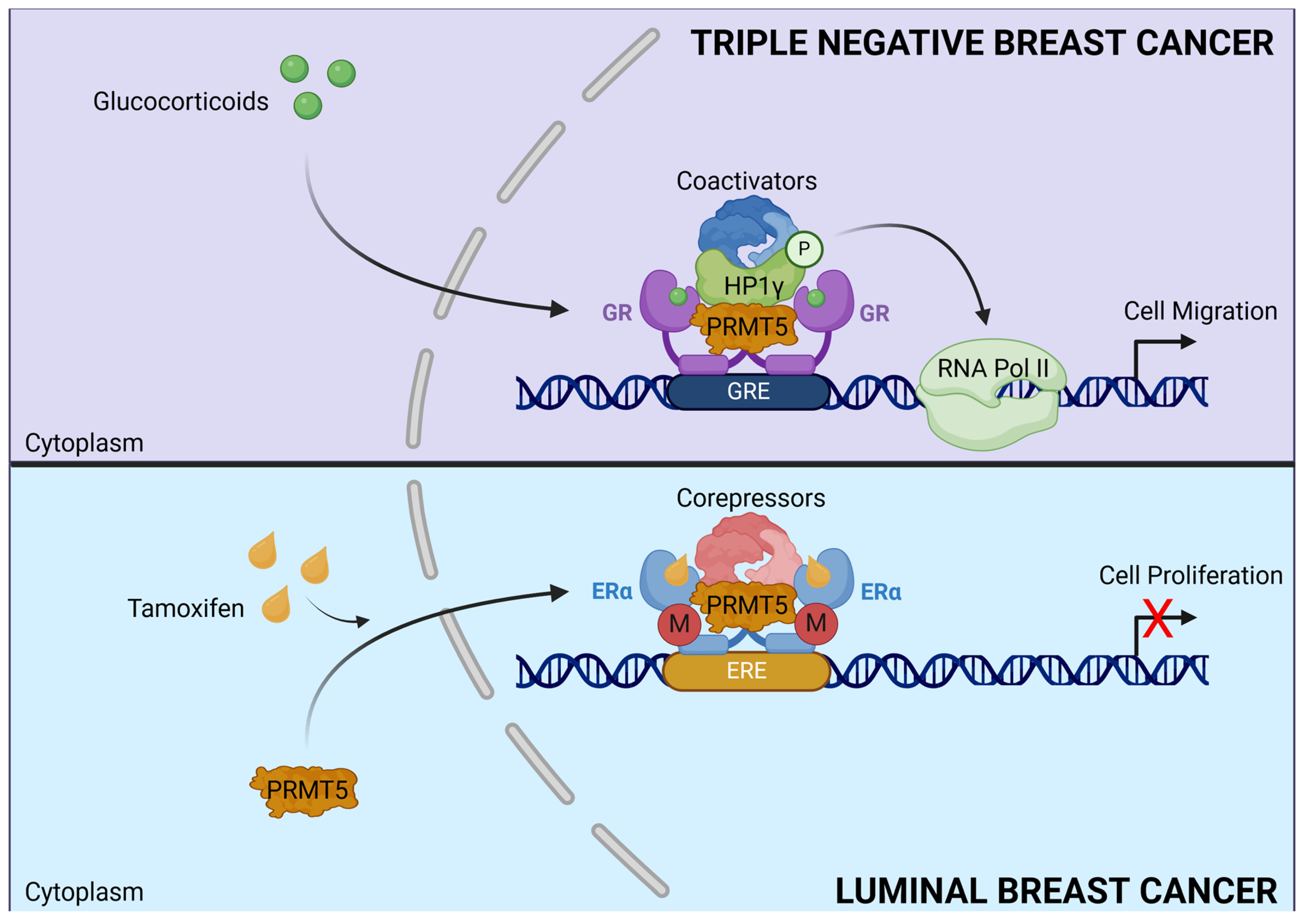
- Poulard, C.; Ha Pham, T.; Drouet, Y.; Jacquemetton, J.; Surmielova, A.; Kassem, L.; Mery, B.; Lasset, C.; Reboulet, J.; Treilleux, I.; et al. Nuclear PRMT5 Is a Biomarker of Sensitivity to Tamoxifen in ERα + Breast Cancer. EMBO Mol. Med. 2023, 15, e17248. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, T.; Hébert, J.; Li, E.; Richard, S. A Mouse PRMT1 Null Allele Defines an Essential Role for Arginine Methylation in Genome Maintenance and Cell Proliferation. Mol. Cell. Biol. 2009, 29, 2982–2996. [Google Scholar] [CrossRef]
- Wei, M.; Tan, C.; Tang, Z.; Lian, Y.; Huang, Y.; Chen, Y.; Chen, C.; Zhou, W.; Cai, T.; Hu, J. Proteome-Wide Alterations of Asymmetric Arginine Dimethylation Associated With Pancreatic Ductal Adenocarcinoma Pathogenesis. Front. Cell Dev. Biol. 2020, 8, 545934. [Google Scholar] [CrossRef]
- Hashimoto, M.; Fukamizu, A.; Nakagawa, T.; Kizuka, Y. Roles of Protein Arginine Methyltransferase 1 (PRMT1) in Brain Development and Disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2021, 1865, 129776. [Google Scholar] [CrossRef]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.J.; et al. Dysregulation of PRMT1 and PRMT6, Type I Arginine Methyltransferases, Is Involved in Various Types of Human Cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar] [CrossRef]
- Bao, J.; Di Lorenzo, A.; Lin, K.; Lu, Y.; Zhong, Y.; Sebastian, M.M.; Muller, W.J.; Yang, Y.; Bedford, M.T. Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res. 2019, 79, 21–32. [Google Scholar] [CrossRef]
- Morettin, A.; Baldwin, R.M.; Côté, J. Arginine Methyltransferases as Novel Therapeutic Targets for Breast Cancer. Mutagenesis 2015, 30, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Eve, L.; Poulard, C.; Le Romancer, M. Structure, Activity, and Function of PRMT1. Life 2021, 11, 1147. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Integrated Analysis of Public Datasets for the Discovery and Validation of Survival-Associated Genes in Solid Tumors. Innovation 2024, 5, 100625. [Google Scholar] [CrossRef]
- Guendel, I.; Carpio, L.; Pedati, C.; Schwartz, A.; Teal, C.; Kashanchi, F.; Kehn-Hall, K. Methylation of the Tumor Suppressor Protein, BRCA1, Influences Its Transcriptional Cofactor Function. PLoS ONE 2010, 5, e11379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jankovic, V.; Gural, A.; Huang, G.; Pardanani, A.; Menendez, S.; Zhang, J.; Dunne, R.; Xiao, A.; Erdjument-Bromage, H.; et al. Methylation of RUNX1 by PRMT1 Abrogates SIN3A Binding and Potentiates Its Transcriptional Activity. Genes Dev. 2008, 22, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; Déry, U.; Masson, J.-Y.; Richard, S. Arginine Methylation of MRE11 by PRMT1 Is Required for DNA Damage Checkpoint Control. Genes Dev. 2005, 19, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zheng, L.; Xu, H.; Dai, H.; Zhou, M.; Pascua, M.R.; Chen, Q.M.; Shen, B. Methylation of FEN1 Suppresses Nearby Phosphorylation and Facilitates PCNA Binding. Nat. Chem. Biol. 2010, 6, 766–773. [Google Scholar] [CrossRef]
- Baldwin, R.M.; Morettin, A.; Côté, J. Role of PRMTs in Cancer: Could Minor Isoforms Be Leaving a Mark? World J. Biol. Chem. 2014, 5, 115–129. [Google Scholar] [CrossRef]
- Suresh, S.; Huard, S.; Brisson, A.; Némati, F.; Dakroub, R.; Poulard, C.; Ye, M.; Martel, E.; Reyes, C.; Silvestre, D.C.; et al. PRMT1 Regulates EGFR and Wnt Signaling Pathways and Is a Promising Target for Combinatorial Treatment of Breast Cancer. Cancers 2022, 14, 306. [Google Scholar] [CrossRef]
- Liu, L.-M.; Sun, W.-Z.; Fan, X.-Z.; Xu, Y.-L.; Cheng, M.-B.; Zhang, Y. Methylation of C/EBPα by PRMT1 Inhibits Its Tumor-Suppressive Function in Breast Cancer. Cancer Res. 2019, 79, 2865–2877. [Google Scholar] [CrossRef]
- Harms, K.L.; Chen, X. The Functional Domains in P53 Family Proteins Exhibit Both Common and Distinct Properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Scoumanne, A.; Chen, X. Protein Methylation: A New Regulator of the P53 Tumor Suppressor. Histol. Histopathol. 2008, 23, 1143–1149. [Google Scholar]
- Yang, E.S.; Xia, F. BRCA1 16 Years Later: DNA Damage-Induced BRCA1 Shuttling. FEBS J. 2010, 277, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-M.; Tang, Q.; Hu, X.; Zhao, J.-J.; Zhang, Y.; Ying, G.-G.; Zhang, F. Arginine Methyltransferase PRMT1 Regulates P53 Activity in Breast Cancer. Life 2021, 11, 789. [Google Scholar] [CrossRef]
- Wang, H.; Yang, E.S.; Jiang, J.; Nowsheen, S.; Xia, F. DNA Damage-Induced Cytotoxicity Is Dissociated from BRCA1’s DNA Repair Function but Is Dependent on Its Cytosolic Accumulation. Cancer Res. 2010, 70, 6258–6267. [Google Scholar] [CrossRef] [PubMed]
- Laulier, C.; Barascu, A.; Guirouilh-Barbat, J.; Pennarun, G.; Le Chalony, C.; Chevalier, F.; Palierne, G.; Bertrand, P.; Verbavatz, J.M.; Lopez, B.S. Bcl-2 Inhibits Nuclear Homologous Recombination by Localizing BRCA1 to the Endomembranes. Cancer Res. 2011, 71, 3590–3602. [Google Scholar] [CrossRef]
- Montenegro, M.F.; González-Guerrero, R.; Sánchez-Del-Campo, L.; Piñero-Madrona, A.; Cabezas-Herrera, J.; Rodríguez-López, J.N. PRMT1-Dependent Methylation of BRCA1 Contributes to the Epigenetic Defense of Breast Cancer Cells against Ionizing Radiation. Sci. Rep. 2020, 10, 13275. [Google Scholar] [CrossRef] [PubMed]
