
Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia
 , and
, and 
Abstract
:1. Introduction
2. LSCs and Relapse
3. Current Treatments
4. Genetic Mutations in AML
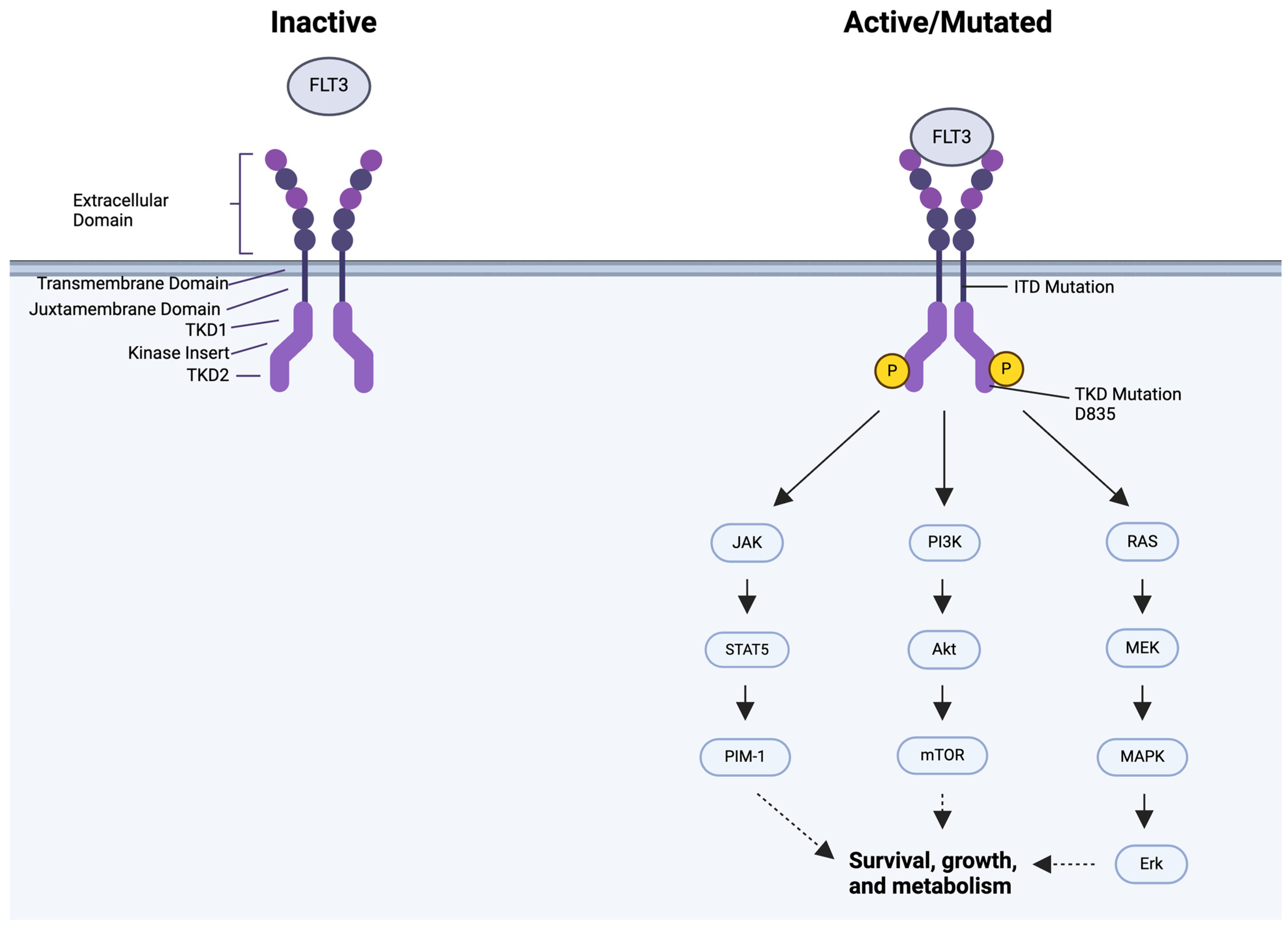
5. FLT3 Mutation
6. Targeting FLT3
6.1. First-Generation FLT3 Inhibitors
6.2. Midostaurin
6.3. Sorafenib
6.4. Second-Generation FLT3 Inhibitors
6.5. Quizartinib
6.6. Gilteritinib
7. TKI Resistance and Disease Relapse
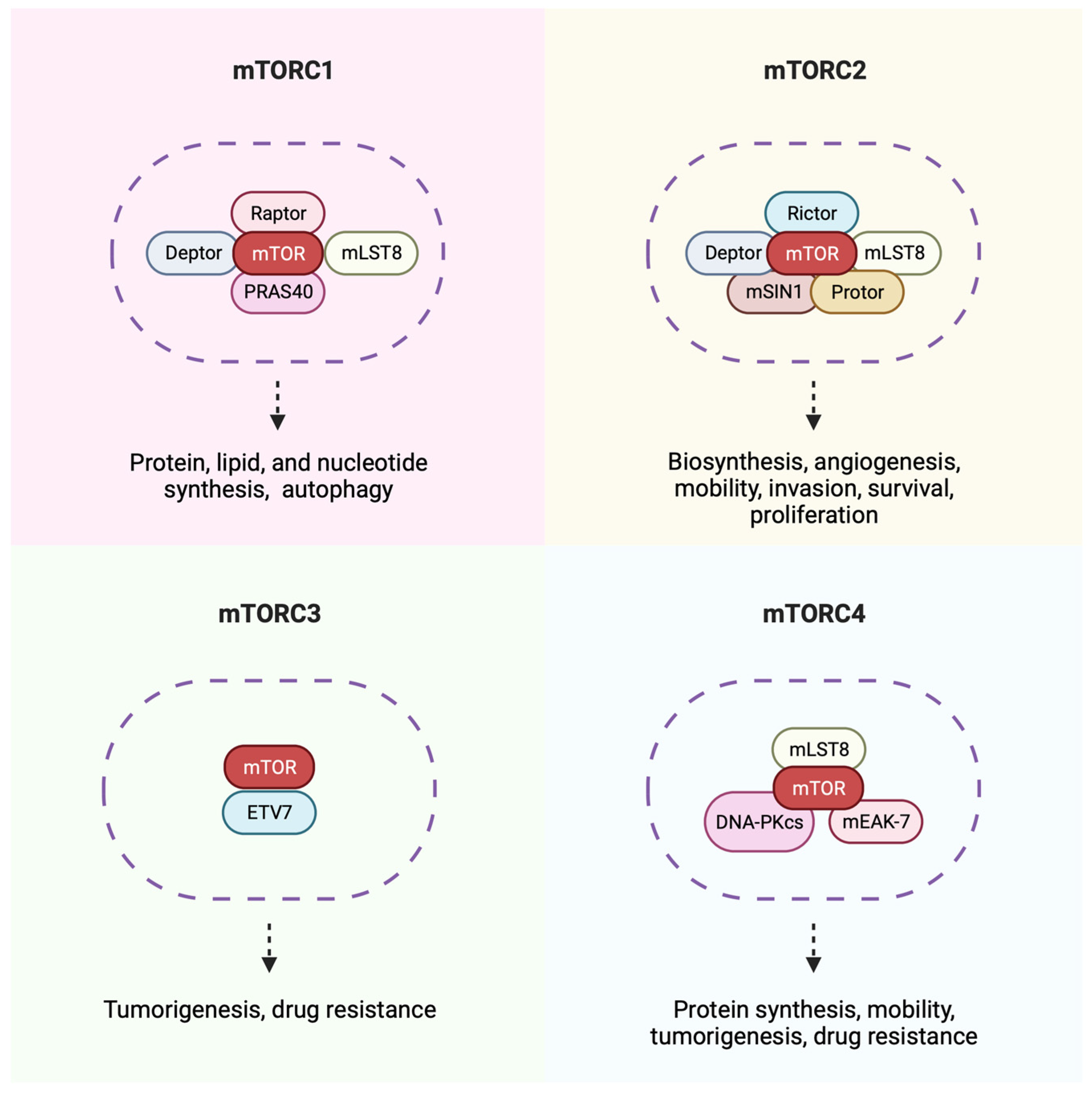
8. The mTOR Pathway
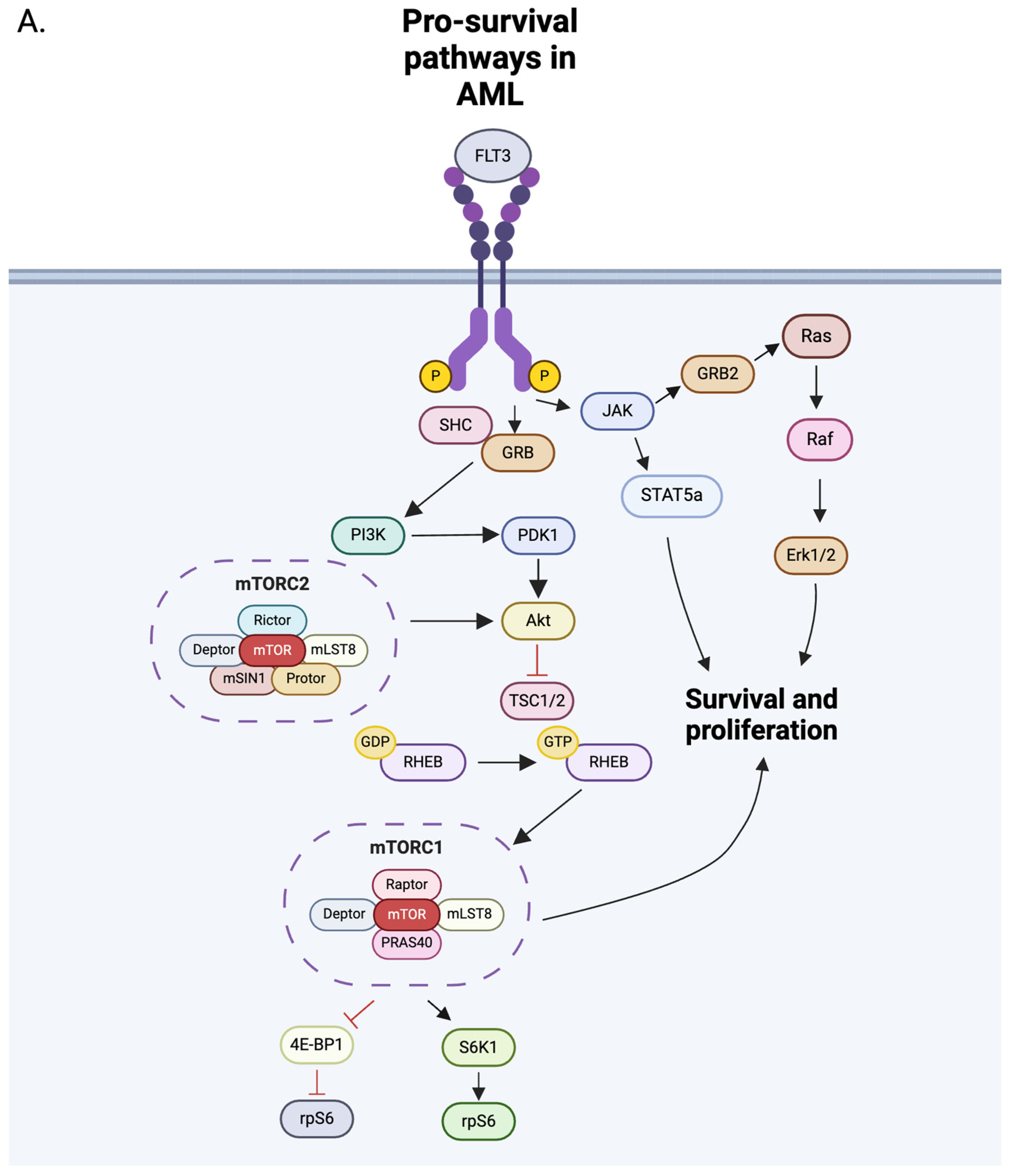
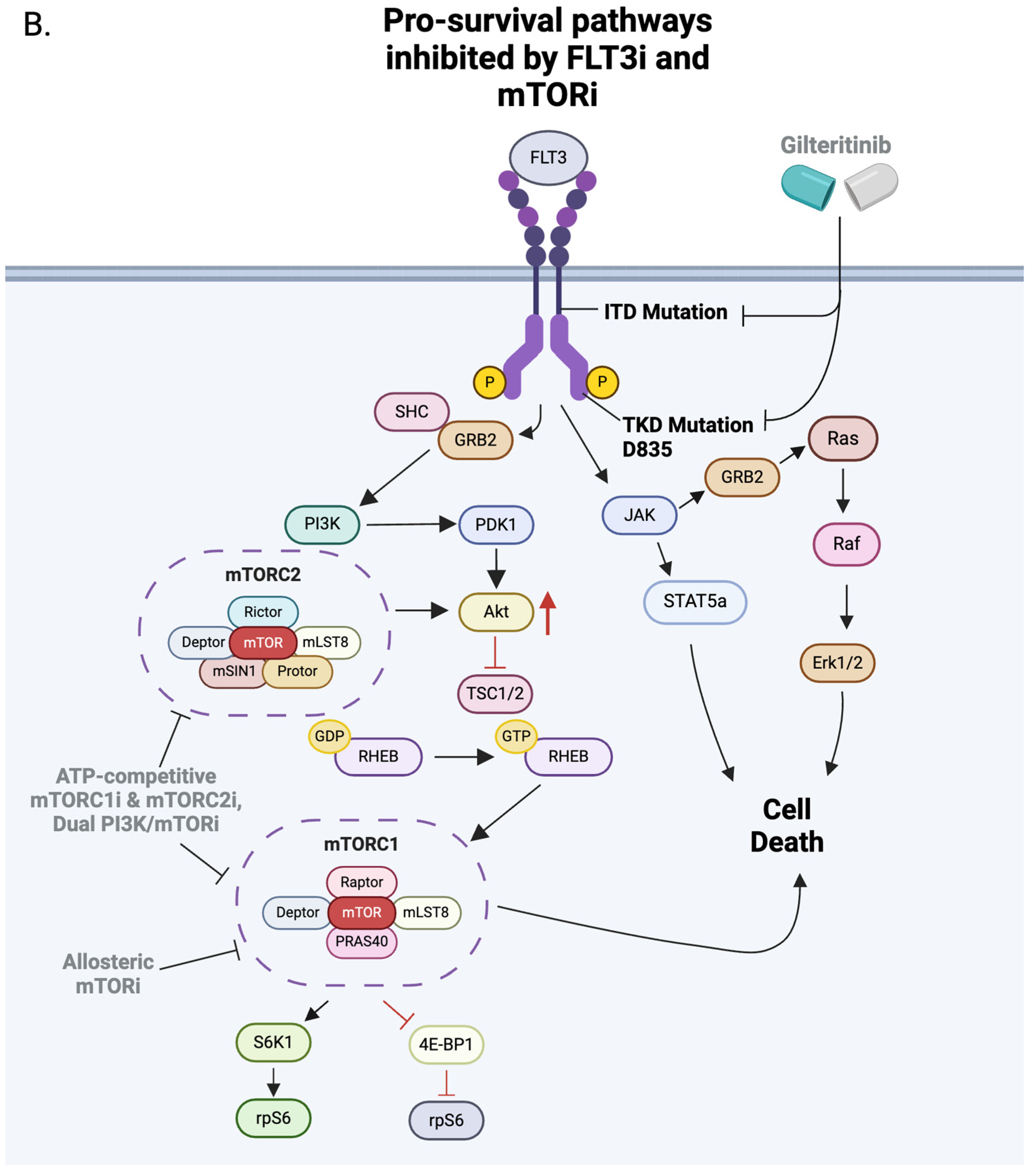
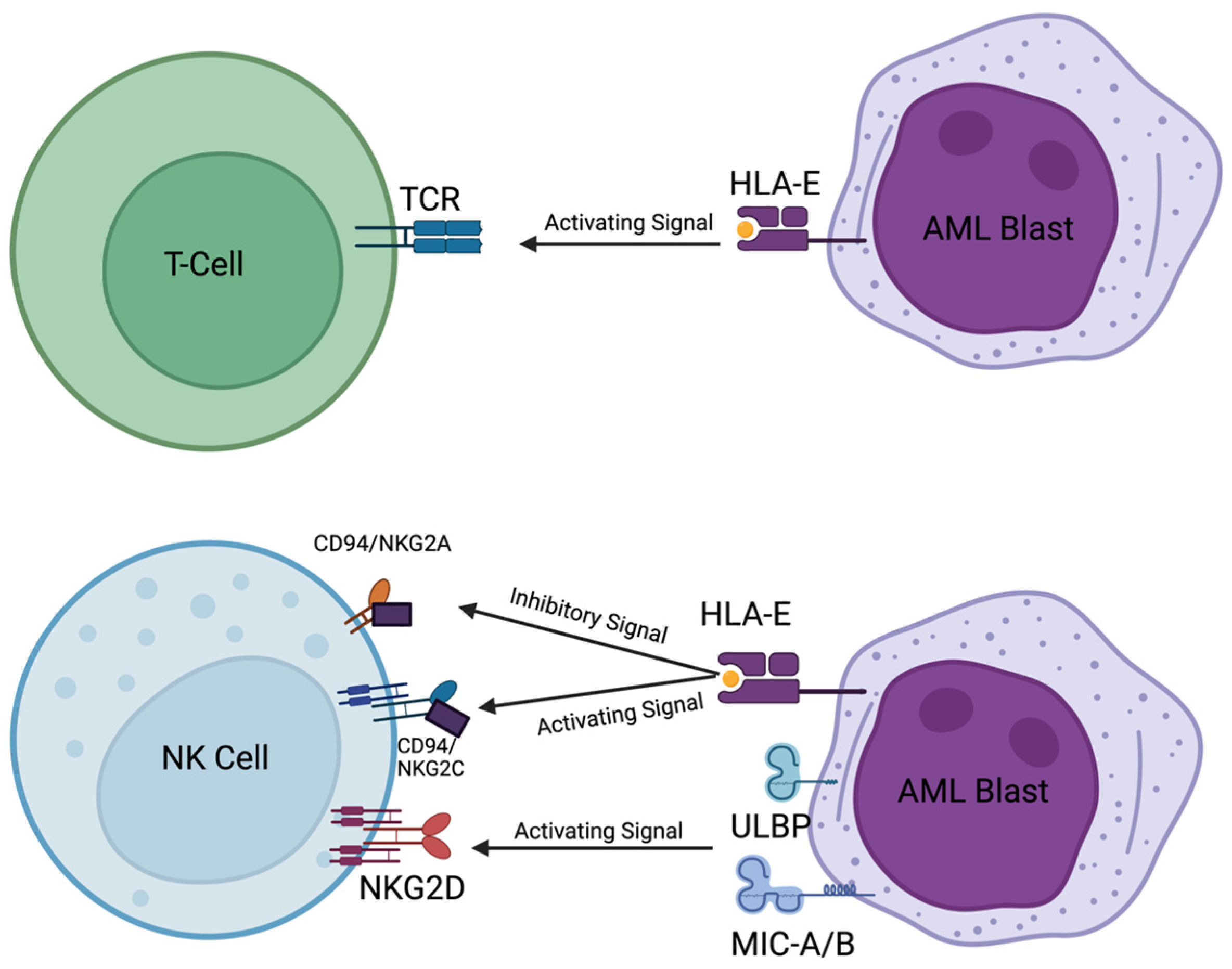
9. NK Cells
10. Engaging Immune Cells with IL-15
11. Engaging Immune Cells by Blocking TGF-β
12. Combination Treatments
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A review. RIMedJ 2020, 103, 38–40. [Google Scholar]
- Grimwade, D.; Ivey, A.; Huntly, B.J.P. Review Series Advances in Acute Myeloid Leukemia Molecular Landscape of Acute Myeloid Leukemia in Younger Adults and Its Clinical Relevance. Blood J. Am. Soc. Hematol. 2016, 127, 29–41. [Google Scholar]
- Turkalj, S.; Radtke, F.A.; Vyas, P. An Overview of Targeted Therapies in Acute Myeloid Leukemia. Hemasphere 2023, 7, E914. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1,612 Patients Entered into the MRC AML 10 Trial. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Ruglioni, M.; Crucitta, S.; Luculli, G.I.; Tancredi, G.; Del Giudice, M.L.; Mechelli, S.; Galimberti, S.; Danesi, R.; Del Re, M. Understanding Mechanisms of Resistance to FLT3 Inhibitors in Adult FLT3-Mutated Acute Myeloid Leukemia to Guide Treatment Strategy. Crit. Rev. Oncol. Hematol. 2024, 201, 104424. [Google Scholar] [CrossRef]
- Levis, M.; Perl, A.E. Gilteritinib: Potent Targeting of FLT3 Mutations in AML. Blood Adv. 2020, 4, 1178–1191. [Google Scholar] [CrossRef]
- Tecik, M.; Adan, A. Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches. OncoTargets Ther. 2022, 15, 1449–1478. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.-P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b Study of 2 Dosing Regimens of Quizartinib Monotherapy in FLT3-ITD–Mutated, Relapsed or Refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of Constitutive Activation of FLT3 with Internal Tandem Duplication in the Juxtamembrane Domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting MTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Sindaco, P.; Pandey, H.; Isabelle, C.; Chakravarti, N.; Brammer, J.E.; Porcu, P.; Mishra, A. The Role of Interleukin-15 in the Development and Treatment of Hematological Malignancies. Front. Immunol. 2023, 14, 1141208. [Google Scholar] [CrossRef]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs Join CARs on the Cancer Immunotherapy Highway. Hum. Vaccin. Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting Natural Killer Cells to Acute Myeloid Leukemia in Vitro with a CD16×33 Bispecific Killer Cell Engager and ADAM17 Inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Whiteley, A.E.; Price, T.T.; Cantelli, G.; Sipkins, D.A. Leukaemia: A Model Metastatic Disease. Nat. Rev. Cancer 2021, 21, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer Stem Cells Revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Woolthuis, C.M.; Stranahan, A.W.; Park, C.Y.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Jordan, C.T. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Thomas, D.; Majeti, R. Review Series Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood J. Am. Soc. Hematol. 2017, 129, 1577–1585. [Google Scholar]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A Trispecific Killer Engager Molecule against CLEC12A Effectively Induces NK-Cell Mediated Killing of AML Cells. Leukemia 2021, 35, 1586–1596. [Google Scholar] [CrossRef]
