Abstract
Our study highlights the apoptosis, cell cycle, DNA ploidy, and autophagy molecular mechanisms network to identify prostate pathogenesis and its prognostic role. Caspase 3/7 expressions, cell cycle, adhesion glycoproteins, autophagy, nuclear shrinkage, and oxidative stress by flow-cytometry analysis are used to study the BPH microenvironment’s heterogeneity. A high late apoptosis expression by caspases 3/7 activity represents an unfavorable prognostic biomarker, a dependent predictor factor for cell adhesion, growth inhibition by arrest in the G2/M phase, and oxidative stress processes network. The heterogeneous aggressive phenotype prostate adenoma primary cell cultures present a high S-phase category (>12%), with an increased risk of death or recurrence due to aneuploid status presence, representing an unfavorable prognostic biomarker, a dependent predictor factor for caspase 3/7 activity (late apoptosis and necrosis), and cell growth inhibition (G2/M arrest)-linked mechanisms. Increased integrin levels in heterogenous BPH cultures suggest epithelial–mesenchymal transition (EMT) that maintains an aggressive phenotype by escaping cell apoptosis, leading to the cell proliferation necessary in prostate cancer (PCa) development. As predictor biomarkers, the biological mechanisms network involved in apoptosis, the cell cycle, and autophagy help to establish patient prognostic survival or target cancer therapy development.
1. Introduction
In prostate pathology cases, benign prostate hyperplasia (BPH), and prostate cancer (PCa), morphological changes in the normal prostate begin with inflammatory transition status. Inflammation and apoptosis mechanisms bring knowledge into the BPH pathogenesis, helping to understand the BPH etiology. Prostatic stroma presents various cells entrapped in an extracellular matrix, such as fibroblasts, smooth-muscle cells, endothelial cells, nerve cells, and T lymphocytes [1].
Apoptosis mechanisms, by cell surface receptors activation or cytochrome c release from mitochondria, determine the caspase cascades, which are divided into initiators (-2, -8, -9, -10, and -12) and effectors (-3, -6, and -7), with roles in cell cycle and apoptosis regulation [2]. Clinical studies have shown the role of inflammation in BPH in a study of 4000 patients by transurethral prostate resection (TURP) or prostatectomy, observing an acute or chronic inflammation in 43.1% of cases. The authors demonstrated a strong correlation between increased prostate volume and inflammation with neutrophil or mononuclear infiltrates [3].
A few in vitro studies showed apoptosis mechanisms in BPH cases. Epithelial cells in BPH overexpress the Bcl-2 level reported in healthy prostate tissue. Increased Bcl-2 expression shows an apoptotic mechanisms deregulation that promotes prostatic hyperplasia [4]. In the epithelial cells, caspase-3 expression in patients with BPH counteracts apoptosis, being implied in mitotic progression regulation. Many studies reported a strong correlation between inflammation, apoptosis, and PCa development. Inflammation and apoptosis processes are essential in BPH and PCa development, and several molecular pathways are known. But the factors implied in prostate carcinogenesis are mostly unknown [5].
Apoptosis, cell-cycle, and oxidative stress pathways in cancer cells represent critical targets in cancer therapy development [6]. Apoptosis represents a tumor growth control mechanism by counterbalancing prostate cell proliferation [7].
Oxidative stress is another important mechanism to study in prostate carcinogenesis because it determines cell-to-cell communication blockage, leading to decreasing cell proliferation and apoptosis [8,9]. Reactive oxygen species (ROS) releasing linked mechanism-DNA damage initiates tumorigenesis. In the functions of cancer cell phenotype and ROS level, there persists a dual role of cell proliferation. In various in vitro human cancer cell cultures, uncontrolled cell proliferation requires different signaling pathways, upregulation, and cell-cycle progression [10,11]. Interactions between tumor cells, platelets, and lymphocytes determine cell adhesion, leading to cancer progression and metastasis [12].
Autophagy is another interesting mechanism that inhibits tumor growth, limits inflammation, and eliminates the damaged mitochondria [13]. In oxidative stress conditions, autophagy determines damaged proteins and organelles accumulation [14,15,16], promoting tumor progression after cancer initiation [17,18,19,20].
Our study presents a major molecular mechanisms network in heterogeneous aggressive phenotype hyperplasia primary cell cultures reported to non-malignant cell cultures, highlighting the BPH pathogenesis and bringing new knowledge to prognostic establishment or targeting cancer therapy development.
Apoptosis deregulation by caspase-3/7 expressions by DEVD-MR/propidium iodide stain and oxidative stress by total reactive oxygen species count was analyzed by flow-cytometry methods. Changes in expressions of G0/G1, S, and G2/M phases of the cell cycle by PI stain highlight the aneuploidy status. Adhesion glycoprotein expression made by CD42b-PE stain reveals platelets and lymphocyte aggregation to tumoral and endothelial cells. The adaptive response to maintain cell survival under stress conditions was analyzed by studying autophagy and nuclear shrinkage mechanisms using Hoechst and acridine orange stain using flow-cytometry techniques. Furthermore, multiple regression was performed to determine the potential biomarkers with prognostic roles in patient survival in heterogeneous aggressive phenotype BHP cell cultures.
2. Results
2.1. Caspases 3/7 Activity
In heterogeneous BPH cell cultures reported to non-malignant adjacent prostate cell cultures (C1, negative control), the effector caspase-3/7 intracellular activity was analyzed by a flow-cytometry technique, being presented in Figure 1A–L.
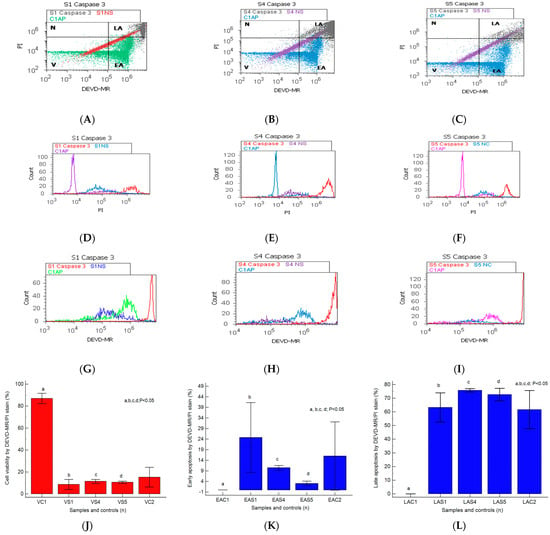
Figure 1.
Caspases-3/7 activity plotted by DEVD-MR/PI stain highlighted to observe viability (V), early and late apoptosis (EA; LA), and necrosis (N) in heterogeneous BPH cell cultures (S1, S4, and S5) reported to non-malignant prostate cell cultures (C1AP). (A–C) V: A—15.48%; B—9.81%; C—9.66%; C1—82.88%; EA: A—40.62%; B—11.42%; C—3.99%; LA: A—42.94; B—76.80%; C—76.61%; N: A—0.94%; B—1.95%; C—9.82%; C1—17.12%. (D–I) Cell apoptosis escape pathway in heterogeneous BPH cell cultures by PI stain (D–F): 20 × 106 (S1); 45 × 106 (S4); 40 × 106 (S5) vs. 12.5 × 105 (C1AP); by DEVD-MR stain (G–I): 65 × 107 (S1); 90 × 107 (S4); 130 × 107 (S5) vs. 35 × 106 (C1AP). (J–L): Caspases 3/7 expression statistics of BPH patients reported to controls (S1; S4; S5; C1; C2); a, b, c, d; p < 0.05 represent significant statistical differences between samples and controls (Mann–Whitney test by MedCalc program). Legend: A—benign prostatic hyperplasia (S1); B—prostate nodular hyperplasia, atrophy, and chronic inflammation (S4); C—prostate adenoma with chronic inflammation (S5); NC—unstained control prostate cells (S1, S4, and S5); C1AP—non-malignant adjacent prostate cells (C1, negative control); C2—positive control (BPH).
The caspase-3/7 activation mechanism induces changes in cell viability, with significant differences between the samples and the control (S1: 8.61 ± 4.60; S4: 11.41 ± 1.85; S5: 10.55 ± 1.02 vs. C1: 86.95% ± 4.69, p < 0.05, Figure 1J–L).
The pro-apoptotic signal highlighted by the biochemical cascade shows significant changes in early apoptosis (EA) in the experimental prostate cell cultures reported to negative control (S1: 24.75 ± 16.50; S4: 10.45 ± 1.12; S5: 3.03 ± 1.10 vs. C1: 0.02 ± 0.01, p < 0.05, Figure 1J–L).
A particular interest is represented by late apoptosis, as a chronic response to injury induction and oxidative stress, which was highlighted by increased significant values in nodular hyperplasia, adenoma, and BHP cell cultures than the control (S4: 75.76 ± 1.20; S5: 72.70 ± 4.50; S1: 63.24 ± 10.65 vs. C1: 0.03 ± 0.01, p < 0.05, Figure 1J–L).
The BPH cell cultures (S1, S4, and S5) adopt modified apoptosis mechanisms with peaks of more than 106 to 107, as shown by the DEVD-MR/PI stain reported for non-malignant prostate cell cultures that present an apoptosis peak of 105 to maximum 106 (C1AP, Figure 1D–I).
2.2. Microenvironment Characterization of Nodular Hyperplasia and Adenoma Samples
The epithelial–mesenchymal transition of pro-inflammatory and profibrogenic phenotypes that characterize the prostate heterogeneous microenvironment of nodular hyperplasia and adenoma cell cultures is highlighted in Figure 2A–L.
