
Clinical Properties and Non-Clinical Testing of Mineralocorticoid Receptor Antagonists in In Vitro Cell Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
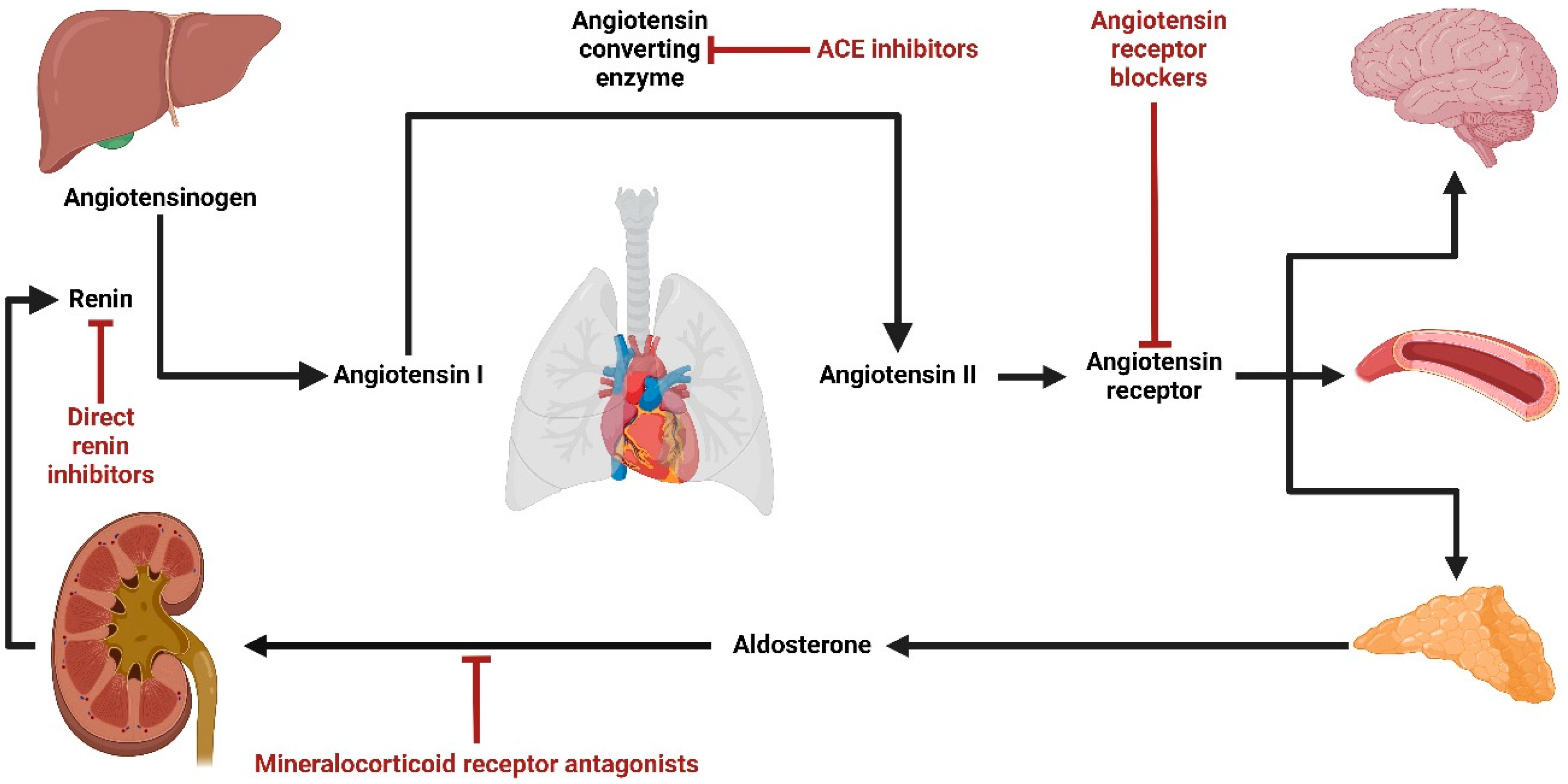
:1. Introduction
2. Methods
3. Overview of the Current Most Commonly Used Mineralocorticoid Receptor Antagonists (MRAs)
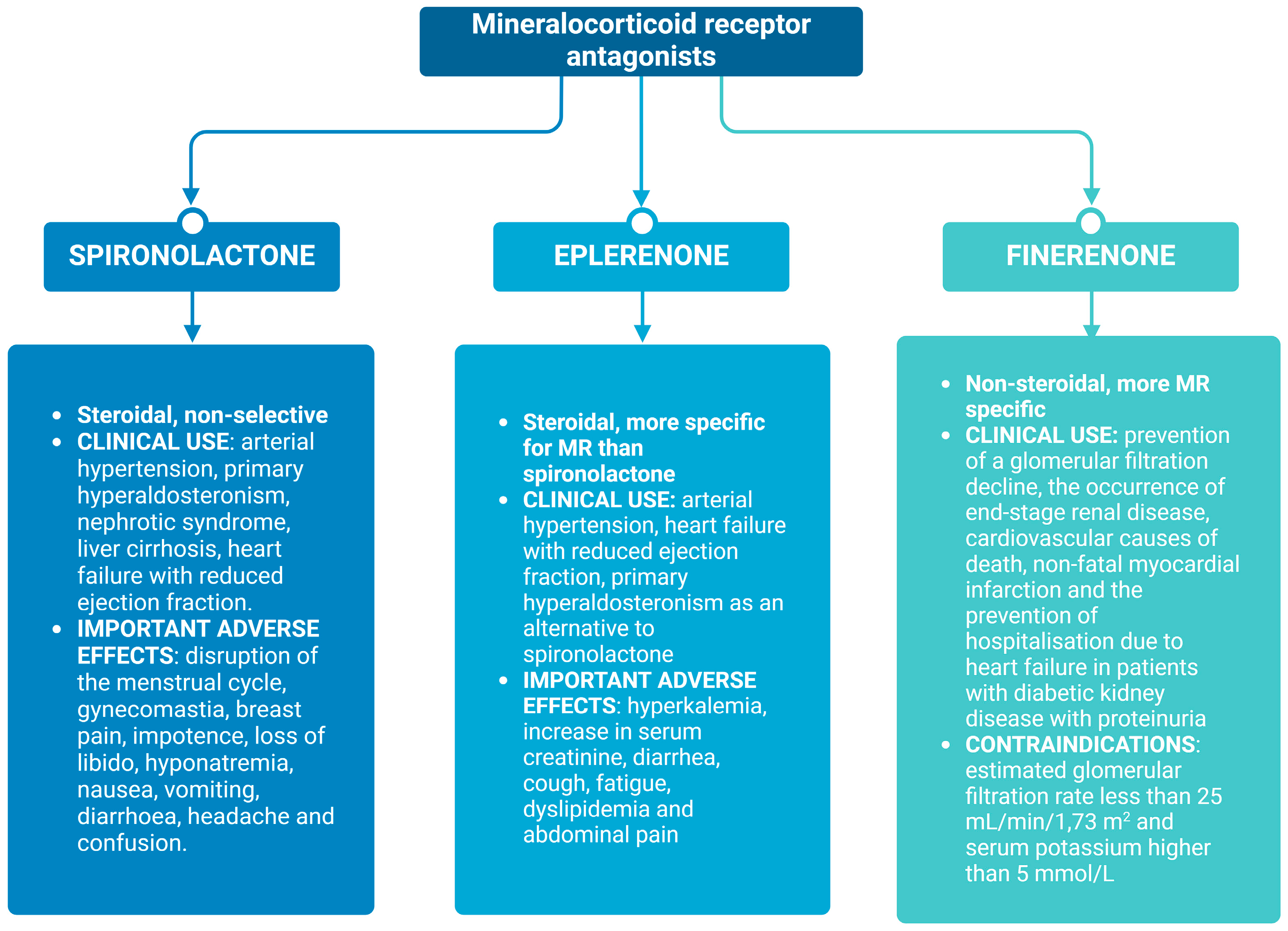
3.1. Spironolactone
3.2. Eplerenone
3.3. Finerenone
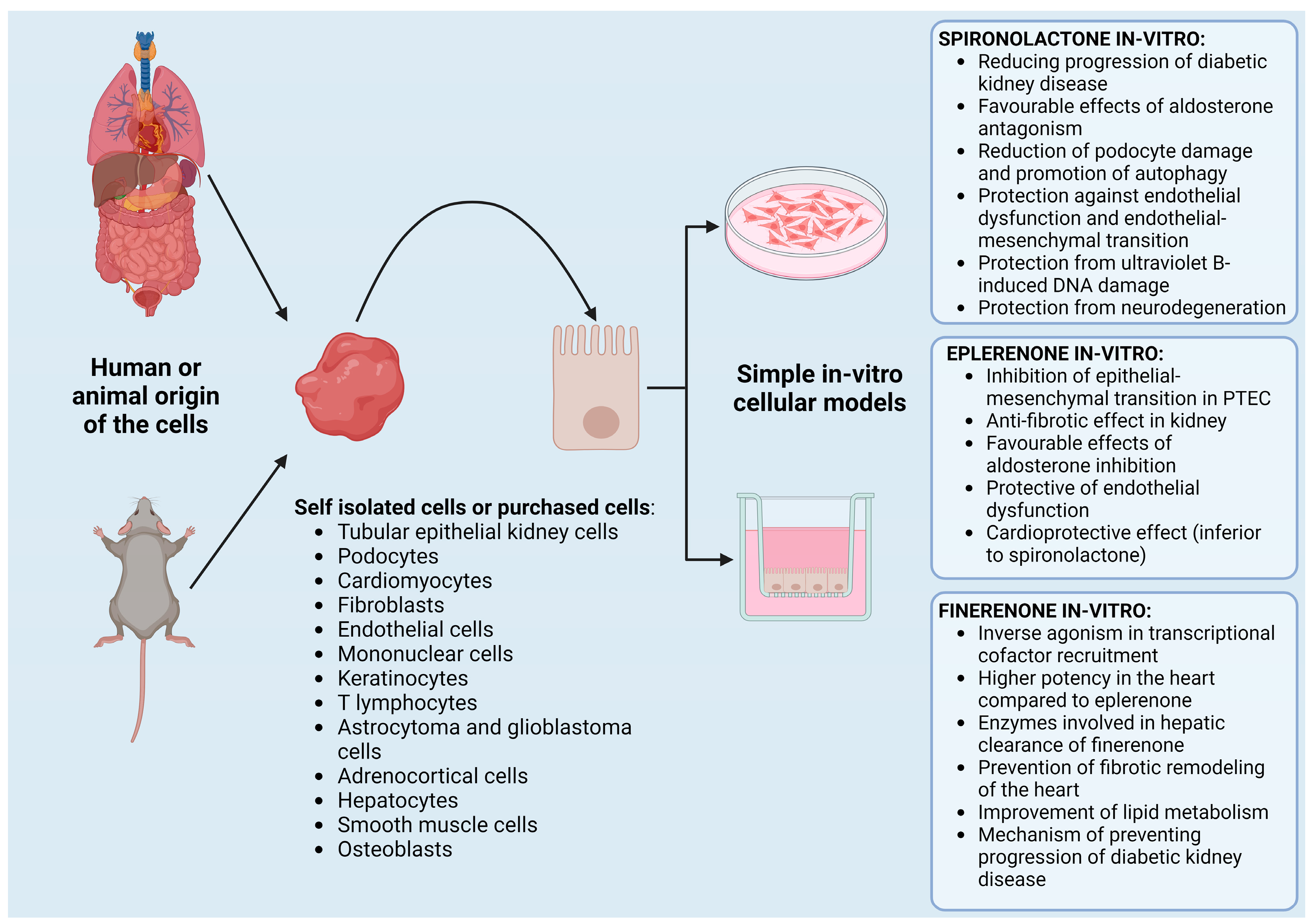
4. MRA Testing on In Vitro Cell Models
4.1. In Vitro Testing of Spironolactone
4.2. In Vitro Testing of Eplerenone
4.3. In Vitro Testing of Finerenone
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Connell, J.M.; Davies, E. The new biology of aldosterone. J. Endocrinol. 2005, 186, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.E.; Johnson, J.P.; Stockand, J.D. Aldosterone. Adv. Physiol. Educ. 2002, 26, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Delyani, J.A. Mineralocorticoid receptor antagonists: The evolution of utility and pharmacology. Kidney Int. 2000, 57, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef]
- Brown, M.J. Direct renin inhibition—A new way of targeting the renin system. J. Renin-Angiotensin-Aldosterone Syst. 2006, 7, S7–S11. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Spat, A.; Hunyady, L. Control of aldosterone secretion: A model for convergence in cellular signaling pathways. Physiol. Rev. 2004, 84, 489–539. [Google Scholar] [CrossRef]
- Cozza, E.N.; Vila, M.D.C.; Acevedo-Duncan, M.; Farese, R.V.; Gómez-Sánchez, C.E. Treatment of primary cultures of calf adrenal glomerulosa cells with adrenocorticotropin (ACTH) and phorbol esters: A comparative study of the effects on aldosterone production and ACTH signaling system. Endocrinology 1990, 126, 2169–2176. [Google Scholar] [CrossRef]
- Goodfriend, T.; Ball, D.; Raff, H.; Bruder, E.; Gardner, H.; Spiteller, G. Oxidized products of linoleic acid stimulate adrenal steroidogenesis. Endocr. Res. 2002, 28, 325–330. [Google Scholar] [CrossRef]
- Namsolleck, P.; Unger, T. Aldosterone synthase inhibitors in cardiovascular and renal diseases. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 1), i62–i68. [Google Scholar] [CrossRef]
- Rossi, F.; Mascolo, A.; Mollace, V. The pathophysiological role of natriuretic peptide-RAAS cross talk in heart failure. Int. J. Cardiol. 2017, 226, 121–125. [Google Scholar] [CrossRef]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13 (Suppl. B), 9–20. [Google Scholar] [CrossRef]
- Aroor, A.R.; DeMarco, V.G.; Jia, G.; Sun, Z.; Nistala, R.; Meininger, G.A.; Sowers, J.R. The role of tissue renin-angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front. Endocrinol. 2013, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Novello, M.; Colussi, G.; Di Fabio, A.; Chiuch, A.; Nadalini, E.; Casanova-Borca, A.; Uzzau, A.; Catena, C. Relationship of plasma renin with a prothrombotic state in hypertension: Relevance for organ damage. Am. J. Hypertens. 2008, 21, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Papademetriou, V. Inhibition of the renin-angiotensin-aldosterone system to prevent ischemic and atherothrombotic events. Am. Heart J. 2009, 157, S24–S30. [Google Scholar] [CrossRef]
- Remuzzi, G.; Perico, N.; Macia, M.; Ruggenenti, P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney Int. 2005, 68, S57–S65. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Fujita, T. Pathophysiology of salt sensitivity hypertension. Ann. Med. 2012, 44 (Suppl. 1), S119–S126. [Google Scholar] [CrossRef]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef]
- Mo, C.; Ke, J.; Zhao, D.; Zhang, B. Role of the renin–angiotensin–aldosterone system in bone metabolism. J. Bone Miner. Metab. 2020, 38, 772–779. [Google Scholar] [CrossRef]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone system and its suppression. J. Vet. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef]