- Viadana, E.; Bross, I.D.J.; Pickren, J.W. An Autopsy of the Metastatic Patterns of Human Leukemias. Oncology 1978, 35, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Stelmach, P.; Trumpp, A. Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Haematologica 2023, 108, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.Y.; Jordan, C.T. Leukemia Stem Cells and Microenvironment: Biology and Therapeutic Targeting. J. Clin. Oncol. 2011, 29, 591–599. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Trumpp, A.; Haas, S. Cancer Stem Cells: The Adventurous Journey from Hematopoietic to Leukemic Stem Cells. Cell 2022, 185, 1266–1270. [Google Scholar] [CrossRef]
- Othus, M.; Sekeres, M.A.; Nand, S.; Garcia-Manero, G.; Appelbaum, F.R.; Erba, H.P.; Estey, E. Relative Survival Following Response to 7 + 3 versus Azacytidine Is Similar in Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndromes: An Analysis of Four SWOG Studies. Leukemia 2019, 33, 371–378. [Google Scholar] [CrossRef]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline Dose Intensification in Acute Myeloid Leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef]
- Imahashi, N.; Suzuki, R.; Fukuda, T.; Kakihana, K.; Kanamori, H.; Eto, T.; Mori, T.; Kobayashi, N.; Iwato, K.; Sakura, T.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for Intermediate Cytogenetic Risk AML in First CR. Bone Marrow Transpl. 2013, 48, 56–62. [Google Scholar] [CrossRef]
- Ustun, C.; Giannotti, F.; Zhang, M.J.; Wang, H.L.; Brunstein, C.; Labopin, M.; Rocha, V.; De Lima, M.; Baron, F.; Sandmaier, B.M.; et al. Outcomes of UCB Transplantation Are Comparable in FLT3+ AML: Results of CIBMTR, EUROCORD and EBMT Collaborative Analysis. Leukemia 2017, 31, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, S.; Yamaguchi, T.; Uchida, N.; Miyawaki, S.; Usuki, K.; Watanabe, M.; Yamashita, T.; Kanamori, H.; Tomiyama, J.; Nawa, Y.; et al. Comparison of Allogeneic Hematopoietic Cell Transplantation and Chemotherapy in Elderly Patients with Non-M3 Acute Myelogenous Leukemia in First Complete Remission. Biol. Blood Marrow Transplant. 2011, 17, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Lazarus, H.M.; Weisdorf, D. To Transplant or Not: A Dilemma for Treatment of Elderly AML Patients in the Twenty-First Century. Bone Marrow Transpl. 2013, 48, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Jäger, P.; Tischer, J.; Fraccaroli, A.; Bug, G.; Hausmann, A.; Baermann, B.-N.; Tressin, P.; Hoelscher, A.; Kasprzak, A.; et al. Smart Conditioning with Venetoclax Enhanced Sequential FLAMSA + RIC in Patients with High-Risk Myeloid Malignancies. Cancers 2023, 16, 532. [Google Scholar] [CrossRef]
- Scott, B.L.; Pasquini, M.C.; Fei, M.; Fraser, R.; Wu, J.; Devine, S.M.; Porter, D.L.; Maziarz, R.T.; Warlick, E.; Fernandez, H.F.; et al. Myeloablative versus Reduced-Intensity Conditioning for Hematopoietic Cell Transplantation in Acute Myelogenous Leukemia and Myelodysplastic Syndromes—Long-Term Follow-Up of the BMT CTN 0901 Clinical Trial. Transpl. Cell Ther. 2021, 27, 483.e1–483.e6. [Google Scholar] [CrossRef]