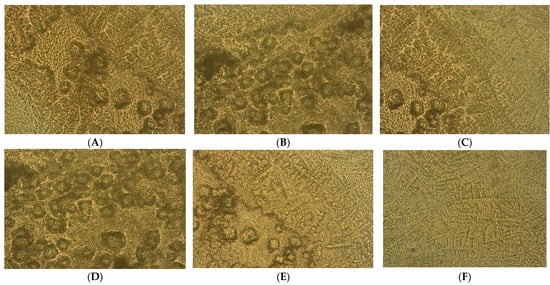

Figure 2.
Heterogeneous primary prostate cell cultures with aggressive phenotype developed from tissue fragments recovered from diagnosed patients with benign prostate hyperplasia (BPH) after 14 days in vitro development by optical microscopy (×40). (A–F) Prostate nodular hyperplasia, atrophy, and chronic inflammation cell cultures (S4) highlighting acini presence with nodular architecture and stromal proliferation; (G–L) prostatic epithelial cells proliferation from adenoma with chronic inflammation cell cultures (S5).
Benign nodular hyperplasia sections used to establish the diagnosis of patients were characterized by acini proliferation with lobulated and nodular architecture. A dilated duct presence surrounded by the small and medium acini sizes was associated with stromal proliferation. Also, p63 and cytokeratin 34 (CK34 BE12) markers have positive reaction in basal cells from acini and Ki67 marker showed a negative expression that is correlated with benign lesion character (Figure 3A–D). Benign adenomatous hyperplasia samples studied in the diagnosed patient’s establishment were characterized by acini proliferation, interglandular distance presence, p63, and CK34 markers have positive reaction in basal acinar cells, and ki67 marker presented a negative expression in epithelial cells (Figure 4A–D).
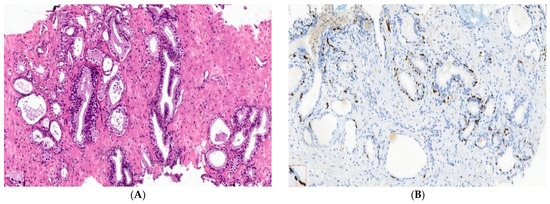
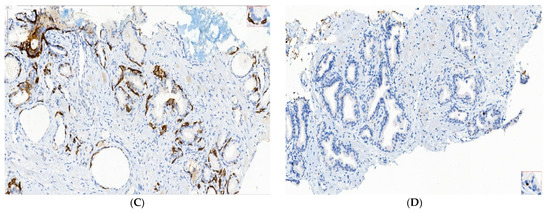
Figure 3.
IHC staining methods by optical microscopy were used to establish the benign prostatic hyperplasia diagnosis in patients. Nodular hyperplasia, atrophy, and chronic inflammation (A–D) tissue-fragment samples were used to develop in vitro heterogeneous primary prostate cell cultures with an aggressive phenotype (S4). Legend: A—benign prostatic hyperplasia acini proliferation with lobulated, nodular architecture, dilated duct presence surrounded by the small and medium acini size, associated with stromal proliferation (HE; ×100); B—benign prostatic hyperplasia p63—marker—positive in basal cells in evaluated prostatic acini (×100); C—HMWCK (clone 34BE12)—continuously positive in basal cells in acini with lobulated architecture (×100); D—Ki67 marker-negative correlated with benign lesion character (×100).
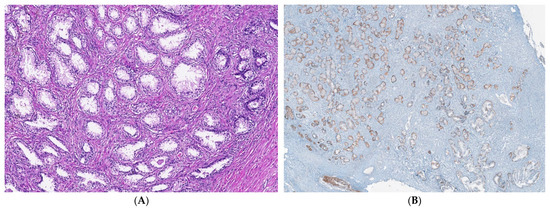
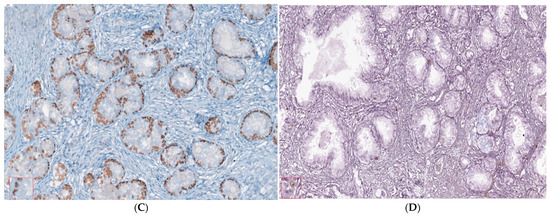
Figure 4.
IHC staining methods by optical microscopy were used to establish benign adenomatous hyperplasia diagnosis in patients (A–D). Prostate adenoma with chronic inflammation tissue-fragment samples were used to develop in vitro heterogeneous primary prostate cell cultures with an aggressive phenotype (S5). Legend: A—benign adenomatous hyperplasia, with prostatic acini proliferation and interglandular distance presence (HE; ×100); B—p63 marker—continuously positive in basal acinar cells (×40); C—HMWCK (clone 34BE12)—continuously positive in basal acinar cells (×100); D—Adenomatous prostatic hyperplasia—ki67 marker-negative in prostatic epithelial cells (×100).
2.3. Cell Cycle
As shown in Figure 5A–F,I by PI staining of the cell-cycle distribution and DNA ploidy, heterogeneous aggressive phenotype BHP cell cultures present cell growth inhibition by cell-cycle arrest in the G2/M phase (S1: 96.03 ± 0.23; S4: 90.99 ± 0.46; S5: 83.59 ± 0.94) reported to negative control (C1: 14.64 ± 2.44, p < 0.05, Figure 3I), characterizing the presence of the aneuploidy status.
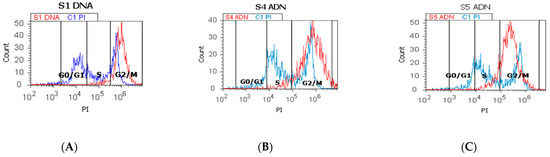
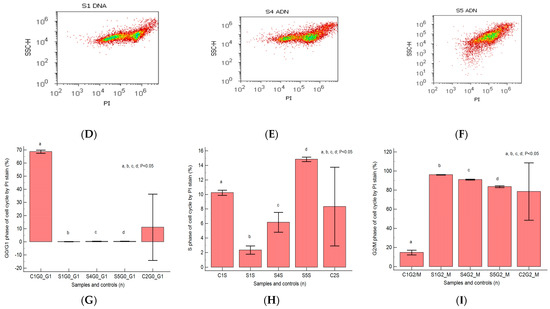
Figure 5.
Cell cycle represented by propidium iodide stain (PI). (A–C): control DNA (C1) is extrapolated on the PI ax; G0/G1 phase = A—0.18%; B—0.44%; C—0.45; C1—67.70%; S phase = A—4.80%; B—4.98%; C—14.57%; C1—10.54%; G2/M phase = A—94.27%; B—90.54%; C—82.78%; C1—12.53%. (D–F): Arrest of G2/M phase in heterogeneous BPH cells (S1, S4, and S5) reported to non-malignant prostate cells (C1PI). (G–I): Cell-cycle statistics of BPH patients reported to controls (S1; S4; S5; C1; C2); a, b, c, d; p < 0.05 represent significant statistical differences between samples and controls (Mann-Whitney test by MedCalc program). Legend: A; D—benign prostatic hyperplasia (S1); B; E—nodular hyperplasia, atrophy, and chronic inflammation (S4); C; F—prostate adenoma with chronic inflammation (S5); C1PI—non-malignant adjacent prostate cells (negative control); C2—positive control (BPH).
The proliferative S phase revealed statistically significant increased values in prostate adenoma (S5: 14.83 ± 0.30) reported to non-malignant prostate-adjacent cell cultures (C1: 10.23 ± 0.35, p < 0.05, Figure 5C,F,H), being included in the high S-phase category (>12%) with a 50% higher risk of death or recurrence (worse prognostic) due aneuploid status presence rather than intermediary or low S-phase categories.
Nodular hyperplasia (S4) and BPH cell cultures (S1) showed statistically significant decreased values of the S phase of the cell-cycle reported to negative control, being included in the low S-phase category (<7%), and having a better prognostic value for the patient survival (S4: 6.16 ± 1.36; S1: 2.34 ± 0.57 vs. C1: 10.23 ± 0.35, p < 0.05, Figure 5A,B,D,E,H).
2.4. Cell Adhesion
A significant increase in CD42b transmembrane glycoprotein levels was observed in heterogeneous BPH cultures (S1: 48.21 ± 8.78; S4: 53.95 ± 6.98; S5: 67.73 ± 2.13) reported to negative control (C2: 26.24 ± 0.50, p < 0.05, Figure 6A–F).
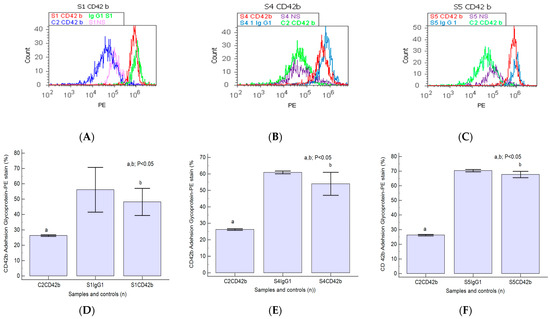
Figure 6.
Integrin expression in platelet aggregation to tumoral cells by highlighting adhesion glycoproteins with CD42b-PE stain in heterogeneous BPH cells (A–C) reported to non-malignant prostate cells (C2). (A–C): CD42b+ expression: A—40.60%; B—47.90%; C—65.88%; C2—25.80%. (D–F): CD42b glycoprotein expressions statistics of BPH patients reported to controls (S1; S4; S5; C2; Ig-G1); a, b; p < 0.05 represent significant statistical differences between samples and controls (Mann–Whitney test by MedCalc program). Legend: A—benign prostatic hyperplasia (S1); B—prostate nodular hyperplasia, atrophy, and chronic inflammation (S4); C—prostate adenoma with chronic inflammation (S5); NS—unstained control prostate cell cultures (S1, S4, and S5); C2—non-malignant adjacent prostate cell cultures (negative control); IgG1—positive control prostate cell cultures (S1, S4, and S5).