- Gery, S.; Tanosaki, S.; Bose, S.; Bose, N.; Vadgama, J.; Koeffler, H.P. Down-Regulation and Growth Inhibitory Role of C/EBPalpha in Breast Cancer. Clin. Cancer Res. 2005, 11, 3184–3190. [Google Scholar] [CrossRef]
- Milde-Langosch, K.; Löning, T.; Bamberger, A.-M. Expression of the CCAAT/Enhancer-Binding Proteins C/EBPalpha, C/EBPbeta and C/EBPdelta in Breast Cancer: Correlations with Clinicopathologic Parameters and Cell-Cycle Regulatory Proteins. Breast Cancer Res. Treat. 2003, 79, 175–185. [Google Scholar] [CrossRef]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt Signaling in Triple-Negative Breast Cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef]
- Merikhian, P.; Eisavand, M.R.; Farahmand, L. Triple-Negative Breast Cancer: Understanding Wnt Signaling in Drug Resistance. Cancer Cell Int. 2021, 21, 419. [Google Scholar] [CrossRef] [PubMed]
- Maubant, S.; Tahtouh, T.; Brisson, A.; Maire, V.; Némati, F.; Tesson, B.; Ye, M.; Rigaill, G.; Noizet, M.; Dumont, A.; et al. LRP5 Regulates the Expression of STK40, a New Potential Target in Triple-Negative Breast Cancers. Oncotarget 2018, 9, 22586–22604. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.D. New Strategies in Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2010, 16, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; de Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-Kinase in Concert with Src Promotes the S-Phase Entry of Oestradiol-Stimulated MCF-7 Cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Integration of the Extranuclear and Nuclear Actions of Estrogen. Mol. Endocrinol. 2005, 19, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef]
- Choucair, A.; Pham, T.H.; Omarjee, S.; Jacquemetton, J.; Kassem, L.; Trédan, O.; Rambaud, J.; Marangoni, E.; Corbo, L.; Treilleux, I.; et al. The Arginine Methyltransferase PRMT1 Regulates IGF-1 Signaling in Breast Cancer. Oncogene 2019, 38, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Author Correction: Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2021, 22, 834. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Das, V.; Bhattacharya, S.; Chikkaputtaiah, C.; Hazra, S.; Pal, M. The Basics of Epithelial-Mesenchymal Transition (EMT): A Study from a Structure, Dynamics, and Functional Perspective. J. Cell. Physiol. 2019, 234, 14535–14555. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Cyclin-Dependent Kinase 1 (CDK1)-Mediated Phosphorylation of Enhancer of Zeste 2 (Ezh2) Regulates Its Stability. J. Biol. Chem. 2011, 286, 28511–28519. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, D.; Lu, J.; Huang, B.; Wang, Y.; Dong, M.; Fan, D.; Li, H.; Gao, Y.; Hou, P.; et al. Methylation of EZH2 by PRMT1 Regulates Its Stability and Promotes Breast Cancer Metastasis. Cell Death Differ. 2020, 27, 3226–3242. [Google Scholar] [CrossRef]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef]
- Liao, H.-W.; Hsu, J.-M.; Xia, W.; Wang, H.-L.; Wang, Y.-N.; Chang, W.-C.; Arold, S.T.; Chou, C.-K.; Tsou, P.-H.; Yamaguchi, H.; et al. PRMT1-Mediated Methylation of the EGF Receptor Regulates Signaling and Cetuximab Response. J. Clin. Investig. 2015, 125, 4529–4543. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.-Y.; Choi, S.-J.; Lee, J.-S.; Jo, J.; Lee, J.; Kim, J.; Cha, H.-J. Loss of E-Cadherin Activates EGFR-MEK/ERK Signaling, Which Promotes Invasion via the ZEB1/MMP2 Axis in Non-Small Cell Lung Cancer. Oncotarget 2013, 4, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Zhang, W.; Chen, D.; Wang, Y. Aberrant Alternative Splicing in Breast Cancer. J. Mol. Cell Biol. 2019, 11, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative Splicing and Cancer: A Systematic Review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef]
- Jackson, C.; Browell, D.; Gautrey, H.; Tyson-Capper, A. Clinical Significance of HER-2 Splice Variants in Breast Cancer Progression and Drug Resistance. Int. J. Cell Biol. 2013, 2013, 973584. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, X.; Zhou, H.; Wu, X.; Tian, J.; Zeng, J.; Yan, L.; Duan, C.; Liu, H.; Li, H.; et al. OncoSplicing: An Updated Database for Clinically Relevant Alternative Splicing in 33 Human Cancers. Nucleic Acids Res. 2022, 50, D1340–D1347. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, F.; Miles, H.N.; Kim, E.-J.; Fields, L.; Xu, W.; Li, L. Proteome-Wide Profiling of Asymmetric Dimethylated Arginine in Human Breast Tumors. J. Am. Soc. Mass Spectrom. 2023, 34, 1692–1700. [Google Scholar] [CrossRef]
- Li, W.-J.; Huang, Y.; Lin, Y.-A.; Zhang, B.-D.; Li, M.-Y.; Zou, Y.-Q.; Hu, G.-S.; He, Y.-H.; Yang, J.-J.; Xie, B.-L.; et al. Targeting PRMT1-Mediated SRSF1 Methylation to Suppress Oncogenic Exon Inclusion Events and Breast Tumorigenesis. Cell Rep. 2023, 42, 113385. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Xu, J.; Cui, B.; Liang, Q.; Zeng, S.; He, B.; Zou, H.; Li, M.; Zhao, H.; Meng, Y.; et al. Oncogenic AURKA-Enhanced N6-Methyladenosine Modification Increases DROSHA mRNA Stability to Transactivate STC1 in Breast Cancer Stem-like Cells. Cell Res. 2021, 31, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Donnella, H.J.; Webber, J.T.; Levin, R.S.; Camarda, R.; Momcilovic, O.; Bayani, N.; Shah, K.N.; Korkola, J.E.; Shokat, K.M.; Goga, A.; et al. Kinome Rewiring Reveals AURKA Limits PI3K-Pathway Inhibitor Efficacy in Breast Cancer. Nat. Chem. Biol. 2018, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA Acquires Kinase-Independent Transactivating Function to Enhance Breast Cancer Stem Cell Phenotype. Nat. Commun. 2016, 7, 10180. [Google Scholar] [CrossRef]