- Lainscak, M.; Pelliccia, F.; Rosano, G.; Vitale, C.; Schiariti, M.; Greco, C.; Speziale, G.; Gaudio, C. Safety profile of mineralocorticoid receptor antagonists: Spironolactone and eplerenone. Int. J. Cardiol. 2015, 200, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W. Mineralocorticoid receptor antagonists: Emerging roles in cardiovascular medicine. Integr. Blood Press. Control 2013, 6, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.J.; Hu, X. Non-steroidal mineralocorticoid receptor antagonists. Expert Opin. Ther. Pat. 2007, 17, 17–23. [Google Scholar] [CrossRef]
- Pandey, A.K.; Bhatt, D.L.; Cosentino, F.; Marx, N.; Rotstein, O.; Pitt, B.; Pandey, A.; Butler, J.; Verma, S. Non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Eur. Heart J. 2022, 43, 2931–2945. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Finerenone: First approval. Drugs 2021, 81, 1787–1794. [Google Scholar] [CrossRef]
- Kintscher, U.; Edelmann, F. The non-steroidal mineralocorticoid receptor antagonist finerenone and heart failure with preserved ejection fraction. Cardiovasc. Diabetol. 2023, 22, 162. [Google Scholar] [CrossRef]
- Duggan, S. Esaxerenone: First Global Approval. Drugs 2019, 79, 477–481. [Google Scholar] [CrossRef]
- Hoshide, S. Is esaxerenone the ultimate mineralocorticoid receptor antagonist? Hypertens. Res. 2023, 46, 516–517. [Google Scholar] [CrossRef]
- Wan, N.; Rahman, A.; Nishiyama, A. Esaxerenone, a novel nonsteroidal mineralocorticoid receptor blocker (MRB) in hypertension and chronic kidney disease. J. Hum. Hypertens. 2021, 35, 148–156. [Google Scholar] [CrossRef]
- Arai, K.; Homma, T.; Morikawa, Y.; Ubukata, N.; Tsuruoka, H.; Aoki, K.; Ishikawa, H.; Mizuno, M.; Sada, T. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist. Eur. J. Pharmacol. 2015, 761, 226–234. [Google Scholar] [CrossRef]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-Induced Nephrotoxicity: Clinical Impact and Preclinical in Vitro Models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Soo, J.Y.C.; Jansen, J.; Masereeuw, R.; Little, M.H. Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat. Rev. Nephrol. 2018, 14, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Patibandla, S.; Heaton, J.; Kyaw, H. Spironolactone. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [PubMed]
- Valdivielso, J.M.; Balafa, O.; Ekart, R.; Ferro, C.J.; Mallamaci, F.; Mark, P.B.; Rossignol, P.; Sarafidis, P.; Del Vecchio, L.; Ortiz, A. Hyperkalemia in chronic kidney disease in the new era of kidney protection therapies. Drugs 2021, 81, 1467–1489. [Google Scholar] [CrossRef]
- Aguilar Medina, D.A.; Cazarín, J.; Magaña, M. Spironolactone in dermatology. Dermatol. Ther. 2022, 35, e15321. [Google Scholar] [CrossRef]
- Carone, L.; Oxberry, S.G.; Twycross, R.; Charlesworth, S.; Mihalyo, M.; Wilcock, A. Spironolactone. J. Pain. Symptom Manag. 2017, 53, 288–292. [Google Scholar] [CrossRef]
- Tawada, M.; Suzuki, Y.; Sakata, F.; Mizuno, M.; Ito, Y. Mineralocorticoid receptor antagonists in dialysis patients. Ren. Replace. Ther. 2016, 2, 64. [Google Scholar] [CrossRef]
- Taheri, S.; Mortazavi, M.; Shahidi, S.; Pourmoghadas, A.; Garakyaraghi, M.; Seirafian, S.; Eshaghian, A.; Ghassami, M. Spironolactone in chronic hemodialysis patients improves cardiac function. Saudi J. Kidney Dis. Transplant. 2009, 20, 392–397. [Google Scholar]
- Feniman-De-Stefano, G.M.M.; Zanati-Basan, S.G.; De Stefano, L.M.; Xavier, P.S.; Castro, A.D.; Caramori, J.C.T.; Barretti, P.; Jorge da Silva Franco, R.; Martin, L.C. Spironolactone is secure and reduces left ventricular hypertrophy in hemodialysis patients. Ther. Adv. Cardiovasc. Dis. 2015, 9, 158–167. [Google Scholar] [CrossRef]