Nodular prostate hyperplasia (S4) and adenoma heterogeneous primary cell cultures (S5) present increased CD42b-adhesion glycoprotein expressions reported for the BPH cell cultures and controls (Figure 6A–C), indicating platelets and lymphocytes recruiting to inflammation site due to the chronic inflammation presence in BPH patients. Increased integrin levels in BPH cell cultures suggest epithelial–mesenchymal transition (EMT), acquiring and maintaining an aggressive phenotype by escaping cell apoptosis and cell proliferation, representing a base for PCa development.
2.5. Autophagy and Nuclear Shrinkage
The heterogeneous BPH cell cultures highlighted in Figure 7A–E present pyknotic nuclei by Hoechst 33342 stain and lysosomal activity by acridine orange (AO) stain. In prostate heterogeneous phenotype adenoma cell cultures (S5) were observed statistically significantly higher values of H+ nuclear shrinkage and AO+ autophagy expressions than in the positive control (H+: 77.15 ± 3.74 vs. C1: 54.28 ± 16.12, p < 0.05; AO+: 92.08 ± 0.30 vs. C1: 76.13 ± 19.59, p < 0.05; Figure 7C,F,H,K,L).
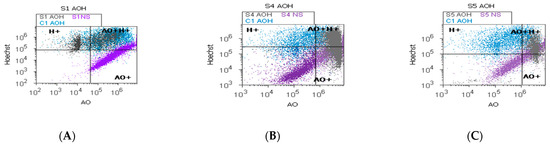
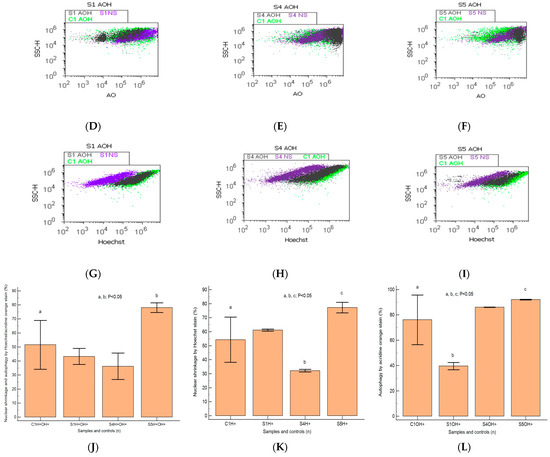
Figure 7.
Nuclear shrinkage and autophagy plotted by Hoechst–acridine orange (AO) dual stain. (A–C): AO+H+: A—38.28%; B—44.43%; C—81.03%; C1—48.18%; H+: A—60.47%; B—31.34%; C—80.40%; C1—61.79%; AO+: A—42.08%; B—85.95%; C—91.82%; C1—37.09%. Lysosomal activity (D–F) and nuclear membrane integrity (G–I) changes in heterogeneous BPH cell cultures (S1, S4, and S5) reported to positive control prostate cell cultures (C1) and unstained control prostate cell cultures (S1NS; S4NS; S5NS); (J–L) nuclear shrinkage and autophagy statistics of BPH patients reported to controls (S1; S4; S5; C1); a, b, c; p < 0.05 represent significant statistical differences between samples and control (Mann–Whitney test by MedCalc program). Legend: A; D; G—benign prostatic hyperplasia (S1); B; E; H—nodular hyperplasia, atrophy, and chronic inflammation (S4); C; F; I—prostate adenoma with chronic inflammation (S5); NC—unstained control prostate cell cultures (S1, S4, and S5); C1—positive control prostate cell cultures.
In prostate nodular hyperplasia with aggressive phenotype heterogeneity (S4, atrophy, and chronic inflammation), cell cultures were observed a decreased significant statistical value of H+ nuclear shrinkage than the positive control (32.19 ± 0.98 vs. C1: 54.28 ± 16.12, p < 0.05, Figure 7B,I,K) and an increased AO+ autophagy expression than the positive control (AO+: 86.05 ± 0.11 vs. C1: 76.13 ± 19.59, p ≥ 0.05; Figure 7B,E,L).
A particular interest was represented by the BPH cell populations with double positively expressed by acridine orange and Hoechst dual stain (AO+H+). Most valuable observations were highlighted in the cell apoptosis program for the prostate primary heterogeneous adenoma cell culture (S5), being represented by increasing nuclear shrinkage and lysosomal activity reported to positive control (S5: 78.07 ± 3.41 vs. C1: 51.58 ± 17.41, p < 0.05, Figure 7C,J).
2.6. Oxidative Stress
Changes in the oxidative cellular stress mechanism in heterogeneous BPH cell cultures were analyzed by the total ROS flow-cytometry method, as presented in Figure 8A–G.
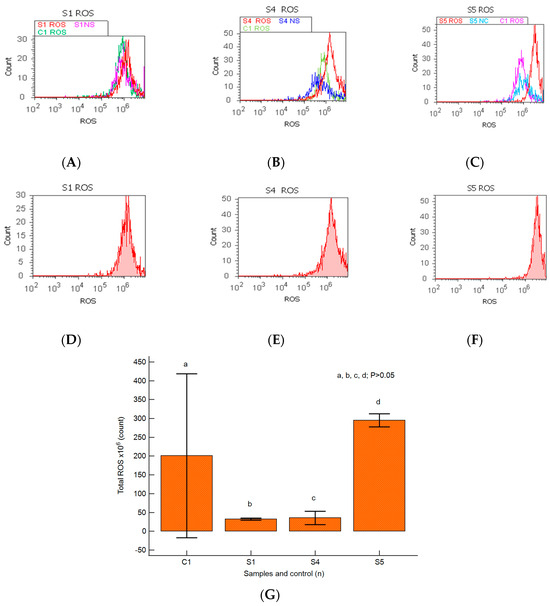
Figure 8.
Total reactive oxygen species pattern (ROS). (A–F) Oxidative stress changes in heterogeneous BPH cells (S1, S4, and S5). ROS: D-30 × 106; E-51 × 106; F-28 × 107; C1-35 × 106. (A–C) Positive control prostate cells (C1ROS) and unstained control prostate cells (S1NS, S4NS, and S5NS) are extrapolated on the ROS ax. (G) Oxidative stress pattern statistics of BPH patients reported for controls (S1; S4; S5; C1); a, b, c, d; p > 0.05 represent non-significant statistical differences between samples and controls (Mann–Whitney test by MedCalc program). Legend: A; D—benign prostatic hyperplasia (S1); B; E—nodular hyperplasia, atrophy, and chronic inflammation (S4); C; F—prostate adenoma with chronic inflammation (S5); NS—unstained control prostate cell cultures (S1, S4, and S5); C1—positive control BPH cell cultures.
A linked apoptosis–oxidative stress mechanism was observed in primary BPH cell cultures with an aggressive phenotype, highlighting the lower ROS values reported for the positive control (S1: 32.50 ± 2.88 × 106; S4: 35.50 ± 17.89 × 106 vs. C1: 234.50 ± 224.92 × 106, p ≥ 0.05, Figure 8A,B,D,E,G).
In prostate heterogeneous aggressive phenotype adenoma cell cultures (S5), the highest peak of ROS was observed and reported to control cell cultures (S5: 295.00 ± 17.32 × 106 vs. C1: 234.50 ± 224.92 × 106, p ≥ 0.05, Figure 8C,F,G).
2.7. Correlations between Apoptosis, Cell Cycle, Adhesion Glycoprotein Expression, Autophagy, and Nuclear Shrinkage in Different BPH Cell Cultures with Aggressive Phenotype
The relationships between molecular mechanisms in heterogeneous BPH cell cultures are presented in Figure 9A–D. Cell adhesion represented by CD42b+ glycoprotein expression was negatively correlated with the G0/G1 phase of the cell cycle (r = −0.57, p < 0.05) and viability (r = −0.69, p < 0.05) and positively correlated with immunoglobulin positive expression (IgG1+, r = 0.78, p < 0.01, Figure 9A).
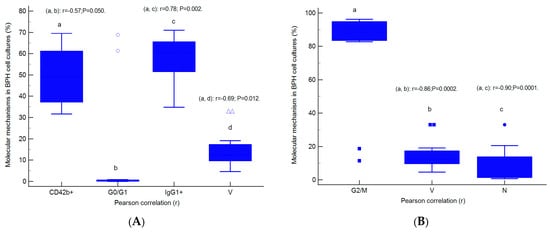
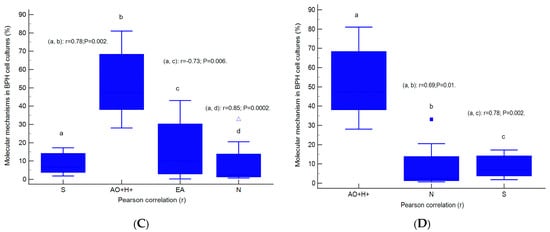
Figure 9.
Parametrical correlations (r) between molecular mechanisms in heterogeneous BPH cell cultures with an aggressive phenotype (A–D). Legend: CD42b+—adhesion glycoprotein positive expression; IgG1+—immunoglobulin positive expression; G0/G1; G2/M; S—cell-cycle phases; V—viability; N—necrosis; EA—early apoptosis; AO+H+—positive cell populations expressed for autophagy and nuclear shrinkage; a, b, c, d; p < 0.01 represent significant statistical differences between samples (Pearson test, r, by MedCalc program).