- Cai, R.L.; Yan-Neale, Y.; Cueto, M.A.; Xu, H.; Cohen, D. HDAC1, a Histone Deacetylase, Forms a Complex with Hus1 and Rad9, Two G2/M Checkpoint Rad Proteins. J. Biol. Chem. 2000, 275, 27909–27916. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Vlaeminck-Guillem, V.; Trédan, O.; Poulard, C.; Le Romancer, M. Non-Genomic Signaling of Steroid Receptors in Cancer. Mol. Cell. Endocrinol. 2021, 538, 111453. [Google Scholar] [CrossRef]
- Grimm, S.L.; Hartig, S.M.; Edwards, D.P. Progesterone Receptor Signaling Mechanisms. J. Mol. Biol. 2016, 428, 3831–3849. [Google Scholar] [CrossRef]
- Knutson, T.P.; Lange, C.A. Tracking Progesterone Receptor-Mediated Actions in Breast Cancer. Pharmacol. Ther. 2014, 142, 114–125. [Google Scholar] [CrossRef]
- Malbeteau, L.; Poulard, C.; Languilaire, C.; Mikaelian, I.; Flamant, F.; Le Romancer, M.; Corbo, L. PRMT1 Is Critical for the Transcriptional Activity and the Stability of the Progesterone Receptor. iScience 2020, 23, 101236. [Google Scholar] [CrossRef]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Poulard, C.; Treilleux, I.; Lavergne, E.; Bouchekioua-Bouzaghou, K.; Goddard-Léon, S.; Chabaud, S.; Trédan, O.; Corbo, L.; Le Romancer, M. Activation of Rapid Oestrogen Signalling in Aggressive Human Breast Cancers. EMBO Mol. Med. 2012, 4, 1200–1213. [Google Scholar] [CrossRef]
- Poulard, C.; Jacquemetton, J.; Trédan, O.; Cohen, P.A.; Vendrell, J.; Ghayad, S.E.; Treilleux, I.; Marangoni, E.; Le Romancer, M. Oestrogen Non-Genomic Signalling Is Activated in Tamoxifen-Resistant Breast Cancer. Int. J. Mol. Sci. 2019, 20, 2773. [Google Scholar] [CrossRef] [PubMed]
- Jacquemetton, J.; Kassem, L.; Poulard, C.; Dahmani, A.; De Plater, L.; Montaudon, E.; Sourd, L.; Morisset, L.; El Botty, R.; Chateau-Joubert, S.; et al. Analysis of Genomic and Non-Genomic Signaling of Estrogen Receptor in PDX Models of Breast Cancer Treated with a Combination of the PI3K Inhibitor Alpelisib (BYL719) and Fulvestrant. Breast Cancer Res. 2021, 23, 57. [Google Scholar] [CrossRef]
- Malbeteau, L.; Jacquemetton, J.; Languilaire, C.; Corbo, L.; Le Romancer, M.; Poulard, C. PRMT1, a Key Modulator of Unliganded Progesterone Receptor Signaling in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9509. [Google Scholar] [CrossRef]
- Brobbey, C.; Liu, L.; Yin, S.; Gan, W. The Role of Protein Arginine Methyltransferases in DNA Damage Response. Int. J. Mol. Sci. 2022, 23, 9780. [Google Scholar] [CrossRef] [PubMed]
- Auclair, Y.; Richard, S. The Role of Arginine Methylation in the DNA Damage Response. DNA Repair 2013, 12, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hébert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR Motif Regulates DNA Double-Strand Break Processing and ATR Activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef]
- Sanchez-Bailon, M.P.; Choi, S.-Y.; Dufficy, E.R.; Sharma, K.; McNee, G.S.; Gunnell, E.; Chiang, K.; Sahay, D.; Maslen, S.; Stewart, G.S.; et al. Arginine Methylation and Ubiquitylation Crosstalk Controls DNA End-Resection and Homologous Recombination Repair. Nat. Commun. 2021, 12, 6313. [Google Scholar] [CrossRef]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A Mechanism for the Suppression of Homologous Recombination in G1 Cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Ting, X.; Xia, L.; Yang, J.; He, L.; Si, W.; Shang, Y.; Sun, L. USP11 Acts as a Histone Deubiquitinase Functioning in Chromatin Reorganization during DNA Repair. Nucleic Acids Res. 2019, 47, 9721–9740. [Google Scholar] [CrossRef]
- Nishi, R.; Wijnhoven, P.; le Sage, C.; Tjeertes, J.; Galanty, Y.; Forment, J.V.; Clague, M.J.; Urbé, S.; Jackson, S.P. Systematic Characterization of Deubiquitylating Enzymes for Roles in Maintaining Genome Integrity. Nat. Cell Biol. 2014, 16, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.R.; Apgar, S.; Dolios, G.; Wang, R.; Aaronson, S.A. BRCA2 Is Ubiquitinated in Vivo and Interacts with USP11, a Deubiquitinating Enzyme That Exhibits Prosurvival Function in the Cellular Response to DNA Damage. Mol. Cell. Biol. 2004, 24, 7444–7455. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.A.; Baek, C.; Estrada, M.V.; Tysl, T.; Bennett, E.J.; Yang, J.; Chang, J.T. USP11 Enhances TGFβ-Induced Epithelial-Mesenchymal Plasticity and Human Breast Cancer Metastasis. Mol. Cancer Res. 2018, 16, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Mirman, Z.; de Lange, T. 53BP1: A DSB Escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Paiano, J.; Zolnerowich, N.; Wu, W.; Pavani, R.; Wang, C.; Li, H.; Zheng, L.; Shen, B.; Sleckman, B.P.; Chen, B.-R.; et al. Role of 53BP1 in End Protection and DNA Synthesis at DNA Breaks. Genes Dev. 2021, 35, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.-M.; Rhie, A.; Richard, S.; Doherty, A.J. The GAR Motif of 53BP1 Is Arginine Methylated by PRMT1 and Is Necessary for 53BP1 DNA Binding Activity. Cell Cycle 2005, 4, 1834–1841. [Google Scholar] [CrossRef]
