

Cell Death: Mechanisms and Potential Targets in Breast Cancer Therapy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
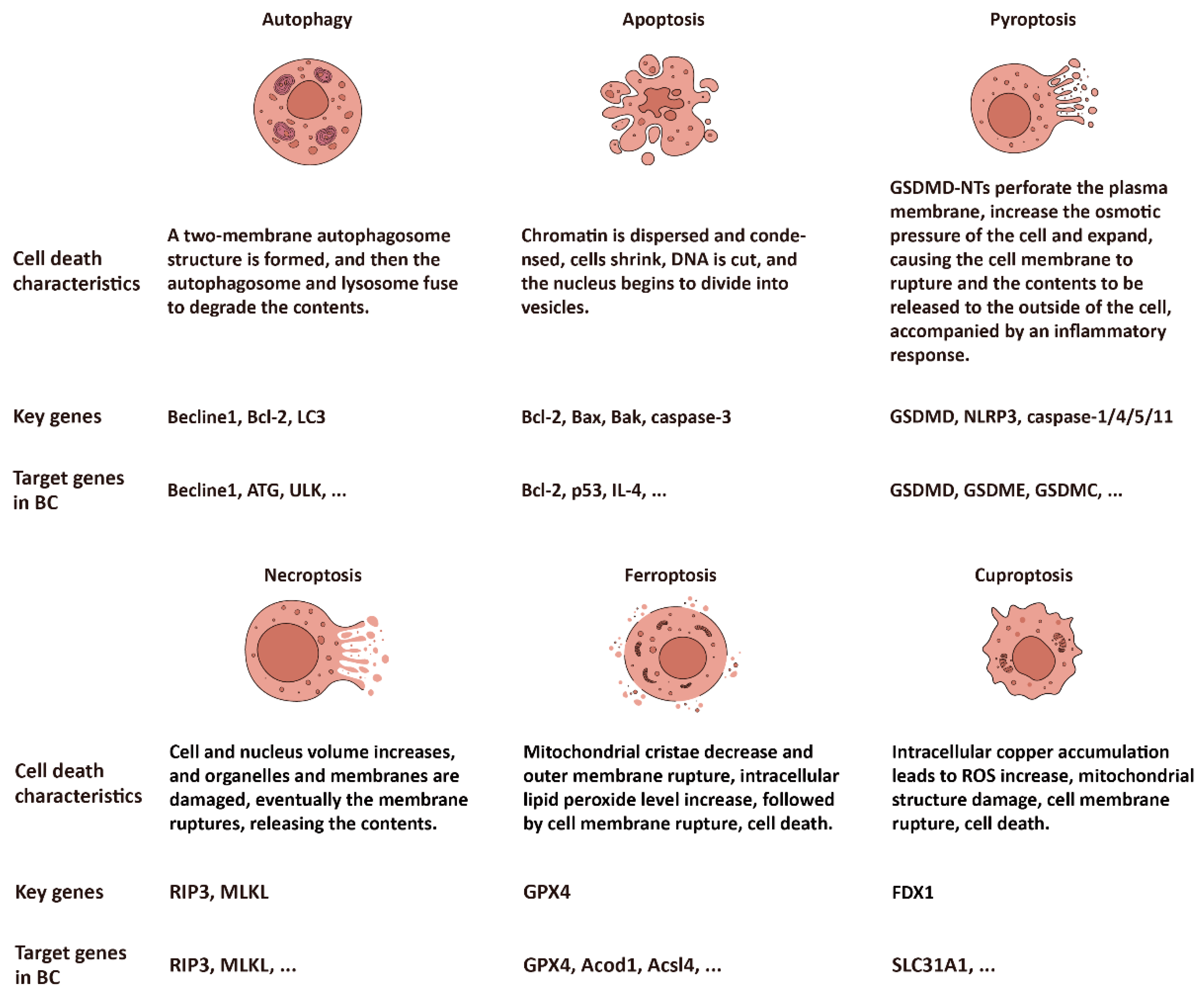
2. Autophagy
2.1. Autophagy: A PCD Mediated by Lysosomes
2.2. Autophagy-Related Genes Dysregulation and Therapy in BC
3. PANoptosis: A Cross-Regulated Mode of Cell Death
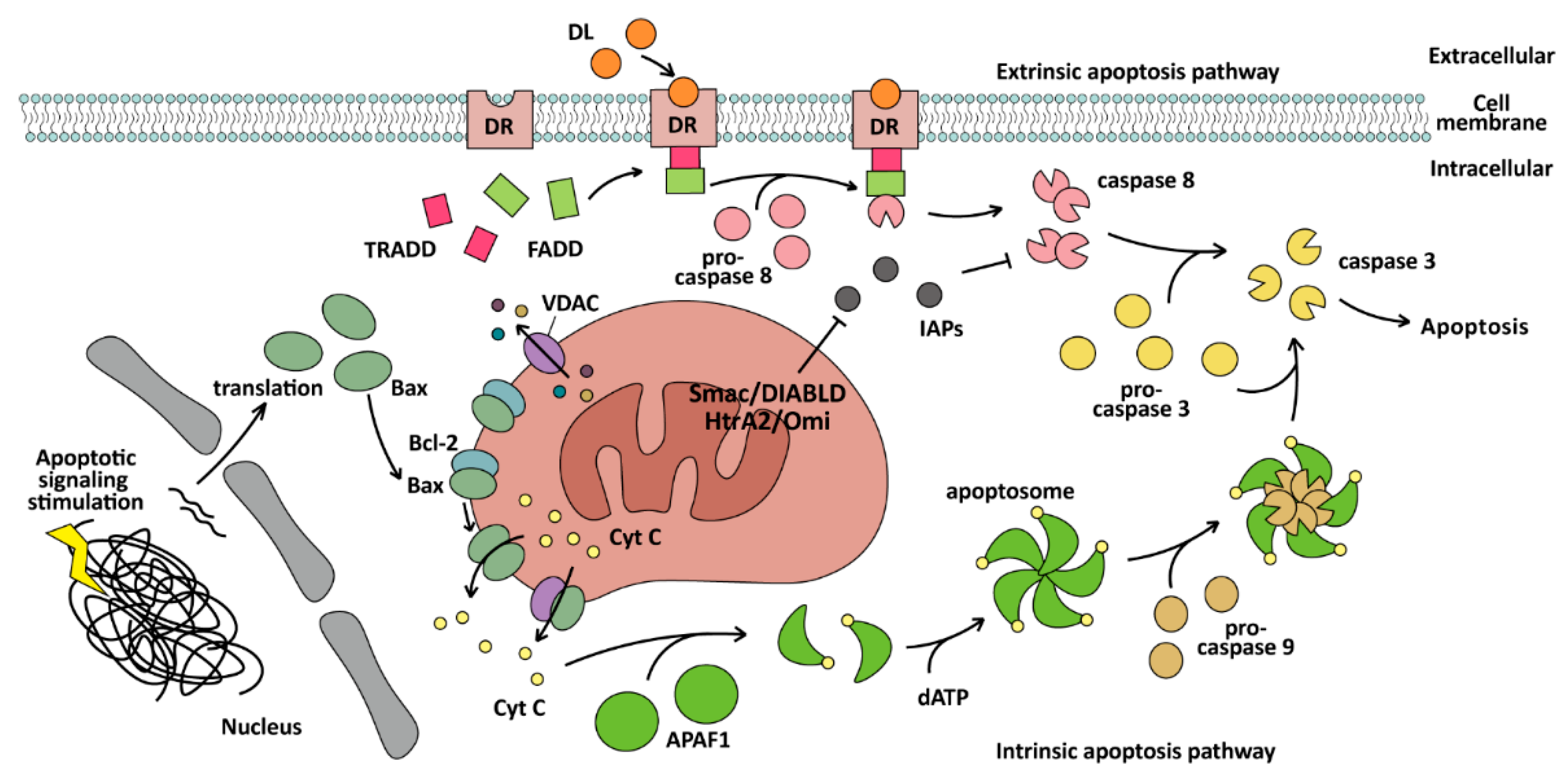
3.1. Apoptosis
3.1.1. Apoptosis: A PCD Induced by Caspases
3.1.2. Apoptosis-Related Genes Dysregulation and Therapy in BC
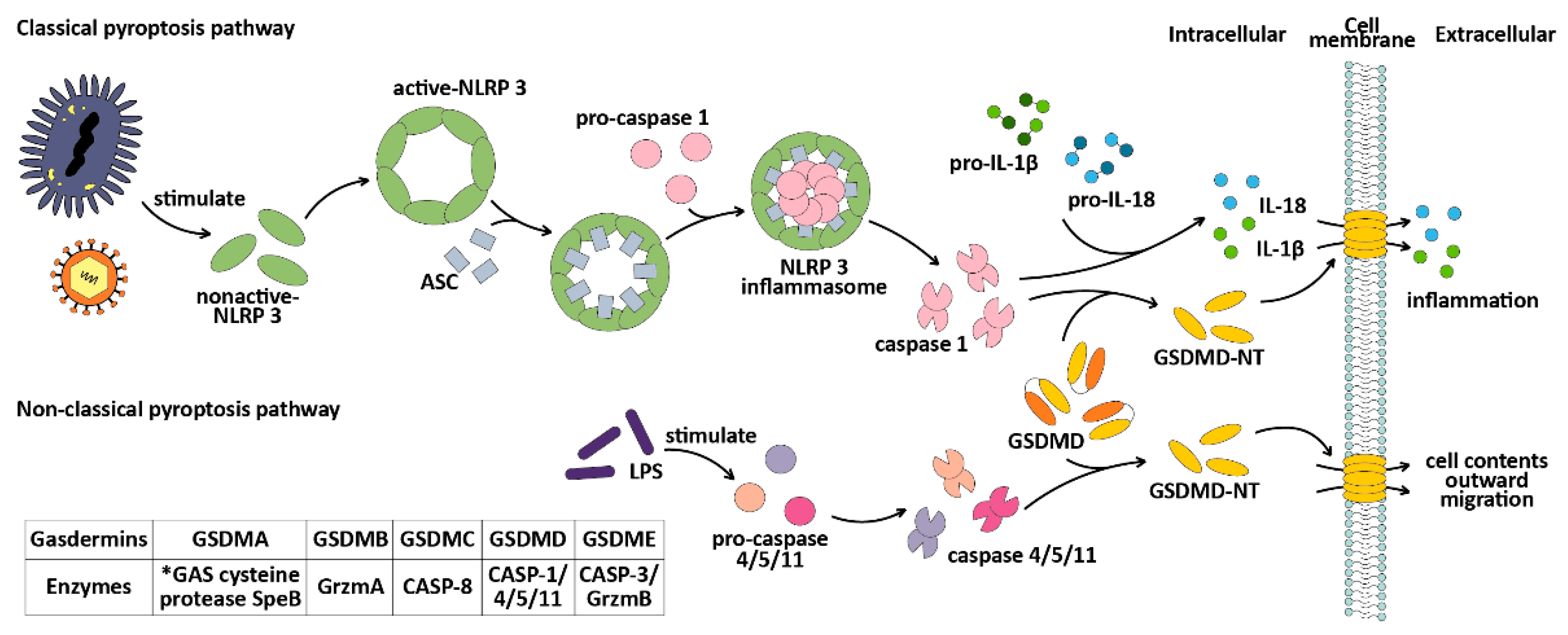
3.2. Pyroptosis
3.2.1. Pyroptosis: A PCD Induced by the GSDMs
3.2.2. Pyroptosis-Related Genes Dysregulation and Therapy in BC
3.3. Necroptosis
3.3.1. Necroptosis: A PCD Induced by RIP and MLKL
3.3.2. Necroptosis and Breast Cancer Therapy
4. Cell Death Modulated by Metal Ions in the Microenvironment
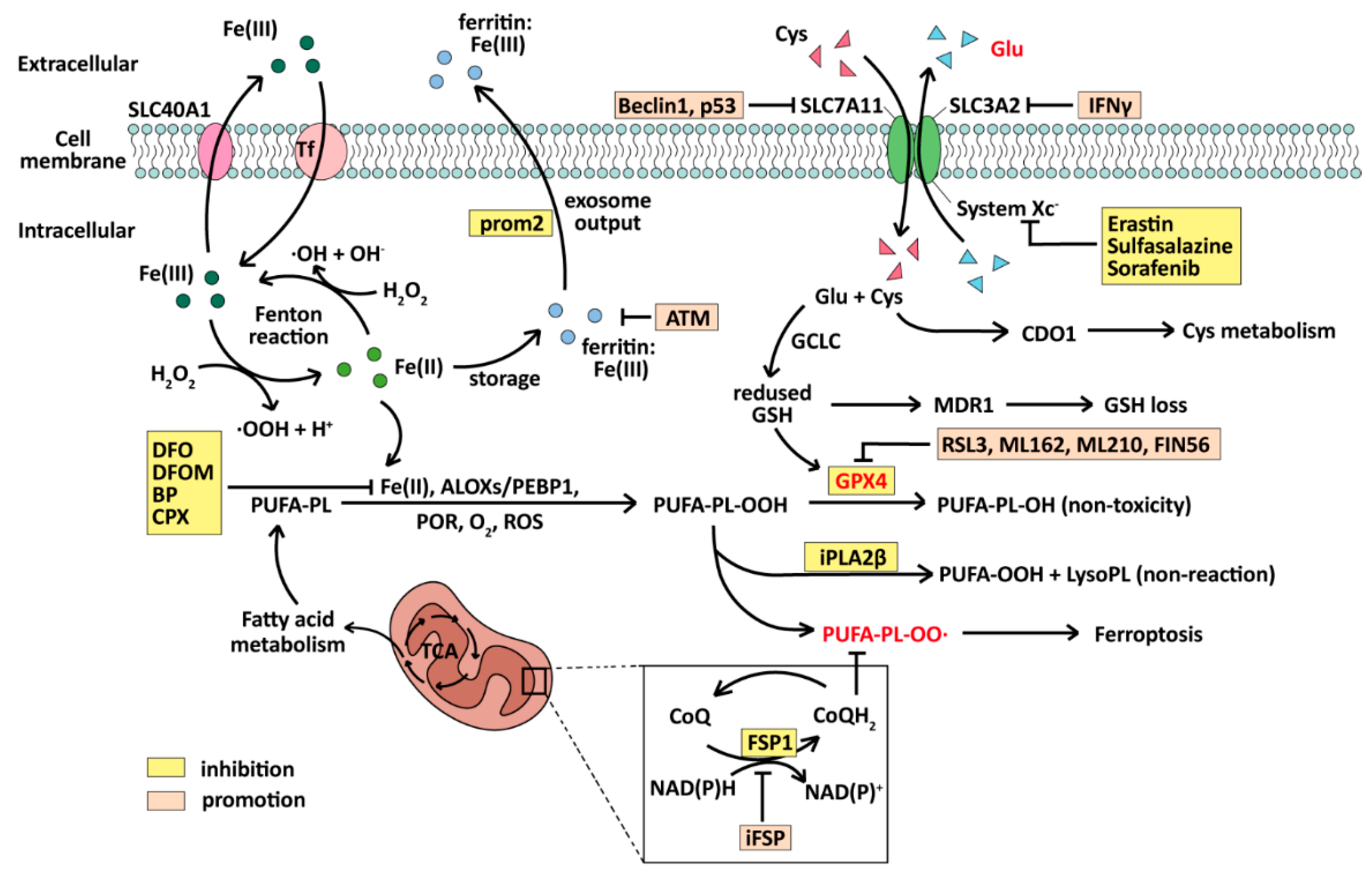
4.1. Ferroptosis
4.1.1. Ferroptosis: A PCD Induced by Fe(II) and ROS
4.1.2. Ferroptosis-Related Genes Dysregulation and Therapy in BC
4.2. Cuproptosis
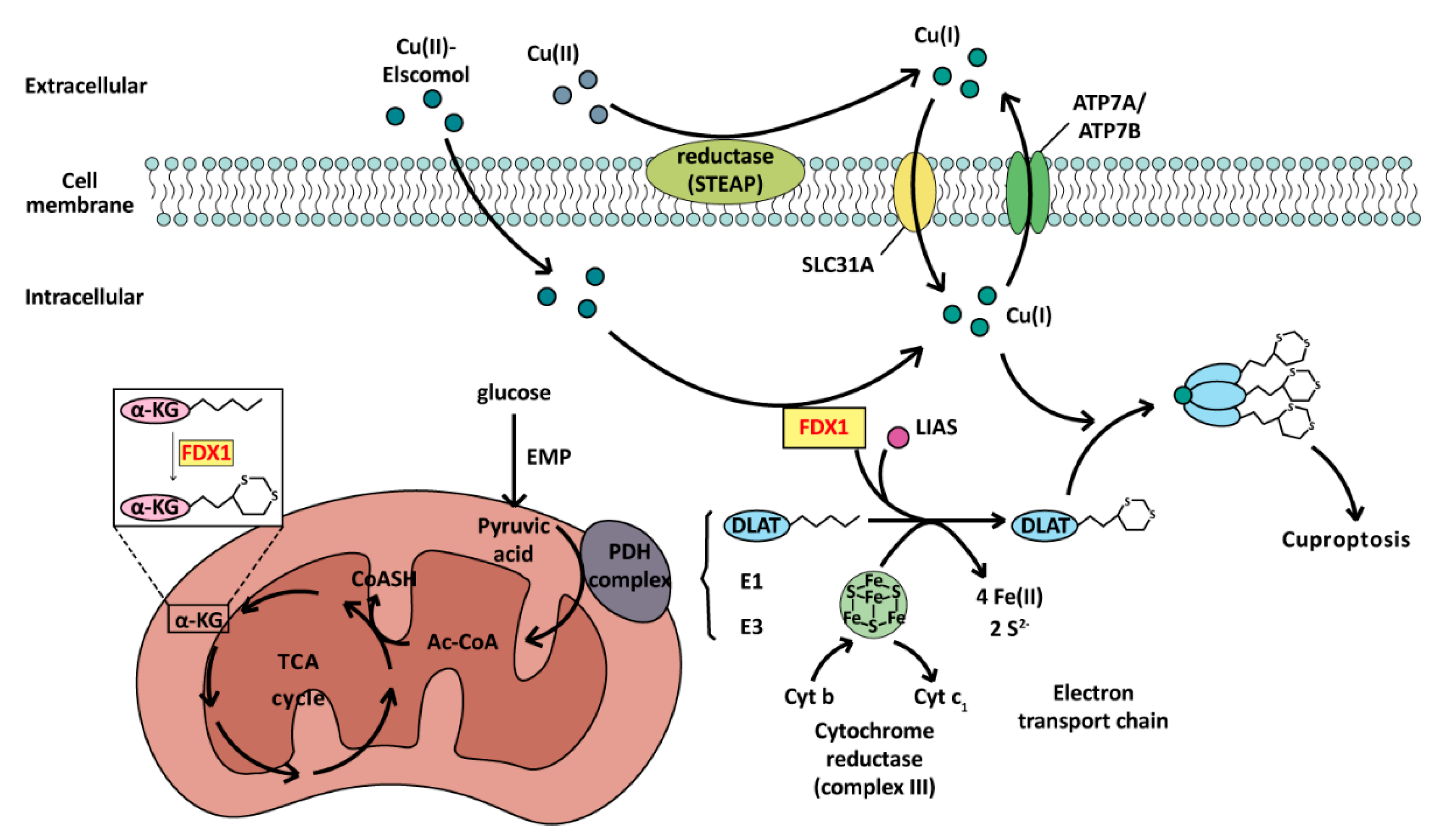
4.2.1. Cuproptosis: A PCD Induced by Cu(I)
4.2.2. Cuproptosis-Related Genes Dysregulation and Therapy in BC
5. Prospect
5.1. Beyond PANoptosis, the Other Crosstalk of Cell Death in BC
5.2. The Complexity of Cell Death in BC: Mutations and Drug Resistance
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA: A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.D.B.; Morgan, E.; Aguilar, A.d.L.; Mafra, A.; Shah, R.; Giusti, F.; Vignat, J.; Znaor, A.; Musetti, C.; Yip, C.-H.; et al. Global Stage Distribution of Breast Cancer at Diagnosis. JAMA Oncol. 2024, 10, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: a retrospective of the last decade. J. Pathol. 2009, 220, 263–280. [Google Scholar] [CrossRef]
- Prat, A.; Cheang, M.C.U.; Martín, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic Significance of Progesterone Receptor–Positive Tumor Cells Within Immunohistochemically Defined Luminal A Breast Cancer. J. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, F.; Mei, X.; Yang, R.; Lu, B.; Wang, Z.; Ji, L. Toosendanin and isotoosendanin suppress triple-negative breast cancer growth via inducing necrosis, apoptosis and autophagy. Chem. Interactions 2021, 351, 109739. [Google Scholar] [CrossRef]
- Abdullah, M.L.; Al-Shabanah, O.; Hassan, Z.K.; Hafez, M.M. Eugenol-Induced Autophagy and Apoptosis in Breast Cancer Cells via PI3K/AKT/FOXO3a Pathway Inhibition. Int. J. Mol. Sci. 2021, 22, 9243. [Google Scholar] [CrossRef]
- Ma, S.; Fu, X.; Liu, L.; Liu, Y.; Feng, H.; Jiang, H.; Liu, X.; Liu, R.; Liang, Z.; Li, M.; et al. Iron-Dependent Autophagic Cell Death Induced by Radiation in MDA-MB-231 Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, X.; Liu, C.; Ge, W.; Wang, Q.; Hao, X.; Wang, M.; Chen, Y.; Zhang, Q. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J. Hematol. Oncol. 2021, 14, 1–21. [Google Scholar] [CrossRef]
- Lee, S.Y.; Seo, J.; Kim, S.; Hwang, C.; Jeong, D.I.; Park, J.; Yang, M.; Huh, J.W.; Cho, H. Cuproptosis-Inducible Chemotherapeutic/Cascade Catalytic Reactor System for Combating with Breast Cancer. Small 2023, 19, e2301402. [Google Scholar] [CrossRef]
- D’arcy, M.S. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, D.; Liu, G.; Zhang, M.; Pan, F. Caspase-Linked Programmed Cell Death in Prostate Cancer: From Apoptosis, Necroptosis, and Pyroptosis to PANoptosis. Biomolecules 2023, 13, 1715. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Deter, R.L.; de Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Yamamoto, H.; Matsui, T. Molecular Mechanisms of Macroautophagy, Microautophagy, and Chaperone-Mediated Autophagy. J. Nippon. Med Sch. 2024, 91, 2–9. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Cocco, S.; Leone, A.; Piezzo, M.; Caputo, R.; Di Lauro, V.; Di Rella, F.; Fusco, G.; Capozzi, M.; di Gioia, G.; Budillon, A.; et al. Targeting Autophagy in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7836. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Author Correction: Autophagy maintains tumour growth through circulating arginine. Nature 2018, 565, E3. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Cesare, A.J.; Rivera, T.; Karlseder, J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature 2015, 522, 492–496. [Google Scholar] [CrossRef]
- Nassour, J.; Radford, R.; Correia, A.; Fusté, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef]