Inhibition of cell growth represented by G2/M phase arrest presents negative strong correlations with viability (r = −0.86, p < 0.01) and the necrosis (r = −0.90, p < 0.01) mechanisms highlighted in heterogeneous BPH cell cultures with an aggressive phenotype (Figure 9B).
Cell proliferation, represented by the S proliferative phase of the cell cycle, was directly correlated with linked autophagy–nuclear shrinkage mechanisms (AO+H+, r = 0.78, p < 0.01) and necrosis (r = 0.85, p < 0.01), and negatively correlated with early apoptosis (r = −0.73, p < 0.01, Figure 9C).
Double positive BPH cell culture expression for lysosomal activity and nuclear shrinkage present a positive correlation with necrosis (r = 0.69, p < 0.01) and the S phase of the cell cycle (r = 0.78, p < 0.01, Figure 9D).
2.8. Prognostic Role of Molecular Mechanisms Network in Heterogeneous BPH Cell Cultures
Furthermore, multivariate analysis regression analyzed the linked molecular mechanisms of late apoptosis and the S proliferative phase of cell-cycle expressions to observe their potential prognostic roles in heterogeneous aggressive phenotype BHP cell cultures (Figure 10A,B). Our analysis observed that late apoptosis and the S phase of the cell cycle represent unfavorable prognostic biomarkers, being dependent predictor factors for the molecular mechanisms network in heterogeneous aggressive phenotype BHP cell cultures.
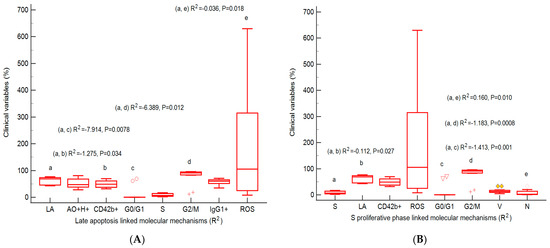
Figure 10.
Predictor factors represented by the coefficient of multiple regression (R2) for linked molecular mechanisms in heterogeneous aggressive phenotype BHP cell cultures. (A) Late apoptosis: AO+H+: R2 = 0.180 ± 0.189, p = 0.394; CD42b+: R2 = −1.275 ± 0.403 *, p = 0.034; G0/G1 phase: R2 = −7.914 ± 1.603 **, p = 0.0078; S phase: R2 = −3.422 ± 1.326, p = 0.061; G2/M phase: −6.389 ± 1.468 *, p = 0.012; IgG1+: R2 = 0.135 ± 0.348, p = 0.717; ROS: R2 = −0.036 ± 0.009 *, p = 0.018. (B) S proliferative phase of cell cycle: LA: R2 = −0.112 ± 0.033 *, p = 0.027; CD42b+: R2 = −0.099 ± 0.039, p = 0.064; ROS: R2 = −0.002 ± 0.001, p = 0.129; G0/G1 phase: R2 = −1.413 ± 0.167 **, p = 0.001; G2/M phase: R2 = −1.183 ± 0.129 **, p = 0.0008; V: R2 = 0.078 ± 0.052; p = 0.2136; N: R2 = 0.160 ± 0.035 **; p = 0.010. Legend: a, b, c, d, e; * p < 0.05 and ** p < 0.01 represent statistically significant differences between variables made by Least squares multiple regression (R2) by MedCalc v20.111 Software Ltd., Ostend, Belgium. AO+H+—positive cell populations expressed for autophagy and nuclear shrinkage; CD42b+—adhesion glycoprotein positive expression; G0/G1; G2/M; S—cell-cycle phases; IgG1+—immunoglobulin positive expression; ROS—reactive oxygen species; LA—late apoptosis; V—viability; N—necrosis.
3. Discussion
Our study analyzed cell death, DNA content, and the autophagy molecular mechanisms network by flow-cytometry methods to highlight prostate pathogenesis and its prognostic role in heterogeneous aggressive phenotype BPH primary cell cultures reported to non-malignant cell cultures.
Cell-death escape presents importance to target cancer therapies [21], involving molecular events by major apoptotic pathways, including the death receptor pathway (extrinsic) and the mitochondrial pathway (intrinsic) [22]. The essential caspases involved in cell-death mechanisms are initiators (caspase-2,-8,-9,-10) and effectors (caspase-3,-6,-7) [23,24,25].
Our study presented a pro-apoptotic signal highlighted by a caspase-3/7 cascade showing low changes in early apoptosis in heterogeneous BHP cell cultures. Increased late apoptosis by the caspases-3/7 pattern as a chronic response by injury induction and oxidative stress presents a particular interest in heterogeneous aggressive phenotype BPH cell cultures. This is because modified cells adopt modified apoptosis mechanisms with peaks of more than 106 to 107 by DEVD-MR/PI stain reported for non-malignant prostate primary cells with an apoptosis peak of 105 to a maximum of 106. The epithelial –mesenchymal transition by pro-inflammatory and profibrogenic phenotypes characterizes the prostate heterogeneous microenvironment of nodular hyperplasia and adenoma primary cell cultures.
In heterogeneous aggressive phenotype BHP cell cultures, a multivariate regression analysis showed that a higher late apoptosis status by caspases-3/7 cascade is an unfavorable prognostic biomarker, a dependent predictor factor for network molecular mechanisms by cell adhesion by CD42b+ glycoprotein expression, cell-cycle blockage by G2/M phase arrest, and oxidative stress by total ROS count. An apoptosis–oxidative stress mechanisms network was highlighted by the highest peak of ROS reported to control in prostate heterogeneous aggressive phenotype adenoma cell cultures.
A recent study that highlighted prostate cancer progression by cell proliferation and apoptosis imbalance reported a silent increase in early apoptosis by FITC/PI stain in PCa patients and a higher necrosis pattern in BPH patients than in healthy patients [26]. Apoptosis results after activation mechanisms of initiator and executioner caspases [27], leading to chromatin condensation, cell adhesion loss, cell shrinkage, membrane blebbing, and DNA fragmentation. Mutations of caspase-3 activity were identified in different human cancers, such as colon and stomach cancer, lymphomas, and hepato-carcinoma [28,29,30,31,32,33]. In breast and prostate cancers, caspase-3 activity was identified, but without mutations [31,32,33]. On the other hand, caspase-7 expression presents mutations in human cancers [33,34]. Lower caspase-7 activity was observed in gastric carcinoma cases, but caspase-3 showed a higher pattern than non-malignant mucosa samples [35,36]. In acute myelogenous leukemia (AML) patients were observed increased caspase-3 levels reported to patients with normal peripheral blood lymphocytes [37]. Caspase-3 activity, expressed in breast and prostate carcinoma, is positively correlated with tumor progression [28,29]. Macrophage infiltration in breast cancer cells determines tumor progression [38]. The associated macrophages of tumor progression were reported in human cancer [39], and cancer apoptotic cells represent a source of chemo-attractants by stimulating macrophage infiltration [40]. In a dormant tumor, the continuous disappearance of cells by apoptosis allows a proliferation degree [41], but accumulated mutations lead to increased cell malignancy [42]. In both apoptotic ways, tumor progression initiation depends on the functional caspases, suggesting a caspase-dependent mechanism to release chemo-attractant factors from apoptotic cells with roles in macrophage infiltration [43]. Errors in the signaling pathways of cellular differentiation stimulate the cell proliferative program, which depends on non-apoptotic caspase activities. In many reports, it was found that caspase activity is implied in cellular proliferation. Caspase-3 activity is directly correlated with the cell cycle in HeLa cells, having an inhibition peak in the G2/M stage [44]. Caspase-7 expressions showed activated mitosis in tumor cell lines, leading to mitotic arrest, suggesting their role in tumor proliferation [45]. Another study reported that caspase-3 activity lost in transgenic mice models induces pancreatic beta cell hyper-proliferation by the c-myc signaling pathway, showing their role in the cell cycle of tumor cells [46].
The modified cell apoptosis mechanism in tumorigenesis is fundamental because acquired apoptosis resistance during therapy represents a major obstacle to cancer treatment. Many reports showed that tumor cells adapt to evading apoptosis by losing caspase function in cancer cell cultures [37]. Biochemical cascades highlighted in apoptosis are represented by caspase-3, -6, and -7 activity. Cytochrome c, released from mitochondria and its subsequent binding to caspase-9, determines caspase-3 activation, which is the executioner caspase essential for the nuclear changes associated with apoptosis, including chromatin condensation. A central role of the biochemical caspase cascade, implied in immune response and prostate cancer cell apoptosis, was studied to caspase-1, -3, and -9 expressions in normal and malignant human prostate samples [32].