- Adams, M.M.; Wang, B.; Xia, Z.; Morales, J.C.; Lu, X.; Donehower, L.A.; Bochar, D.A.; Elledge, S.J.; Carpenter, P.B. 53BP1 Oligomerization Is Independent of Its Methylation by PRMT1. Cell Cycle 2005, 4, 1854–1861. [Google Scholar] [CrossRef]
- Hsu, W.-J.; Chen, C.-H.; Chang, Y.-C.; Cheng, C.-H.; TsaI, Y.-H.; Lin, C.-W. PRMT1 Confers Resistance to Olaparib via Modulating MYC Signaling in Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 1009. [Google Scholar] [CrossRef] [PubMed]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor Cell Kill by C-MYC Depletion: Role of MYC-Regulated Genes That Control DNA Double-Strand Break Repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bu, X.; Chu, C.; Dai, X.; Asara, J.M.; Sicinski, P.; Freeman, G.J.; Wei, W. PRMT1 Mediated Methylation of cGAS Suppresses Anti-Tumor Immunity. Nat. Commun. 2023, 14, 2806. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Shang, M.; Han, Y.; Liu, J.; Liu, J.; Kong, S.H.; Hou, J.; Huang, B.; Lu, J.; Zhang, Y. EZH2-CCF-cGAS Axis Promotes Breast Cancer Metastasis. Int. J. Mol. Sci. 2022, 23, 1788. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Li, Z.; Wang, D.; Wang, W.; Chen, X.; Tang, A.; Hou, P.; Li, M.; Zheng, J.; Bai, J. Macrophages-Stimulated PRMT1-Mediated EZH2 Methylation Promotes Breast Cancer Metastasis. Biochem. Biophys. Res. Commun. 2020, 533, 679–684. [Google Scholar] [CrossRef]
- Sakamaki, J.; Daitoku, H.; Ueno, K.; Hagiwara, A.; Yamagata, K.; Fukamizu, A. Arginine Methylation of BCL-2 Antagonist of Cell Death (BAD) Counteracts Its Phosphorylation and Inactivation by Akt. Proc. Natl. Acad. Sci. USA 2011, 108, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-H.; Lee, M.-K.; Yoon, K.W.; Lee, J.; Cho, S.-G.; Choi, E.-J. Arginine Methylation-Dependent Regulation of ASK1 Signaling by PRMT1. Cell Death Differ. 2012, 19, 859–870. [Google Scholar] [CrossRef]
- Hatai, T.; Matsuzawa, A.; Inoshita, S.; Mochida, Y.; Kuroda, T.; Sakamaki, K.; Kuida, K.; Yonehara, S.; Ichijo, H.; Takeda, K. Execution of Apoptosis Signal-Regulating Kinase 1 (ASK1)-Induced Apoptosis by the Mitochondria-Dependent Caspase Activation. J. Biol. Chem. 2000, 275, 26576–26581. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 Is Required for Sustained Activations of JNK/P38 MAP Kinases and Apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and P38 Signaling Pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The Role of PARP in DNA Repair and Its Therapeutic Exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takano, N.; Ishiwata, K.; Ohmura, M.; Nagahata, Y.; Matsuura, T.; Kamata, A.; Sakamoto, K.; Nakanishi, T.; Kubo, A.; et al. Reduced Methylation of PFKFB3 in Cancer Cells Shunts Glucose towards the Pentose Phosphate Pathway. Nat. Commun. 2014, 5, 3480. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hayashida, T.; Masugi, Y.; Oshikawa, K.; Hayakawa, N.; Itoh, M.; Nishime, C.; Suzuki, M.; Nagayama, A.; Kawai, Y.; et al. PRMT1 Sustains de Novo Fatty Acid Synthesis by Methylating PHGDH to Drive Chemoresistance in Triple-Negative Breast Cancer. Cancer Res. 2024, 84, 1065–1083. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional Genomics Reveal That the Serine Synthesis Pathway Is Essential in Breast Cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. The Structure and Functions of PRMT5 in Human Diseases. Life 2021, 11, 1074. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 Arginine Methyltransferase: Many Roles in Development, Cancer and Beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFβ-PRMT5-MEP50 Axis Regulates Cancer Cell Invasion through Histone H3 and H4 Arginine Methylation Coupled Transcriptional Activation and Repression. Oncogene 2017, 36, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Pollack, B.P.; Kotenko, S.V.; He, W.; Izotova, L.S.; Barnoski, B.L.; Pestka, S. The Human Homologue of the Yeast Proteins Skb1 and Hsl7p Interacts with Jak Kinases and Contains Protein Methyltransferase Activity. J. Biol. Chem. 1999, 274, 31531–31542. [Google Scholar] [CrossRef] [PubMed]
- Branscombe, T.L.; Frankel, A.; Lee, J.H.; Cook, J.R.; Yang, Z.; Pestka, S.; Clarke, S. PRMT5 (Janus Kinase-Binding Protein 1) Catalyzes the Formation of Symmetric Dimethylarginine Residues in Proteins. J. Biol. Chem. 2001, 276, 32971–32976. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. PRMT5 Function and Targeting in Cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Tarighat, S.S.; Santhanam, R.; Frankhouser, D.; Radomska, H.S.; Lai, H.; Anghelina, M.; Wang, H.; Huang, X.; Alinari, L.; Walker, A.; et al. The Dual Epigenetic Role of PRMT5 in Acute Myeloid Leukemia: Gene Activation and Repression via Histone Arginine Methylation. Leukemia 2016, 30, 789–799. [Google Scholar] [CrossRef] [PubMed]
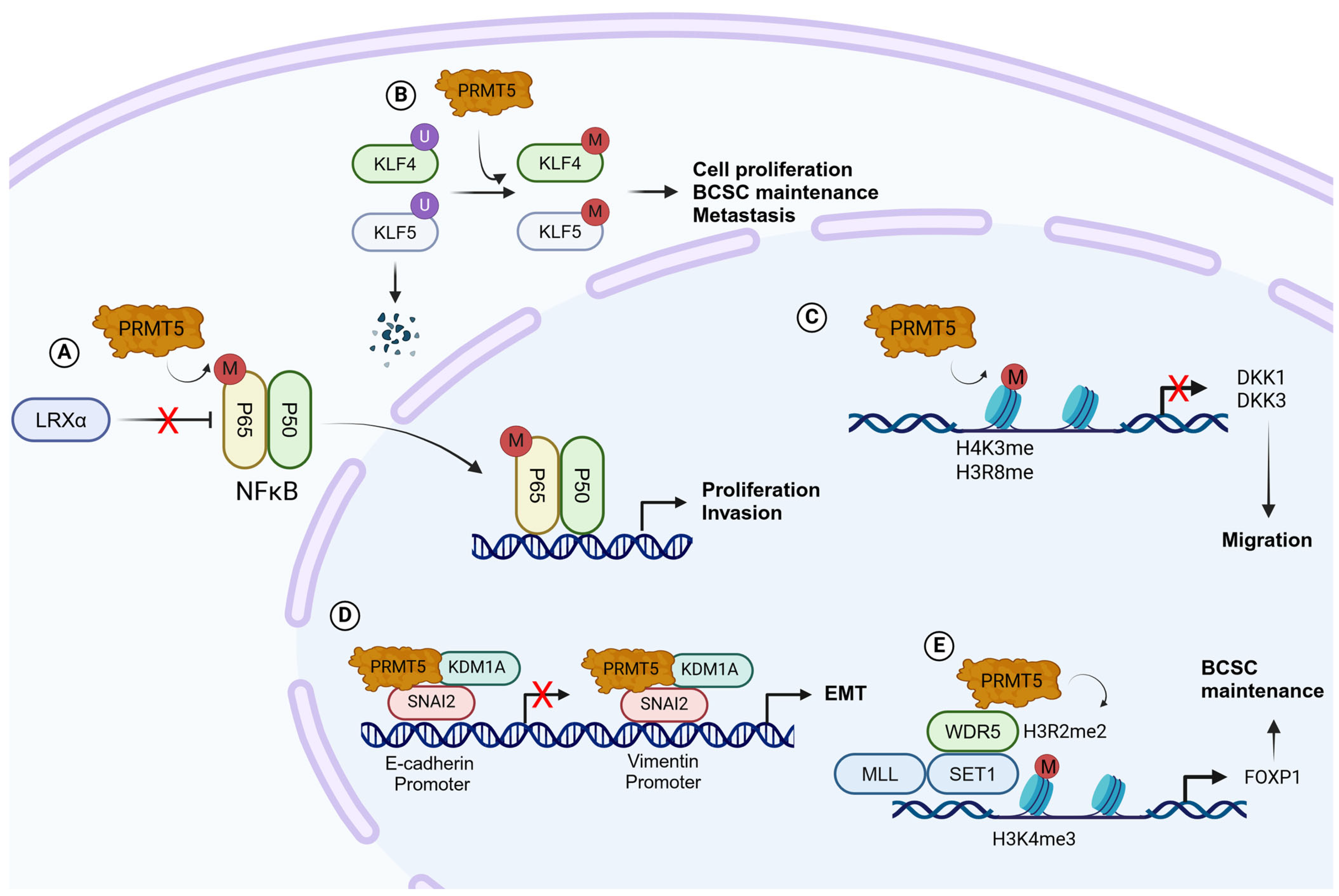
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef]
- Fu, T.; Lv, X.; Kong, Q.; Yuan, C. A Novel SHARPIN-PRMT5-H3R2me1 Axis Is Essential for Lung Cancer Cell Invasion. Oncotarget 2017, 8, 54809–54820. [Google Scholar] [CrossRef]