- Bornhäuser, M.; Kienast, J.; Trenschel, R.; Burchert, A.; Hegenbart, U.; Stadler, M.; Baurmann, H.; Schäfer-Eckart, K.; Holler, E.; Kröger, N.; et al. Reduced-Intensity Conditioning versus Standard Conditioning before Allogeneic Haemopoietic Cell Transplantation in Patients with Acute Myeloid Leukaemia in First Complete Remission: A Prospective, Open-Label Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 1035–1044. [Google Scholar] [CrossRef]
- Kröger, N.; Iacobelli, S.; Franke, G.-N.; Platzbecker, U.; Uddin, R.; Hübel, K.; Scheid, C.; Weber, T.; Robin, M.; Stelljes, M.; et al. Dose-Reduced Versus Standard Conditioning Followed by Allogeneic Stem-Cell Transplantation for Patients with Myelodysplastic Syndrome: A Prospective Randomized Phase III Study of the EBMT (RICMAC Trial). J. Clin. Oncol. 2017, 35, 2157–2164. [Google Scholar] [CrossRef]
- Scott, B.L. Long-Term Follow up of BMT CTN 0901, a Randomized Phase III Trial Comparing Myeloablative (MAC) to Reduced Intensity Conditioning (RIC) Prior to Hematopoietic Cell Transplantation (HCT) for Acute Myeloid Leukemia (AML) or Myelodysplasia (MDS) (MAvRIC Trial). Biol. Blood Marrow Transplant. 2020, 26, S11. [Google Scholar] [CrossRef]
- Leotta, S.; Condorelli, A.; Sciortino, R.; Milone, G.A.; Bellofiore, C.; Garibaldi, B.; Schininà, G.; Spadaro, A.; Cupri, A.; Milone, G. Prevention and Treatment of Acute Myeloid Leukemia Relapse after Hematopoietic Stem Cell Transplantation: The State of the Art and Future Perspectives. J. Clin. Med. 2022, 11, 253. [Google Scholar] [CrossRef]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Capanni, M.; Carotti, A.; Aloisi, T.; Aversa, F.; Martelli, M.F.; Velardi, A. NK Cell Alloreactivity and Allogeneic Hematopoietic Stem Cell Transplantation. Blood Cells Mol. Dis. 2008, 40, 84–90. [Google Scholar] [CrossRef]
- Makkar, H.; Kumar Majhi, R.; Goel, H.; Kumar Gupta, A.; Chopra, A.; Tanwar, P.; Seth, R. Acute Myeloid Leukemia: Novel Mutations and Their Clinical Implications. Am. J. Blood Res. 2023, 13, 12–27. [Google Scholar]
- Dinardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and Therapeutic Implications. Hematol. Am. Soc. Hematol. Educ. Program 2016, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, Y.; Zhang, D.; Wan, D.; Jiang, Z. Clinical Implications of Recurrent Gene Mutations in Acute Myeloid Leukemia. Exp. Hematol. Oncol. 2020, 9, 4. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-AKT-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed]
- Gary Gilliland, D.; Griffin, J.D. The Roles of FLT3 in Hematopoiesis and Leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Scholl, S.; Fleischmann, M.; Schnetzke, U.; Heidel, F.H. Molecular Mechanisms of Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia: Ongoing Challenges and Future Treatments. Cells 2020, 9, 2493. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The Structural Basis for Autoinhibition of FLT3 by the Juxtamembrane Domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Tvedt, T.H.A.; Reikvam, H.; Bruserud, Ø. Clonal Heterogeneity Reflected by Pi3k-Akt-Mtor Signaling in Human Acute Myeloid Leukemia Cells and Its Association with Adverse Prognosis. Cancers 2018, 10, 332. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Aasebø, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of Insulin and Pathway Inhibitors on the PI3K-Akt-MTOR Phosphorylation Profile in Acute Myeloid Leukemia Cells. Signal Transduct. Target. Ther. 2019, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional Proteomic Profiling of AML Predicts Response and Survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.H.; Eom, J.I.; Cheong, J.W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive Phosphorylation of Akt/PKB Protein in Acute Myeloid Leukemia: Its Significance as a Prognostic Variable. Leukemia 2003, 17, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Drakos, E.; Grammatikakis, I.; Schlette, E.J.; Li, J.; Leventaki, V.; Staikou-Drakopoulou, E.; Patsouris, E.; Panayiotidis, P.; Medeiros, L.J.; et al. MTOR Signaling Is Activated by FLT3 Kinase and Promotes Survival of FLT3-Mutated Acute Myeloid Leukemia Cells. Mol. Cancer 2010, 9, 292. [Google Scholar] [CrossRef]
- Wang, J.; Tomlinson, B.; Lazarus, H.M. Update on Small Molecule Targeted Therapies for Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2023, 24, 770–801. [Google Scholar] [CrossRef]
- Ambinder, A.J.; Levis, M. Potential Targeting of FLT3 Acute Myeloid Leukemia. Haematologica 2021, 106, 671–681. [Google Scholar] [CrossRef]
- Zhao, J.C.; Agarwal, S.; Ahmad, H.; Amin, K.; Bewersdorf, J.P.; Zeidan, A.M. A Review of FLT3 Inhibitors in Acute Myeloid Leukemia. Blood Rev. 2022, 52, 100905. [Google Scholar] [CrossRef]
- Senapati, J.; Kadia, T.M. Which FLT3 Inhibitor for Treatment of AML? Curr. Treat. Options Oncol. 2022, 23, 359–380. [Google Scholar] [CrossRef]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I Study of Sorafenib in Patients with Refractory or Relapsed Acute Leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Stone, R.M.; Heaney, M.L.; Nimer, S.D.; Paquette, R.L.; Klisovic, R.B.; Caligiuri, M.A.; Cooper, M.R.; Lecerf, J.M.; Karol, M.D.; et al. Phase 1 Clinical Results with Tandutinib (MLN518), a Novel FLT3 Antagonist, in Patients with Acute Myelogenous Leukemia or High-Risk Myelodysplastic Syndrome: Safety, Pharmacokinetics, and Pharmacodynamics. Blood 2006, 108, 3674–3681. [Google Scholar] [CrossRef]
- Fischer, T.; Stone, R.M.; DeAngelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB Trial of Oral Midostaurin (PKC412), the FMS-like Tyrosine Kinase 3 Receptor (FLT3) and Multi-Targeted Kinase Inhibitor, in Patients with Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome with Either Wild-Type or Mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a Randomized Trial of Salvage Chemotherapy Followed by Lestaurtinib for Patients with FLT3 Mutant AML in First Relapse. Blood J. Am. Soc. Hematol. 2011, 117, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Serve, H.; Dö, H.; Schwittay, M.; Ottmann, O.G.; O’farrell, A.-M.; Bello, C.L.; Allred, R.; Manning, W.C.; Cherrington, J.M.; et al. A Phase 1 Study of SU11248 in the Treatment of Patients with Refractory or Resistant Acute Myeloid Leukemia (AML) or Not Amenable to Conventional Therapy for the Disease. Blood 2005, 105, 986–993. [Google Scholar] [CrossRef]
- Food and Drug Administration. RYDAPT (Midostaurin) Capsules, for Oral Use; Novartis Pharmaceuticals Corporation: East Hanover, NJ, USA, 2017.
