
A Splice Form of VEGF, a Potential Anti-Angiogenetic Form of Head and Neck Squamous Cell Cancer Inhibition



Abstract
:1. Introduction
- Heterogeneous response: Tumors can vary greatly in their response to anti-angiogenic therapy, not only between different cancers but also between patients with the same cancer. This variability makes it difficult to establish universal criteria for efficacy and treatment [18].
- The dynamic nature of tumor angiogenesis: Tumors can adapt to anti-angiogenic therapy over time, either by activating alternative angiogenic pathways or by adopting less angiogenesis-dependent mechanisms. This ability to adapt means that the efficacy of therapy may decrease with time or that initial responses may not be sustained, complicating the assessment of long-term efficacy [19].
- Lack of direct biomarkers: There is a lack of direct, reliable biomarkers that can accurately reflect changes in angiogenesis due to therapy. Most current methods assess tumor size or growth rates using imaging techniques such as MRI (magnetic resonance imaging) or CT (computed tomography). However, these indicators may not sensitively or specifically reflect changes in angiogenesis, especially in the early stages of treatment [20].
- Difficulty in measuring microenvironment changes: Anti-angiogenic therapies not only affect tumor cells, but also have a significant impact on the tumor microenvironment, including altering vascular permeability, interstitial pressure, and hypoxia. These changes are difficult to measure directly and quantitatively in patients [21].
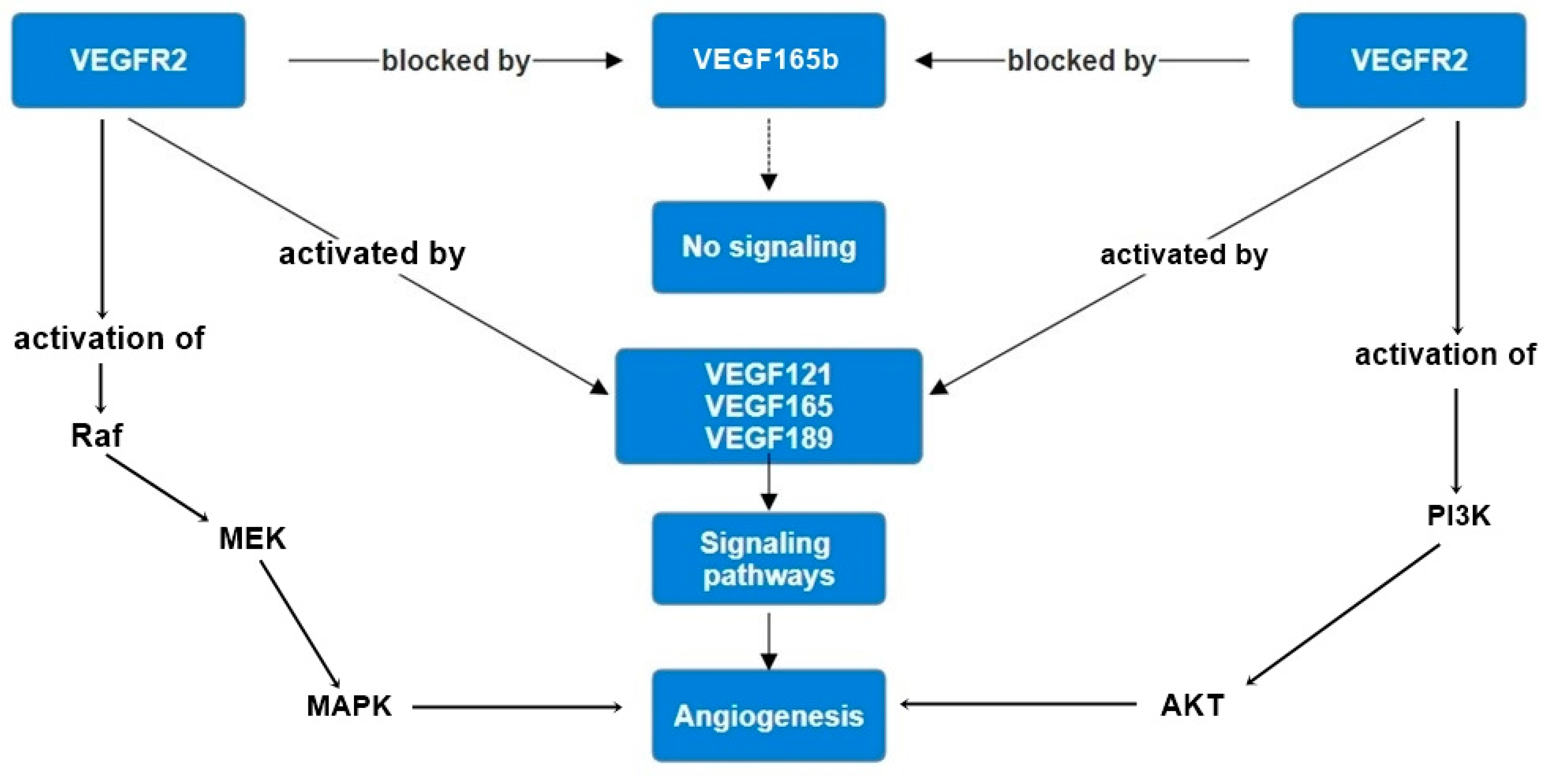
2. Understanding the Splice Forms of VEGF
2.1. The Biology of VEGF Splicing
2.2. Molecular Perspective
3. Experimental Evidence of VEGF Splice Variants in Angiogenesis Inhibition
3.1. Preclinical Studies
3.2. Clinical Perspectives on VEGF165 as a Treatment for HNSCC
3.3. Potential Limitations and Future Directions
4. Pro-Angiogenic vs. Anti-Angiogenic VEGF Variants in HNSCC: Clinical Impact
5. Resistance Mechanisms and Overcoming Therapeutic Challenges
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Z.; Anderson, K.S. Therapeutic Targeting of FGFR Signaling in Head and Neck Cancer. Cancer J. (Sudbury Mass.) 2022, 28, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Davis, G.E. Angiogenesis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Novacescu, D.; Cut, T.G.; Cumpanas, A.A.; Latcu, S.C.; Bardan, R.; Ferician, O.; Secasan, C.-C.; Rusmir, A.; Raica, M. Evaluating Established Roles, Future Perspectives and Methodological Heterogeneity for Wilms’ Tumor 1 (WT1) Antigen Detection in Adult Renal Cell Carcinoma, Using a Novel N-Terminus Targeted Antibody (Clone WT49). Biomedicines 2022, 10, 912. [Google Scholar] [CrossRef] [PubMed]
- Novacescu, D.; Cut, T.G.; Cumpanas, A.A.; Bratosin, F.; Ceausu, R.A.; Raica, M. Novel Expression of Thymine Dimers in Renal Cell Carcinoma, Demonstrated through Immunohistochemistry. Biomedicines 2022, 10, 2673. [Google Scholar] [CrossRef]