- McGill, R.L.; Biederman, R.W.; Getts, R.T.; Hazlett, S.M.; Sharma, S.B.; Duran, J.; Brandys, D.E.; Sysak, J.C.; Sureshkumar, K.K.; Sandroni, S.E. Cardiac magnetic resonance imaging in hemodialysis patients. JN J. Nephrol. 2009, 22, 367. [Google Scholar] [PubMed]
- Flevari, P.; Kalogeropoulou, S.; Drakou, A.; Leftheriotis, D.; Panou, F.; Lekakis, J.; Kremastinos, D.; Vlahakos, D.V. Spironolactone improves endothelial and cardiac autonomic function in non heart failure hemodialysis patients. J. Hypertens. 2013, 31, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Vukusich, A.; Kunstmann, S.; Varela, C.; Gainza, D.; Bravo, S.; Sepulveda, D.; Cavada, G.; Michea, L.; Marusic, E.T. A randomized, double-blind, placebo-controlled trial of spironolactone on carotid intima-media thickness in nondiabetic hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Mori, Y.; Kageyama, S.; Arihara, K.; Sugiyama, T.; Ohmura, H.; Yakushigawa, T.; Sugiyama, H.; Shimada, Y.; Nojima, Y. Spironolactone reduces cardiovascular and cerebrovascular morbidity and mortality in hemodialysis patients. J. Am. Coll. Cardiol. 2014, 63, 528–536. [Google Scholar] [CrossRef]
- Papadimitriou, M.; Vyzantiadis, A.; Milionis, A.; Memmos, D.; Metaxas, P. The effect of spironolactone in hypertensive patients on regular haemodialysis and after renal allotransplantation. Life Support Syst. J. Eur. Soc. Artif. Organs 1983, 1, 197–205. [Google Scholar]
- Brest, U. ALdosterone Antagonist Chronic HEModialysis Interventional Survival Trial (ALCHEMIST); National Library of Medicine: Bethesda, MD, USA. Available online: https://clinicaltrials.gov/study/NCT01848639 (accessed on 10 August 2024).
- Tyrwhitt, J. Aldosterone BloCkade for Health Improvement EValuation in End-Stage Renal Disease (ACHIEVE). 2017. Available online: https://clinicaltrials.gov/study/NCT03020303?cond=%20NCT03020303&rank=1 (accessed on 10 August 2024).
- Taheri, S.; Mortazavi, M.; Pourmoghadas, A.; Seyrafian, S.; Alipour, Z.; Karimi, S. A prospective double-blind randomized placebo-controlled clinical trial to evaluate the safety and efficacy of spironolactone in patients with advanced congestive heart failure on continuous ambulatory peritoneal dialysis. Saudi J. Kidney Dis. Transplant. 2012, 23, 507–512. [Google Scholar]
- Ito, Y.; Mizuno, M.; Suzuki, Y.; Tamai, H.; Hiramatsu, T.; Ohashi, H.; Ito, I.; Kasuga, H.; Horie, M.; Maruyama, S. Long-term effects of spironolactone in peritoneal dialysis patients. J. Am. Soc. Nephrol. 2014, 25, 1094–1102. [Google Scholar] [CrossRef]
- Vazquez-Rangel, A.; Soto, V.; Escalona, M.; Toledo, R.G.; Castillo, E.A.; Flores, N.A.P.; Falcon-Chavez, I.; Madero, M. Spironolactone to prevent peritoneal fibrosis in peritoneal dialysis patients: A randomized controlled trial. Am. J. Kidney Dis. 2014, 63, 1072–1074. [Google Scholar] [CrossRef]
- Brown, N.J. Eplerenone: Cardiovascular protection. Circulation 2003, 107, 2512–2518. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef]
- De Gasparo, M.; Joss, U.; Ramjoue, H.; Whitebread, S.; Haenni, H.; Schenkel, L.; Kraehenbuehl, C.; Biollaz, M.; Grob, J.; Schmidlin, J. Three new epoxy-spirolactone derivatives: Characterization in vivo and in vitro. J. Pharmacol. Exp. Ther. 1987, 240, 650–656. [Google Scholar] [PubMed]
- Weinberger, M.H.; Roniker, B.; Krause, S.L.; Weiss, R.J. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am. J. Hypertens. 2002, 15, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Struthers, A.; Krum, H.; Williams, G.H. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clin. Cardiol. Int. Index. Peer-Rev. J. Adv. Treat. Cardiovasc. Dis. 2008, 31, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Kreutz, R.; Brunström, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension Endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA). J. Hypertens. 2023, 41, 1874–2071. [Google Scholar] [CrossRef]