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; SträngStr, E.; Theis, F.; Jahn, N.; Panina, E.; Bïatte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal Evolution of Acute Myeloid Leukemia with FLT3-ITD Mutation under Treatment with Midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Strati, P.; Kantarjian, H.; Ravandi, F.; Nazha, A.; Borthakur, G.; Daver, N.; Kadia, T.; Estrov, Z.; Garcia-Manero, G.; Konopleva, M.; et al. Phase I/II Trial of the Combination of Midostaurin (PKC412) and 5-Azacytidine for Patients with Acute Myeloid Leukemia and Myelodysplastic Syndrome. Am. J. Hematol. 2015, 90, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.W.; Kindwall-Keller, T.L.; Craig, M.D.; Creger, R.J.; Hamadani, M.; Tse, W.W.; Lazarus, H.M. A Phase i Study of Midostaurin and Azacitidine in Relapsed and Elderly AML Patients. Clin. Lymphoma Myeloma Leuk. 2015, 15, 428–432.e2. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Weber, D.; Fiedler, W.; Salih, H.R.; Wulf, G.; Salwender, H.; Schroeder, T.; Kindler, T.; Heuser, M.; Thol, F.; et al. Midostaurin Added to Chemotherapy and Continued Single-Agent Maintenance Therapy in Acute Myeloid Leukemia with FLT3-ITD. Blood 2019, 133, 840–851. [Google Scholar] [CrossRef]
- Maziarz, R.T.T.; Patnaik, M.M.; Scott, B.L.; Mohan, S.R.; Deol, A.; Rowley, S.D.; Kim, D.; Haines, K.; Bonifacio, G.J.; Rine, P.; et al. Radius: A Phase 2 Randomized Trial Investigating Standard of Care ± Midostaurin after Allogeneic Stem Cell Transplant in FLT3-ITD-Mutated AML. Blood 2018, 132, 662. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 Inhibitors in Acute Myeloid Leukemia: Ten Frequently Asked Questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Ganguly, S.; Cortes, J.E.; Krämer, A.; Levis, M.J.; Martinelli, G.; Perl, A.E.; Russell, N.H.; Arunachalam, M.; Santos, C.D.; Gammon, G.; et al. Clinical Outcomes in Patients with FLT3-ITD-Mutated Relapsed/Refractory Acute Myelogenous Leukemia Undergoing Hematopoietic Stem Cell Transplantation after Quizartinib or Salvage Chemotherapy in the QuANTUM-R Trial. Transplant. Cell Ther. 2021, 27, 153–162. [Google Scholar] [CrossRef]
- Fei, X.; Zhang, S.; Gu, J.; Wang, J. FLT3 Inhibitors as Maintenance Therapy Post Allogeneic Hematopoietic Stem Cell Transplantation in Acute Myeloid Leukemia Patients with FLT3 Mutations: A Meta-Analysis. Cancer Med. 2023, 12, 6877–6888. [Google Scholar] [CrossRef]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Outcomes in Patients with FLT3-Mutated Relapsed/Refractory Acute Myelogenous Leukemia Who Underwent Transplantation in the Phase 3 ADMIRAL Trial of Gilteritinib versus Salvage Chemotherapy. Transplant. Cell Ther. 2023, 29, 265.e1–265.e10. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, C.; Zhu, X. FLT3 Inhibitors in Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Ottone, T.; Zhdanovskaya, N.; Molica, M. How Acute Myeloid Leukemia (AML) Escapes from FMS-Related Tyrosine Kinase 3 (FLT3) Inhibitors? Still an Overrated Complication? Cancer Drug Resist. 2023, 6, 223–238. [Google Scholar] [CrossRef]
- Aydin, S.; Passera, R.; Scaldaferri, M.; Dellacasa, C.M.; Poggiu, M.; Cattel, F.; Zallio, F.; Brunello, L.; Giaccone, L.; Dogliotti, I.; et al. Sorafenib Maintenance after Hematopoietic Stem Cell Transplantation Improves Outcome of FLT3–ITD-Mutated Acute Myeloid Leukemia. Int. J. Hematol. 2022, 116, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, U.; Stelljes, M.; Wollmer, E.; BornhäuserBornh, M.; Berg, T.; Lang, F.; Ehninger, G.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia with FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Röllig, C.; Serve, H.; Hüttmann, A.; Noppeney, R.; Müller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of Sorafenib versus Placebo to Standard Therapy in Patients Aged 60 Years or Younger with Newly Diagnosed Acute Myeloid Leukaemia (SORAML): A Multicentre, Phase 2, Randomised Controlled Trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Altman, J.K.; Foran, J.M.; Pratz, K.W.; Trone, D.; Cortes, J.E.; Tallman, M.S. Phase 1 Study of Quizartinib in Combination with Induction and Consolidation Chemotherapy in Patients with Newly Diagnosed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, 213–221. [Google Scholar] [CrossRef]
- Sandmaier, B.M.; Khaled, S.; Oran, B.; Gammon, G.; Trone, D.; Frankfurt, O. Results of a Phase 1 Study of Quizartinib as Maintenance Therapy in Subjects with Acute Myeloid Leukemia in Remission Following Allogeneic Hematopoietic Stem Cell Transplant. Am. J. Hematol. 2018, 93, 222–231. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I Study of Quizartinib Administered Daily to Patients with Relapsed or Refractory Acute Myeloid Leukemia Irrespective of FMS-like Tyrosine Kinase 3-Internal Tandem Duplication Status. J. Clin. Oncol. 2013, 31, 3681–3867. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 Inhibitor, as Monotherapy in Patients with Relapsed or Refractory Acute Myeloid Leukaemia: An Open-Label, Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Kropp, E.M.; Li, Q. Mechanisms of resistance to targeted therapies for relapsed or refractory acute myeloid leukemia. Exp. Hematol. 2022, 111, 13–24. [Google Scholar] [CrossRef]
- Erba, H.P.; Montesinos, P.; Kim, H.-J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.-N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus Chemotherapy in Newly Diagnosed Patients with FLT3-Internal-Tandem-Duplication-Positive Acute Myeloid Leukaemia (QuANTUM-First): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Quizartinib (VANFLYTA®) for acute myeloid leukemia. Med. Lett. Drugs Ther. 2023, 65. Available online: https://secure.medicalletter.org/TML-article-1687d (accessed on 30 January 2024).