- Novacescu, D.; Feciche, B.O.; Cumpanas, A.A.; Bardan, R.; Rusmir, A.V.; Bitar, Y.A.; Barbos, V.I.; Cut, T.G.; Raica, M.; Latcu, S.C. Contemporary Clinical Definitions, Differential Diagnosis, and Novel Predictive Tools for Renal Cell Carcinoma. Biomedicines 2022, 10, 2926. [Google Scholar] [CrossRef]
- Baderca, F.; Alexa, A.; Lighezan, R.; Izvernariu, D.; Raica, M. The diagnostic value of VEGF expression in the renal parenchyma tumors. Rom. J. Morphol. Embryol. 2011, 52, 581–586. [Google Scholar]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 789–791. [Google Scholar] [CrossRef]
- Miller, J.W. VEGF: From Discovery to Therapy: The Champalimaud Award Lecture. Transl. Vis. Sci. Technol. 2016, 5, 9. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef]
- Matei, S.C.; Matei, M.; Anghel, F.M.; Derban, M.D.; Olariu, A.; Olariu, S. Impact of statin treatment on patients diagnosed with chronic venous disease. Morphological analysis of the venous wall and clinical implications. Phlebology 2022, 37, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Matei, S.C.; Matei, M.; Anghel, F.M.; Olariu, A.; Olariu, S. Great saphenous vein giant aneurysm. Acta Phlebol. 2022, 23, 87–92. [Google Scholar] [CrossRef]
- Cheng, C.; Nguyen, M.N.; Nayernama, A.; Jones, S.C.; Brave, M.; Agrawal, S.; Amiri-Kordestani, L.; Woronow, D. Arterial aneurysm and dissection with systemic vascular endothelial growth factor inhibitors: A review of cases reported to the FDA Adverse Event Reporting System and published in the literature. Vasc. Med. 2021, 26, 526–534. [Google Scholar] [CrossRef]
- Saba, N.F.; Vijayvargiya, P.; Vermorken, J.B.; Rodrigo, J.P.; Willems, S.M.; Zidar, N.; de Bree, R.; Mäkitie, A.; Wolf, G.T.; Argiris, A.; et al. Targeting Angiogenesis in Squamous Cell Carcinoma of the Head and Neck: Opportunities in the Immunotherapy Era. Cancers 2022, 14, 1202. [Google Scholar] [CrossRef] [PubMed]
- Micaily, I.; Johnson, J.; Argiris, A. An update on angiogenesis targeting in head and neck squamous cell carcinoma. Cancers Head. Neck 2020, 5, 5. [Google Scholar] [CrossRef]
- Li, D.; Finley, S.D. The impact of tumor receptor heterogeneity on the response to anti-angiogenic cancer treatment. Integr Biol (Camb) 2018, 10, 253–269. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Hilbe, W.; Heidegger, I.; Drevs, J.; Tichelli, A.; Medinger, M. Biomarkers in tumor angiogenesis and anti-angiogenic therapy. Int. J. Mol. Sci. 2011, 12, 7077–7099. [Google Scholar] [CrossRef]
- Ma, S.; Pradeep, S.; Hu, W.; Zhang, D.; Coleman, R.; Sood, A. The role of tumor microenvironment in resistance to anti-angiogenic therapy. F1000Res 2018, 7, 326. [Google Scholar] [CrossRef] [PubMed]
- Maae, E.; Nielsen, M.; Steffensen, K.D.; Jakobsen, E.H.; Jakobsen, A.; Sørensen, F.B. Estimation of immunohistochemical expression of VEGF in ductal carcinomas of the breast. J. Histochem. Cytochem. 2011, 59, 750–760. [Google Scholar] [CrossRef]