- Hughes, J.C.; Cassagnol, M. Eplerenone. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Muldowney Iii, J.A.; Schoenhard, J.A.; Benge, C.D. The clinical pharmacology of eplerenone. Expert Opin. Drug Metab. Toxicol. 2009, 5, 425–432. [Google Scholar] [CrossRef]
- Shavit, L.; Neykin, D.; Lifschitz, M.; Slotki, I. Effect of eplerenone on blood pressure and the renin-angiotensin-aldosterone system in oligo-anuric chronic hemodialysis patients-a pilot study. Clin. Nephrol. 2011, 76, 388–395. [Google Scholar] [CrossRef]
- Walsh, M.; Manns, B.; Garg, A.X.; Bueti, J.; Rabbat, C.; Smyth, A.; Tyrwhitt, J.; Bosch, J.; Gao, P.; Devereaux, P. The safety of eplerenone in hemodialysis patients: A noninferiority randomized controlled trial. Clin. J. Am. Soc. Nephrol. 2015, 10, 1602–1608. [Google Scholar] [CrossRef]
- Donderski, R.Ł.; Stróżecki, P. Eplerenone add-on treatment in resistant hypertension in peritoneal dialysis patient. A case report. Med. Res. J. 2023, 8, 81–84. [Google Scholar] [CrossRef]
- Sato, A.; Hayashi, K.; Naruse, M.; Saruta, T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension 2003, 41, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hayashi, K.; Saruta, T. Antiproteinuric effects of mineralocorticoid receptor blockade in patients with chronic renal disease. Am. J. Hypertens. 2005, 18, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Rossing, K.; Schjoedt, K.J.; Smidt, U.M.; Boomsma, F.; Parving, H.-H. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: A randomized, double-masked, cross-over study. Diabetes Care 2005, 28, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Clarisse, D.; Deng, L.; De Bosscher, K.; Lother, A. Approaches towards tissue-selective pharmacology of the mineralocorticoid receptor. Br. J. Pharmacol. 2022, 179, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef]
- Kolkhof, P.; Bärfacker, L. 30 years of the mineralocorticoid receptor: Mineralocorticoid receptor antagonists: 60 years of research and development. J. Endocrinol. 2017, 234, T125–T140. [Google Scholar] [CrossRef]
- Yang, J.; Young, M.J. Mineralocorticoid receptor antagonists—Pharmacodynamics and pharmacokinetic differences. Curr. Opin. Pharmacol. 2016, 27, 78–85. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Agarwal, R. The Nonsteroidal Mineralocorticoid-Receptor-Antagonist Finerenone in Cardiorenal Medicine: A State-of-the-Art Review of the Literature. Am. J. Hypertens. 2023, 36, 135–143. [Google Scholar] [CrossRef]
- Marcath, L.A. Finerenone. Clin. Diabetes 2021, 39, 331–332. [Google Scholar] [CrossRef]
- Rico-Mesa, J.S.; White, A.; Ahmadian-Tehrani, A.; Anderson, A.S. Mineralocorticoid receptor antagonists: A comprehensive review of finerenone. Curr. Cardiol. Rep. 2020, 22, 140. [Google Scholar] [CrossRef]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Filippatos, G.; Pitt, B.; Anker, S.D.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Gebel, M.; Ruilope, L.M. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: The FIDELITY pooled analysis. Eur. Heart J. 2022, 43, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Heart J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef]
- Agarwal, R.; Pitt, B.; Palmer, B.F.; Kovesdy, C.P.; Burgess, E.; Filippatos, G.; Małyszko, J.; Ruilope, L.M.; Rossignol, P.; Rossing, P. A comparative post hoc analysis of finerenone and spironolactone in resistant hypertension in moderate-to-advanced chronic kidney disease. Clin. Kidney J. 2023, 16, 293–302. [Google Scholar] [CrossRef]
- Duric, L.F.; Vujicic, B.; Gulin, T.; Gulin, M. The Impact of Finerenone on Changes in Pulse Wave Velocity, Arterial Pressure and Heart Related Deaths in Hemodialysis Patients—Study Perspective. Open J. Nephrol. 2024, 14, 216–225. [Google Scholar] [CrossRef]
