
Transcriptome Analysis and Identification of Genes Associated with Cotton Seed Size

Abstract
:1. Introduction
2. Results
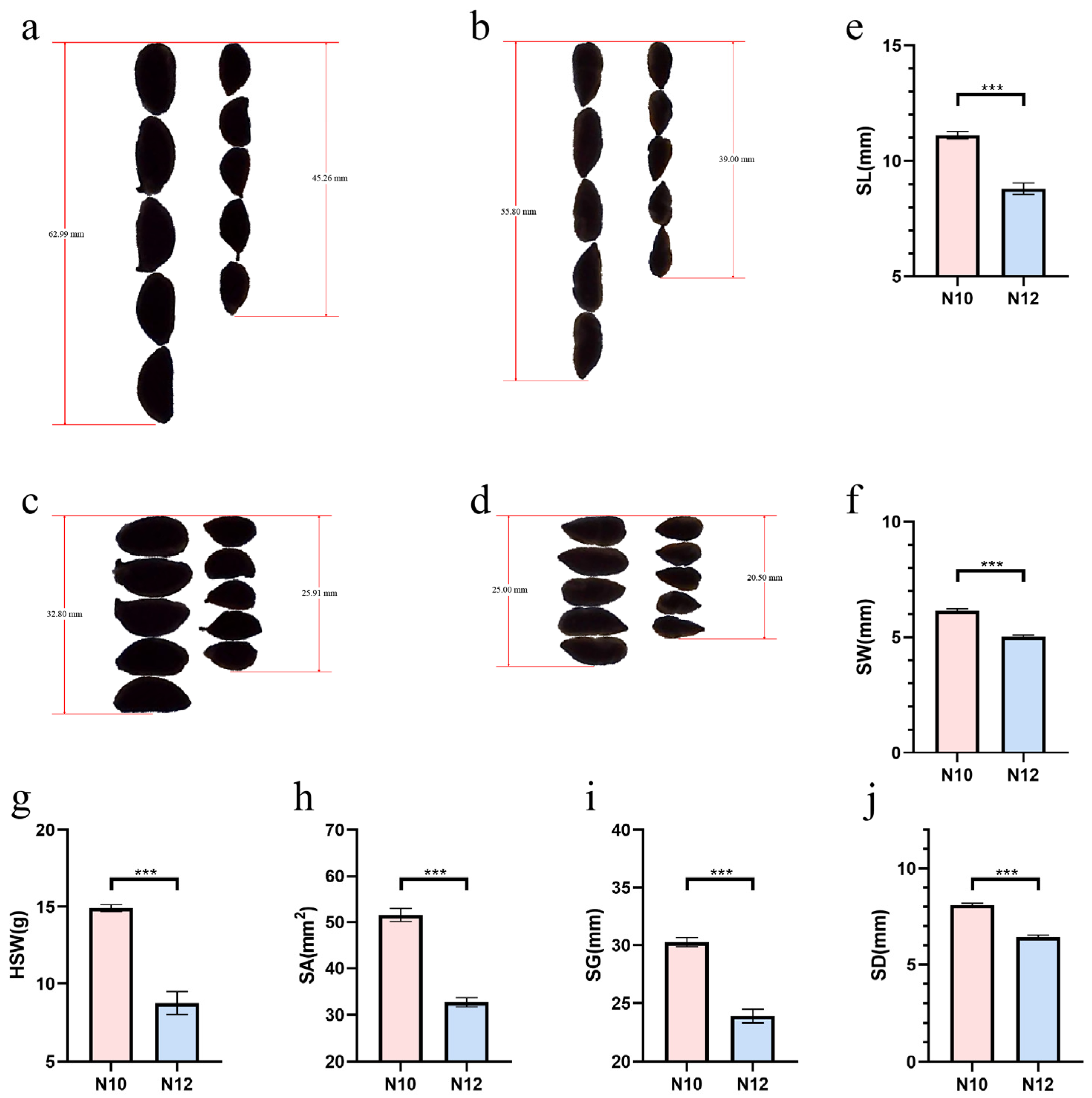
2.1. Obtaining and Phenotypic Analysis of Cotton Seed Size Materials
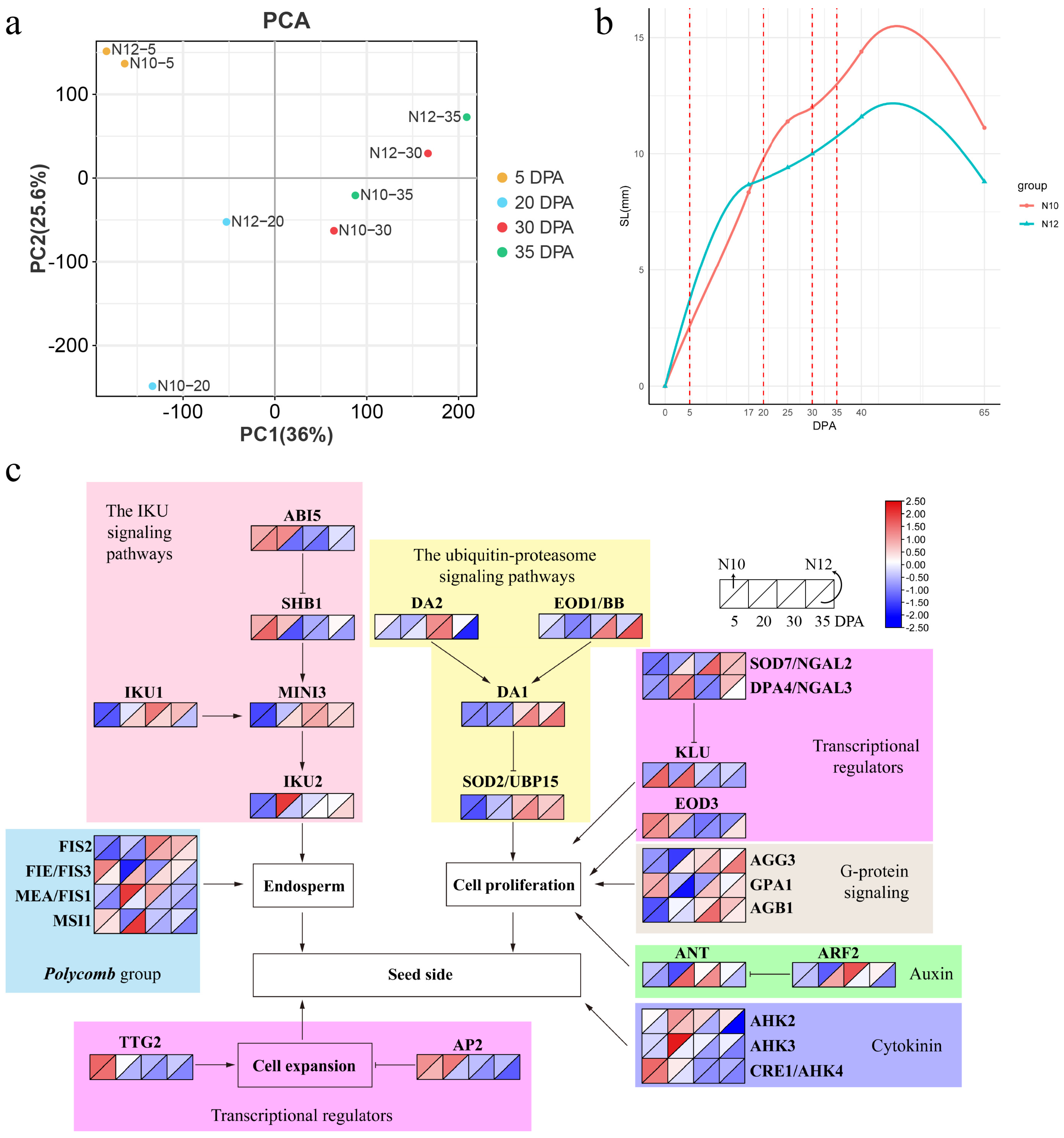
2.2. Transcriptome Assembly and Sample Clustering
2.3. Cottonseed Growth Curve and Gene Expression Network
2.4. Transcriptome Differences between N10 and N12 during Seed Development
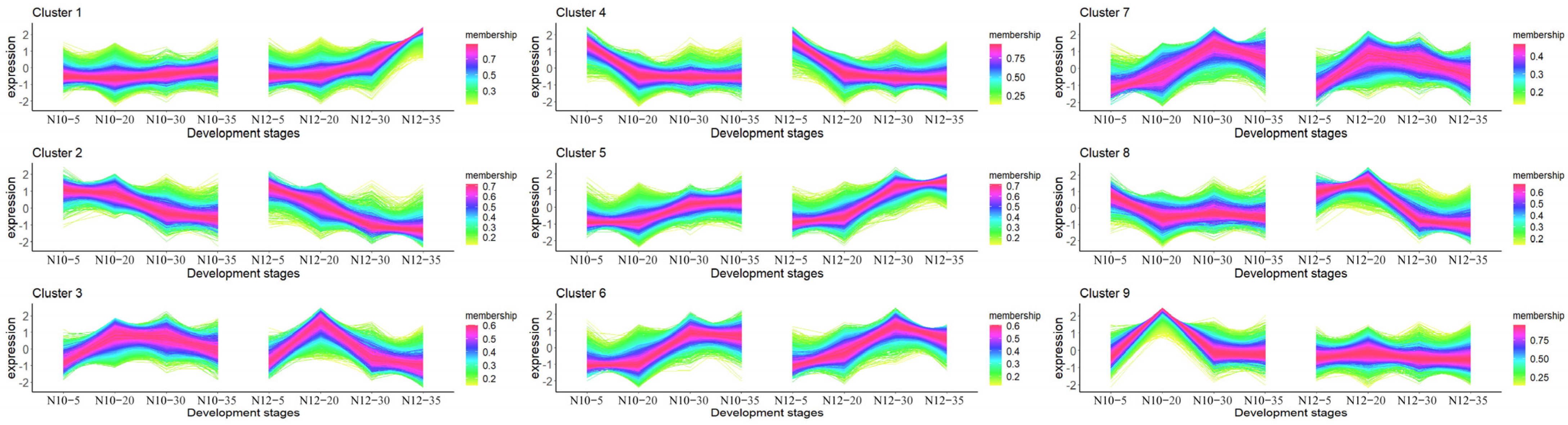
2.5. Temporal Pattern Clusters of N10 and N12
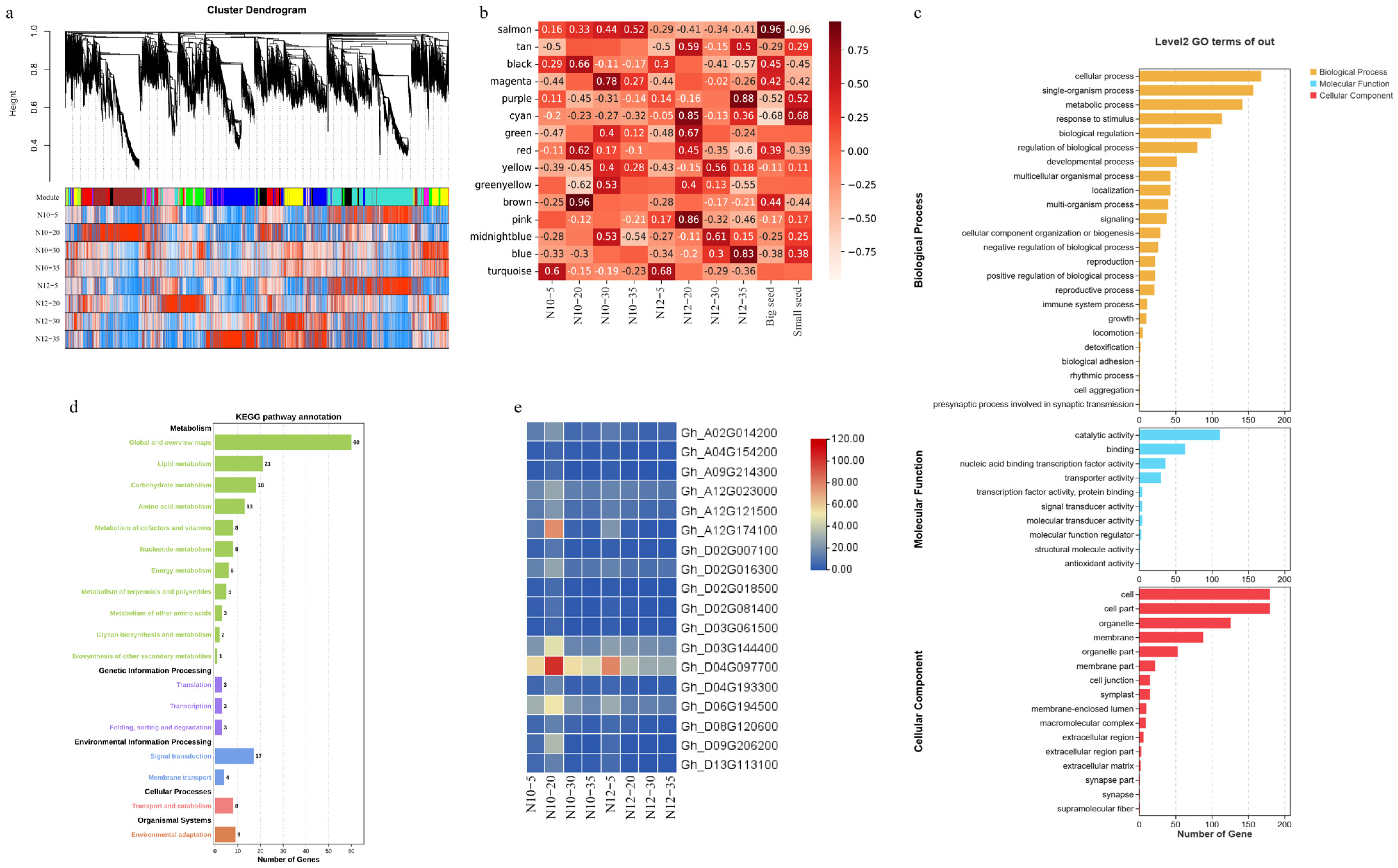
2.6. Establishment of Weighed Gene Co-Expression Network Analysis (WGCNA)
2.7. Cottonseed Size Candidate Gene Screening
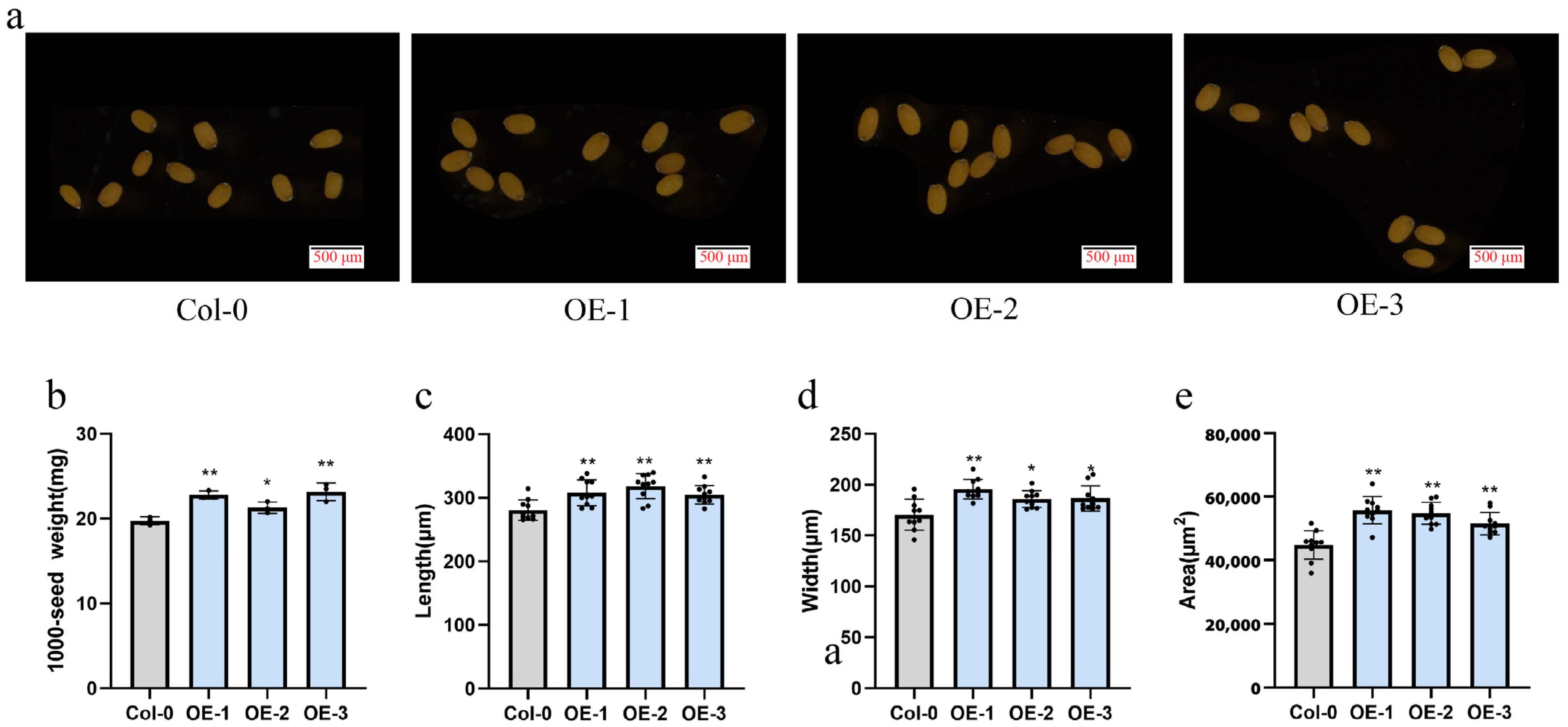
2.8. GhUXS5 Is a Candidate Gene That Affects Seed Size Function
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sampling
