
Integrating Genomics and Transcriptomics to Identify Candidate Genes for Egg Production in Taihe Black-Bone Silky Fowls (Gallus gallus domesticus Brisson)


Abstract
1. Introduction
2. Results
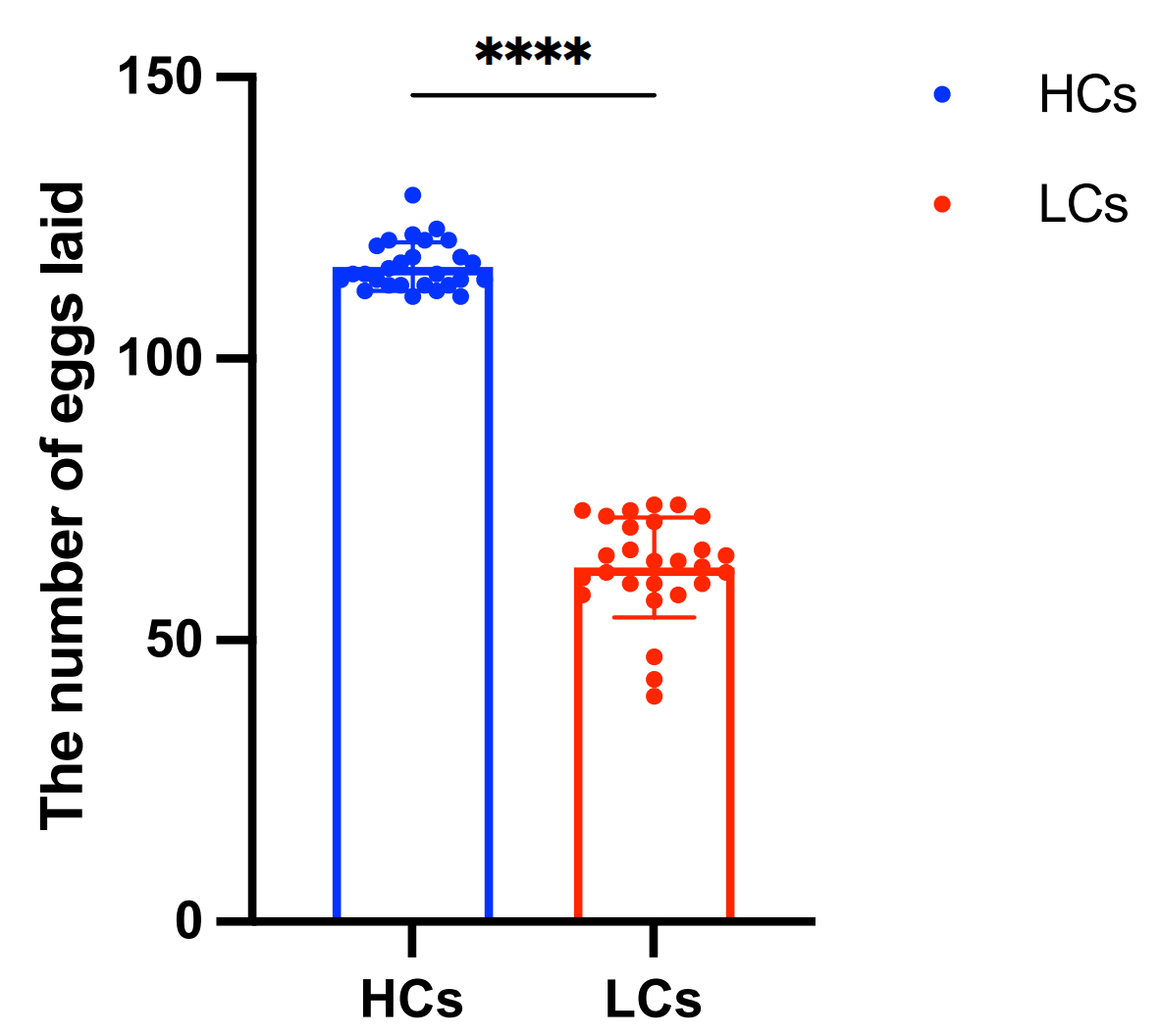
2.1. Phenotypic Statistics and Genomic Variation
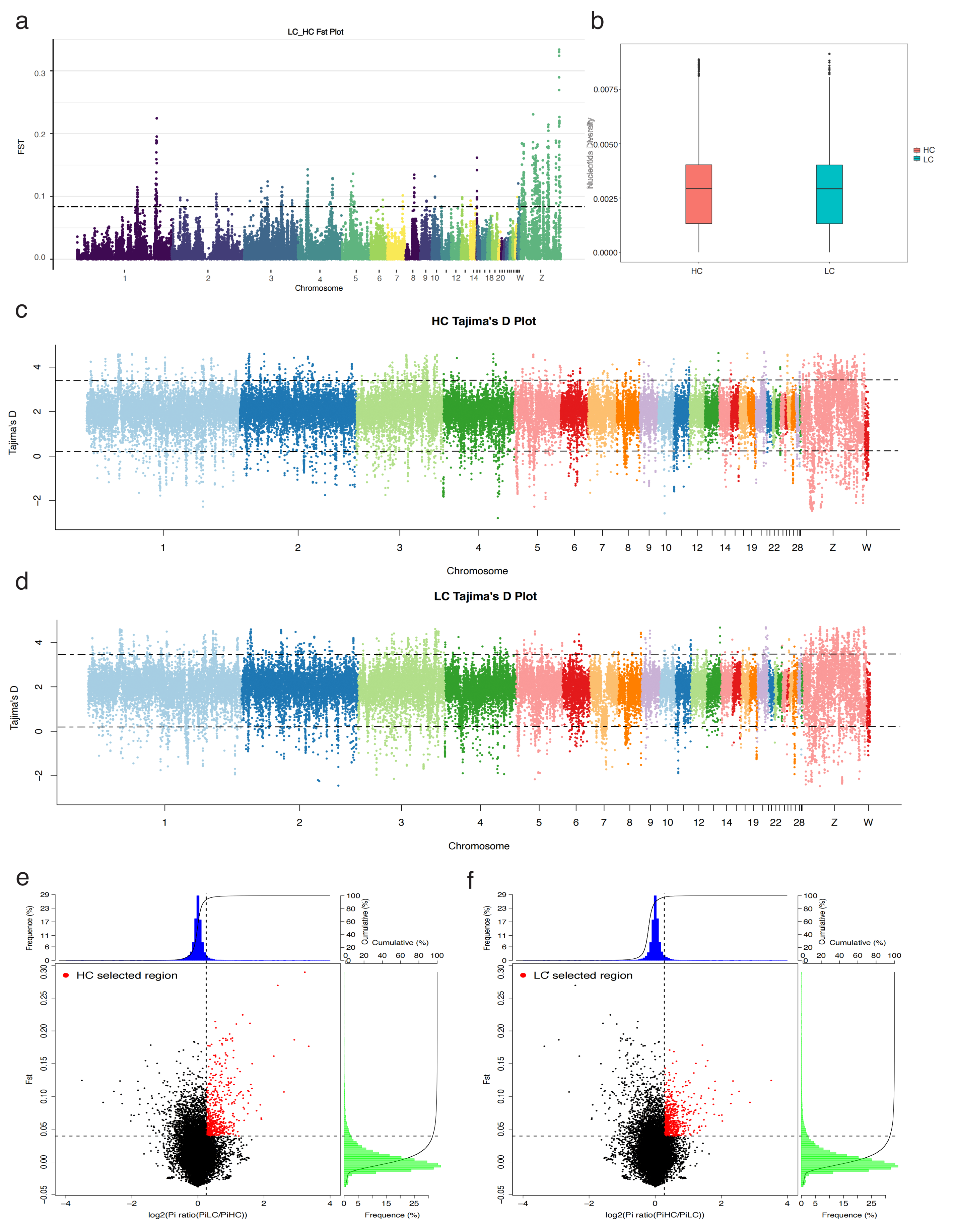
2.2. Single-Nucleotide Polymorphism (SNP) Detection
2.3. Genetics of the Population and Selection Signal Analysis (SSA)
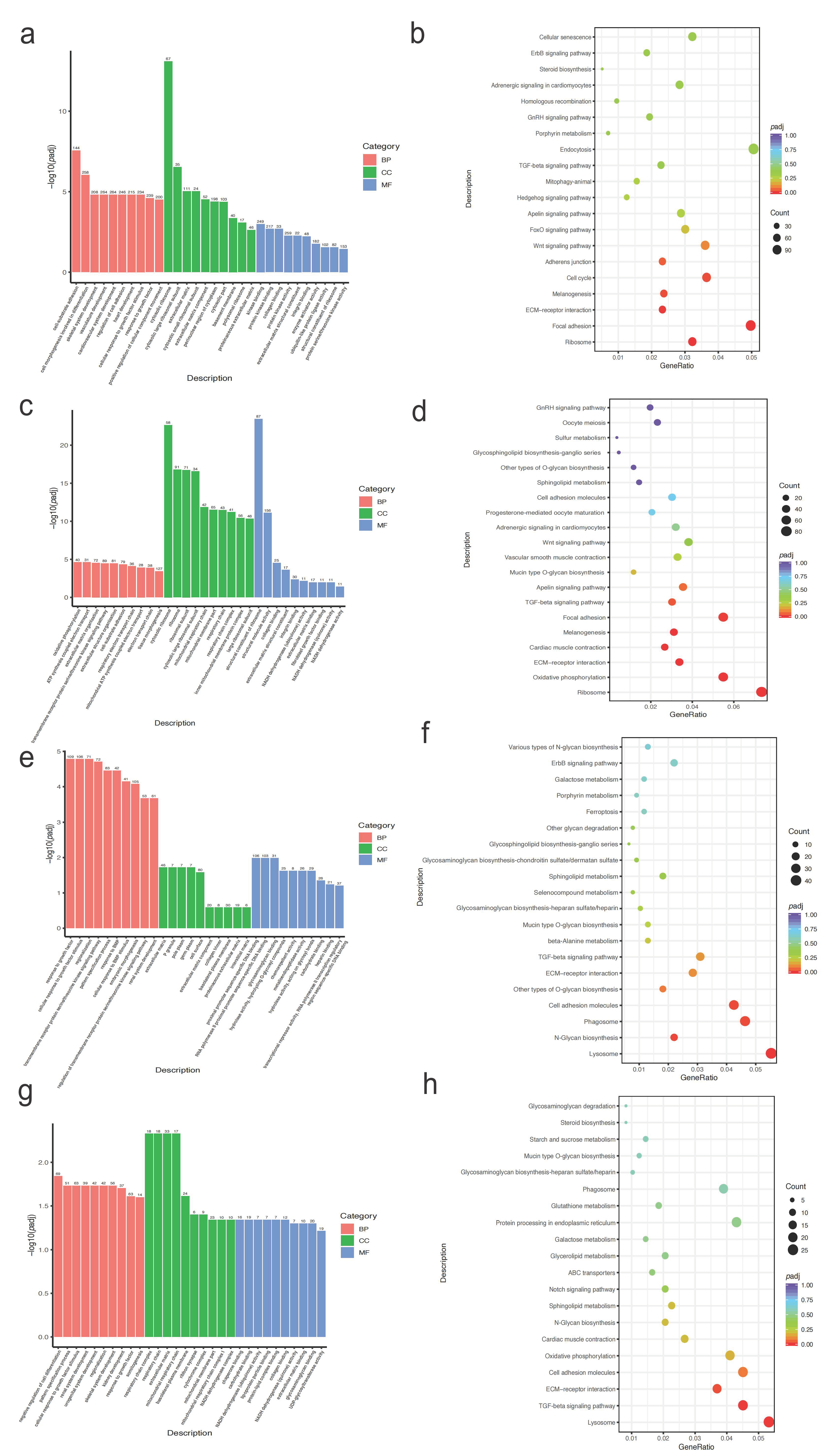
2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis on Genes in the Selected Regions
2.5. Transcriptome Sequencing and Alignment Analysis
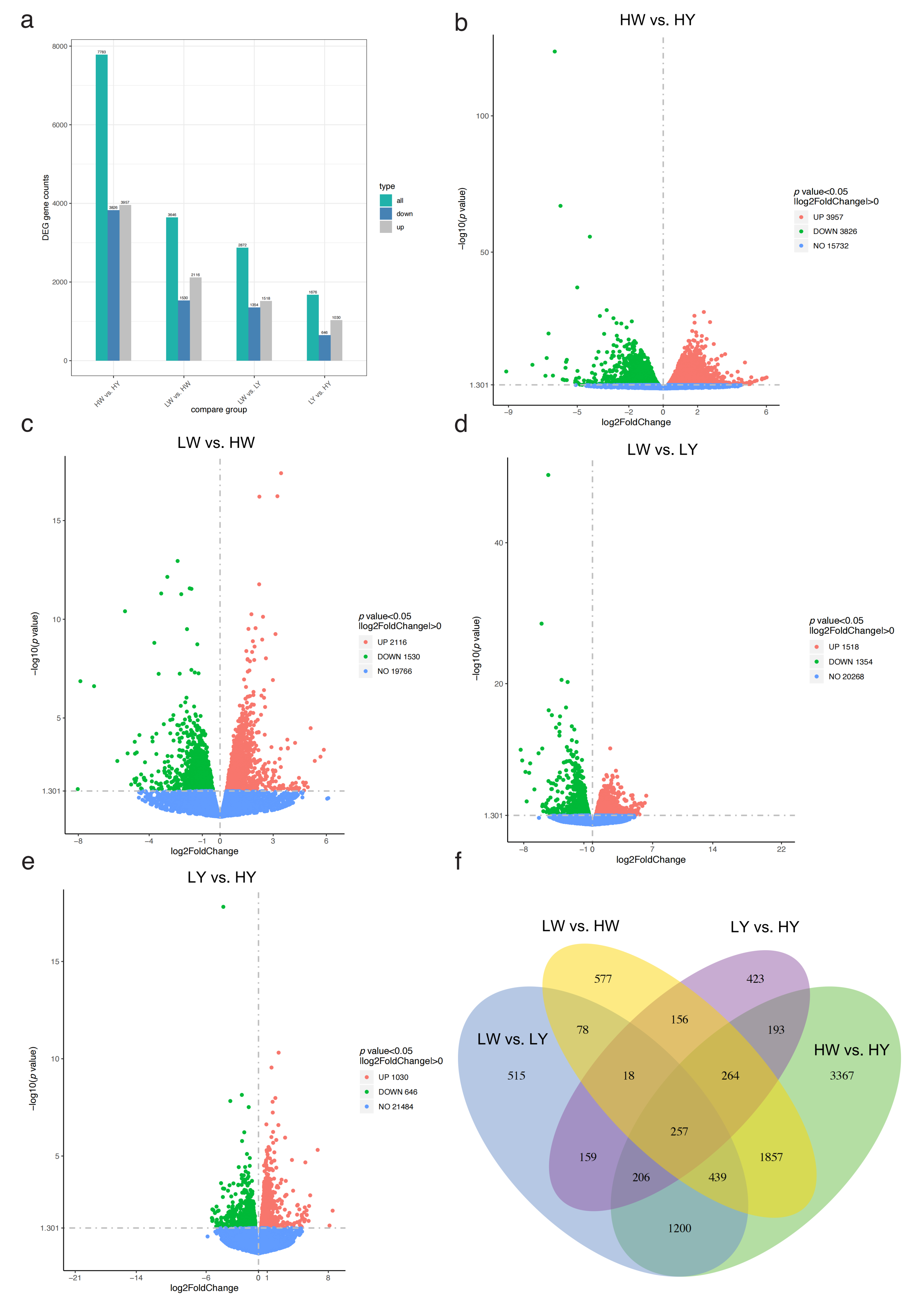
2.6. Analysis of Differentially Expressed Genes (DEGs)
2.7. GO and KEGG Analysis for DEGs
2.8. Joint Analysis of WGS and RNA-Seq to Explore the Candidate Genes of Egg Production Performance between HCs and LCs
2.9. Validation of RNA-Seq by Quantitative Real-Time PCR (qRT-PCR)
3. Discussion
4. Materials and Methods
4.1. Chicken and Samples
4.2. Whole-Genome Resequencing (WGS) and Quality Control
4.3. Detection and Annotation of Genomic Variants
4.4. Population Analysis and Detection of Selective Signatures
4.5. Transcriptome Data Comparison and Expression Analysis
4.6. Functional Enrichment Analysis of Selected Genes and Differentially Expressed Genes
4.7. Joint Analysis of WGS and RNA-Seq
4.8. Quantitative Real-Time PCR
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, W.; Yang, Y.; Wang, L.; Lv, X.; Li, J.; Cui, H.; Tang, C.; Zhao, Q.; Jia, Y.; Qin, Y.; et al. Comparative Characterization of Flavor Precursors and Volatiles of Taihe Black-Boned Silky Fowl and Hy-Line Brown Yolks Using Multiomics and GC-O-MS-Based Volatilomics. Food Res. Int. 2023, 172, 113168. [Google Scholar] [CrossRef]