Various studies propose two different types of cell-death mechanisms: accidental cell death (ACD) and regulated cell death (RCD) [47,48]. Physical and chemical stress factors exceeding the cell’s ability to restore their functions determine ACD, characterized by morphological events such as cell membrane alterations and cellular content release [49]. RCD is regulated by specific signaling pathways that determine biochemical, morphological, and immunological consequences in cells. The most studied ACD mechanism based on cell morphology, gene expression, and biochemical properties is autophagy [50,51,52,53,54,55]. In the meantime, regulatory molecules from RCD pathways represent biomarkers that are useful as potential therapeutic targets [56]. PCa is the second maligned affection and the fifth global cause of cancer death in men [57]. In apoptosis, reactive oxygen species (ROS) production causes a pro-inflammatory environment. Apoptosis-inducing factor (AIF), Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) are death-inducing biomarkers that act on FasL/TNF receptor (TNFR)1, inducing apoptosis dependent upon caspase activity [54]. Apoptosis is visualized in different tissue samples using acridine orange (AO) [58,59], highlighted in areas with lysosomal and phagocytosis activity. Another study showed that the necrotic-like cell phenotypes required by gene activation and protein synthesis are programmed cell-death forms [60] with morphological characteristics of necrosis and apoptosis [61,62]. Apoptosis directly correlates with the cell cycle, plasma membrane changes, chromatin condensation, and DNA fragmentation. Damage-related pattern molecules (DAMPs) and mitochondrial DNA are released into the extracellular environment. Apoptosis is characterized by limited DMAP release. Membrane death receptor activation via the TNF, TRAIL, and FasL pathways and intracellular stimulation via genetic damage, hypoxia, and oxidative stress trigger apoptosis [63]. Mitochondrial apoptosis induced by cytochrome c, SMAC/DIABLO, HrtA2/Omi, and AIF factors determines apoptosis. Cytotoxic factors activate the BH3 proteins that determine BAX and BAK activation with increasing mitochondrial permeability. Cytoplasmic cytochrome c interacts with apoptotic protease activating factor 1 (APAF1), leading to apoptosomes initiation that stimulates pro-caspase-9, -3, and -7 activities, determining the dysfunction of cellular components and apoptosis [54,58,64].
Because BPH heterogeneity limits treatment response, understanding the molecular mechanisms involved in patient prognostic survival is necessary. Our study presents cell cycle, aneuploidy status, and cell adhesion by flow-cytometric analysis to highlight the heterogeneously microenvironment with aggressive phenotype BPH primary cell cultures reported to non-malignant cell cultures. Cell growth inhibition by cell-cycle arrest in the G2/M phase characterizes the aneuploidy status of DNA. The proliferative S phase of the cell cycle in prostate adenoma cell cultures is included in the high S-phase category (>12%) with a higher risk of death or recurrence (worse prognostic) due to aneuploid status presence. Also, nodular hyperplasia and BPH primary cell cultures with S phases included in the low S-phase category (<7%) present a better prognostic for patient survival. Inhibition of cell growth represented by G2/M phase arrest presents negatively strong correlations with viability and necrosis. In the meantime, cell proliferation, represented by the S phase of the cell cycle, was positively correlated with autophagy–nuclear shrinkage and necrosis, and negatively correlated with early apoptosis in BPH cell cultures. In our multivariate regression analysis, the S proliferative phase represents an unfavorable prognostic biomarker, being a dependent predictor factor for the molecular mechanisms network represented by caspase-3/7 activity (late apoptosis and necrosis) and cell growth inhibition (G2/M arrest) in heterogeneous aggressive phenotype BHP cell cultures.
Aneuploidy status is a tumorigenesis driver, representing a prognostic biomarker of tumor progression [65,66]. Aneuploidy cells are found in various tumors, often indicating higher malignancy [67]. Aneuploidy flow-cytometric analysis was used as a prognostic indicator in prostate, colon, and breast tumors [68]. The authors reported that cell distribution in the G0/G1, S, and G2/M phases by flow cytometry represents a rapid and efficient method for prognostic analysis [69,70]. The S phase has a prognostic role in patients, being divided into low (<7.0%), intermediate (7.0–11.9%), and high (≥12%) categories. The risk of death or recurrence for aneuploid cases is increased for the high S-phase category than for the intermediary or low categories. Different techniques or samples do not affect the cell cycle because a high S phase remains correlated with a worse tumor grade in breast cancer tissue samples [71].
The regulatory proteins implied in cell cycle guarantees damaged DNA elimination from cells to assure the homeostasis. Cell-cycle regulators represent targets for cancer therapies [72]. Cell-cycle regulation is guaranteed by the retinoblastoma tumor suppressor protein (Rb), p53, p27, or p21 being described in cancer [73,74,75]. In the cell cycle, Rb regulates the G1 to S phase’s transition [76]. In oxidative stress and cell damage, the p53 protein (TP53 gene) represents the control point as a tumor suppressor and pleiotropic transcription factor determining cell-cycle arrest. When cells do not repair DNA damage, p53 induces an apoptosis process mediated by the Bcl-2 family [77]. DNA damage causes the p21 and p53 response, determining the cell-cycle arrest in the G1 and G2 phases [78]. In phase S, p21 inhibits the cyclin A-CDK1 complex by blocking the transition to the G2M phase [79]. The p21 protein represents the independent anti-proliferative effector reported for the p53 pathway [80]. A higher p21 level induces prostate cancer aggressiveness. In the meantime, its lower values act against prostate tumorigenesis [81]. The p27 protein is a member of the CIP/KIP family. P27 regulates the G1 to S phase transition by cyclin-dependent kinases (CDKs) inhibition [82]. P27 regulates apoptosis. A lower p27 level is associated with aggressive tumor behavior [83,84].
Our study presents increased CD42b+ transmembrane glycoprotein levels in prostate hyperplasia heterogeneous primary cell cultures, indicating platelets and lymphocytes recruiting to the inflammation site due to the chronic inflammation presence in BPH patients. Increased integrin levels in heterogeneous BPH cultures suggest epithelial–mesenchymal transition (EMT), acquiring and maintaining an aggressive phenotype by escaping cell apoptosis and cell proliferation, leading to PCa development. Cell adhesion represented by CD42b+ glycoprotein expression was negatively correlated with the G0/G1 phase of the cell cycle and viability and positively correlated with immunoglobulin-positive expression (IgG1+). Integrins have an essential role in cancer progression. Modified cell adhesion biomarkers lead to cell proliferation, migration, and metastasis, being correlated with tumoral stages and outcomes (recurrence and survival) [85,86,87,88,89,90]. In another study, the CD42b+ adhesion glycoprotein pattern was increased in PCa cases [26]. Other reports presented CD42b glycoproteins as having essential roles in cell signaling networks, growth, differentiation, and survival [91,92,93].
Autophagy, another essential mechanism in our study, was presented in heterogeneous aggressive phenotype BHP cell cultures by a double positive expressed by acridine orange and Hoechst stain (AO+H+). Significant changes in the apoptosis program by increasing nuclear shrinkage and lysosomal activity reported to non-malignant cell cultures were observed in heterogeneous primary prostate adenoma cell cultures. Double positive heterogeneous BPH cell culture expression for lysosomal activity and nuclear shrinkage presents a positive correlation with necrosis and the proliferative S phase of the cell cycle.
Autophagy transports damaged proteins and organelles at the lysosomes for degradation. In prostate cancer cells, when the mitochondrial dysfunction is present, cytochrome c is released from the mitochondria in the cytoplasm, determining the caspase-3 activation and cell apoptosis. ROS generation activates the cell-death processes [47,94,95]. Autophagy promotes cellular adaptation to survival in stressful conditions [96]. The anticancer factors induce autophagy in different cancer cells [97,98]. GnRH-II antagonist Trp-1 determines the inhibition of cell proliferation and death signaling pathway activation in human prostate cancer cell cultures by mitochondrial dysfunction and autophagy [99,100]. Autophagy presents autophagosome formation, fusion with the lysosome, autophagy body breakdown, and recycling. Similar settings were observed between autophagy and apoptosis pathways [101]. The difference between apoptotic pathways depends on the physiologic status, developmental stage, and tissue type [102].
Mitochondria represent the principal organelles that play a role in apoptosis and autophagy [103]. Akt (protein kinase B) and p70S6 kinase regulation involve similar signaling pathways in apoptosis and autophagy. The cell death–autophagy network includes DAPk, Beclin 1, and protymosin-α [104]. Genetic alterations in the cell proliferation/apoptosis regulation pathways in cancer development suggest an autophagy–apoptosis network [103]. In tumor progression, autophagy pathway deregulation determines autophagy activity reduction, highlighted in its function as a safeguard mechanism in uncontrolled cell growth restriction, a protective mechanism against apoptosis.
An autophagy vesicle’s presence in dying cells reflects an adaptive response to maintain cell survival under stress conditions. In cancer cells, apoptosis and autophagy pathways depend on response severity, cell constituents, and other signaling pathways. Apoptosis and autophagy are not mutually exclusive, but various morphologic changes are still attributed, in part, to distinct biochemical and molecular events [103,104]. In certain cells, autophagy morphological features occur prior to apoptosis. Controversy exists when autophagy is constantly increased because it is necessary to elucidate the molecular basis in cell death by studying new biomarkers. BPH and cancer influence homeostasis, assuring an unstable report between proliferation and cell death [105]. Significant changes in the mitosis–apoptosis balance affect homeostasis, exposing patients to immune disorders, degenerative diseases, and cancer [106].
Future directions in the research area highlight the central molecular mechanisms network involved in apoptosis, the cell cycle, and autophagy as predictor biomarkers useful for establishing patient prognostic survival in different malignant affections or targeting cancer therapy development.
4. Materials and Methods
4.1. Materials
Various tissue samples excised by transurethral resection of the prostate (TURP), recovered from patients with benign prostatic hyperplasia (BPH, who signed an informed consent form, agreeing to participate in this study, n = 12) from the Clinical Service of Pathology, Sf. Apostol Andrei Clinical Emergency County Hospital in Constanta, Romania, were mechanically homogenized with TissueRuptor II (Qiagen, Hilden, Germany).
4.2. Prostate Primary Cell Cultures
Different cells and small tissue fragments are placed in a tissue-culture flask with a growth area (25 cm2) cultured in DMEM High Glucose and mixed in humidity conditions of 5% CO2 at 37 °C for 14 days. Prostate cells were distributed in tissue-culture test plate 6 and growth-enhanced treated (ThermoFisher Scientific Inc., Waltham, MA, USA). After 14 days, caspase-3/7 activity, cell cycle, cell adhesion, nuclear shrinkage, lysosomal activity, and oxidative stress were performed by flow-cytometry methods in the Cell Biology Department, CEDMOG, Ovidius University of Constanta, Romania.