- Baragaño Raneros, A.; López-Larrea, C.; Suárez-Álvarez, B. Acute Myeloid Leukemia and NK Cells: Two Warriors Confront Each Other. Oncoimmunology 2019, 8, e1539617. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1–2 Study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Terao, T.; Matsuoka, K.I.; Ueda, H.; Matsumura, A.; Matsubara, C.; Kondo, K.; Kondo, T.; Fujiwara, H.; Asada, N.; Ennishi, D.; et al. Early Initiation of Low-Dose Gilteritinib Maintenance Improves Posttransplant Outcomes in Patients with R/R FLT3mut AML. Blood Adv. 2023, 7, 681–686. [Google Scholar] [CrossRef]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of Patients with R/R FLT3-Mutation–Positive Treated with Gilteritinib in the Phase 3 Trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Biavasco, F.; Zeiser, R. FLT3-Inhibitor Therapy for Prevention and Treatment of Relapse after Allogeneic Hematopoietic Cell Transplantation. Int. J. Hematol. 2022, 116, 341–350. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Maiti, A.; Rausch, C.R.; Pemmaraju, N.; Naqvi, K.; Daver, N.G.; Kadia, T.M.; Borthakur, G.; Ohanian, M.; Alvarado, Y.; et al. 10-Day Decitabine with Venetoclax for Newly Diagnosed Intensive Chemotherapy Ineligible, and Relapsed or Refractory Acute Myeloid Leukaemia: A Single-Centre, Phase 2 Trial. Lancet Haematol. 2020, 7, e724–e736. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3 -Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2021, 2, 125–134. [Google Scholar] [CrossRef]
- Zhang, Z.; Hasegawa, Y.; Hashimoto, D.; Senjo, H.; Kikuchi, R.; Chen, X.; Yoneda, K.; Sekiguchi, T.; Kawase, T.; Tsuzuki, H.; et al. Gilteritinib Enhances Graft-versus-Leukemia Effects against FLT3-ITD Mutant Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation. Bone Marrow Transpl. 2022, 57, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Müller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib Promotes Graft-versus-Leukemia Activity in Mice and Humans through IL-15 Production in FLT3-ITD-Mutant Leukemia Cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Interleukin-15 Enhances Natural Killer Cell Cytotoxicity in Patients with Acute Myeloid Leukemia by Upregulating the Activating NK Cell Receptors. Cancer Immunol. Immunother. 2010, 59, 73–79. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Houghton, P.J.; Williams, T.M.; Shen, C. Regulation of DNA Duplication by the MTOR Signaling Pathway. Cell Cycle 2021, 20, 742–751. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Harwood, F.C.; Klein Geltink, R.I.; O’hara, B.P.; Cardone, M.; Janke, L.; Finkelstein, D.; Entin, I.; Paul, L.; Houghton, P.J.; Grosveld, G.C. ETV7 Is an Essential Component of a Rapamycin-Insensitive MTOR Complex in Cancer. Sci. Adv. 2018, 4, eaar3938. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonça, D.B.; Kim, J.K.; Krebsbach, P.H. MEAK-7 Forms an Alternative MTOR Complex with DNA-PKcs in Human Cancer. iScience 2019, 17, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonça, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mtor signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, eaao5838. [Google Scholar] [CrossRef]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb Governs Growth Factor-Induced MTORC1 Activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt Regulates Growth by Directly Phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Erratum: Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control MTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb (Current Biology: CB (2003) 13 15 (1259–1268) PII: S0960-9822(22)00038-0). Curr. Biol. 2022, 32, 733–734. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. Ptdins(3,4,5) P3 -Dependent Activation of the MTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–11209. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Bum Shin, T.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A Mammalian Protein Targeted by G1-Arresting Rapamycin–Receptor Complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. MTORC2 Promotes Type I Insulin-like Growth Factor Receptor and Insulin Receptor Activation through the Tyrosine Kinase Activity of MTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef]
- Guenzle, J.; Akasaka, H.; Joechle, K.; Reichardt, W.; Venkatasamy, A.; Hoeppner, J.; Hellerbrand, C.; Fichtner-Feigl, S.; Lang, S.A. Pharmacological Inhibition of Mtorc2 Reduces Migration and Metastasis in Melanoma. Int. J. Mol. Sci. 2021, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jørgensen, H.G.; Marmiroli, S.; Huang, X. Targeting Pi3k/Akt/Mtor in Aml: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Wei, J.; Hui, A.M. Role of Biomarkers in FLT3 AML. Cancers 2022, 14, 1164. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.J.; Lee, K.M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20, e14. [Google Scholar] [CrossRef]
- Yang, L.; Feng, Y.; Wang, S.; Jiang, S.; Tao, L.; Li, J.; Wang, X. Siglec-7 Is an Indicator of Natural Killer Cell Function in Acute Myeloid Leukemia. Int. Immunopharmacol. 2021, 99, 107965. [Google Scholar] [CrossRef]
- Lion, E.; Willemen, Y.; Berneman, Z.N.; Van Tendeloo, V.F.I.; Smits, E.L.J. Natural Killer Cell Immune Escape in Acute Myeloid Leukemia. Leukemia 2012, 26, 2019–2026. [Google Scholar] [CrossRef]
- Davis, Z.B.; Cogswell, A.; Scott, H.; Mertsching, A.; Boucau, J.; Wambua, D.; Le Gall, S.; Planelles, V.; Campbell, K.S.; Barker, E. A Conserved HIV-1-Derived Peptide Presented by HLA-E Renders Infected T-Cells Highly Susceptible to Attack by NKG2A/CD94-Bearing Natural Killer Cells. PLoS Pathog. 2016, 12, e1005421. [Google Scholar] [CrossRef]
- Aktas, E.; Kucuksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between CD107a Expression and Cytotoxic Activity. Cell Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef]
- Nguyen, S.; Beziat, V.; Dhedin, N.; Kuentz, M.; Vernant, J.P.; Debre, P.; Vieillard, V. HLA-E Upregulation on IFN-γ-Activated AML Blasts Impairs CD94/NKG2A-Dependent NK Cytolysis after Haplo-Mismatched Hematopoietic SCT. Bone Marrow Transplant. 2009, 43, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Weisdorf, D.J.; Miller, J.S. NK Cell Education after Allogeneic Transplantation: Dissociation between Recovery of Cytokine-Producing and Cytotoxic Functions. Blood 2011, 118, 2784–2792. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.; Dührsen, U.; Hüttmann, A.; Nückel, H.; Michita, R.T.; Rohn, H.; Schramm, S.; Horn, P.A.; Rebmann, V. Genetic Variants of the Nkg2c/Hla-e Receptor– Ligand Axis Are Determinants of Progression-Free Survival and Therapy Outcome in Aggressive b-Cell Lymphoma. Cancers 2020, 12, 3429. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful Adoptive Transfer and in Vivo Expansion of Human Haploidentical NK Cells in Patients with Cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef]
- Bachanova, V.; Cooley, S.; Defor, T.E.; Verneris, M.R.; Zhang, B.; Mckenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lewis, D.; Hippen, K.; et al. Clearance of Acute Myeloid Leukemia by Haploidentical Natural Killer Cells Is Improved Using IL-2 Diphtheria Toxin Fusion Protein. Blood 2014, 123, 3855–3863. [Google Scholar] [CrossRef]
- Cooley, S.; He, F.; Bachanova, V.; Vercellotti, G.M.; DeFor, T.E.; Curtsinger, J.M.; Robertson, P.; Grzywacz, B.; Conlon, K.C.; Waldmann, T.A.; et al. First-in-Human Trial of RhIL-15 and Haploidentical Natural Killer Cell Therapy for Advanced Acute Myeloid Leukemia. Blood Adv. 2019, 3, 1970–1980. [Google Scholar] [CrossRef]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 Delays Tumor Growth and Promotes Intratumoral CTL and Dendritic Cell Accumulation by a Cytokine Network Involving XCL1, IFN-γ, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, e000599. [Google Scholar] [CrossRef]