- Madorran, E.; Stožer, A.; Bevc, S.; Maver, U. In vitro toxicity model: Upgrades to bridge the gap between preclinical and clinical research. Bosn. J. Basic. Med. Sci. 2020, 20, 157–168. [Google Scholar] [CrossRef]
- Davies, J. Engineered renal tissue as a potential platform for pharmacokinetic and nephrotoxicity testing. Drug Discov. Today 2014, 19, 725–729. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Mihevc, M.; Petreski, T.; Maver, U.; Bevc, S. Renal proximal tubular epithelial cells: Review of isolation, characterization, and culturing techniques. Mol. Biol. Rep. 2020, 47, 9865–9882. [Google Scholar] [CrossRef]
- Bejoy, J.; Qian, E.S.; Woodard, L.E. Tissue Culture Models of AKI: From Tubule Cells to Human Kidney Organoids. J. Am. Soc. Nephrol. 2022, 33, 487–501. [Google Scholar] [CrossRef]
- Liu, K.-H.; Zhou, Q.-L.; Ao, X.; Tang, T.-F.; Hong, X.-M.; Bao, R.-L. Effect of spironolactone on the expression of Toll-like receptor 4 in renal tubular epithelia cells exposed to high glucose. Zhongguo Dang Dai Er Ke Za Zhi/Chin. J. Contemp. Pediatr. 2010, 12, 280–283. [Google Scholar]
- Koszegi, S.; Molnar, A.; Lenart, L.; Hodrea, J.; Balogh, D.B.; Lakat, T.; Szkibinszkij, E.; Hosszu, A.; Sparding, N.; Genovese, F. RAAS inhibitors directly reduce diabetes-induced renal fibrosis via growth factor inhibition. J. Physiol. 2019, 597, 193–209. [Google Scholar] [CrossRef]
- Patni, H.; Mathew, J.T.; Luan, L.; Franki, N.; Chander, P.N.; Singhal, P.C. Aldosterone promotes proximal tubular cell apoptosis: Role of oxidative stress. Am. J. Physiol.-Ren. Physiol. 2007, 293, F1065–F1071. [Google Scholar] [CrossRef] [PubMed]
- Drumm, K.; Kress, T.R.; Gassner, B.; Krug, A.W.; Gekle, M. Aldosterone stimulates activity and surface expression of NHE3 in human primary proximal tubule epithelial cells (RPTEC). Cell. Physiol. Biochem. 2006, 17, 21–28. [Google Scholar] [CrossRef]
- Good, D.W. Nongenomic actions of aldosterone on the renal tubule. Hypertension 2007, 49, 728–739. [Google Scholar] [CrossRef]
- Xu, G.; Liu, A.; Liu, X. Aldosterone induces collagen synthesis via activation of extracellular signal-regulated kinase 1 and 2 in renal proximal tubules. Nephrology 2008, 13, 694–701. [Google Scholar] [CrossRef]
- Pinto, V.; Pinho, M.J.; Hopfer, U.; Jose, P.A.; Soares-da-Silva, P. Oxidative stress and the genomic regulation of aldosterone-stimulated NHE1 activity in SHR renal proximal tubular cells. Mol. Cell. Biochem. 2008, 310, 191–201. [Google Scholar] [CrossRef]
- Iwazu, Y.; Muto, S.; Hirahara, I.; Fujisawa, G.; Takeda, S.-I.; Kusano, E. Matrix metalloproteinase 2 induces epithelial-mesenchymal transition in proximal tubules from the luminal side and progresses fibrosis in mineralocorticoid/salt-induced hypertensive rats. J. Hypertens. 2011, 29, 2440–2453. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, Y. The effect of tubular epithelial cells activated by aldosterone on renal interstitial fibroblasts in co-culture system. Zhonghua Yi Xue Za Zhi 2005, 85, 2070–2075. [Google Scholar]
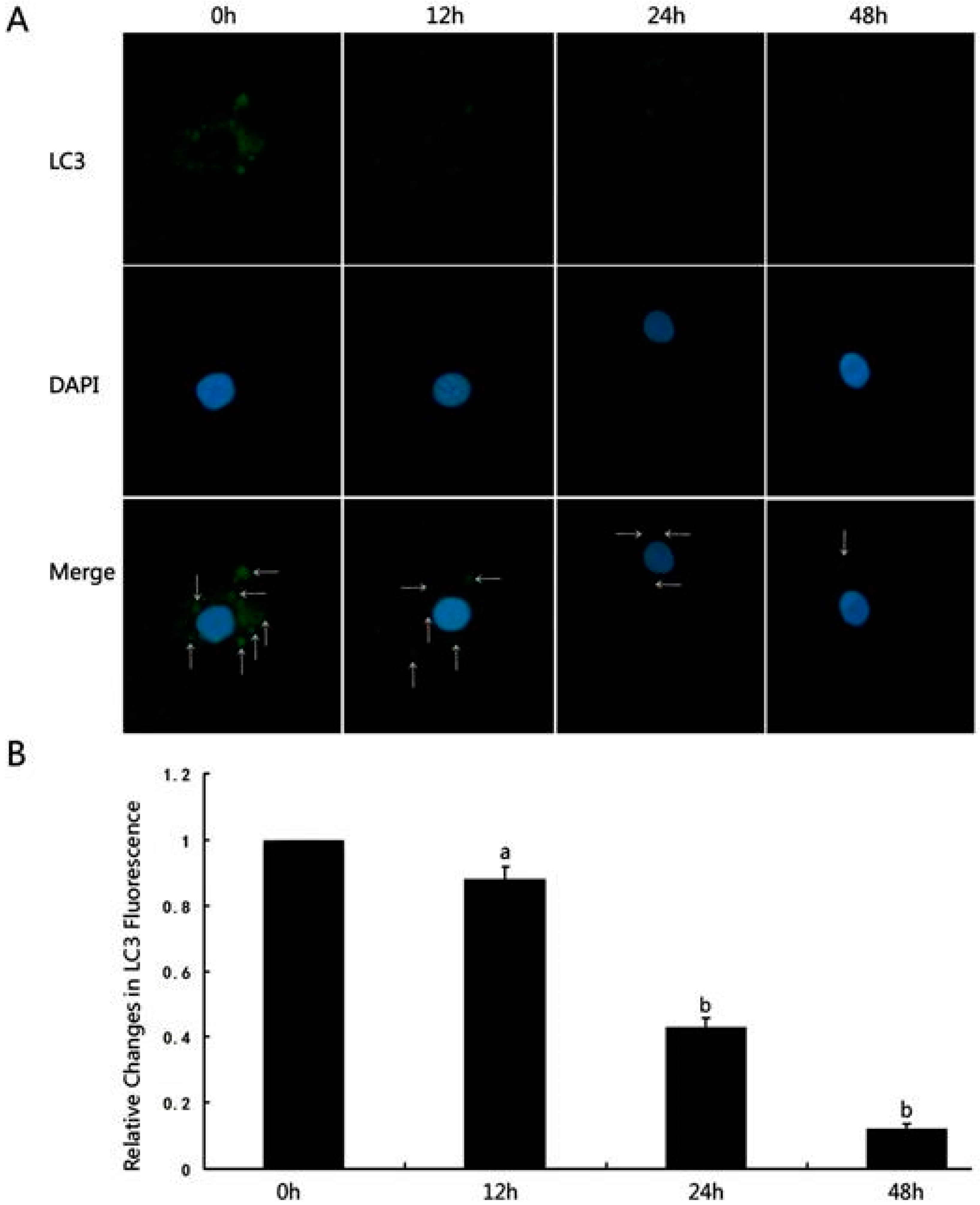
- Li, D.; Lu, Z.; Xu, Z.; Ji, J.; Zheng, Z.; Lin, S.; Yan, T. Spironolactone promotes autophagy via inhibiting PI3K/AKT/mTOR signalling pathway and reduce adhesive capacity damage in podocytes under mechanical stress. Biosci. Rep. 2016, 36, e00355. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Yang, J.; Zhang, Z. LC3 fluorescent puncta in autophagosomes or in protein aggregates can be distinguished by FRAP analysis in living cells. Autophagy 2013, 9, 756–769. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Sønder, S.; Nersting, J.; Bendtzen, K. Spironolactone induces apoptosis in human mononuclear cells. Association between apoptosis and cytokine suppression. Apoptosis 2006, 11, 573–579. [Google Scholar] [CrossRef]