4.2. RNA Extraction, Library Construction, and RNA-Seq Analysis
4.3. Principal Component Analysis, Ovule In Vitro Culture, and Growth Curves
4.4. Gene Expression Network
4.5. Differential Expression Analysis
4.6. Clusters of Temporal Patterns
4.7. WGCNA Pipeline
4.8. Phenotypic Identification of Arabidopsis Overexpressed GhUXS5
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Liu, Y.; Chen, Y.; Zhang, Y.; Zan, X. Design and preparation of polysulfide flexible polymers based on cottonseed oil and its derivatives. Polymers 2020, 12, 1858. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L. Recent advances in understanding cotton fibre and seed development. Seed Sci. Res. 2005, 15, 269–280. [Google Scholar] [CrossRef]
- Dowd, M.K.; Pelitire, S.M.; Delhom, C.D. Seed-Fiber Ratio, Seed Index, and Seed Tissue and Compositional Properties of Current Cotton Cultivars. J. Cotton Sci. 2018, 22, 60–74. [Google Scholar] [CrossRef]
- Zhao, J.; Bai, W.Q.; Zeng, Q.W.; Song, S.Q.; Zhang, M.; Li, X.B.; Hou, L.; Xiao, Y.H.; Luo, M.; Li, D.M.; et al. Moderately enhancing cytokinin level by down-regulation of GhCKX expression in cotton concurrently increases fiber and seed yield. Mol. Breed. 2015, 35, 11. [Google Scholar] [CrossRef]
- Rehman, A.U.; Kamran, M.; Afzal, I. Production and processin of quality cotton seed. In Cotton Production and Uses; Springer: Singapore, 2020; pp. 547–570. [Google Scholar]
- Silvertown, J. The paradox of seed size and adaptation. Trends Ecol. Evol. 1989, 4, 24–26. [Google Scholar] [CrossRef]
- Chaudhury, A.M.; Koltunow, A.; Payne, T.; Luo, M.; Tucker, M.R.; Dennis, E.; Peacock, W. Control of early seed development. Annu. Rev. Cell Dev. Biol. 2001, 17, 677–699. [Google Scholar] [CrossRef]
- Figueiredo, D.D.; Köhler, C. Signalling events regulating seed coat development. Biochem. Soc. Trans. 2014, 42, 358–363. [Google Scholar] [CrossRef]