- Cho, E.-C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.-C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine Methylation Controls Growth Regulation by E2F-1. EMBO J. 2012, 31, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Gao, J.; Yang, Y.; Qiu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Hou, Y.; Wang, S.; Leng, S.; et al. PHD Finger Protein 1 (PHF1) Is a Novel Reader for Histone H4R3 Symmetric Dimethylation and Coordinates with PRMT5–WDR77/CRL4B Complex to Promote Tumorigenesis. Nucleic Acids Res. 2018, 46, 6608–6626. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, J.; Wu, Y.; Zhang, J.; Wang, T.; Li, N.; Fan, J.; Wang, H.; Zhang, J.; Ling, R. PRMT5 Determines the Sensitivity to Chemotherapeutics by Governing Stemness in Breast Cancer. Breast Cancer Res. Treat. 2018, 168, 531–542. [Google Scholar] [CrossRef] [PubMed]
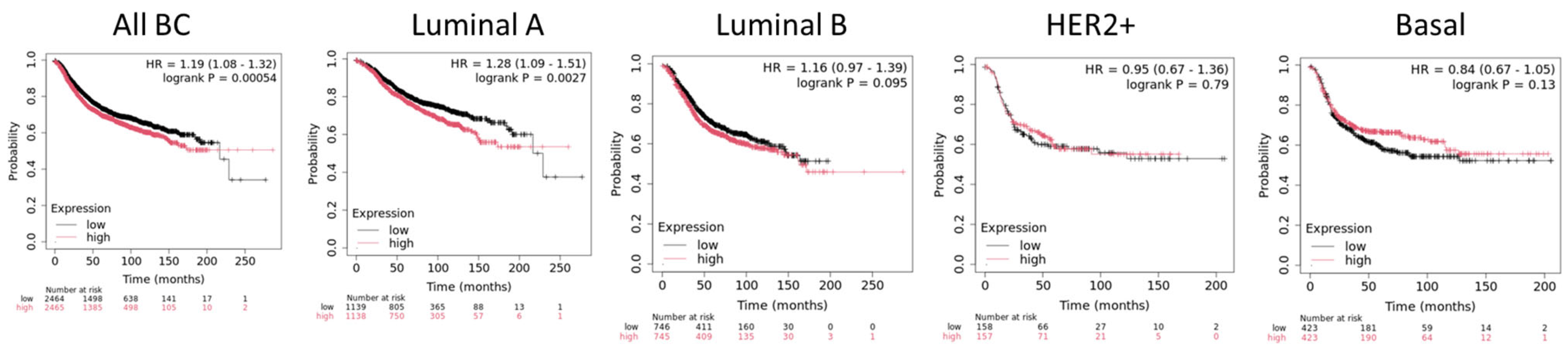
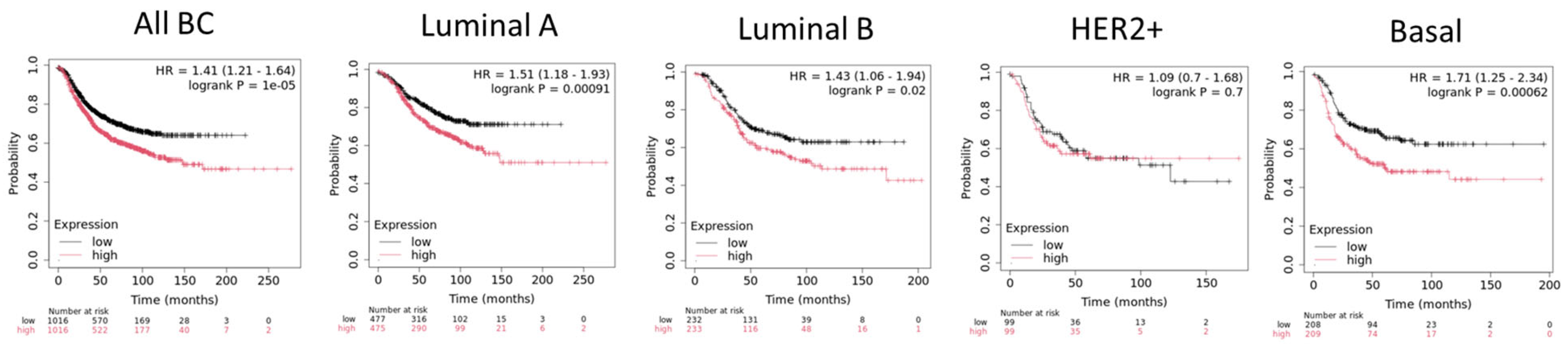
- Wu, Y.; Wang, Z.; Zhang, J.; Ling, R. Elevated Expression of Protein Arginine Methyltransferase 5 Predicts the Poor Prognosis of Breast Cancer. Tumor Biol. 2017, 39, 1010428317695917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, X.; Zhou, Y.; Chen, L.; Rao, H. The PRMT5-LSD1 Axis Confers Slug Dual Transcriptional Activities and Promotes Breast Cancer Progression. J. Exp. Clin. Cancer Res. 2022, 41, 191. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.; Hulse, M.; Sivakumar, M.; Burtell, J.; Thodima, V.; Wang, M.; Agarwal, A.; Vykuntam, K.; Spruance, J.; Bhagwat, N.; et al. PRMT5 Inhibitors Regulate DNA Damage Repair Pathways in Cancer Cells and Improve Response to PARP Inhibition and Chemotherapies. Cancer Res. Commun. 2023, 3, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Shailesh, H.; Siveen, K.S.; Sif, S. Protein Arginine Methyltransferase 5 (PRMT5) Activates WNT/Β-catenin Signalling in Breast Cancer Cells via Epigenetic Silencing of DKK1 and DKK3. J. Cell. Mol. Med. 2021, 25, 1583–1600. [Google Scholar] [CrossRef] [PubMed]
- Girardot, M.; Hirasawa, R.; Kacem, S.; Fritsch, L.; Pontis, J.; Kota, S.K.; Filipponi, D.; Fabbrizio, E.; Sardet, C.; Lohmann, F.; et al. PRMT5-Mediated Histone H4 Arginine-3 Symmetrical Dimethylation Marks Chromatin at G + C-Rich Regions of the Mouse Genome. Nucleic Acids Res. 2014, 42, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Rowland, B.D.; Peeper, D.S. KLF4, P21 and Context-Dependent Opposing Forces in Cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef]
- Tetreault, M.-P.; Yang, Y.; Katz, J.P. Krüppel-like Factors in Cancer. Nat. Rev. Cancer 2013, 13, 701–713. [Google Scholar] [CrossRef]
- Foster, K.W.; Frost, A.R.; McKie-Bell, P.; Lin, C.Y.; Engler, J.A.; Grizzle, W.E.; Ruppert, J.M. Increase of GKLF Messenger RNA and Protein Expression during Progression of Breast Cancer. Cancer Res. 2000, 60, 6488–6495. [Google Scholar] [PubMed]
- Pandya, A.Y.; Talley, L.I.; Frost, A.R.; Fitzgerald, T.J.; Trivedi, V.; Chakravarthy, M.; Chhieng, D.C.; Grizzle, W.E.; Engler, J.A.; Krontiras, H.; et al. Nuclear Localization of KLF4 Is Associated with an Aggressive Phenotype in Early-Stage Breast Cancer. Clin. Cancer Res. 2004, 10, 2709–2719. [Google Scholar] [CrossRef] [PubMed]
- Gamper, A.M.; Qiao, X.; Kim, J.; Zhang, L.; DeSimone, M.C.; Rathmell, W.K.; Wan, Y. Regulation of KLF4 Turnover Reveals an Unexpected Tissue-Specific Role of pVHL in Tumorigenesis. Mol. Cell 2012, 45, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Gur, M.; Zhou, Z.; Gamper, A.; Hung, M.-C.; Fujita, N.; Lan, L.; Bahar, I.; Wan, Y. Interplay between Arginine Methylation and Ubiquitylation Regulates KLF4-Mediated Genome Stability and Carcinogenesis. Nat. Commun. 2015, 6, 8419. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zheng, H.-Q.; Zhou, Z.; Dong, J.-T.; Chen, C. KLF5 Promotes Breast Cell Survival Partially through Fibroblast Growth Factor-Binding Protein 1-pERK-Mediated Dual Specificity MKP-1 Protein Phosphorylation and Stabilization. J. Biol. Chem. 2009, 284, 16791–16798. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Czerwenka, K.; Heinze, G.; Ryffel, M.; Schuster, E.; Witt, A.; Leodolter, S.; Zeillinger, R. Expression of KLF5 Is a Prognostic Factor for Disease-Free Survival and Overall Survival in Patients with Breast Cancer. Clin. Cancer Res. 2006, 12, 2442–2448. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, T.; Wu, Y.; Yang, C.; Li, Y.; Du, G.; He, Y.; Liu, W.; Liu, R.; Chen, C.; et al. Arginine Methyltransferase PRMT5 Methylates and Stabilizes KLF5 via Decreasing Its Phosphorylation and Ubiquitination to Promote Basal-like Breast Cancer. Cell Death Differ. 2021, 28, 2931–2945. [Google Scholar] [CrossRef]
- Nasir Kansestani, A.; Mansouri, K.; Hemmati, S.; Zare, M.E.; Moatafaei, A. High Glucose-Reduced Apoptosis in Human Breast Cancer Cells Is Mediated by Activation of NF-κB. Iran. J. Allergy Asthma Immunol. 2019, 18, 153–162. [Google Scholar] [CrossRef]
- Han, X.; Wei, L.; Wu, B. PRMT5 Promotes Aerobic Glycolysis and Invasion of Breast Cancer Cells by Regulating the LXRα/NF-κBp65 Pathway. OncoTargets Ther. 2020, 13, 3347–3357. [Google Scholar] [CrossRef]