- Drüppel, V.; Kusche-Vihrog, K.; Grossmann, C.; Gekle, M.; Kasprzak, B.; Brand, E.; Pavenstädt, H.; Oberleithner, H.; Kliche, K. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. 2013, 27, 3652–3659. [Google Scholar] [CrossRef]
- Taye, A.; Morawietz, H. Spironolactone inhibits NADPH oxidase-induced oxidative stress and enhances eNOS in human endothelial cells. Iran. J. Pharm. Res. IJPR 2011, 10, 329. [Google Scholar]
- Chen, X.; Cai, J.; Zhou, X.; Chen, L.; Gong, Y.; Gao, Z.; Zhang, H.; Huang, W.; Zhou, H. Protective effect of spironolactone on endothelial-to-mesenchymal transition in HUVECs via notch pathway. Cell. Physiol. Biochem. 2015, 36, 191–200. [Google Scholar] [CrossRef]
- Kemp, M.G.; Krishnamurthy, S.; Kent, M.N.; Schumacher, D.L.; Sharma, P.; Excoffon, K.J.; Travers, J.B. Spironolactone depletes the XPB protein and inhibits DNA damage responses in UVB-irradiated human skin. J. Investig. Dermatol. 2019, 139, 448–454. [Google Scholar] [CrossRef]
- Lacombe, B.; Morel, M.; Margottin-Goguet, F.; Ramirez, B.C. Specific inhibition of HIV infection by the action of spironolactone in T cells. J. Virol. 2016, 90, 10972–10980. [Google Scholar] [CrossRef]
- Keshavarz, M.; Amirinezhadfard, E.; Mehdipour, M. Effects of spironolactone and fludrocortisone on neuronal and glial toxicity induced by N-methyl-D-Aspartate and chloroquine in cell culture. Iran. J. Pharmacol. Ther. 2019, 17, 1–6. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Dicarbonyls Linked to Damage in the Powerhouse: Glycation of Mitochondrial Proteins and Oxidative Stress; Portland Press Ltd.: London, UK, 2008. [Google Scholar]
- Park, S.Y.; Suh, K.S.; Jung, W.-W.; Chin, S.O. Spironolactone attenuates methylglyoxal-induced cellular dysfunction in MC3T3-E1 osteoblastic cells. J. Korean Med. Sci. 2021, 36, e265. [Google Scholar] [CrossRef]
- Yuan, Y.; Chen, Y.; Zhang, P.; Huang, S.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic. Biol. Med. 2012, 53, 30–43. [Google Scholar] [CrossRef]
- Wei, J.; Li, Z.; Ma, C.; Zhan, F.; Wu, W.; Han, H.; Huang, Y.; Li, W.; Chen, D.; Peng, Y. Rho kinase pathway is likely responsible for the profibrotic actions of aldosterone in renal epithelial cells via inducing epithelial–mesenchymal transition and extracellular matrix excretion. Cell Biol. Int. 2013, 37, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liu, L.; Liu, Z.; Chen, G.; Xiong, Y.; Wang, X.; Ma, X.; Xu, Q. Aldosterone Induces the Proliferation of Renal Tubular Epithelial Cells In Vivo but Not In Vitro. J. Renin-Angiotensin-Aldosterone Syst. 2021, 2021, 9943848. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C.; Chan, L.Y.; Tang, S.C.; Lam, M.-F.; Chow, C.-W.; Lim, A.-I.; Lai, K.-N. Oxidative damages in tubular epithelial cells in IgA nephropathy: Role of crosstalk between angiotensin II and aldosterone. J. Transl. Med. 2011, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Chen, W.; Lu, Y.; Zhang, X.; Fu, C.; Yan, Z.; Zhang, Z.; Ye, Z. Crosstalk between peroxisome proliferator-activated receptor-γ and mineralcorticoid receptor in TNF-α activated renal tubular cell. Inflamm. Res. 2015, 64, 603–614. [Google Scholar] [CrossRef]
- Hillebrand, U.; Schillers, H.; Riethmüller, C.; Stock, C.; Wilhelmi, M.; Oberleithner, H.; Hausberg, M. Dose-dependent endothelial cell growth and stiffening by aldosterone: Endothelial protection by eplerenone. J. Hypertens. 2007, 25, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Yamashita, T.; Pollock, D.M.; Sasano, H.; Rainey, W.E. Contrasting effects of eplerenone and spironolactone on adrenal cell steroidogenesis. Horm. Metab. Res. 2009, 41, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Hermidorff, M.M.; Faria, G.d.O.; Amâncio, G.d.C.S.; de Assis, L.V.M.; Isoldi, M.C. Non-genomic effects of spironolactone and eplerenone in cardiomyocytes of neonatal Wistar rats: Do they evoke cardioprotective pathways? Biochem. Cell Biol. 2015, 93, 83–93. [Google Scholar] [CrossRef]