- Mi, S.; Shang, K.; Jia, W.; Zhang, C.; Li, X.; Fan, Y.; Wang, H. Characterization and Discrimination of Taihe Black-Boned Silky Fowl (Gallus gallus domesticus Brisson) Muscles Using LC/MS-Based Lipidomics. Food Res. Int. 2018, 109, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, N.; Yan, Y.; Li, G.; Liu, A.; Wu, G.; Sun, C. Genome-Wide Association Analysis of Egg Production Performance in Chickens across the Whole Laying Period. BMC Genet. 2019, 20, 67. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Y.; Wu, Q.; Lin, R.; Chen, H.; Zhang, M.; Lin, J.; Xu, E.; Li, M.; Cai, Y.; et al. Whole-Genome Sequencing Identifies Potential Candidate Genes for Egg Production Traits in Laying Ducks (Anas platyrhynchos). Sci. Rep. 2023, 13, 1821. [Google Scholar] [CrossRef]
- Gautron, J.; Dombre, C.; Nau, F.; Feidt, C.; Guillier, L. Review: Production Factors Affecting the Quality of Chicken Table Eggs and Egg Products in Europe. Animal 2022, 16, 100425. [Google Scholar] [CrossRef]
- El-Sabrout, K.; Aggag, S.; Mishra, B. Advanced Practical Strategies to Enhance Table Egg Production. Scientifica 2022, 2022, 1393392. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.E.; Wilson, P.B. Progress and Opportunities through Use of Genomics in Animal Production. Trends Genet. 2022, 38, 1228–1252. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Molecular Signatures of Natural Selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef]
- Bello, S.F.; Lawal, R.A.; Adeola, A.C.; Nie, Q. The Study of Selection Signature and Its Applications on Identification of Candidate Genes Using Whole Genome Sequencing Data in Chicken—A Review. Poult. Sci. 2023, 102, 102657. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.; Liu, G.; Xu, Y.; Lv, F.; Yang, H.; Yang, Y.; Liu, C.; et al. Whole-Genome Resequencing of Wild and Domestic Sheep Identifies Genes Associated with Morphological and Agronomic Traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Choi, J.W.; Chung, W.H.; Lee, K.T.; Cho, E.S.; Lee, S.W.; Choi, B.H.; Lee, S.H.; Lim, W.; Lim, D.; Lee, Y.G.; et al. Whole-Genome Resequencing Analyses of Five Pig Breeds, Including Korean Wild and Native, and Three European Origin Breeds. DNA Res. 2015, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, J.; Xiao, C.; Sun, L.; Xiang, W.; Chen, N.; Lei, C.; Lei, H.; Long, Y.; Long, T.; et al. Whole-Genome Resequencing of Xiangxi Cattle Identifies Genomic Diversity and Selection Signatures. Front. Genet. 2022, 13, 816379. [Google Scholar] [CrossRef]
- Li, L.; Quan, J.; Gao, C.; Liu, H.; Yu, H.; Chen, H.; Xia, C.; Zhao, S. Whole-Genome Resequencing to Unveil Genetic Characteristics and Selection Signatures of Specific Pathogen-Free Ducks. Poult. Sci. 2023, 102, 102748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, G.; Mu, X.; Li, X.; Wang, J.; Zhao, M.; Zhang, G.; Ji, R.; Chen, C.; Gao, G.; et al. Genome-Wide Selective Signatures Mining the Candidate Genes for Egg Laying in Goose. BMC Genom. 2023, 24, 750. [Google Scholar] [CrossRef]
- Li, D.; Ning, C.; Zhang, J.; Wang, Y.; Tang, Q.; Kui, H.; Wang, T.; He, M.; Jin, L.; Li, J.; et al. Dynamic Transcriptome and Chromatin Architecture in Granulosa Cells during Chicken Folliculogenesis. Nat. Commun. 2022, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Q.; Liu, Z.; Guo, X.; Du, Y.; Yuan, Z.; Guo, M.; Kang, L.; Sun, Y.; Jiang, Y. Transcriptome Analysis on Single Small Yellow Follicles Reveals That Wnt4 Is Involved in Chicken Follicle Selection. Front. Endocrinol. 2017, 8, 317. [Google Scholar] [CrossRef]