- Isaac-Lam, M.F.; DeMichael, K.M. Calorie restriction and breast cancer treatment: a mini-review. J. Mol. Med. 2022, 100, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Li, Z.; Chen, B.; Wu, Y.; Jin, F.; Xia, Y.; Liu, X. Genetic and epigenetic silencing of the beclin 1gene in sporadic breast tumors. BMC Cancer 2010, 10, 98. [Google Scholar] [CrossRef]
- Wang, M.-C.; Wu, A.-G.; Huang, Y.-Z.; Shao, G.-L.; Ji, S.-F.; Wang, R.-W.; Yuan, H.-J.; Fan, X.-L.; Zheng, L.-H.; Jiao, Q.-L. Autophagic regulation of cell growth by altered expression of Beclin 1 in triple-negative breast cancer. Int. J. Clin. Exp. Med. 2015, 8, 7049–7058. [Google Scholar] [PubMed]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Esparza-Garrido, R.R.; Torres-Márquez, M.E.; Viedma-Rodríguez, R.; Velázquez-Wong, A.C.; Salamanca-Gómez, F.; Rosas-Vargas, H.; Velázquez-Flores, M. Breast cancer cell line MDA-MB-231 miRNA profile expression after BIK interference: BIK involvement in autophagy. Tumor Biol. 2015, 37, 6749–6759. [Google Scholar] [CrossRef]
- Soni, M.; Patel, Y.; Markoutsa, E.; Jie, C.; Liu, S.; Xu, P.; Chen, H. Autophagy, Cell Viability, and Chemoresistance Are Regulated By miR-489 in Breast Cancer. Mol. Cancer Res. 2018, 16, 1348–1360. [Google Scholar] [CrossRef]
- Ueda, S.; Takanashi, M.; Sudo, K.; Kanekura, K.; Kuroda, M. miR-27a ameliorates chemoresistance of breast cancer cells by disruption of reactive oxygen species homeostasis and impairment of autophagy. Mod. Pathol. 2020, 100, 863–873. [Google Scholar] [CrossRef]
- Tian, W.; Alsaadi, R.; Guo, Z.; Kalinina, A.; Carrier, M.; Tremblay, M.-E.; Lacoste, B.; Lagace, D.; Russell, R.C. An antibody for analysis of autophagy induction. Nat. Methods 2019, 17, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Mo, J.; Yang, Y.; Wang, Y.; Lu, H. Crucial Roles of LncRNAs-Mediated Autophagy in Breast Cancer. Int. J. Med Sci. 2022, 19, 1082–1092. [Google Scholar] [CrossRef]
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein–lncRNA interaction. Briefings Bioinform. 2015, 17, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; A Yang, Y.; Zhang, A.; Fong, K.-W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 2015, 35, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qian, J.; Li, J.; Zhu, C. Knockdown of lncRNA-HOTAIR downregulates the drug-resistance of breast cancer cells to doxorubicin via the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2019, 18, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, N.; Liu, Y.; Su, P.; Liang, Y.; Li, Y.; Wang, X.; Chen, T.; Song, X.; Sang, Y.; et al. Epigenetic Regulation of NAMPT by NAMPT-ASDrives Metastatic Progression in Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 3347–3359. [Google Scholar] [CrossRef]
- Zhang, X.H.; Li, B.F.; Ding, J.; Shi, L.; Ren, H.M.; Liu, K.; Huang, C.C.; Ma, F.X.; Wu, X.Y. LncRNA DANCR-miR-758-3p-PAX6 Molecular Network Regulates Apoptosis and Autophagy of Breast Cancer Cells. Cancer Manag. Res. 2020, ume 12, 4073–4084. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 1–21. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Yee, Y.H.; Chong, S.J.F.; Kong, L.R.; Goh, B.C.; Pervaiz, S. Sustained IKKβ phosphorylation and NF-κB activation by superoxide-induced peroxynitrite-mediated nitrotyrosine modification of B56γ3 and PP2A inactivation. Redox Biol. 2020, 41, 101834. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxidants Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Nössing, C.; Ryan, K.M. 50 years on and still very much alive: ‘Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics’. Br. J. Cancer 2022, 128, 426–431. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Gores, G.J. Death receptor-mediated apoptosis and the liver. J. Hepatol. 2002, 37, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, J.D.; Gamage, K.K.; Heffron, D.S.; LeBlanc, A.C.; Deppmann, C.D.; Mandell, J.W. Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury. Acta Neuropathol. Commun. 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Johnson, P.W.; Packham, G. Mcl-1. Int. J. Biochem. Cell Biol. 2004, 37, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Tahzib, N.; Ransom, N.; Reitsamer, H.; McKinnon, S. Alpha-fodrin is cleaved by caspase-3 in a chronic ocular hypertensive (COH) rat model of glaucoma. Brain Res. Bull. 2003, 62, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Liu, X.M.; Kreis, W.; Budman, D.R. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother. Pharmacol. 1999, 44, 355–361. [Google Scholar] [CrossRef]