- Łabuzek, K.; Liber, S.; Bułdak, Ł.; Machnik, G.; Liber, J.; Okopień, B. Eplerenone promotes alternative activation in human monocyte-derived macrophages. Pharmacol. Rep. 2013, 65, 226–234. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Łabuzek, K.; Liber, S.; Bułdak, Ł.; Krupej-Kędzierska, J.; Machnik, G.; Bobrzyk, M.; Okopień, B. Eplerenone mimics features of the alternative activation in macrophages obtained from patients with heart failure and healthy volunteers. Eur. J. Pharmacol. 2014, 726, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Estrela, G.R.; Lechner, S.M.; Giraud, S.; El Moghrabi, S.; Kaaki, S.; Kolkhof, P.; Hauet, T.; Jaisser, F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018, 93, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- González-Blázquez, R.; Somoza, B.; Gil-Ortega, M.; Martín Ramos, M.; Ramiro-Cortijo, D.; Vega-Martín, E.; Schulz, A.; Ruilope, L.M.; Kolkhof, P.; Kreutz, R. Finerenone attenuates endothelial dysfunction and albuminuria in a chronic kidney disease model by a reduction in oxidative stress. Front. Pharmacol. 2018, 9, 1131. [Google Scholar] [CrossRef]
- Lattenist, L.; Lechner, S.M.; Messaoudi, S.; Le Mercier, A.; El Moghrabi, S.; Prince, S.; Bobadilla, N.A.; Kolkhof, P.; Jaisser, F.; Barrera-Chimal, J. Nonsteroidal Mineralocorticoid Receptor Antagonist Finerenone Protects Against Acute Kidney Injury–Mediated Chronic Kidney Disease: Role of Oxidative Stress. Hypertension 2017, 69, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Grune, J.; Benz, V.; Brix, S.; Salatzki, J.; Blumrich, A.; Höft, B.; Klopfleisch, R.; Foryst-Ludwig, A.; Kolkhof, P.; Kintscher, U. Steroidal and Nonsteroidal Mineralocorticoid Receptor Antagonists Cause Differential Cardiac Gene Expression in Pressure Overload-induced Cardiac Hypertrophy. J. Cardiovasc. Pharmacol. 2016, 67, 402–411. [Google Scholar] [CrossRef]
- Gueret, A.; Harouki, N.; Favre, J.; Galmiche, G.; Nicol, L.; Henry, J.P.; Besnier, M.; Thuillez, C.; Richard, V.; Kolkhof, P.; et al. Vascular Smooth Muscle Mineralocorticoid Receptor Contributes to Coronary and Left Ventricular Dysfunction After Myocardial Infarction. Hypertension 2016, 67, 717–723. [Google Scholar] [CrossRef]
- Grune, J.; Beyhoff, N.; Smeir, E.; Chudek, R.; Blumrich, A.; Ban, Z.; Brix, S.; Betz, I.R.; Schupp, M.; Foryst-Ludwig, A. Selective mineralocorticoid receptor cofactor modulation as molecular basis for finerenone’s antifibrotic activity. Hypertension 2018, 71, 599–608. [Google Scholar] [CrossRef]
- Amazit, L.; Le Billan, F.; Kolkhof, P.; Lamribet, K.; Viengchareun, S.; Fay, M.R.; Khan, J.A.; Hillisch, A.; Lombès, M.; Rafestin-Oblin, M.-E. Finerenone impedes aldosterone-dependent nuclear import of the mineralocorticoid receptor and prevents genomic recruitment of steroid receptor coactivator-1. J. Biol. Chem. 2015, 290, 21876–21889. [Google Scholar] [CrossRef]
- Desimine, V.L.; Ghandour, J.; Cora, N.; Pollard, C.M.; Valiente, R.; Ferraino, K.E.; Pereyra, J.; Noa, D.P.; Duarte, Y.; Martinez, Y. GRK5 is an Essential Co-repressor for Cardiac Mineralocorticoid Receptor Antagonism induced by Finerenone but not Eplerenone. Preprints 2020, 2020090374. [Google Scholar] [CrossRef]
- Heinig, R.; Gerisch, M.; Bairlein, M.; Nagelschmitz, J.; Loewen, S. Results from drug–drug interaction studies in vitro and in vivo investigating the effect of finerenone on the pharmacokinetics of comedications. Eur. J. Drug Metab. Pharmacokinet. 2020, 45, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Heinig, R.; Gerisch, M.; Engelen, A.; Nagelschmitz, J.; Loewen, S. Pharmacokinetics of the novel, selective, non-steroidal mineralocorticoid receptor antagonist finerenone in healthy volunteers: Results from an absolute bioavailability study and drug–drug interaction studies in vitro and in vivo. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 715–727. [Google Scholar] [CrossRef]