- Zhu, G.; Fang, C.; Li, J.; Mo, C.; Wang, Y.; Li, J. Transcriptomic Diversification of Granulosa Cells during Follicular Development in Chicken. Sci. Rep. 2019, 9, 5462. [Google Scholar] [CrossRef]
- Hlokoe, V.R.; Tyasi, T.L.; Gunya, B. Chicken Ovarian Follicles Morphology and Growth Differentiation Factor 9 Gene Expression in Chicken Ovarian Follicles: Review. Heliyon 2022, 8, e08742. [Google Scholar] [CrossRef]
- Song, X.; Wang, D.; Zhou, Y.; Sun, Y.; Ao, X.; Hao, R.; Gao, M.; Xu, Y.; Li, P.; Jia, C.; et al. Yolk Precursor Synthesis and Deposition in Hierarchical Follicles and Effect on Egg Production Performance of Hens. Poult. Sci. 2023, 102, 102756. [Google Scholar] [CrossRef]
- Johnson, A.L. Ovarian Follicle Selection and Granulosa Cell Differentiation1. Poult. Sci. 2015, 94, 781–785. [Google Scholar] [CrossRef]
- Sun, X.; Chen, X.; Zhao, J.; Ma, C.; Yan, C.; Liswaniso, S.; Xu, R.; Qin, N. Transcriptome Comparative Analysis of Ovarian Follicles Reveals the Key Genes and Signaling Pathways Implicated in Hen Egg Production. BMC Genom. 2021, 22, 899. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; Woods, D.C. Dynamics of Avian Ovarian Follicle Development: Cellular Mechanisms of Granulosa Cell Differentiation. Gen. Comp. Endocrinol. 2009, 163, 12–17. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Yuan, J.; Wang, Y.; Sun, Y.; Ni, A.; Li, Y.; Ma, H.; Ma, T.; Chen, J. Integrated Transcriptomic Analysis on Chicken Ovary Reveals CYP21A1 Affects Follicle Granulosa Cell Development and Steroid Hormone Synthesis. Poult. Sci. 2024, 103, 103589. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Guo, R.; Zeng, T.; Sun, H.; Tian, Y.; Han, X.; Cao, Y.; Xu, L.; Duan, M.; Lu, L.; et al. Analysis of Transcriptomic Differences in the Ovaries of High- and Low-Laying Ducks. Genes 2024, 15, 181. [Google Scholar] [CrossRef]