- Bao, P.; Kodra, A.; Tomic-Canic, M.; Golinko, M.S.; Ehrlich, H.P.; Brem, H. The role of vascular endothelial growth factor in wound healing. J. Surg. Res. 2009, 153, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes. Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Kuppuswamy, S.; Annex, B.H.; Ganta, V.C. Targeting Anti-Angiogenic VEGF165b-VEGFR1 Signaling Promotes Nitric Oxide Independent Therapeutic Angiogenesis in Preclinical Peripheral Artery Disease Models. Cells 2022, 11, 2676. [Google Scholar] [CrossRef]
- Catena, R.; Larzabal, L.; Larrayoz, M.; Molina, E.; Hermida, J.; Agorreta, J.; Montes, R.; Pio, R.; Montuenga, L.M.; Calvo, A. VEGF121b and VEGF165b are weakly angiogenic isoforms of VEGF-A. Mol. Cancer 2010, 31, 320. [Google Scholar] [CrossRef]
- Smith, G.A.; Fearnley, G.W.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci. Rep. 2015, 35, 00253. [Google Scholar] [CrossRef]
- Biselli-Chicote, P.M.; Biselli, J.M.; Cunha, B.R.; Castro, R.; Maniglia, J.V.; Neto, D.S.; Tajara, E.H.; Góis Filho, J.F.; Fukuyama, E.E.; Pavarino, É.C.; et al. Overexpression of Antiangiogenic Vascular Endothelial Growth Factor Isoform and Splicing Regulatory Factors in Oral, Laryngeal and Pharyngeal Squamous Cell Carcinomas. Asian Pac. J. Cancer Prev. 2017, 18, 2171–2177. [Google Scholar] [CrossRef]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef]
- Peach, C.J.; Kilpatrick, L.E.; Friedman-Ohana, R.; Zimmerman, K.; Robers, M.B.; Wood, K.V.; Woolard, J.; Hill, S.J. Real-Time Ligand Binding of Fluorescent VEGF-A Isoforms that Discriminate between VEGFR2 and NRP1 in Living Cells. Cell Chem. Biol. 2018, 25, 1208–1218.e5. [Google Scholar] [CrossRef]
- Boudria, A.; Abou, F.C.; Jia, T.; Gout, S.; Keramidas, M.; Didier, C.; Lemaître, N.; Manet, S.; Coll, J.L.; Toffart, A.C.; et al. VEGF165b, a splice variant of VEGF-A, promotes lung tumor progression and escape from anti-angiogenic therapies through a β1 integrin/VEGFR autocrine loop. Oncogene 2019, 38, 1050–1066. [Google Scholar] [CrossRef]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation. 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [CrossRef]
- Lee, C.; Chen, R.; Sun, G.; Liu, X.; Lin, X.; He, C.; Xing, L.; Liu, L.; Jensen, L.D.; Kumar, A.; et al. VEGF-B prevents excessive angiogenesis by inhibiting FGF2/FGFR1 pathway. Signal Transduct. Target. Ther. 2023, 8, 305. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef]
- Zheng, X.; Peng, Q.; Wang, L.; Zhang, X.; Huang, L.; Wang, J.; Qin, Z. Serine/arginine-rich splicing factors: The bridge linking alternative splicing and cancer. Int. J. Biol. Sci. 2020, 16, 2442–2453. [Google Scholar] [CrossRef]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Ntellas, P.; Mavroeidis, L.; Gkoura, S.; Gazouli, I.; Amylidi, A.-L.; Papadaki, A.; Zarkavelis, G.; Mauri, D.; Karpathiou, G.; Kolettas, E.; et al. Old Player-New Tricks: Non Angiogenic Effects of the VEGF/VEGFR Pathway in Cancer. Cancers 2020, 12, 3145. [Google Scholar] [CrossRef]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef]
- Peiris-Pagès, M. The role of VEGF 165b in pathophysiology. Cell Adh Migr. 2012, 6, 561–568. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef]