- Lin, S.Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.C. Beta-Catenin, a Novel Prognostic Marker for Breast Cancer: Its Roles in Cyclin D1 Expression and Cancer Progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhang, M.; Zhou, F.; Zhang, L.; Meng, X. The Breast Cancer Stem Cells Traits and Drug Resistance. Front. Pharmacol. 2021, 11, 599965. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting Methyltransferase PRMT5 Eliminates Leukemia Stem Cells in Chronic Myelogenous Leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [PubMed]
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5–PTEN Molecular Pathway Regulates Senescence and Self-Renewal of Primary Glioblastoma Neurosphere Cells. Oncogene 2017, 36, 263–274. [Google Scholar] [CrossRef]
- Choi, E.J.; Seo, E.J.; Kim, D.K.; Lee, S.I.; Kwon, Y.W.; Jang, I.H.; Kim, K.-H.; Suh, D.-S.; Kim, J.H. FOXP1 Functions as an Oncogene in Promoting Cancer Stem Cell-like Characteristics in Ovarian Cancer Cells. Oncotarget 2015, 7, 3506–3519. [Google Scholar] [CrossRef]
- Huang, J.; Zheng, Y.; Zheng, X.; Qian, B.; Yin, Q.; Lu, J.; Lei, H. PRMT5 Promotes EMT Through Regulating Akt Activity in Human Lung Cancer. Cell Transplant. 2021, 30, 9636897211001772. [Google Scholar] [CrossRef]
- Zheng, Y.; Ji, H.; Yi, W.; Chen, Z.; Hu, X.; Zhou, J.; Wang, Y.; Zheng, X. PRMT5 Facilitates Angiogenesis and EMT via HIF-1α/VEGFR/Akt Signaling Axis in Lung Cancer. Aging 2023, 15, 6163–6178. [Google Scholar] [CrossRef]
- Ge, L.; Wang, H.; Xu, X.; Zhou, Z.; He, J.; Peng, W.; Du, F.; Zhang, Y.; Gong, A.; Xu, M. PRMT5 Promotes Epithelial-mesenchymal Transition via EGFR-β-catenin Axis in Pancreatic Cancer Cells. J. Cell. Mol. Med. 2020, 24, 1969–1979. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A Day in the Life of the Spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Will, C.L.; Lührmann, R. Spliceosome Structure and Function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Pesiridis, G.S.; Diamond, E.; Van Duyne, G.D. Role of pICLn in Methylation of Sm Proteins by PRMT5. J. Biol. Chem. 2009, 284, 21347–21359. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Eggert, C.; Bühler, D.; Brahms, H.; Kambach, C.; Fischer, U. Methylation of Sm Proteins by a Complex Containing PRMT5 and the Putative U snRNP Assembly Factor pICln. Curr. Biol. 2001, 11, 1990–1994. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.Y.; Pignata, L.; Goy, P.-A.; Kawabata, K.C.; Lee, S.C.-W.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, Y.; Yang, J.; Hu, G.; Lin, Y.; Ran, T.; Peng, B.; Xie, B.; Huang, M.; Gao, X.; et al. Profiling PRMT Methylome Reveals Roles of hnRNPA1 Arginine Methylation in RNA Splicing and Cell Growth. Nat. Commun. 2021, 12, 1946. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 Methylome Profiling Uncovers a Direct Link to Splicing Regulation in Acute Myeloid Leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of Constitutive and Alternative Splicing by PRMT5 Reveals a Role for Mdm4 Pre-mRNA in Sensing Defects in the Spliceosomal Machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.-P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the P53-MDM4 Regulatory Axis Defines the Anti-Tumour Response to PRMT5 Inhibition through Its Role in Regulating Cellular Splicing. Sci. Rep. 2018, 8, 9711. [Google Scholar] [CrossRef]
- Szewczyk, M.M.; Luciani, G.M.; Vu, V.; Murison, A.; Dilworth, D.; Barghout, S.H.; Lupien, M.; Arrowsmith, C.H.; Minden, M.D.; Barsyte-Lovejoy, D. PRMT5 Regulates ATF4 Transcript Splicing and Oxidative Stress Response. Redox Biol. 2022, 51, 102282. [Google Scholar] [CrossRef] [PubMed]
- Rengasamy, M.; Zhang, F.; Vashisht, A.; Song, W.-M.; Aguilo, F.; Sun, Y.; Li, S.; Zhang, W.; Zhang, B.; Wohlschlegel, J.A.; et al. The PRMT5/WDR77 Complex Regulates Alternative Splicing through ZNF326 in Breast Cancer. Nucleic Acids Res. 2017, 45, 11106–11120. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Wagner, M.; Koyasu, S. Cancer Immunoediting by Innate Lymphoid Cells. Trends Immunol. 2019, 40, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 Control of cGAS/STING and NLRC5 Pathways Defines Melanoma Response to Antitumor Immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Ji, M.Q.; Zhu, F.; Xiao, Y.; Tanaka, Y.; Kambayashi, T.; Fujimoto, S.; Goldberg, M.M.; Zhang, H.; Li, B.; et al. PRMT5 Associates With the FOXP3 Homomer and When Disabled Enhances Targeted p185erbB2/Neu Tumor Immunotherapy. Front. Immunol. 2019, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T Cells, Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T Cells in Tumor Immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, R.; Hou, N.; Zhang, J.; Wang, T.; Fan, P.; Ji, C.; Zhang, B.; Liu, L.; Wang, Y.; et al. PRMT5 Reduces Immunotherapy Efficacy in Triple-Negative Breast Cancer by Methylating KEAP1 and Inhibiting Ferroptosis. J. Immunother. Cancer 2023, 11, e006890. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.-C.; Chiang, S.-K.; Chen, S.-E.; Yu, Y.-L.; Chou, R.-H.; Chang, W.-C. Heme Oxygenase-1 Mediates BAY 11-7085 Induced Ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Tinsley, S.L.; Asberry, A.M.; Deng, X.; Huang, J.; Li, C.; Wan, J.; Hu, C.-D. PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 2020, 23, 100750. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 Inhibition Disrupts Splicing and Stemness in Glioblastoma. Nat. Commun. 2021, 12, 979. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Z.; Han, L.; Guo, Z.; Yan, B.; Guo, L.; Zhao, H.; Wei, M.; Hou, N.; Ye, J.; et al. PRMT5 Regulates RNA m6A Demethylation for Doxorubicin Sensitivity in Breast Cancer. Mol. Ther. 2022, 30, 2603–2617. [Google Scholar] [CrossRef]