- He, Z.; Ouyang, Q.; Chen, Q.; Song, Y.; Hu, J.; Hu, S.; He, H.; Li, L.; Liu, H.; Wang, J. Molecular Mechanisms of Hypothalamic-Pituitary-Ovarian/Thyroid Axis Regulating Age at First Egg in Geese. Poult. Sci. 2024, 103, 103478. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, C.; Gao, Y.; Cui, X.; Zhang, Y.; Gu, L.; He, Y.; Li, J.; Gao, S.; Gao, R.; et al. Transcriptome Dynamics and Cell Dialogs Between Oocytes and Granulosa Cells in Mouse Follicle Development. Genom. Proteom. Bioinform. 2024, 22, qzad001. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Ren, P.; Zhang, S.; Liu, Y.; Zhu, M. Integrating Genomics and Transcriptomics to Identify Candidate Genes for High Egg Production in Wulong Geese (Anser cygnoides orientalis). BMC Genom. 2023, 24, 481. [Google Scholar] [CrossRef]
- Cai, D.; Wang, Z.; Zhou, Z.; Lin, D.; Ju, X.; Nie, Q. Integration of Transcriptome Sequencing and Whole Genome Resequencing Reveal Candidate Genes in Egg Production of Upright and Pendulous-Comb Chickens. Poult. Sci. 2023, 102, 102504. [Google Scholar] [CrossRef]
- Fulton, J.E. Genomic Selection for Poultry Breeding. Anim. Front. 2012, 2, 30–36. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the Lysosome: A Control Centre for Cellular Clearance and Energy Metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- MacDonald, J.A.; Takai, Y.; Ishihara, O.; Seki, H.; Woods, D.C.; Tilly, J.L. Extracellular Matrix Signaling Activates Differentiation of Adult Ovary-Derived Oogonial Stem Cells in a Species-Specific Manner. Fertil. Steril. 2019, 111, 794–805. [Google Scholar] [CrossRef]
- Gifford, J.A.H. The Role of WNT Signaling in Adult Ovarian Folliculogenesis. Reproduction 2015, 150, R137–R148. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef] [PubMed]