4.3. Samples and Controls
Experimental heterogeneous BPH primary cell cultures with an aggressive phenotype were divided in three types of groups: 1-BPH (S1–S3; S6–S12); 2-nodular BPH, atrophy, and chronic inflammation (S4); 3-prostate adenoma with chronic inflammation (S5), being studied the biological processes network implied in tumorigenesis. Experimental BPH primary cell cultures were reported to negative and positive controls. The negative control is represented by prostate primary cell cultures developed from non-malignant adjacent tissue samples recovered from diagnosed patients with BPH. The positive control is represented by prostate hyperplasia primary cell cultures developed from prostatic tissue samples recovered from diagnosed patients with BPH.
4.4. Nodular Hyperplasia and Adenoma Microenvironment Characterization by IHC Methods
After the macroscopic description, prostate tissue samples were fixed in 10% formaldehyde, paraffin-embedded, sectioned, and stained in hematoxylin–eosin (HE) [26]. Monoclonal antibodies from immunohistochemical methods (IHC) were applied to selected sections of the study case. The antibody anti-Ki67 (Master Diagnostica, clone SP6, Sao Paolo, Brazil) is used to evaluate nuclear expression. HMWCK (clone 34BE12) and p63 (Master Diagnostica, clone 4A4, Sao Paolo, Brazil) are used to observe basal cell layer expressions, being basal cell markers. The basal cell layer is present in the histologically normal prostate and in benign lesions, while it is absent in prostatic carcinoma. Also, Ki67 expression represents a nuclear proliferation marker, being negative in benign lesions (BPH) and positive in prostate cancer (PCa).
4.5. Equipment
Our analysis was performed on an Attune Acoustic focusing cytometer (Applied Biosystems, Waltham, MA, USA). The flow cytometer was set using Attune performance-tracking beads, labeling, and detection (Life Technologies, Europe BV, Bleiswijk, The Netherlands) [107]. More than 10,000 cells per sample for each analysis were gated using the flow cytometer’s forward scatter (FSC) and side scatter (SSC). Data were collected and interpreted using Attune Cytometric Software v.1.2.5, Applied Biosystems, 2010. Heterogeneous prostate hyperplasia cell cultures with an aggressive phenotype (nodular hyperplasia with chronic inflammation and adenoma with chronic inflammation) were analyzed using a Primo Star optical microscope (Zeiss, Gottingen, Germany).
4.6. Methods
4.6.1. Caspase-3/7 Activity
Cell apoptosis was observed using the DEVD-MR/PI methodology (FAM Caspase-3/7 Assay Kit, Abcam, Boston, MA, USA). Prostate primary cell cultures (BPH cells or non-malignant cells-negative control-C1, 200 μL) were transferred in tubes, and 20 μL of DEVD-MR solution and 20 μL of PI, were added, mixed, and incubated for 30 min at room temperature in darkness. Also, an intern control tub with experimental unstained BPH cells (NS) was made. One mL FCB (eBioscienceTM, Life Technologies Europe BV, The Netherlands) is added. Viability, early and late apoptosis, and necrosis were analyzed by BL3 channel (DEVD-MR) and BL2 channel (PI) [108].
4.6.2. Cell Cycle
Two hundred µL of prostate primary cell cultures (BPH cells or non-malignant cells-negative control-C1) were fixed with 200 µL absolute ethanol for 30 min. Then, the fixed prostate cells were treated with 10 µL of PI (20 mg/mL) and kept in darkness at room temperature for 30 min. After adding 1 mL FCB, the cell-cycle phases were analyzed using the BL2 channel (PI) [109].
4.6.3. CD42b Adhesion Glycoprotein
Anti-CD42b-PE (HIP1, Invitrogen, eBioscience, Life Technologies Corp, Carlsbad, CA, USA) monoclonal antibodies conjugated with phycoerythrin (PE), were used to assess the GPIba platelet glycoprotein in different experimental samples. Prostate cell cultures (100 µL) were spread for each tube, including 1.—experimental CD42b-PE stain; 2.—positive control-IgG 1 stain; 3.—negative controls (C2-prostate non-malignant cells and NS-unstained BPH cells). In experimental tubes with BPH cells, 5 µL of CD42b-PE were added. A positive control tube, with BPH cells and 5 µL mouse IgG 1, was made. Another two tubes as negative controls were prepared, one tube with prostate non-malignant cell cultures (C2) with 5 µL of CD42b-PE and another tube with unstained BPH cells (NS) to report the experimental tubes. All tubes were vortexed and incubated in darkness for 25 min at 37 °C. One ml of flow-cytometry stain buffer (FCB) was added and vortexed before analysis. To identify the CD42b adhesion glycoprotein by size and specificity to flow cytometer was used the BL2 channel (PE) [26].
4.6.4. Nuclear Shrinkage and Lysosomal Activity
In primary prostate cell cultures (BPH cells or positive control, 300 μL), two μL of Hoechst 33342 stain, and 50 μL of acridine orange (1.0 μM, AO) were added into the tubes, mixed, and incubated in darkness at room temperature for 30 min. Also, an intern control tub with experimental unstained BPH cells (NS) was made. Cells were analyzed by flow cytometry after 0.5 mL FCB addition using UV excitation to the VL2 channel (Hoechst) and BL1 channel (AO) [109].
4.6.5. Total Reactive Oxygen Species (ROS)
The total reactive oxygen species (ROS Assay Kit 520 nm, Invitrogen, USA) was used to identify the linked cell signaling pathway. After 60 min at 37 °C and 5% CO2 incubation, stained cells (1 mL of BPH cells or positive control) with two μL 500X ROS stock solution were analyzed in the BL-1 channel. Also, an intern control tube with experimental unstained BPH cells (NS) was made [110].
4.7. Data Analysis
Our results were presented as means ± standard deviation (SD), representing flow-cytometry methods as caspase-3/7 activity (%), cell cycle (%), nuclear shrinkage (%), autophagy (%), adhesion glycoprotein (%, integrin CD42b), and oxidative stress count (×106). To identify the normal distribution of parameters, the Kolmogorov–Smirnov test was used. The Mann–Whitney test was applied to establish the differences between samples and controls, and p < 0.05 was considered statistically significant, made by MedCalc v20.111 Software Ltd. (Ostend, Belgium). Parametrical correlations (Pearson test, r) between apoptosis, cell cycle, adhesion glycoprotein expression, autophagy, and nuclear shrinkage in different BPH cell cultures were analyzed. Furthermore, multiple regression was performed by MedCalc v20.111 Software Ltd. (Ostend, Belgium) to determine the potential prognostic values of the linked mechanisms of late apoptosis and the S proliferative phase of the cell cycle in heterogeneous phenotypically BHP cell cultures. Figure 1A–I, Figure 5A–F, Figure 6A–C, Figure 7A–H and Figure 8A–F were made with Attune Cytometric Software v.1.2.5, Applied Biosystems, 2010 (Bedford, MA, USA) and Figure 1J–L, Figure 5G–I, Figure 6D–F, Figure 7J–L, Figure 8G, Figure 9A–D and Figure 10A,B with MedCalc v20.111 Software Ltd. (Ostend, Belgium).
5. Conclusions
Different factors limit a treatment response in aggressive phenotype BHP patients, so studying and understanding the molecular mechanisms involved in prognostic survival is necessary. Our study observed that a high late apoptosis expression, a high proliferative S phase of the cell-cycle category, and aneuploidy status represent unfavorable prognostic biomarkers and dependent predictor factors for the major biological mechanisms network in heterogeneous aggressive phenotype BHP primary cell cultures.
Author Contributions
Conceptualization, E.M., M.E., M.C.R., A.C.I. and M.A.; methodology, E.M., M.E. and M.C.R.; software, E.M.; validation, M.E. and A.C.; formal analysis, A.C.I. and M.E.; investigation F.V., M.E. and A.C.I.; resources, M.E., M.C.R., G.I.B., A.-A.N., A.F.M., A.C. and M.D.; data curation, M.E.; writing—original draft preparation, E.M., M.E., M.C.R. and A.C.I.; writing—review and editing, E.M., G.I.B., A.-A.N., A.F.M., M.D., F.V., A.C. and M.E.; visualization, M.A., G.I.B., A.-A.N., A.F.M. and M.C.R.; supervision, M.A. and M.D.; project administration, M.E.; funding acquisition, M.E. All authors have read and agreed to the published version of the manuscript.
Funding
This study is part of the result indicators assumed within the FOXPROS grant titled “The predictive role of FOXP3 and ROS factors, involved in the immune response and oxidative stress in prostate cancer”, contract number 15557/27 November 2023. This grant was won in the biomedical competition organized into the CNFIS-FDI-2023-F-0391 project entitled “Excellence, performance, and competitiveness in biomedical research at the Ovidius University of Constanta”.
Institutional Review Board Statement
All aspects of ethics approval and consent to participate for patients in this study are made in conformity with the Declaration of Helsinki 2000 and finally approved by the Research and Development Ethics Committee from “Ovidius” University of Constanta, Romania, 16 July 2024.
Informed Consent Statement
Informed consent was obtained from patients involved in the research activity.
Data Availability Statement
Data and their interpretation are contained in the research article.