- Mathios, D.; Park, C.K.; Marcus, W.D.; Alter, S.; Rhode, P.R.; Jeng, E.K.; Wong, H.C.; Pardoll, D.M.; Lim, M. Therapeutic Administration of IL-15 Superagonist Complex ALT-803 Leads to Long-Term Survival and Durable Antitumor Immune Response in a Murine Glioblastoma Model. Int. J. Cancer 2016, 138, 187–194. [Google Scholar] [CrossRef]
- Xu, W.; Jones, M.; Liu, B.; Zhu, X.; Johnson, C.B.; Edwards, A.C.; Kong, L.; Jeng, E.K.; Han, K.; Marcus, W.D.; et al. Efficacy and Mechanism-of-Action of a Novel Superagonist Interleukin-15: Interleukin-15 Receptor ASu/Fc Fusion Complex in Syngeneic Murine Models of Multiple Myeloma. Cancer Res. 2013, 73, 3075–3086. [Google Scholar] [CrossRef]
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waitz, R.; Fasso, M.; Allison, J.P.; Waldmann, T.A. Simultaneous Inhibition of Two Regulatory T-Cell Subsets Enhanced Interleukin-15 Efficacy in a Prostate Tumor Model. Proc. Natl. Acad. Sci. USA 2012, 109, 6187–6192. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waldmann, T.A. Simultaneous Blockade of Multiple Immune System Inhibitory Checkpoints Enhances Antitumor Activity Mediated by Interleukin-15 in a Murine Metastatic Colon Carcinoma Model. Clin. Cancer Res. 2010, 16, 6019–6028. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.; Watson, D.C.; Waldmann, T.A.; Valentin, A.; Bergamaschi, C.; Felber, B.K.; Peer, C.J.; Figg, W.D.; Potter, E.L.; Roederer, M.; et al. Phase I Study of Single Agent NIZ985, a Recombinant Heterodimeric IL-15 Agonist, in Adult Patients with Metastatic or Unresectable Solid Tumors. J. Immunother. Cancer 2021, 9, e003388. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production during First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients with Cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lake Potter, E.; Pittaluga, S.; Lee, C.C.R.; Miljkovic, M.D.; Fleisher, T.A.; Dubois, S.; Bryant, B.R.; Petrus, M.; Perera, L.P.; et al. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin. Cancer Res. 2019, 25, 4945–4954. [Google Scholar] [CrossRef]
- Cai, M.; Huang, X.; Huang, X.; Ju, D.; Zhu, Y.Z.; Ye, L. Research progress of interleukin-15 in cancer immunotherapy. Front. Pharmacol. 2023, 14, 1184703. [Google Scholar] [CrossRef]
- Margolin, K.; Morishima, C.; Velcheti, V.; Miller, J.S.; Lee, S.M.; Silk, A.W.; Holtan, S.G.; Lacroix, A.M.; Fling, S.P.; Kaiser, J.C.; et al. Phase I Trial of ALT-803, a Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 5552–5561. [Google Scholar] [CrossRef]
- Miller, J.S.; Morishima, C.; McNeel, D.G.; Patel, M.R.; Kohrt, H.E.K.; Thompson, J.A.; Sondel, P.M.; Wakelee, H.A.; Disis, M.L.; Kaiser, J.C.; et al. A First-in-Human Phase I Study of Subcutaneous Outpatient Recombinant Human IL15 (RhIL15) in Adults with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1525–1535. [Google Scholar] [CrossRef]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; Defor, T.E.; et al. First-in-Human Phase 1 Clinical Study of the IL-15 Superagonist Complex ALT-803 to Treat Relapse after Transplantation. Blood J. Am. Soc. Hematol. 2018, 131, 2515–2527. [Google Scholar] [CrossRef]
- Stellas, D.; Karaliota, S.; Stravokefalou, V.; Angel, M.; Nagy, B.A.; Goldfarbmuren, K.C.; Bergamaschi, C.; Felber, B.K.; Pavlakis, G.N. Tumor Eradication by HetIL-15 Locoregional Therapy Correlates with an Induced Intratumoral CD103intCD11b+ Dendritic Cell Population. Cell Rep. 2023, 42, 112501. [Google Scholar] [CrossRef]
- Chiu, E.; Felices, M.; Cichocki, F.; Davis, Z.; Wang, H.; Tuninga, K.; Vallera, D.A.; Lee, T.; Bjordahl, R.; Malmberg, K.J.; et al. Anti-NKG2C/IL-15/Anti-CD33 Killer Engager Directs Primary and IPSC-Derived NKG2C+ NK Cells to Target Myeloid Leukemia. Mol. Ther. 2021, 29, 3410–3421. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dincheva-Vogel, L.; Ayemoba, C.E.; Fung, J.P.; Bergamaschi, C.; Pavlakis, G.N.; Farzaneh, F.; Gaensler, K.M.L. IL-15/IL-15Ra/CD80-Expressing AML Cell Vaccines Eradicate Minimal Residual Disease in Leukemic Mice. Blood Adv. 2018, 2, 3177–3192. [Google Scholar] [CrossRef] [PubMed]
- Berrien-Elliott, M.M.; Becker-Hapak, M.; Cashen, A.F.; Jacobs, M.; Wong, P.; Foster, M.; McClain, E.; Desai, S.; Pence, P.; Cooley, S.; et al. Systemic IL-15 Promotes Allogeneic Cell Rejection in Patients Treated with Natural Killer Cell Adoptive Therapy. Blood 2022, 139, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, in Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 Bispecific Killer Cell Engager (BiKE) Activates NK Cells against Primary MDS and MDSC CD33 1 Targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.L.; Flavell, R.A. Transforming Growth Factor-β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Delvig, A.A.; Lee, J.J.; Chrzanowska-Lightowlers, Z.M.A.; Robinson, J.H. TGF-Beta1 and IFN-Gamma Cross-Regulate Antigen Presentation to CD4 T Cells by Macrophages. J. Leukoc. Biol. 2002, 72, 163–166. [Google Scholar] [CrossRef]
- Nandan, D.; Reiner, N.E. TGF-Beta Attenuates the Class II Transactivator and Reveals an Accessory Pathway of IFN-Gamma Action. J. Immunol. 1997, 158, 1095–1101. [Google Scholar] [CrossRef]
- Chen, W.J.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Immunology Cutting Edge: TGF-Induces a Regulatory Phenotype in CD4 CD25 T Cells through Foxp3 Induction and Down-Regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Papaspyridonos, M.; Matei, I.; Huang, Y.; Do Rosario Andre, M.; Brazier-Mitouart, H.; Waite, J.C.; Chan, A.S.; Kalter, J.; Ramos, I.; Wu, Q.; et al. Id1 Suppresses Anti-Tumour Immune Responses and Promotes Tumour Progression by Impairing Myeloid Cell Maturation. Nat. Commun. 2015, 6, 6840. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.-G.; Becker, D.; Geisert, A.; Lyght, M.; Flanderst, K.C.; Robertst, A.B.; Spornt, M.B.; Wardt, J.M.; Karlsson, S. Transforming Growth Factor 181 Null Mutation in Mice Causes Excessive Inflammatory Response and Early Death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted Disruption of the Mouse Transforming Growth Factor-Β1 Gene Results in Multifocal Inflammatory Disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK Cells and Cancer Immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Immune-Mediated Eradication of Tumors through the blockade of Transforming Growth Factor-Beta Signaling in T Cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Donkor, M.K.; Sarkar, A.; Savage, P.A.; Franklin, R.A.; Johnson, L.K.; Jungbluth, A.A.; Allison, J.P.; Li, M.O. T Cell Surveillance of Oncogene-Induced Prostate Cancer Is Impeded by T Cell-Derived TGF-Β1 Cytokine. Immunity 2011, 35, 123–134. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-Β1 Attenuates the Acquisition and Expression of Effector Function by Tumor Antigen-Specific Human Memory CD8 T Cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef]