- antamarina-Fojo, S.; Dugi, K.A. Structure, Function and Role of Lipoprotein Lipase in Lipoprotein Metabolism. Curr. Opin. Lipidol. 1994, 5, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Ning, Z.; Deng, X.; Du, X.; Amevor, F.K.; Liu, L.; Kang, X.; Tian, Y.; Wang, Y.; Li, D.; et al. Integrated Proteomic and Metabolomic Analyses of Chicken Ovary Revealed the Crucial Role of Lipoprotein Lipase on Lipid Metabolism and Steroidogenesis During Sexual Maturity. Front. Physiol. 2022, 13, 885030. [Google Scholar] [CrossRef]
- Divers, S.L.; McQuillan, H.J.; Matsubara, H.; Todo, T.; Lokman, P.M. Effects of Reproductive Stage and 11-Ketotestosterone on LPL mRNA Levels in the Ovary of the Shortfinned Eel. J. Lipid Res. 2010, 51, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- van Eck, L.M.; Enting, H.; Carvalhido, I.J.; Chen, H.; Kwakkel, R.P. Lipid Metabolism and Body Composition in Long-Term Producing Hens. World’s Poult. Sci. J. 2023, 79, 243–264. [Google Scholar] [CrossRef]
- Whitlock, J.H.; Wilk, E.J.; Howton, T.C.; Clark, A.D.; Lasseigne, B.N. The Landscape of SETBP1 Gene Expression and Transcription Factor Activity across Human Tissues. PLoS ONE 2024, 19, e0296328. [Google Scholar] [CrossRef]
- Qiao, H.; Zhang, Q.; Wang, J.; Jiang, J.; Huyan, L.; Yan, J.; Li, C.; Wang, H. TRIM29 Regulates the SETBP1/SET/PP2A Axis via Transcription Factor VEZF1 to Promote Progression of Ovarian Cancer. Cancer Lett. 2022, 529, 85–99. [Google Scholar] [CrossRef]
- Piazza, R.; Magistroni, V.; Redaelli, S.; Mauri, M.; Massimino, L.; Sessa, A.; Peronaci, M.; Lalowski, M.; Soliymani, R.; Mezzatesta, C.; et al. SETBP1 Induces Transcription of a Network of Development Genes by Acting as an Epigenetic Hub. Nat. Commun. 2018, 9, 2192. [Google Scholar] [CrossRef]