- Du, W.; Amarachintha, S.; Erden, O.; Wilson, A.; Pang, Q. The Fanconi Anemia Pathway Controls Oncogenic Response in Hematopoietic Stem and Progenitor Cells by Regulating PRMT5-Mediated P53 Arginine Methylation. Oncotarget 2016, 7, 60005–60020. [Google Scholar] [CrossRef]
- Zhou, Z.; Feng, Z.; Hu, D.; Yang, P.; Gur, M.; Bahar, I.; Cristofanilli, M.; Gradishar, W.J.; Xie, X.; Wan, Y. A Novel Small-Molecule Antagonizes PRMT5-Mediated KLF4 Methylation for Targeted Therapy. eBioMedicine 2019, 44, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Checa-Rodríguez, C.; Cepeda-García, C.; Ramón, J.; López-Saavedra, A.; Balestra, F.R.; Domínguez-Sánchez, M.S.; Gómez-Cabello, D.; Huertas, P. Methylation of the Central Transcriptional Regulator KLF4 by PRMT5 Is Required for DNA End Resection and Recombination. DNA Repair 2020, 94, 102902. [Google Scholar] [CrossRef]
- Du, C.; Li, S.W.; Singh, S.X.; Roso, K.; Sun, M.A.; Pirozzi, C.J.; Yang, R.; Li, J.-L.; He, Y. Epigenetic Regulation of Fanconi Anemia Genes Implicates PRMT5 Blockage as a Strategy for Tumor Chemosensitization. Mol. Cancer Res. 2021, 19, 2046–2056. [Google Scholar] [CrossRef]
- Hamard, P.-J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- O’Brien, S.; Butticello, M.; Thompson, C.; Wilson, B.; Wyce, A.; Mahajan, V.; Kruger, R.; Mohammad, H.; Fedoriw, A. Inhibiting PRMT5 Induces DNA Damage and Increases Anti-Proliferative Activity of Niraparib, a PARP Inhibitor, in Models of Breast and Ovarian Cancer. BMC Cancer 2023, 23, 775. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 Is Essential for Early Mouse Development and Acts in the Cytoplasm to Maintain ES Cell Pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef]
- Ancelin, K.; Lange, U.C.; Hajkova, P.; Schneider, R.; Bannister, A.J.; Kouzarides, T.; Surani, M.A. Blimp1 Associates with Prmt5 and Directs Histone Arginine Methylation in Mouse Germ Cells. Nat. Cell Biol. 2006, 8, 623–630. [Google Scholar] [CrossRef]
- Gu, Z.; Li, Y.; Lee, P.; Liu, T.; Wan, C.; Wang, Z. Protein Arginine Methyltransferase 5 Functions in Opposite Ways in the Cytoplasm and Nucleus of Prostate Cancer Cells. PLoS ONE 2012, 7, e44033. [Google Scholar] [CrossRef]
- Vinet, M.; Suresh, S.; Maire, V.; Monchecourt, C.; Némati, F.; Lesage, L.; Pierre, F.; Ye, M.; Lescure, A.; Brisson, A.; et al. Protein Arginine Methyltransferase 5: A Novel Therapeutic Target for Triple-negative Breast Cancers. Cancer Med. 2019, 8, 2414–2428. [Google Scholar] [CrossRef]
- Lattouf, H.; Kassem, L.; Jacquemetton, J.; Choucair, A.; Poulard, C.; Trédan, O.; Corbo, L.; Diab-Assaf, M.; Hussein, N.; Treilleux, I.; et al. LKB1 Regulates PRMT5 Activity in Breast Cancer. Int. J. Cancer 2019, 144, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.M.; Ablain, J.; Surmieliova-Garnès, A.; Jacquemetton, J.; Pham, T.H.; Marangoni, E.; Schnitzler, A.; Bieche, I.; Badran, B.; Trédan, O.; et al. PRMT5 Triggers Glucocorticoid-Induced Cell Migration in Triple-Negative Breast Cancer. Life Sci. Alliance 2023, 6, e202302009. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.-P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J. The LIM Protein AJUBA Recruits Protein Arginine Methyltransferase 5 to Mediate SNAIL-Dependent Transcriptional Repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Liu, L.; Wen, C.; Jiang, H.; Ye, T.; Ma, S.; Liu, X. Clinicopathological and Prognostic Significance of PRMT5 in Cancers: A System Review and Meta-Analysis. Cancer Control 2021, 28, 10732748211050583. [Google Scholar] [CrossRef] [PubMed]
- Kaniskan, H.Ü.; Martini, M.L.; Jin, J. Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev. 2018, 118, 989–1068. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Protein Methyltransferase Inhibitors as Precision Cancer Therapeutics: A Decade of Discovery. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170080. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Clarke, J.; Neff, T.; Crossman, T.; Ratia, N.; Rathi, C.; Noto, P.; Tarkar, A.; Garrido-Laguna, I.; Calvo, E.; et al. Phase 1 Study of GSK3368715, a Type I PRMT Inhibitor, in Patients with Advanced Solid Tumors. Br. J. Cancer 2023, 129, 309–317. [Google Scholar] [CrossRef]
- Feustel, K.; Falchook, G.S. Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A Review. J. Immunother. Precis. Oncol. 2022, 5, 58–67. [Google Scholar] [CrossRef]