- Gerisch, M.; Heinig, R.; Engelen, A.; Lang, D.; Kolkhof, P.; Radtke, M.; Platzek, J.; Lovis, K.; Rohde, G.; Schwarz, T. Biotransformation of finerenone, a novel nonsteroidal mineralocorticoid receptor antagonist, in dogs, rats, and humans, in vivo and in vitro. Drug Metab. Dispos. 2018, 46, 1546–1555. [Google Scholar] [CrossRef]
- Dutzmann, J.; Musmann, R.-J.; Haertlé, M.; Daniel, J.-M.; Sonnenschein, K.; Schäfer, A.; Kolkhof, P.; Bauersachs, J.; Sedding, D.G. The novel mineralocorticoid receptor antagonist finerenone attenuates neointima formation after vascular injury. PLoS ONE 2017, 12, e0184888. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Jacobs, N.; Mahfoud, F.; Kolkhof, P.; Böhm, M.; Laufs, U. The non-steroidal mineralocorticoid receptor antagonist finerenone prevents cardiac fibrotic remodeling. Biochem. Pharmacol. 2019, 168, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Feraco, A.; Gorini, S.; Mammi, C.; Marrese, C.; Mularoni, V.; Boitani, C.; Lombès, M.; Kolkhof, P.; Ciriolo, M.R. The novel non-steroidal MR antagonist finerenone improves metabolic parameters in high-fat diet-fed mice and activates brown adipose tissue via AMPK-ATGL pathway. FASEB J. 2020, 34, 12450–12465. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Fu, X.; Liu, M.; An, F. Finerenone attenuates myocardial apoptosis, metabolic disturbance and myocardial fibrosis in type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2023, 15, 87. [Google Scholar] [CrossRef]
- Le Billan, F.; Perrot, J.; Carceller, E.; Travers, S.; Viengchareun, S.; Kolkhof, P.; Lombès, M.; Fagart, J. Antagonistic effects of finerenone and spironolactone on the aldosterone-regulated transcriptome of human kidney cells. FASEB J. 2021, 35, e21314. [Google Scholar] [CrossRef]
- Zhao, X.P.; Chang, S.Y.; Liao, M.C.; Lo, C.S.; Chenier, I.; Luo, H.; Chiasson, J.L.; Ingelfinger, J.R.; Chan, J.S.D.; Zhang, S.L. Hedgehog Interacting Protein Promotes Fibrosis and Apoptosis in Glomerular Endothelial Cells in Murine Diabetes. Sci. Rep. 2018, 8, 5958. [Google Scholar] [CrossRef]
- Moritoh, Y.; Oka, M.; Yasuhara, Y.; Hozumi, H.; Iwachidow, K.; Fuse, H.; Tozawa, R. Inositol Hexakisphosphate Kinase 3 Regulates Metabolism and Lifespan in Mice. Sci. Rep. 2016, 6, 32072. [Google Scholar] [CrossRef]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J. Cell Physiol. 2019, 234, 3043–3057. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ren, D.; Xu, G. Long noncoding RNA MALAT1 mediates high glucose-induced glomerular endothelial cell injury by epigenetically inhibiting klotho via methyltransferase G9a. IUBMB Life 2019, 71, 873–881. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varda, L.; Ekart, R.; Lainscak, M.; Maver, U.; Bevc, S. Clinical Properties and Non-Clinical Testing of Mineralocorticoid Receptor Antagonists in In Vitro Cell Models. Int. J. Mol. Sci. 2024, 25, 9088. https://doi.org/10.3390/ijms25169088
Varda L, Ekart R, Lainscak M, Maver U, Bevc S. Clinical Properties and Non-Clinical Testing of Mineralocorticoid Receptor Antagonists in In Vitro Cell Models. International Journal of Molecular Sciences. 2024; 25(16):9088. https://doi.org/10.3390/ijms25169088
Chicago/Turabian StyleVarda, Luka, Robert Ekart, Mitja Lainscak, Uroš Maver, and Sebastjan Bevc. 2024. "Clinical Properties and Non-Clinical Testing of Mineralocorticoid Receptor Antagonists in In Vitro Cell Models" International Journal of Molecular Sciences 25, no. 16: 9088. https://doi.org/10.3390/ijms25169088