- Milisits, G.; Szentirmai, E.; Donkó, T.; Budai, Z.; Ujvári, J.; Áprily, S.; Bajzik, G.; Sütő, Z. Effect of Starting Body Fat Content and Genotype of Laying Hens on the Changes in Their Live Weight, Body Fat Content, Egg Production and Egg Composition during the First Egg-Laying Period. Br. Poult. Sci. 2015, 56, 666–672. [Google Scholar] [CrossRef]
- Wei, W.; Qin, F.; Gao, J.; Chang, J.; Pan, X.; Jiang, X.; Che, L.; Zhuo, Y.; Wu, D.; Xu, S. The Effect of Maternal Consumption of High-Fat Diet on Ovarian Development in Offspring. Anim. Reprod. Sci. 2023, 255, 107294. [Google Scholar] [CrossRef]
- Gonnella, F.; Konstantinidou, F.; Di Berardino, C.; Capacchietti, G.; Peserico, A.; Russo, V.; Barboni, B.; Stuppia, L.; Gatta, V. A Systematic Review of the Effects of High-Fat Diet Exposure on Oocyte and Follicular Quality: A Molecular Point of View. Int. J. Mol. Sci. 2022, 23, 8890. [Google Scholar] [CrossRef]
- Dupont, J.; Reverchon, M.; Cloix, L.; Froment, P.; Ramé, C. Involvement of Adipokines, AMPK, PI3K and the PPAR Signaling Pathways in Ovarian Follicle Development and Cancer. Int. J. Dev. Biol. 2013, 56, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ File Format for Sequences with Quality Scores, and the Solexa/Illumina FASTQ Variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ma, Y.; Cao, X.; Li, H.; Han, W.; Qu, L.; Lamont, S.J. PTEN Regulated by Gga-miR-20a-5p Is Involved in Chicken Macrophages Inflammatory Response to APEC Infection via Autophagy. Poult. Sci. 2024, 103, 104170. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Accession Number | Forward Primer (5->3) | Reverse Primer (5->3) | Product Length (bp) | Tm (°C) |
|---|---|---|---|---|---|
| β-actin [53] | NM_205518.2 | CAGCCAGCCATGGATGATGA | ACCAACCATCACACCCTGAT | 147 | 60 |