Acknowledgments
Technical support (laboratory materials, work kits) was provided by UOC grants, and the authors’ contributions assured the financial support.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Lee, C.; Cockett, A.; Cussenot, O.; Griffiths, K.; Isaacs, W.; Schalken, J. Regulation of prostate growth. In Fifth International Consultation on Benign Prostatic Hyperplasia; Chatelain, C., Denis, L., Foo, J.K.T., Eds.; Health Publications: Plymouth, UK, 2001; pp. 81–106. [Google Scholar]
- Degterev, A.; Boyce, M.; Yuan, J.A. decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed]
- Di Silverio, F.; Gentile, V.; De Matteis, A.; Mariotti, G.; Giuseppe, V.; Luigi, P.A.; Sciarra, A. Distribution of inflammation, pre-malignant lesions, incidental carcinoma in histologically confirmed benign prostatic hyperplasia: A retrospective analysis. Eur. Urol. 2003, 23, 164–175. [Google Scholar] [CrossRef]
- Novara, G.; Galfano, A.; Berto, R.B.; Ficarra, V.; Navarrete, R.V.; Artibani, W. Inflammation, Apoptosis, and BPH: What is the Evidence? Eur. Urol. Suppl. 2006, 5, 401–409. [Google Scholar] [CrossRef]
- De Marzo, A.M.; DeWeese, T.L.; Platz, E.A.; Meeker, A.K.; Nakayama, M.; Epstein, J.I.; Isaacs, W.B.; Nelson, W.G. Pathological and molecular mechanisms of prostate carcinogenesis: Implications for diagnosis, detection, prevention, and treatment. J. Cell Biochem. 2004, 91, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.; Korsmeyer, S. Cell death: Critical control points. Cell Prolif. 2004, 116, 205–219. [Google Scholar]
- Thangapazham, R.L.; Gaddipati, J.P.; Rajeshkumar, N.V.; Sharma, A.; Singh, A.K.; Ives, J.A.; Maheshwari, R.K.; Jonas, W.B. Homeopathic Medicines Do Not Alter Growth and Gene Expression in Prostate and Breast Cancer Cells In Vitro. Integr. Cancer Ther. 2006, 5, 356–361. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M.; Hocevar, B.A. Oxidative stress and oxidative damage in carcinogenesis. Toxicol. Pathol. 2010, 38, 96–109. [Google Scholar] [CrossRef]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. 2001, 477, 7–21. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Bendas, G.; Borsig, L. Cancer Cell Adhesion and Metastasis: Selectins, Integrins, and the Inhibitory Potential of Heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes. Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Jin, S.; White, E. Role of autophagy in cancer: Management of metabolic stress. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef]
- Lorin, S.; Hamai, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes. Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Nakazawa, M.; Paller, C.; Kyprianou, N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Rai, N.K.; Tripathi, K.; Sharma, D.; Shukla, V.K. Apoptosis: A basic physiologic process in wound healing. Int. J. Low. Extrem. Wounds 2005, 4, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Koenig, U.; Eckhart, L.; Tschachler, E. Evidence that caspase-13 is not a human but a bovine gene. Biochem. Biophys. Res. Commun. 2001, 285, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Wang, S.; Kuida, K.; Yuan, J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002, 9, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, C.-A.; Aschie, M.; Matei, E.; Cozaru, G.C.; Deacu, M.; Mitroi, A.F.; Baltatescu, G.I.; Nicolau, A.-A.; Mazilu, L.; Tuta, L.A.; et al. Characterization of the Tumor Microenvironment and the Biological Processes with a Role in Prostatic Tumorigenesis. Biomedicines 2022, 10, 1672. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Anwar, S.; Ambros, R.A.; Jennings, T.A.; Ross, J.S.; Beza, A.; Mian, B.; Nazeer, T. Expression of cysteine protease protein 32 in prostatic adenocarcinoma correlates with tumor grade. Arch. Pathol. Lab. Med. 2004, 128, 649–652. [Google Scholar] [CrossRef]
- O’Donovan, N.; Crown, J.; Stunell, H.; Hill, A.D.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Caspase 3 in breast cancer. Clin. Cancer Res. 2003, 9, 738–742. [Google Scholar]
- O’Neill, A.J.; Boran, S.A.; O’Keane, C.; Coffey, R.N.; Hegarty, N.J.; Hegarty, P.; Gaffney, E.F.; Fitzpatrick, J.M.; Watson, R.W. Caspase 3 expression in benign prostatic hyperplasia and prostate carcinoma. Prostate 2001, 47, 183–188. [Google Scholar] [CrossRef]
- Sohn, J.H.; Kim, D.H.; Choi, N.G.; Park, Y.E.; Ro, J.Y. Caspase-3/CPP32 immunoreactivity and its correlation with frequency of apoptotic bodies in human prostatic carcinomas and benign nodular hyperplasias. Histopathology 2000, 37, 555–560. [Google Scholar]
- Winter, R.N.; Kramer, A.; Borkowski, A.; Kyprianou, N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001, 61, 1227–1232. [Google Scholar]
- Soung, Y.H.; Lee, J.W.; Kim, H.S.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Park, J.Y.; Cho, Y.G.; Kim, C.J.; Park, Y.G.; et al. Inactivating mutations of CASPASE-7 gene in human cancers. Oncogene 2003, 22, 8048–8052. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Soung, Y.H.; Lee, S.H.; Kim, K.M.; Lee, S.H. Absence of CASP7 and CASP8 mutation in gastrointestinal lymphomas. Eur. J. Haematol. 2007, 79, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.S.; Kim, S.Y.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Lee, S.H. Stomach cancer highly expresses both initiator and effector caspases; an immunohistochemical study. Apmis 2002, 110, 825–832. [Google Scholar] [CrossRef]
- Yoo, N.J.; Lee, J.W.; Kim, Y.J.; Soung, Y.H.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Lee, J.Y.; Lee, S.H. Loss of caspase-2, -6 and -7 expression in gastric cancers. Apmis 2004, 112, 330–335. [Google Scholar]
- Jäger, R.; Zwacka, M.R. The Enigmatic Roles of Caspases in Tumor Development. Cancers 2010, 2, 1952–1979. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Caraban, B.M.; Matei, E.; Cozaru, G.C.; Aşchie, M.; Deacu, M.; Enciu, M.; Bălţătescu, G.I.; Chisoi, A.; Dobrin, N.; Petcu, L.; et al. PD-L1, CD4+, and CD8+ Tumor-Infiltrating Lymphocytes (TILs) Expression Profiles in Melanoma Tumor Microenvironment Cells. J. Pers. Med. 2023, 13, 221. [Google Scholar] [CrossRef]
- Enderling, H.; Anderson, A.R.; Chaplain, M.A.; Beheshti, A.; Hlatky, L.; Hahnfeldt, P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res. 2009, 69, 8814–8821. [Google Scholar] [CrossRef]
- Gurova, K.V.; Gudkov, A.V. Paradoxical role of apoptosis in tumor progression. J. Cell Biochem. 2003, 88, 128–137. [Google Scholar] [CrossRef]
- Lauber, K.; Bohn, E.; Krober, S.M.; Xiao, Y.J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.L.; Yu, C.T.; Yin, S.C.; Tang, M.J.; Tien, A.C.; Wu, Y.M.; Huang, C.Y. Caspase 3, periodically expressed and activated at G2/M transition, is required for nocodazole-induced mitotic checkpoint. Apoptosis 2006, 11, 765–771. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamauchi, L.; Hunter, T.; Kikkawa, U.; Kamada, S. Possible involvement of caspase-7 in cell cycle progression at mitosis. Genes. Cells 2008, 13, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Radziszewska, A.; Schroer, S.A.; Choi, D.; Tajmir, P.; Radulovich, N.; Ho, J.C.; Wang, L.; Liadis, N.; Hakem, R.; Tsao, M.S.; et al. Absence of caspase-3 protects pancreatic {beta}-cells from c-Myc-induced apoptosis without leading to tumor formation. J. Biol. Chem. 2009, 284, 10947–10956. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qi, F.; Li, L.; Qin, Z.; Li, X.; Wang, X. Autophagy-related genes are potential diagnostic and prognostic biomarkers in prostate cancer. Transl. Androl. Urol. 2020, 9, 2616–2628. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Choi, J.J.; Reich, C.F.; Pisetsky, D.S. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 2004, 60, 159–166. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 2018, 24, 97–108.e104. [Google Scholar] [CrossRef]
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood J. Am. Soc. Hematol. 2016, 127, 139–148. [Google Scholar] [CrossRef]
- Haberzettl, P.; Hill, B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol. 2013, 1, 56–64. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Jiang, N. Autophagy provides a conceptual therapeutic framework for bone metastasis from prostate cancer. Cell Death Dis. 2021, 12, 909. [Google Scholar] [CrossRef] [PubMed]