- Rosen, A.; Casciola-Rosen, L. Macromolecular substrates for the ICE-like proteases during apoptosis. J. Cell. Biochem. 1997, 64, 50–54. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef]
- Chin, H.S.; Li, M.X.; Tan, I.K.L.; Ninnis, R.L.; Reljic, B.; Scicluna, K.; Dagley, L.F.; Sandow, J.J.; Kelly, G.L.; Samson, A.L.; et al. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat. Commun. 2018, 9, 4976. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Chandna, S. Putative partners in Bax mediated cytochrome-c release: ANT, CypD, VDAC or none of them? Mitochondrion 2008, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Singh, G.; Guibao, C.D.; Seetharaman, J.; Aggarwal, A.; Grace, C.R.; McNamara, D.E.; Vaithiyalingam, S.; Waddell, M.B.; Moldoveanu, T. Structural basis of BAK activation in mitochondrial apoptosis initiation. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Kashyap, D.; Garg, V.K.; Goel, N. Chapter Four - Intrinsic and extrinsic pathways of apoptosis: Role in cancer development and prognosis. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Dordrecht, The Netherlands, 2021; Volume 125, pp. 73–120. [Google Scholar]
- Yang, Q.-H.; Church-Hajduk, R.; Ren, J.; Newton, M.L.; Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003, 17, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Parton, M.; Dowsett, M.; Smith, I. Studies of apoptosis in breast cancer. BMJ 2001, 322, 1528–1532. [Google Scholar] [CrossRef]
- Merino, D.; Lok, S.W.; E Visvader, J.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2015, 35, 1877–1887. [Google Scholar] [CrossRef]
- Alipour, M.; Sheikhnejad, R.; Fouani, M.H.; Bardania, H.; Hosseinkhani, S. DNAi-peptide nanohybrid smart particles target BCL-2 oncogene and induce apoptosis in breast cancer cells. Biomed. Pharmacother. 2023, 166, 115299. [Google Scholar] [CrossRef]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.E.; Smyth, G.K.; Christie, M.; Phillipson, L.J.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 Mimetic ABT-199 in Estrogen Receptor-Positive Breast Cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef]
- Ezzati, M.; Yousefi, B.; Velaei, K.; Safa, A. A review on anti-cancer properties of Quercetin in breast cancer. Life Sci 2020, 248, 117463. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Wang, D.Q.; Sui, C.G.; Meng, F.D.; Sun, S.L.; Zheng, J.; Jiang, Y.H. Oleandrin induces apoptosis via activating endoplasmic reticulum stress in breast cancer cells. Biomed Pharmacother 2020, 124, 109852. [Google Scholar] [CrossRef]
- Ding, W.; Chen, C.; Li, J.; Geng, X.; Zhang, H.; Sun, Y. Quercus acutissima Carruth. root extract triggers apoptosis, autophagy and inhibits cell viability in breast cancer cells. J. Ethnopharmacol. 2022, 289, 115039. [Google Scholar] [CrossRef] [PubMed]
- Stan, S.D.; Abtahi, M. Diallyl Trisulfide Induces Apoptosis in Breast Ductal Carcinoma In Situ Derived and Minimally Invasive Breast Cancer Cells. Nutrients 2022, 14, 1455. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.L.; Christy, B.; Yee, D. STAT6 Mediates Interleukin-4 Growth Inhibition in Human Breast Cancer Cells. Neoplasia 2002, 4, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Doseff, A.I.; Baker, J.H.; Bourgeois, T.A.; Wewers, M.D. Interleukin-4–Induced Apoptosis Entails Caspase Activation and Suppression of Extracellular Signal–Regulated Kinase Phosphorylation. Am. J. Respir. Cell Mol. Biol. 2003, 29, 367–374. [Google Scholar] [CrossRef]
- Kolben, T.; Jeschke, U.; Reimer, T.; Karsten, N.; Schmoeckel, E.; Semmlinger, A.; Mahner, S.; Harbeck, N.; Kolben, T.M. Induction of apoptosis in breast cancer cells in vitro by Fas ligand reverse signaling. J. Cancer Res. Clin. Oncol. 2017, 144, 249–256. [Google Scholar] [CrossRef]
- Wang, S.; Xu, Y.; Li, C.; Tao, H.; Wang, A.; Sun, C.; Zhong, Z.; Wu, X.; Li, P.; Wang, Y. Gambogic acid sensitizes breast cancer cells to TRAIL-induced apoptosis by promoting the crosstalk of extrinsic and intrinsic apoptotic signalings. Food Chem. Toxicol. 2018, 119, 334–341. [Google Scholar] [CrossRef]
- Breunig, C.; Pahl, J.; Küblbeck, M.; Miller, M.; Antonelli, D.; Erdem, N.; Wirth, C.; Will, R.; Bott, A.; Cerwenka, A.; et al. MicroRNA-519a-3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells. Cell Death Dis. 2017, 8, e2973. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- Campbell, M.J.; Carlberg, C.; Koeffler, H.P. A Role for the PPARγ in Cancer Therapy. PPAR Res. 2008, 2008, 314974. [Google Scholar] [CrossRef] [PubMed]