| LPL | NM_205282.2 | ACTGAAACTTTTTCGCCGCTG | TTCATCTCAGCTTCGGGATCG | 127 | 60 |
| SETBP1 | XM_046937440.1 | TGGCGAGGGATTGAAACCG | GAGATCAGGTCTGCCACCAT | 147 | 60 |
| BRDT | XM_040705075.1 | AGGCTGTTCCAGAGGTATCCA | TTGTGACAGTACATACTCTGCTCT | 163 | 60 |
| RNF152 | NM_001305034.1 | ATGTCGTGCATTTCCAAGCG | CGGCCAAAGTTGCAGTGAAT | 146 | 60 |
| SCD | NM_204890.2 | CGGATGCAGACCCTCACAAT | GGGCTTGTAGTATCTCCGCTG | 164 | 60 |
| WIF1 | NM_001199607.3 | TGTCCTTGCGCTCTTTGGAT | AACCCAACCTGAACCACTGA | 103 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.; Huang, X.; Xu, C.; Huang, Y.; Li, S.; Yin, Z. Integrating Genomics and Transcriptomics to Identify Candidate Genes for Egg Production in Taihe Black-Bone Silky Fowls (Gallus gallus domesticus Brisson). Int. J. Mol. Sci. 2024, 25, 9373. https://doi.org/10.3390/ijms25179373
Tan Y, Huang X, Xu C, Huang Y, Li S, Yin Z. Integrating Genomics and Transcriptomics to Identify Candidate Genes for Egg Production in Taihe Black-Bone Silky Fowls (Gallus gallus domesticus Brisson). International Journal of Molecular Sciences. 2024; 25(17):9373. https://doi.org/10.3390/ijms25179373
Chicago/Turabian StyleTan, Yuting, Xuan Huang, Chunhui Xu, Yunyan Huang, Shibao Li, and Zhaozheng Yin. 2024. "Integrating Genomics and Transcriptomics to Identify Candidate Genes for Egg Production in Taihe Black-Bone Silky Fowls (Gallus gallus domesticus Brisson)" International Journal of Molecular Sciences 25, no. 17: 9373. https://doi.org/10.3390/ijms25179373
APA StyleTan, Y., Huang, X., Xu, C., Huang, Y., Li, S., & Yin, Z. (2024). Integrating Genomics and Transcriptomics to Identify Candidate Genes for Egg Production in Taihe Black-Bone Silky Fowls (Gallus gallus domesticus Brisson). International Journal of Molecular Sciences, 25(17), 9373. https://doi.org/10.3390/ijms25179373