- Bhandari, K.; Ding, W.-Q. Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy. Int. J. Mol. Sci. 2024, 25, 3958. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, J.; Ren, Y.; Dong, L.; Wu, H.; Hong, W.; Huang, H.; Yang, X.; Pang, Z.; Wang, H. Discovery of 2,4-Diphenyl-Substituted Thiazole Derivatives as PRMT1 Inhibitors and Investigation of Their Anti-Cervical Cancer Effects. Biozorg. Med. Chem. 2023, 92, 117436. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Jang, J.; Park, J.G.; Kim, K.-H.; Yoon, K.; Yoo, B.C.; Cho, J.Y. Protein Arginine Methyltransferase 1 (PRMT1) Selective Inhibitor, TC-E 5003, Has Anti-Inflammatory Properties in TLR4 Signaling. Int. J. Mol. Sci. 2020, 21, 3058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tao, H.; Yu, L.; Zhou, L.; Zhu, C. Developing Protein Arginine Methyltransferase 1 (PRMT1) Inhibitor TC-E-5003 as an Antitumor Drug Using INEI Drug Delivery Systems. Drug Deliv. 2020, 27, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.-H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective Inhibition of Protein Arginine Methyltransferase 5 Blocks Initiation and Maintenance of B-Cell Transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.-V.; Wang, B.; Mundade, R.; Jiang, G.; Sun, M.; Wei, H.; Sun, S.; Liu, Y.; Lu, T. PRMT5-Mediated Methylation of YBX1 Regulates NF-κB Activity in Colorectal Cancer. Sci. Rep. 2020, 10, 15934. [Google Scholar] [CrossRef] [PubMed]
- Demetriadou, C.; Pavlou, D.; Mpekris, F.; Achilleos, C.; Stylianopoulos, T.; Zaravinos, A.; Papageorgis, P.; Kirmizis, A. NAA40 Contributes to Colorectal Cancer Growth by Controlling PRMT5 Expression. Cell Death Dis. 2019, 10, 236. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A Selective Inhibitor of PRMT5 with in Vivo and in Vitro Potency in MCL Models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Targeted CRISPR Screening Identifies PRMT5 as Synthetic Lethality Combinatorial Target with Gemcitabine in Pancreatic Cancer Cells|PNAS. Available online: https://www.pnas.org/doi/full/10.1073/pnas.2009899117 (accessed on 1 July 2024).
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-Mediated Arginine Methylation Activates AKT Kinase to Govern Tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef]
- Smith, C.R.; Aranda, R.; Bobinski, T.P.; Briere, D.M.; Burns, A.C.; Christensen, J.G.; Clarine, J.; Engstrom, L.D.; Gunn, R.J.; Ivetac, A.; et al. Fragment-Based Discovery of MRTX1719, a Synthetic Lethal Inhibitor of the PRMT5•MTA Complex for the Treatment of MTAP-Deleted Cancers. J. Med. Chem. 2022, 65, 1749–1766. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Residue | Functional Impact | Refs. |
|---|---|---|---|
| H4 | R3 | Transcriptional regulation | [41,42] |
| P53 | N/A | Inhibits p53 transcriptional activity | [43] |
| C/EBPα | R35, R156, R165 | Promotes C/EBPα transcriptional activity | [44] |
| EGFR | R198, R200 | Enhances receptor signaling | [40,42] |
| EZH2 | R342 | Inhibits EZH2 transcriptional activity | [45] |
| Erα | R260 | Promotes MAPK and AKT signaling | [46] |
| PR | R637 | Regulates stability and transcriptional activity | [47,48] |
| MRE11 | R587 | Promotes DNA end resection | [37] |
| BRCA1 | R610 | Regulates BRCA1 subcellular localization | [49] |
| USP11 | R433 | RPA foci formation and RAD51 regulation | [50] |
| 53BP1 | ND | Enhances DNA repair process | [51] |
| BAD | R94, R96 | Inhibits BAD anti-apoptotic activity | [52] |
| ASK1 | R78, R80 | Anti-apoptotic function and drug resistance | [31] |
| Substrate | Residue | Functional Impact | Refs. |
|---|---|---|---|
| H3 | R8, R2 | Transcriptional regulation | [94,103,104,105] |
| H4 | R3 | Transcriptional regulation | [103,105] |
| NF-κB (p65) | R30 | Enhances p65 binding to DNA and transcriptional activity | [106] |
| KLF4 | R376, R377 | Protein stabilization | [107] |
| KLF5 | R57 | Protein stabilization | [108] |
| GR | ND | Promotes GR transcriptional activity | [109] |
| ERα | ND | Inhibits ERα transcriptional activity | [23] |
| Target | Drug | Mechanism of Action | Phase/Status | Intended Use |
|---|---|---|---|---|
| Type I PRMTs | GSK3368715 | Substrate competitive | Phase I; terminated | Single agent |
| PRMT1 | ZJG51 | Substrate competitive | Preclinical models | Single agent |
| TC-E 5003 | Substrate competitive | Preclinical models | Single agent | |
| PRMT5 | GSK3326595 | Substrate competitive | Phase II; terminated | Single agent |
| Phase I; active | Combination with pembrolizumab | |||
| Preclinical models | Combination with palbociclib | |||
| JNJ-64619178 | Dual SAM/substrate competitive | Phase I; active | Single agent | |
| CMP5 | SAM-competitive | Preclinical models | Single agent | |
| EPZ015666 | Substrate competitive | Preclinical models | Single agent | |
| Preclinical models | Combination with erlotinib | |||
| Preclinical models | Combination with paclitaxel | |||
| MRTX1719 | PRMT5-MTA inhibitor | Phase I/II; recruiting | Single agent |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, S.; Sentis, S.; Poulard, C.; Trédan, O.; Le Romancer, M. Role of PRMT1 and PRMT5 in Breast Cancer. Int. J. Mol. Sci. 2024, 25, 8854. https://doi.org/10.3390/ijms25168854
Martinez S, Sentis S, Poulard C, Trédan O, Le Romancer M. Role of PRMT1 and PRMT5 in Breast Cancer. International Journal of Molecular Sciences. 2024; 25(16):8854. https://doi.org/10.3390/ijms25168854
Chicago/Turabian StyleMartinez, Sébastien, Stéphanie Sentis, Coralie Poulard, Olivier Trédan, and Muriel Le Romancer. 2024. "Role of PRMT1 and PRMT5 in Breast Cancer" International Journal of Molecular Sciences 25, no. 16: 8854. https://doi.org/10.3390/ijms25168854