- Elstner, E.; Müller, C.; Koshizuka, K.; Williamson, E.A.; Park, D.; Asou, H.; Shintaku, P.; Said, J.W.; Heber, D.; Koeffler, H.P. Ligands for peroxisome proliferator-activated receptorγ and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc. Natl. Acad. Sci. 1998, 95, 8806–8811. [Google Scholar] [CrossRef]
- Preet, R.; Siddharth, S.; Satapathy, S.R.; Das, S.; Nayak, A.; Das, D.; Wyatt, M.D.; Kundu, C.N. Chk1 inhibitor synergizes quinacrine mediated apoptosis in breast cancer cells by compromising the base excision repair cascade. Biochem. Pharmacol. 2016, 105, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Q.; Shao, Y.; Wang, W.; Zhao, C. CD155 downregulation synergizes with adriamycin to induce breast cancer cell apoptosis. Apoptosis 2018, 23, 512–520. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Pan, T.-C.; Ruth, J.; Feng, Y.; Zhou, A.; Pant, D.; Grimley, J.S.; Wandless, T.J.; DeMichele, A.; Chodosh, L.A. Par-4 Downregulation Promotes Breast Cancer Recurrence by Preventing Multinucleation following Targeted Therapy. Cancer Cell 2013, 24, 30–44. [Google Scholar] [CrossRef]
- Van Laer, L.; Huizing, E.H.; Verstreken, M.; van Zuijlen, D.; Wauters, J.G.; Bossuyt, P.J.; Van de Heyning, P.; McGuirt, W.T.; Smith, R.J.; Willems, P.J.; et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 1998, 20, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Runkel, F.; Marquardt, A.; Stoeger, C.; Kochmann, E.; Simon, D.; Kohnke, B.; Korthaus, D.; Wattler, F.; Fuchs, H.; de Angelis, M.H.; et al. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics 2004, 84, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kang, M.; Kim, B.; Kwon, J.; Song, Y.; Choi, W.; Shin, Y.; Hong, S. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr. Pulmonol. 2011, 46, 701–708. [Google Scholar] [CrossRef]
- Li, T.; Zheng, G.; Li, B.; Tang, L. Pyroptosis: A promising therapeutic target for noninfectious diseases. Cell Prolif. 2021, 54, e13137. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Sarrió, D.; Molina-Crespo, Á.; Megias, D.; Mota, A.; Rojo-Sebastian, A.; García-Sanz, P.; Morales, S.; Abril, S.; Cano, A.; et al. Gasdermin-B Promotes Invasion and Metastasis in Breast Cancer Cells. PLOS ONE 2014, 9, e90099. [Google Scholar] [CrossRef]
- Saeki, N.; Usui, T.; Aoyagi, K.; Kim, D.H.; Sato, M.; Mabuchi, T.; Yanagihara, K.; Ogawa, K.; Sakamoto, H.; Yoshida, T.; et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes, Chromosom. Cancer 2008, 48, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Saeki, N.; Kim, D.H.; Usui, T.; Aoyagi, K.; Tatsuta, T.; Aoki, K.; Yanagihara, K.; Tamura, M.; Mizushima, H.; Sakamoto, H.; et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-β-dependent apoptotic signalling. Oncogene 2007, 26, 6488–6498. [Google Scholar] [CrossRef]
- Zou, J.; Zheng, Y.; Huang, Y.; Tang, D.; Kang, R.; Chen, R. The Versatile Gasdermin Family: Their Function and Roles in Diseases. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Saeki, N.; Kuwahara, Y.; Sasaki, H.; Satoh, H.; Shiroishi, T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome 2000, 11, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Tanaka, S.; Fujii, T.; Aoki, A.; Komiyama, H.; Ezawa, K.; Sumiyama, K.; Sagai, T.; Shiroishi, T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 2007, 89, 618–629. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Broz, P. Caspase target drives pyroptosis. Nature 2015, 526, 642–643. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Monack, D.M.; Navarre, W.W.; Falkow, S. -induced macrophage death: The role of caspase-1 in death and inflammation. Microbes Infect. 2001, 3, 1201–1212. [Google Scholar] [CrossRef]
- Johnson, A.G.; Wein, T.; Mayer, M.L.; Duncan-Lowey, B.; Yirmiya, E.; Oppenheimer-Shaanan, Y.; Amitai, G.; Sorek, R.; Kranzusch, P.J. Bacterial gasdermins reveal an ancient mechanism of cell death. Science 2022, 375, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, Y.-H.; Chen, N.-H.; Wang, H.-B. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 2018, 67, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN- γ production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of Interferon-γ Inducing Factor Mediated by Interleukin-1β Converting Enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Shi, X.; Sun, Q.; Hou, Y.; Zeng, H.; Cao, Y.; Dong, M.; Ding, J.; Shao, F. Recognition and maturation of IL-18 by caspase-4 noncanonical inflammasome. Nature 2023, 624, 442–450. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Xia, S.; Pan, X.; Ye, K.; Li, Z.; Li, H.; Tang, X.; Sahni, N.; Yi, S.S.; Liu, X.; et al. Alternative splicing of GSDMB modulates killer lymphocyte–triggered pyroptosis. Sci. Immunol. 2023, 8, eadg3196. [Google Scholar] [CrossRef]