- Rennel, E.S.; Harper, S.J.; Bates, D.O. Therapeutic potential of manipulating VEGF splice isoforms in oncology. Future Oncol. 2009, 5, 703–712. [Google Scholar] [CrossRef]
- Bessa, C.; Matos, P.; Jordan, P.; Gonçalves, V. Alternative Splicing: Expanding the Landscape of Cancer Biomarkers and Therapeutics. Int. J. Mol. Sci. 2020, 21, 9032. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef]
- Albuquerque, R.J.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G.; et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023–1030. [Google Scholar] [CrossRef]
- Abou Faycal, C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative splicing in lung carcinoma cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef]
- Moens, S.; Goveia, J.; Stapor, P.C.; Cantelmo, A.R.; Carmeliet, P. The multifaceted activity of VEGF in angiogenesis—Implications for therapy responses. Cytokine Growth Factor. Rev. 2014, 25, 473–482. [Google Scholar] [CrossRef]
- Zhang, H.; Jia, E.; Xia, W.; Lu, C.; Zhu, W. VEGF165b mutant with a prolonged half-life and enhanced anti-tumor potency in a mouse model. J. Biotechnol. 2018, 284, 84–90. [Google Scholar] [CrossRef]
- Kim, M.; Jang, K.; Miller, P.; Picon-Ruiz, M.; Yeasky, T.M.; El-Ashry, D.; Slingerland, J.M. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene 2017, 36, 5199–5211. [Google Scholar] [CrossRef]
- Koyama, S.; Matsunaga, S.; Imanishi, M.; Maekawa, Y.; Kitano, H.; Takeuchi, H.; Tomita, S. Tumour blood vessel normalisation by prolyl hydroxylase inhibitor repaired sensitivity to chemotherapy in a tumour mouse model. Sci. Rep. 2017, 7, 45621. [Google Scholar] [CrossRef] [PubMed]
- Vassilakopoulou, M.; Psyrri, A.; Argiris, A. Targeting angiogenesis in head and neck cancer. Oral. Oncol. 2015, 51, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, O. Targeting vascular endothelial growth factor (VEGF) pathway in iodine-refractory differentiated thyroid carcinoma (DTC): From bench to bedside. Crit. Rev. Oncol. Hematol. 2015, 94, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Ashina, K.; Tsubosaka, Y.; Kobayashi, K.; Omori, K.; Murata, T. VEGF-induced blood flow increase causes vascular hyper-permeability in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 590–595. [Google Scholar] [CrossRef]
- Florek, K.; Mendyka, D.; Gomułka, K. Vascular Endothelial Growth Factor (VEGF) and Its Role in the Cardiovascular System. Biomedicines 2024, 12, 1055. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Montemagno, C.; Durivault, J.; Gastaldi, C.; Dufies, M.; Vial, V.; He, X.; Ambrosetti, D.; Kamenskaya, A.; Negrier, S.; Bernhard, J.C.; et al. A group of novel VEGF splice variants as alternative therapeutic targets in renal cell carcinoma. Mol. Oncol. 2023, 17, 1379–1401. [Google Scholar] [CrossRef]
- Guyot, M.; Pagès, G. VEGF Splicing and the Role of VEGF Splice Variants: From Physiological-Pathological Conditions to Specific Pre-mRNA Splicing. Methods Mol. Biol. 2015, 1332, 3–23. [Google Scholar] [CrossRef]