- Zaffaroni, N.; Beretta, G.L. Nanoparticles for Ferroptosis Therapy in Cancer. Pharmaceutics 2021, 13, 1785. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy RIPs into cell death. Cell Cycle 2016, 15, 3014–3015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heidaryan, F.; Bamehr, H.; Babaabasi, B.; Emamvirdizadeh, A.; Mohammadzadeh, N.; Khalili, A. The Trend of ripk1/ripk3 and mlkl Mediated Necroptosis Pathway in Patients with Different Stages of Prostate Cancer as Promising Progression Biomarkers. Clin. Lab. 2020, 66. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Zucker, R.M.; Hunter, E.S.; Rogers, J.M. Confocal laser scanning microscopy of morphology and apoptosis in organogenesis-stage mouse embryos. Methods Mol. Biol. 2000, 135, 191–202. [Google Scholar]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Orlandini, S.Z. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicologic Pathology. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z.; Halicka, H.D.; Zhao, H. Analysis of cellular DNA content by flow and laser scanning cytometry. Adv. Exp. Med. Biol. 2010, 676, 137–147. [Google Scholar] [PubMed]
- Duesberg, P. Does aneuploidy or mutation start cancer? Science 2005, 307, 41. [Google Scholar] [CrossRef]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 57–162. [Google Scholar] [CrossRef]
- Zimmet, J.; Ravid, K. Polyploidy: Occurrence in nature, mechanisms, and significance for the megakaryocyte-platelet system. Exp. Hematol. 2000, 28, 3–16. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Traganos, F.; Zhao, H.; Halicka, H.D.; Li, J. Cytometry of DNA replication and RNA synthesis: Historical perspective and recent advances based on “click chemistry”. Cytom. Part. A 2011, 79, 328–337. [Google Scholar] [CrossRef]
- López-Otero, A.; Ruiz-Delgado, G.J.; Hernández-Arizpe, A.; Ruiz-Argüelles, A.; Ruiz-Argüelles, G.J. The flow-cytometric DNA content of the plasma cells of patients with multiple myeloma is a prognostic factor: A single institution experience. Hematology 2010, 15, 378–381. [Google Scholar] [CrossRef]
- Gerashchenko, B.I.; Huna, A.; Erenpreisa, J. Characterization of breast cancer DNA content profiles as a prognostic tool. Exp. Oncol. 2014, 36, 219–225. [Google Scholar]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Garcıa-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. Role of tumor necrosis factor-alpha and its receptors in human benign breast lesions and tumors (in situ and infiltrative). Cancer Sci. 2006, 97, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Royuela, M.; de Miguel, M.P.; Ruiz, A.; Fraile, B.; Arenas, M.I.; Romo, E.; Paniagua, R. Interferongamma and its functional receptors overexpression in benign prostatic hyperplasia and prostatic carcinoma: Parallelism with c-myc and p53 expression. Eur. Cytokine Netw. 2000, 11, 119–127. [Google Scholar]
- Royuela, M.; de Miguel, M.P.; Bethencourt, F.R.; Sánchez-Chapado, M.; Fraile, B.; Arenas, M.I.; Paniagua, R. Estrogen receptors alpha and beta in the normal, hyperplastic and carcinomatous human prostate. J. Endocrinol. 2001, 168, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Levesque, A.A.; Eastman, A. p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis 2007, 28, 13–20. [Google Scholar] [CrossRef]
- Gartel, A.L. Is p21 an oncogene? Mol. Cancer Ther. 2006, 5, 1385–1386. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Jain, A.K.; Raina, K.; Agarwal, R. Deletion of p21/Cdkn1a confers protective effect against prostate tumorigenesis in transgenic adenocarcinoma of the mouse prostate model. Cell Cycle 2013, 12, 1598–1604. [Google Scholar] [CrossRef]
- Slingerland, J.; Pagano, M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J. Cell Physiol. 2000, 183, 10–17. [Google Scholar] [CrossRef]
- Garrett-Engele, C.M.; Tasch, M.A.; Hwang, H.C.; Fero, M.L.; Perlmutter, R.M.; Clurman, B.E.; Roberts, J.M. A mechanism mis regulating p27 in tumors discovered in a functional genomic screen. PLoS Genet. 2007, 3, e219. [Google Scholar] [CrossRef] [PubMed]
- Torrealba, N.; Rodríguez-Berriguete, G.; Vera, R.; Fraile, B.; Olmedilla, G.; Martínez-Onsurbe, P.; Sánchez-Chapado, M.; Paniagua, R.; Royuela, M. Homeostasis: Apoptosis and cell cycle in normal and pathological prostate. Aging Male 2020, 23, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Svenson, K.B.; Longmate, W.M.; Gkirtzimanaki, K.; Sadej, R.; Wang, X.; Zhao, J.; Eliopoulos, A.G.; Berditchevski, F.; Dipersio, C.M. Suppression of integrin alpha 3 beta 1 in breast cancer cells reduces cyclooxygenase-2 gene expression and inhibits tumorigenesis, invasion, and crosstalk to endothelial cells. Cancer Res. 2010, 70, 6359–6367. [Google Scholar] [CrossRef] [PubMed]
- Pontes-Junior, J.; Reis, S.T.; Dall’Oglio, M.; Neves de Oliveira, L.C.; Cury, J.; Carvalho, P.A.; Ribeiro-Filho, L.A.; Moreira Leite, K.R.; Srougi, M. Evaluation of the expression of integrins and cell adhesion molecules through tissue microarray in lymph node metastases of prostate cancer. J. Carcinog. 2009, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Rolli, M.; Fransvea, E.; Pilch, J.; Saven, A.; Felding-Habermann, B. Activated integrin alpha v beta 3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha (V) beta (3) and alpha (V) beta (5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar]
- Ramirez, N.E.; Zhang, Z.; Madamanchi, A.; Boyd, K.L.; O’Rear, L.D.; Nashabi, A.; Li, Z.; Dupont, W.D.; Zijlstra, A.; Zutter, M.M. The alpha (2) beta (1) integrin is a metastasis suppressor in mouse models and human cancer. J. Clin. Investig. 2011, 121, 226–237. [Google Scholar] [CrossRef]
- Hall, C.L.; Dubyk, C.W.; Riesenberger, T.A.; Shein, D.; Keller, E.T.; van Golen, K.L. Type I collagen receptor (alpha 2 beta 1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia 2008, 10, 797–803. [Google Scholar] [CrossRef]
- Goel, H.; Li, J. Integrins in Prostate Cancer Progression. Endocr. Relat. Cancer 2008, 15, 657–664. [Google Scholar] [CrossRef]
- Juan-Rivera, M.C.; Martínez-Ferrer, M. Integrin Inhibitors in Prostate Cancer. Cancers 2018, 10, 44. [Google Scholar] [CrossRef]
- Ryu, S.; Park, K.M.; Lee, S.H. Gleditsia Sinensis Thorn Attenuates the Collagen-Based Migration of PC3 Prostate Cancer Cells through the Suppression of Integrin Expression. Int. J. Mol. Sci. 2016, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, R.L.; Ryan, K.M. Autophagy in tumour cell death. Semin. Cancer Biol. 2013, 23, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Abou-Kheir, W.; Hynes, P.G.; Martin, P.; Yin, J.J.; Liu, Y.N.; Seng, V.; Lake, R.; Spurrier, J.; Kelly, K. Self-renewing Pten-/- TP53-/- protospheres produce metastatic adenocarcinoma cell lines with multipotent progenitor activity. PLoS ONE 2011, 6, e26112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef]
- Maiti, K.; Oh, D.Y.; Moon, J.S.; Acharjee, S.; Li, J.H.; Bai, D.G.; Park, H.; Lee, K.; Lee, Y.C.; Jung, N.C.; et al. Differential effects of gonadotropin-releasing hormone (GnRH)-I and GnRH-II on prostate cancer cell signaling and death. J. Clin. Endocrinol. Metab. 2005, 90, 4287–4298. [Google Scholar] [CrossRef][Green Version]
- Kim, D.K.; Yang, J.S.; Maiti, K.; Hwang, J.I.; Kim, K.; Seen, D.; Ahn, Y.; Lee, C.; Kang, B.C.; Kwon, H.B.; et al. A gonadotropin releasing hormone-II antagonist induces autophagy of prostate cancer cells. Cancer Res. 2009, 69, 923–931. [Google Scholar] [CrossRef]
- Piacentini, M.; Evangelisti, C.; Mastroberardino, P.G.; Nardacci, R.; Kroemer, G. Does prothymosin-alpha act as molecular switch between apoptosis and autophagy? Cell Death Differ. 2003, 10, 937–939. [Google Scholar] [CrossRef]
- Zeiss, C.J. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar] [CrossRef]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Plati, J.; Bucur, O.; Khosravi, F.R. Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. 2011, 3, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar]
- Matei, E.; Aschie, M.; Mitroi, A.F.; Ghinea, M.M.; Gheorghe, E.; Petcu, L.; Dobrin, N.; Chisoi, A.; Manea, M. Biomarkers involved in evaluation of platelets function in South-Eastern Romanian patients with hematological malignancies subtypes. Medicine 2021, 100, e25944. [Google Scholar] [CrossRef]
- Popovici, V.; Musuc, A.M.; Matei, E.; Karampelas, O.; Ozon, E.A.; Cozaru, G.C.; Schröder, V.; Bucur, L.; Aricov, L.; Anastasescu, M.; et al. ROS-Induced DNA-Damage and Autophagy in Oral Squamous Cell Carcinoma by Usnea barbata Oil Extract—An In Vitro Study. Int. J. Mol. Sci. 2022, 23, 14836. [Google Scholar] [CrossRef]
- Popovici, V.; Matei, E.; Cozaru, G.C.; Bucur, L.; Gîrd, C.E.; Schröder, V.; Ozon, E.A.; Mitu, M.A.; Musuc, A.M.; Petrescu, S.; et al. Design, Characterization, and Anticancer and Antimicrobial Activities of Mucoadhesive Oral Patches Loaded with Usnea barbata (L.) F. H. Wigg Ethanol Extract F-UBE-HPMC. Antioxidants 2022, 11, 1801. [Google Scholar] [CrossRef]
- Dumitru, A.; Matei, E.; Cozaru, G.C.; Chisoi, A.; Alexandrescu, L.; Popescu, R.C.; Pundiche Butcaru, M.; Dumitru, E.; Rugină, S.; Tocia, C. Endotoxin Inflammatory Action on Cells by Dysregulated Immunological-Barrier-Linked ROS-Apoptosis Mechanisms in Gut–Liver Axis. Int. J. Mol. Sci. 2024, 25, 2472. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).