- Fernández-Messina, L.; Ashiru, O.; Boutet, P.; Agüera-González, S.; Skepper, J.N.; Reyburn, H.T.; Valés-Gómez, M. Differential Mechanisms of Shedding of the Glycosylphosphatidylinositol (GPI)-Anchored NKG2D Ligands. J. Biol. Chem. 2010, 285, 8543–8551. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural Killer Cell Cytotoxicity Is Suppressed by Exposure to the Human NKG2D Ligand MICA*008 That Is Shed by Tumor Cells in Exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Goehlsdorf, D.; Gieseke, F.; Weinschenk, T.; Wittenbrink, M.; Ludwig, A.; Stevanovic, S.; Rammensee, H.G.; Steinle, A. Tumor-Associated MICA Is Shed by ADAM Proteases. Cancer Res. 2008, 68, 6368–6376. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Rammensee, H.-G.; Steinle, A. Cutting Edge: Down-Regulation of MICA on Human Tumors by Proteolytic Shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. Proteolytic Release of Soluble UL16-Binding Protein 2 from Tumor Cells. Cancer Res. 2006, 66, 2520–2526. [Google Scholar] [CrossRef]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New Prospects on the NKG2D/NKG2DL System for Oncology. Oncoimmunology 2013, 2, e26097. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; De Lima, M.; Wald, D.N. Inhibiting TGF-Beta Signaling Preserves the Function of Highly Activated, in Vitro Expanded Natural Killer Cells in AML and Colon Cancer Models. PLoS ONE 2018, 13, e0191358. [Google Scholar] [CrossRef]
- Tran, H.C.; Wan, Z.; Sheard, M.A.; Sun, J.; Jackson, J.R.; Malvar, J.; Xu, Y.; Wang, L.; Sposto, R.; Kim, E.S.; et al. TGFβR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (Ch14.18) with Natural Killer Cells. Clin. Cancer Res. 2017, 23, 804–813. [Google Scholar] [CrossRef]
- Song, H.; Hur, D.Y.; Kim, K.E.; Park, H.; Kim, T.; Kim, C.W.; Bang, S.; Cho, D.H. IL-2/IL-18 Prevent the down-Modulation of NKG2D by TGF-β in NK Cells via the c-Jun N-Terminal Kinase (JNK) Pathway. Cell Immunol. 2006, 242, 39–45. [Google Scholar] [CrossRef]
- Fujii, R.; Jochems, C.; Tritsch, S.R.; Wong, H.C.; Schlom, J.; Hodge, J.W. An IL-15 Superagonist/IL-15Rα Fusion Complex Protects and Rescues NK Cell-Cytotoxic Function from TGF-Β1-Mediated Immunosuppression. Cancer Immunol. Immunother. 2018, 67, 675–689. [Google Scholar] [CrossRef]
- Bollard, C.M.; Rö, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.-J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a Transforming Growth Factor-Related Tumor Protection Strategy to Enhance Antitumor Immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord Blood Natural Killer Cells Expressing a Dominant Negative TGF-β Receptor: Implications for Adoptive Immunotherapy for Glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef]
- Bollard, C.M.; Tripic, T.; Russell Cruz, C.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients with Relapsed Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-Cell Responses to Soluble Factors with Chimeric Antigen Receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-β signals. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 Is Dispensable for Normal Pancreas Development yet Critical in Progression and Tumor Biology of Pancreas Cancer. Genes. Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [PubMed]
- Malkoski, S.P.; Wang, X.J. Two Sides of the Story? Smad4 Loss in Pancreatic Cancer versus Head-and-Neck Cancer. FEBS Lett. 2012, 586, 1984–1992. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, J.; Yi, P.; Dong, W.; Saultz, J.; Wang, Y.; Wang, H.; Scoville, S.; Zhang, J.; Wu, L.C.; et al. SMAD4 Promotes TGF-β-Independent NK Cell Homeostasis and Maturation and Antitumor Immunity. J. Clin. Investig. 2018, 128, 5123–5136. [Google Scholar] [CrossRef]
- Wang, L.; Gu, S.; Chen, F.; Yu, Y.; Cao, J.; Li, X.; Gao, C.; Chen, Y.; Yuan, S.; Liu, X.; et al. Imatinib Blocks Tyrosine Phosphorylation of Smad4 and Restores TGF-β Growth-Suppressive Signaling in BCR-ABL1-Positive Leukemia. Signal Transduct. Target. Ther. 2023, 8, 120. [Google Scholar] [CrossRef]
- Pratz, K.W.; Cherry, M.; Altman, J.K.; Cooper, B.W.; Podoltsev, N.A.; Cruz, J.C.; Lin, T.L.; Schiller, G.J.; Jurcic, J.G.; Asch, A.; et al. Gilteritinib in Combination with Induction and Consolidation Chemotherapy and as Maintenance Therapy: A Phase IB Study in Patients With Newly Diagnosed AML. J. Clin. Oncol. 2023, 41, 4236–4246. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; Dinardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; Mccloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.D.; Song, Y.; Yang, H.; Freimark, J.; Shah, M.V. Post-Transplant Maintenance Therapy in Patients with FLT3-Mutated Acute Myeloid Leukemia: Real-World Treatment Patterns and Outcomes. Eur. J. Haematol. 2021, 107, 553–565. [Google Scholar] [CrossRef]
- Fleischmann, M.; Schnetzke, U.; Schrenk, K.G.; Schmidt, V.; Sayer, H.G.; Hilgendorf, I.; Hochhaus, A.; Scholl, S. Outcome of FLT3-ITD-Positive Acute Myeloid Leukemia: Impact of Allogeneic Stem Cell Transplantation and Tyrosine Kinase Inhibitor Treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 337–345. [Google Scholar] [CrossRef]
- Chase Doyle Gilteritinib Maintenance Therapy after Transplantation in Patients with FLT3-ITD AML Who Have Measurable Residual Disease. Available online: https://ascopost.com/issues/august-25-2023/gilteritinib-maintenance-therapy-after-transplantation-in-patients-with-flt3-itd-aml-who-have-measurable-residual-disease/ (accessed on 30 January 2024).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Type | Targets | Adverse | Outcome |
|---|---|---|---|---|
| First Generation | ||||
| Midostaurin | Type I | Other kinases (KIT, PDGFR, RAS/RAF/MEK, JAK) | Cytopenias, flu-like symptoms, cardiac failure (<5%), nausea, vomiting, diarrhea | OS, RFS |
| Sorafenib | Type II | Other kinases (KIT, PDGFR, RAS/RAF/MEK, JAK) | Cytopenias, flu-like symptoms, hypertension, cardiac ischemia (<5%), diarrhea, rash, hand-foot | OS, RFS, CIR, NRM, GVHD, AE |
| Second Generation | ||||
| Gilteritinib | Type I | Higher potency, more specific, less off-target | Cytopenias, diarrhea, pancreatitis, headaches | OS, RFS, CIR, AE |
| Quizartinib | Type II | Higher potency, more specific, less off-targe | Cytopenias, flu-like symptoms, cardiac arrythmia, nausea, anorexia | OS, RFS, AE |
| Trial | Developmental Phase | |
|---|---|---|
| First Generation Inhibitors | ||
| Midostaurin + 7+3 chemotherapy | RATIFY (NCT00651261) | Phase III |
| Midostaurin + Azacitidine | Internal Trial at MD Anderson Cancer Center | Phase I/II |
| ±Midostaurin + Allo-HCT | RADIUS (NCT01883362) | Phase II |
| Sorafenib | SORMAIN (DRKS00000591) | Phase II |
| Sorafenib + 7+3 chemotherapy | SORAML (NCT00893373) | Phase II |
| Second Generation Inhibitors | ||
| Quizartinib + Cytarabine/Daunorubicin | NCT 01390337 | Phase I |
| Quizartinib + Allo-HCT | 2689-CL-0011 | Phase I |
| Quizartinib + Allo-HCT | NCT00462761 | Phase I |
| Quizartinib + 7+3 chemotherapy | QuANTUM-First (NCT02668653) | Phase III |
| Gilteritinib or salvage chemotherapy | QuANTUM-First (NCT02668653) | Phase III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leifheit, M.E.; Johnson, G.; Kuzel, T.M.; Schneider, J.R.; Barker, E.; Yun, H.D.; Ustun, C.; Goldufsky, J.W.; Gupta, K.; Marzo, A.L. Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 9448. https://doi.org/10.3390/ijms25179448
Leifheit ME, Johnson G, Kuzel TM, Schneider JR, Barker E, Yun HD, Ustun C, Goldufsky JW, Gupta K, Marzo AL. Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2024; 25(17):9448. https://doi.org/10.3390/ijms25179448
Chicago/Turabian StyleLeifheit, Malia E., Gunnar Johnson, Timothy M. Kuzel, Jeffrey R. Schneider, Edward Barker, Hyun D. Yun, Celalettin Ustun, Josef W. Goldufsky, Kajal Gupta, and Amanda L. Marzo. 2024. "Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia" International Journal of Molecular Sciences 25, no. 17: 9448. https://doi.org/10.3390/ijms25179448
APA StyleLeifheit, M. E., Johnson, G., Kuzel, T. M., Schneider, J. R., Barker, E., Yun, H. D., Ustun, C., Goldufsky, J. W., Gupta, K., & Marzo, A. L. (2024). Enhancing Therapeutic Efficacy of FLT3 Inhibitors with Combination Therapy for Treatment of Acute Myeloid Leukemia. International Journal of Molecular Sciences, 25(17), 9448. https://doi.org/10.3390/ijms25179448