- Touyz, R.M.; Herrmann, S.M.S.; Herrmann, J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J. Am. Soc. Hypertens. 2018, 12, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef] [PubMed]
- Titchenell, P.M.; Antonetti, D.A. Using the past to inform the future: Anti-VEGF therapy as a road map to develop novel therapies for diabetic retinopathy. Diabetes 2013, 62, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Walimbe, T.; Dehghani, T.; Casella, A.; Lin, J.; Wang, A.; Panitch, A. Proangiogenic Collagen-Binding Glycan Therapeutic Promotes Endothelial Cell Angiogenesis. ACS Biomater. Sci. Eng. 2021, 7, 3281–3292. [Google Scholar] [CrossRef] [PubMed]
- Marsters, P.; Alhamdan, R.; Campbell, B.K. Cell density-mediated pericellular hypoxia and the local dynamic regulation of VEGF-a splice variants in ovine ovarian granulosa cells. Biol. Reprod. 2014, 91, 35. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.M.; MacDonald, B.T.; Mizoguchi, T.; Chaffin, M.; Leed, A.; Arduini, A.; Malolepsza, E.; Lage, K.; Kaushik, V.K.; Kathiresan, S.; et al. Endothelial ARHGEF26 is an angiogenic factor promoting VEGF signalling. Cardiovasc. Res. 2022, 118, 2833–2846. [Google Scholar] [CrossRef]
- Jeltsch, M.; Kaipainen, A.; Joukov, V.; Meng, X.; Lakso, M.; Rauvala, H.; Swartz, M.; Fukumura, D.; Jain, R.K.; Alitalo, K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 1997, 276, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Mäkinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2022, 62, 4123–4131. [Google Scholar]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- Al Kawas, H.; Saaid, I.; Jank, P.; Westhoff, C.C.; Denkert, C.; Pross, T.; Weiler, K.B.S.; Karsten, M.M. How VEGF-A and its splice variants affect breast cancer development-clinical implications. Cell Oncol. (Dordr) 2022, 45, 227–239. [Google Scholar] [CrossRef]
- Mamer, S.B.; Wittenkeller, A.; Imoukhuede, P.I. VEGF-A splice variants bind VEGFRs with differential affinities. Sci. Rep. 2020, 10, 14413. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.C.; Patel, N.G.; Moore, L.S.; McGee, A.S.; Ahn, J.C.; Willey, C.D.; Carroll, W.R.; Rosenthal, E.L.; Warram, J.M. Adjuvant anti-angiogenic therapy enhances chemotherapeutic uptake in a murine model of head and neck cancer. J. Drug Target. 2019, 27, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Dong, R.; Jin, T.; Jin, Q.; Chen, X. Anti-PD-1 Monoclonal Antibody Combined With Anti-VEGF Agent Is Safe and Effective in Patients With Recurrent/Metastatic Head and Neck Squamous Cancer as Second-Line or Beyond Treatment. Front. Oncol. 2022, 12, 781348. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| VEGF Splice | Variant | Receptor Binding | Biological Activity | Role in Angiogenesis |
|---|---|---|---|---|
| VEGF121 | Binds | VEGFR1 and VEGFR2 | Initiates cellular events leading to new vessel formation | Pro-angiogenic |
| VEGF165 | Binds | VEGFR1 and VEGFR2 | Promotes proliferation and migration of endothelial cells | Pro-angiogenic |
| VEGF189 | Binds | VEGFR1 and VEGFR2 | Influences extracellular matrix affecting vascular growth | Pro-angiogenic |