- Wang, S.; Chang, C.-W.; Huang, J.; Zeng, S.; Zhang, X.; Hung, M.-C.; Hou, J. Gasdermin C sensitizes tumor cells to PARP inhibitor therapy in cancer models. J. Clin. Investig. 2024, 134. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.-W.; You, Y.; Hsu, J.-M.; Nie, L.; Chen, Y.; Wang, Y.-C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Ma, L.; Lan, F.; Zheng, Z.; Xie, F.; Wang, L.; Liu, W.; Han, J.; Zheng, F.; Xie, Y.; Huang, Q. Epidermal growth factor (EGF) and interleukin (IL)-1β synergistically promote ERK1/2-mediated invasive breast ductal cancer cell migration and invasion. Mol. Cancer 2012, 11, 79. [Google Scholar] [CrossRef]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar] [CrossRef]
- Han, J.; Bae, S.Y.; Oh, S.; Lee, J.; Lee, J.H.; Lee, H.; Lee, S.K.; Kil, W.H.; Kim, S.W.; Nam, S.J.; et al. Zerumbone Suppresses IL-1β-induced Cell Migration and Invasion by Inhibiting IL-8 and MMP-3 Expression in Human Triple-negative Breast Cancer Cells. Phytotherapy Res. 2014, 28, 1654–1660. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Huang, X.; Li, C.; Guan, N.; Pan, T.; Dong, J.; Li, L. Effect of tumor-associated macrophages on the pyroptosis of breast cancer tumor cells. Cell Commun. Signal. 2023, 21, 197. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Fang, P.; Leng, M.; Shi, Y. Promoting GSDME expression through DNA demethylation to increase chemosensitivity of breast cancer MCF-7 / Taxol cells. PLOS ONE 2023, 18, e0282244. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Li, Y.; Li, W.; Lian, S.; Li, Y.; Wu, C.; Zhang, K.; Zhou, G.; Wang, W.; Xu, H.; et al. Ganoderma lucidum extract promotes tumor cell pyroptosis and inhibits metastasis in breast cancer. Food Chem. Toxicol. 2023, 174, 113654. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, H.; Li, D.; Zhou, X.; Qin, Q.; Zhang, Q. Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J. Cell. Mol. Med. 2021, 25, 8159–8168. [Google Scholar] [CrossRef]
- An, H.; Heo, J.S.; Kim, P.; Lian, Z.; Lee, S.; Park, J.; Hong, E.; Pang, K.; Park, Y.; Ooshima, A.; et al. Tetraarsenic hexoxide enhances generation of mitochondrial ROS to promote pyroptosis by inducing the activation of caspase-3/GSDME in triple-negative breast cancer cells. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Molina-Crespo, Á.; Cadete, A.; Sarrio, D.; Gámez-Chiachio, M.; Martinez, L.; Chao, K.; Olivera, A.; Gonella, A.; Díaz, E.; Palacios, J.; et al. Intracellular Delivery of an Antibody Targeting Gasdermin-B Reduces HER2 Breast Cancer Aggressiveness. Clin. Cancer Res. 2019, 25, 4846–4858. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Sarrio, D.; Molina-Crespo, Á.; Vicario, R.; Bernadó-Morales, C.; Martínez, L.; Rojo-Sebastián, A.; Serra-Musach, J.; Mota, A.; Martínez-Ramírez, Á.; et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 2016, 7, 56295–56308. [Google Scholar] [CrossRef]
- Oltra, S.S.; Colomo, S.; Sin, L.; Pérez-López, M.; Lázaro, S.; Molina-Crespo, A.; Choi, K.-H.; Ros-Pardo, D.; Martínez, L.; Morales, S.; et al. Distinct GSDMB protein isoforms and protease cleavage processes differentially control pyroptotic cell death and mitochondrial damage in cancer cells. Cell Death Differ. 2023, 30, 1366–1381. [Google Scholar] [CrossRef]
- Sarrio, D.; Rojo-Sebastián, A.; Teijo, A.; Pérez-López, M.; Díaz-Martín, E.; Martínez, L.; Morales, S.; García-Sanz, P.; Palacios, J.; Moreno-Bueno, G. Gasdermin-B Pro-Tumor Function in Novel Knock-in Mouse Models Depends on the in vivo Biological Context. Front. Cell Dev. Biol. 2022, 10, 813929. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Chen, R.-X.; Li, J.-Z.; Luo, Z.-X. LINC00511/hsa-miR-573 axis-mediated high expression of Gasdermin C associates with dismal prognosis and tumor immune infiltration of breast cancer. Sci. Rep. 2022, 12, 1–20. [Google Scholar] [CrossRef]
- Cui, Y.-Q.; Meng, F.; Zhan, W.-L.; Dai, Z.-T.; Liao, X. High Expression of GSDMC Is Associated with Poor Survival in Kidney Clear Cell Cancer. BioMed Res. Int. 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Liu, X.; Miao, M.; Sun, J.; Wu, J.; Qin, X. PANoptosis: A potential new target for programmed cell death in breast cancer treatment and prognosis. Apoptosis 2023, 29, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zhang, W.; Yang, T.; He, S.-D. Complex roles of necroptosis in cancer. J. Zhejiang Univ. B 2019, 20, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.-B.; Morgan, M.J.; Lee, D.-G.; Kim, W.-J.; Yoon, J.-H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Cai, Z.; Choksi, S.; Ma, D.; Choe, M.; Kwon, H.-J.; Baik, J.Y.; Rowan, B.G.; Liu, C.; Liu, Z.-G. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018, 28, 868–870. [Google Scholar] [CrossRef]
- Stoll, G.; Ma, Y.; Yang, H.; Kepp, O.; Zitvogel, L.; Kroemer, G. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. OncoImmunology 2017, 6, e1299302. [Google Scholar] [CrossRef]
- Jin, G.; Lan, Y.; Han, F.; Sun, Y.; Liu, Z.; Zhang, M.; Liu, X.; Zhang, X.; Hu, J.; Liu, H.; et al. Smac mimetic-induced caspase-independent necroptosis requires RIP1 in breast cancer. Mol. Med. Rep. 2015, 13, 359–366. [Google Scholar] [CrossRef]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.R.; Mansouri, E.; Khodadadi, A. Quercetin induces apoptosis and necroptosis in MCF-7 breast cancer cells. Bratisl. Med J. 2017, 118, 123–128. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.-M.; Zhou, C.; Li, Q.; Ma, L.; Zhang, Z.; Sun, Y.; Wang, L.; Zhang, X.; Zhu, B.; et al. Non-benzoquinone geldanamycin analogs trigger various forms of death in human breast cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 1–13. [Google Scholar] [CrossRef]
- Flamme, M.; Cressey, P.B.; Lu, C.; Bruno, P.M.; Eskandari, A.; Hemann, M.T.; Hogarth, G.; Suntharalingam, K. Induction of Necroptosis in Cancer Stem Cells using a Nickel(II)-Dithiocarbamate Phenanthroline Complex. Chem. A Eur. J. 2017, 23, 9674–9682. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Tang, Z.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Zhang, D.; Gao, H.; Sun, H.; Zhang, J.; Gu, P. HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 2021, 43, 101971. [Google Scholar] [CrossRef] [PubMed]
- Reyhani, A.; McKenzie, T.G.; Fu, Q.; Qiao, G.G. Fenton-Chemistry-Mediated Radical Polymerization. Macromol. Rapid Commun. 2019, 40, e1900220. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Chen, X.; Linkermann, A.; Jiang, X.; Kang, R.; Kagan, V.E.; Bayir, H.; Yang, W.S.; Garcia-Saez, A.J.; Ioannou, M.S.; et al. A guideline on the molecular ecosystem regulating ferroptosis. Nat. Cell Biol. 2024, 1–11. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef]
- Liu, M.-R.; Zhu, W.-T.; Pei, D.-S. System Xc−: a key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 1986, 261, 2256–2263. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2020, 12, 599–620. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Berghe, T.V.; Augustyns, K. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Z.; Liu, G.; Zhang, Y.; Liu, S.; Gan, D.; Chang, W.; Peng, X.; Sung, E.S.; Gilbert, K.; et al. Neutrophils resist ferroptosis and promote breast cancer metastasis through aconitate decarboxylase 1. Cell Metab. 2023, 35, 1688–1703.e10. [Google Scholar] [CrossRef]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegué, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–75. [Google Scholar] [CrossRef]
- Monaco, M.E.; Creighton, C.J.; Lee, P.; Zou, X.; Topham, M.K.; Stafforini, D.M. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity. Transl. Oncol. 2010, 3, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, B.; Zhao, Y.; Tao, Z.; Wang, Y.; Chen, G.; Hu, X. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J. Hematol. Oncol. 2022, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2016, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xiao, Y.; Ding, J.-H.; Jin, X.; Ma, D.; Li, D.-Q.; Shi, J.-X.; Huang, W.; Wang, Y.-P.; Jiang, Y.-Z.; et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023, 35, 84–100.e8. [Google Scholar] [CrossRef]
- Zou, Y.; Zheng, S.; Xie, X.; Ye, F.; Hu, X.; Tian, Z.; Yan, S.-M.; Yang, L.; Kong, Y.; Tang, Y.; et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Chen, M.-S.; Wang, S.-F.; Hsu, C.-Y.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C.; Tseng, L.-M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef]
- Li, H.; Yang, P.; Wang, J.; Zhang, J.; Ma, Q.; Jiang, Y.; Wu, Y.; Han, T.; Xiang, D. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J. Hematol. Oncol. 2022, 15, 1–6. [Google Scholar] [CrossRef]
- Xue, Q.; Yan, D.; Li, X.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Chen, X.; Tang, D.; Liu, J. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy 2023, 19, 1982–1996. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Balamurugan, K.; Schaffner, W. Copper homeostasis in eukaryotes: Teetering on a tightrope. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2006, 1763, 737–746. [Google Scholar] [CrossRef]