| VEGF165b | Binds | VEGF receptors without activating angiogenic signaling | Inhibits angiogenic pathways, acts as a competitive inhibitor | Anti-angiogenic |
| VEGF Variant | Type (Pro-Angiogenic/Anti-Angiogenic) | Characteristics | Clinical Implications |
|---|---|---|---|
| VEGF-A165 [67] | Pro-angiogenic | Promotes endothelial cell proliferation, migration, and new blood vessel formation by binding to VEGFR1 and VEGFR2. | Associated with tumor progression and metastasis in various cancers, including HNSCC. |
| VEGF-A121 [68] | Pro-angiogenic | Similar to VEGF-A165 but more diffusible due to the lack of heparin-binding domains. | Plays a role in angiogenesis and tumor growth. |
| VEGF-A189 [69] | Pro-angiogenic | Strongly binds to heparin and extracellular matrix components, affecting local angiogenesis. | Influences the angiogenic profile in specific tissue environments. |
| VEGF-A165b [70] | Anti-angiogenic | A splice variant of VEGF-A165 that binds to VEGFR1 and VEGFR2 without activating pro-angiogenic signaling pathways. | Inhibits angiogenesis, offering a potential therapeutic target for reducing tumor growth and angiogenesis in cancers. |
| VEGF-A121b [9] | Anti-angiogenic | A splice variant of VEGF-A121 that also inhibits angiogenesis by preventing VEGFR-mediated signaling. | Potentially reduces angiogenesis and tumor progression, similar to VEGF-A165b. |
| VEGF-B [35] | Pro-angiogenic | Binds primarily to VEGFR1, involved in heart development and fatty acid uptake. | Role in cancer is less clear but may be involved in metabolic regulation and survival of cancer cells. |
| VEGF-C [71] | Pro-angiogenic | Induces lymphangiogenesis and angiogenesis through binding to VEGFR2 and VEGFR3. | Implicated in lymphatic metastasis of solid tumors, including HNSCC, by promoting lymphangiogenesis. |
| VEGF-D [72] | Pro-angiogenic | Similar to VEGF-C, promotes lymphangiogenesis and angiogenesis by binding to VEGFR2 and VEGFR3. | Potential role in lymphatic spread and metastasis of cancer, including implications for HNSCC. |
| PIGF [73] | Pro-angiogenic | Binds to VEGFR1 and NRP1; involved in pathological angiogenesis, inflammation, and recruitment of myeloid cells. | Studied for its potential in cancer therapy and cardiovascular diseases, though with varying implications in different types of cancer. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumitru, C.S.; Raica, M. A Splice Form of VEGF, a Potential Anti-Angiogenetic Form of Head and Neck Squamous Cell Cancer Inhibition. Int. J. Mol. Sci. 2024, 25, 8855. https://doi.org/10.3390/ijms25168855
Dumitru CS, Raica M. A Splice Form of VEGF, a Potential Anti-Angiogenetic Form of Head and Neck Squamous Cell Cancer Inhibition. International Journal of Molecular Sciences. 2024; 25(16):8855. https://doi.org/10.3390/ijms25168855
Chicago/Turabian StyleDumitru, Cristina Stefania, and Marius Raica. 2024. "A Splice Form of VEGF, a Potential Anti-Angiogenetic Form of Head and Neck Squamous Cell Cancer Inhibition" International Journal of Molecular Sciences 25, no. 16: 8855. https://doi.org/10.3390/ijms25168855