- Kim, B.-E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Guo, Z.; Zhao, L.; Wei, Y. The copper age in cancer treatment: From copper metabolism to cuproptosis. Prog. Mater. Sci. 2023, 138. [Google Scholar] [CrossRef]
- Uauy, R.; Olivares, M.; Gonzalez, M. Essentiality of copper in humans. Am. J. Clin. Nutr. 1998, 67, 952S–959S. [Google Scholar] [CrossRef] [PubMed]
- Uriu-Adams, J.Y.; Keen, C.L. Copper, oxidative stress, and human health. Mol. Asp. Med. 2005, 26, 268–298. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Z.; Zhang, S.; Zhang, T.; Liu, Y.; Zhang, L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics 2023, 13, 736–766. [Google Scholar] [CrossRef]
- Tao, X.; Wan, X.; Wu, D.; Song, E.; Song, Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A.1 macrophage. J. Hazard. Mater. 2021, 411, 125134. [Google Scholar] [CrossRef]
- Saporito-Magriñá, C.M.; Musacco-Sebio, R.N.; Andrieux, G.; Kook, L.; Orrego, M.T.; Tuttolomondo, M.V.; Desimone, M.F.; Boerries, M.; Borner, C.; Repetto, M.G. Copper-induced cell death and the protective role of glutathione: The implication of impaired protein folding rather than oxidative stress. Metallomics 2018, 10, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Dreishpoon, M.B.; Bick, N.R.; Petrova, B.; Warui, D.M.; Cameron, A.; Booker, S.J.; Kanarek, N.; Golub, T.R.; Tsvetkov, P. FDX1 regulates cellular protein lipoylation through direct binding to LIAS. J. Biol. Chem. 2023, 299, 105046. [Google Scholar] [CrossRef]
- Ni, M.; Solmonson, A.; Pan, C.; Yang, C.; Li, D.; Notzon, A.; Cai, L.; Guevara, G.; Zacharias, L.G.; Faubert, B.; et al. Functional Assessment of Lipoyltransferase-1 Deficiency in Cells, Mice, and Humans. Cell Rep. 2019, 27, 1376–1386.e6. [Google Scholar] [CrossRef]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, K.; Cymbaluk-Płoska, A. The Role of Zinc and Copper in Gynecological Malignancies. Nutrients 2020, 12, 3732. [Google Scholar] [CrossRef]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel Role of Antioxidant-1 (Atox1) as a Copper-dependent Transcription Factor Involved in Cell Proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clinical cancer research: An official journal of the American Association for Cancer Research 2000, 6, 1–10. [Google Scholar] [PubMed]
- Redman, B.G.; Esper, P.; Pan, Q.; Dunn, R.L.; Hussain, H.K.; Chenevert, T.; Brewer, G.J.; Merajver, S.D. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. Off. J Am. Assoc. Cancer Res. 2003, 9, 1666–1672. [Google Scholar] [PubMed]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Lu, Y.; Pan, Q.; Gao, W.; Pu, Y.; He, B. Reversal of cisplatin chemotherapy resistance by glutathione-resistant copper-based nanomedicineviacuproptosis. J. Mater. Chem. B 2022, 10, 6296–6306. [Google Scholar] [CrossRef]
- Guo, B.; Yang, F.; Zhang, L.; Zhao, Q.; Wang, W.; Yin, L.; Chen, D.; Wang, M.; Han, S.; Xiao, H.; et al. Cuproptosis Induced by ROS Responsive Nanoparticles with Elesclomol and Copper Combined with αPD-L1 for Enhanced Cancer Immunotherapy. Adv. Mater. 2023, 35, e2212267. [Google Scholar] [CrossRef]
- Yang, X.; Deng, L.; Diao, X.; Yang, S.; Zou, L.; Yang, Q.; Li, J.; Nie, J.; Zhao, L.; Jiao, B. Targeting cuproptosis by zinc pyrithione in triple-negative breast cancer. iScience 2023, 26, 108218. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Si, L.; Wu, X.; Chen, Y.; Xiong, H.; Xu, Y.; Liu, W.; Mei, H.; Wang, T.; Li, M. Prognostic analysis of cuproptosis-related gene in triple-negative breast cancer. Front. Immunol. 2022, 13, 922780. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, L.; Sun, Q. High expression of cuproptosis-related SLC31A1 gene in relation to unfavorable outcome and deregulated immune cell infiltration in breast cancer: An analysis based on public databases. BMC Bioinform. 2022, 23, 350. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-H.; Cheng, T.-C.; Zhu, B.; Gao, H.-Y.; Zheng, L.; Chen, W.-X. Identification of cuproptosis-related gene SLC31A1 and upstream LncRNA-miRNA regulatory axis in breast cancer. Sci. Rep. 2023, 13, 1–15. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Liu, N.; Shi, Y.; Liu, Y.; Ouyang, L.; Tam, S.; Xiao, D.; Liu, S.; Wen, F.; et al. A Nuclear Long Non-Coding RNA LINC00618 Accelerates Ferroptosis in a Manner Dependent upon Apoptosis. Mol. Ther. 2021, 29, 263–274. [Google Scholar] [CrossRef]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Glorieux, C.; Liu, S.; Trachootham, D.; Huang, P. Targeting ROS in cancer: Rationale and strategies. Nat. Rev. Drug Discov. 2024, 23, 583–606. [Google Scholar] [CrossRef]
- E Chipuk, J.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, H.; Weng, C.; Jiang, H.; Chen, J. Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Merkel, M.; Goebel, B.; Boll, M.; Adhikari, A.; Maurer, V.; Steinhilber, D.; Culmsee, C. Mitochondrial Reactive Oxygen Species Formation Determines ACSL4/LPCAT2-Mediated Ferroptosis. Antioxidants 2023, 12, 1590. [Google Scholar] [CrossRef]
- Springer, C.; Humayun, D.; Skouta, R. Cuproptosis: Unraveling the Mechanisms of Copper-Induced Cell Death and Its Implication in Cancer Therapy. Cancers 2024, 16, 647. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-F.; Hu, P.-S.; Wang, Y.-Y.; Tan, Y.-T.; Yu, K.; Liao, K.; Wu, Q.-N.; Li, T.; Meng, Q.; Lin, J.-Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, P.; Li, Y.; Zhang, D.; Chu, F.; Yuan, F.; Pan, B.; Gao, F. A Bimetallic Polymerization Network for Effective Increase in Labile Iron Pool and Robust Activation of cGAS/STING Induces Ferroptosis-Based Tumor Immunotherapy. Small 2023, 20, e2308397. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Long, Y.C.; Shen, H.-M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef]
- Kma, L.; Baruah, T.J. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol. Appl. Biochem. 2021, 69, 248–264. [Google Scholar] [CrossRef]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Zhang, C.; Xue, L.; Yang, L. Expression and clinical significance of MAPK and EGFR in triple-negative breast cancer. Oncol. Lett. 2020, 19, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- El Hejjioui, B.; Lamrabet, S.; Joutei, S.A.; Senhaji, N.; Bouhafa, T.; Malhouf, M.A.; Bennis, S.; Bouguenouch, L. New Biomarkers and Treatment Advances in Triple-Negative Breast Cancer. Diagnostics 2023, 13, 1949. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yuan, L.X.; Du, Y.; Esteva, F.J. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: Effects on insulin-like growth factor I signaling. Mol. Cancer Ther. 2007, 6, 667–674. [Google Scholar] [CrossRef]
- Burris, H.A. Dual Kinase Inhibition in the Treatment of Breast Cancer: Initial Experience with the EGFR/ErbB-2 Inhibitor Lapatinib. Oncol. 2004, 9, 10–15. [Google Scholar] [CrossRef]
- Shen, M.; Cao, S.; Long, X.; Xiao, L.; Yang, L.; Zhang, P.; Li, L.; Chen, F.; Lei, T.; Gao, H.; et al. DNAJC12 causes breast cancer chemotherapy resistance by repressing doxorubicin-induced ferroptosis and apoptosis via activation of AKT. Redox Biol. 2024, 70, 103035. [Google Scholar] [CrossRef]
- Wang, L.; Sun, J.; Yin, Y.; Sun, Y.; Ma, J.; Zhou, R.; Chang, X.; Li, D.; Yao, Z.; Tian, S.; et al. Transcriptional coregualtor NUPR1 maintains tamoxifen resistance in breast cancer cells. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Chen, C.; Cheng, Y.; Lei, H.; Feng, X.; Zhang, H.; Qi, L.; Wan, J.; Xu, H.; Zhao, X.; Zhang, Y.; et al. SHP2 potentiates anti-PD-1 effectiveness through intervening cell pyroptosis resistance in triple-negative breast cancer. Biomed. Pharmacother. 2023, 168, 115797. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; He, Z.; Yao, F.; Liao, S.; Sun, K.; Sun, S.; Li, Z.; Wang, Z. Role of Escin in breast cancer therapy: Potential mechanism for inducing ferroptosis and synergistic antitumor activity with cisplatin. Apoptosis 2023, 28, 1154–1167. [Google Scholar] [CrossRef]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, J.; Zhao, L.; Xu, L.; Zhao, J.; Tang, Y.; Yu, M.; Lin, J.; Ding, L.; Cui, Q. Cell Death: Mechanisms and Potential Targets in Breast Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 9703. https://doi.org/10.3390/ijms25179703
Qian J, Zhao L, Xu L, Zhao J, Tang Y, Yu M, Lin J, Ding L, Cui Q. Cell Death: Mechanisms and Potential Targets in Breast Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(17):9703. https://doi.org/10.3390/ijms25179703
Chicago/Turabian StyleQian, Jiangying, Linna Zhao, Ling Xu, Jin Zhao, Yongxu Tang, Min Yu, Jie Lin, Lei Ding, and Qinghua Cui. 2024. "Cell Death: Mechanisms and Potential Targets in Breast Cancer Therapy" International Journal of Molecular Sciences 25, no. 17: 9703. https://doi.org/10.3390/ijms25179703
APA StyleQian, J., Zhao, L., Xu, L., Zhao, J., Tang, Y., Yu, M., Lin, J., Ding, L., & Cui, Q. (2024). Cell Death: Mechanisms and Potential Targets in Breast Cancer Therapy. International Journal of Molecular Sciences, 25(17), 9703. https://doi.org/10.3390/ijms25179703