- Cai, G.Q.; Yang, Q.Y.; Yang, Q.; Zhao, Z.X.; Chen, H.; Wu, J.; Fan, C.C.; Zhou, Y.M. Identification of candidate genes of QTLs for seed weight in Brassica napus through comparative mapping among Arabidopsis and Brassica species. BMC Genet. 2012, 13, 105. [Google Scholar] [CrossRef]
- Kesavan, M.; Song, J.T.; Seo, H.S. Seed size: A priority trait in cereal crops. Physiol. Plant. 2013, 147, 113–120. [Google Scholar] [CrossRef]
- Sun, X.D.; Shantharaj, D.; Kang, X.J.; Ni, M. Transcriptional and hormonal signaling control of Arabidopsis seed development. Curr. Opin. Plant Biol. 2010, 13, 611–620. [Google Scholar] [CrossRef]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.J.; Kang, X.J.; Zhao, X.Y.; Zhang, X.S.; Ni, M. SHORT HYPOCOTYL UNDER BLUE1 Associates with MINISEED3 and HAIKU2 Promoters In Vivo to Regulate Arabidopsis Seed Development. Plant Cell 2009, 21, 106–117. [Google Scholar] [CrossRef]
- Johnson, C.S.; Kolevski, B.; Smyth, D.R. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell 2002, 14, 1359–1375. [Google Scholar] [CrossRef] [PubMed]
- Jofuku, K.D.; Omidyar, P.K.; Gee, Z.; Okamuro, J.K. Control of seed mass and seed yield by the floral homeotic gene APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3117–3122. [Google Scholar] [CrossRef]
- Riefler, M.; Novak, O.; Strnad, M.; Schmulling, T. Arabidopsis cytokinin receptor mutants reveal functions in shoot growth, leaf senescence, seed size, germination, root development, and cytokinin metabolism. Plant Cell 2006, 18, 40–54. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Li, Y.H. Molecular Networks of Seed Size Control in Plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef]
- Ryser, U.; Schorderet, M.; Jauch, U.; Meier, H. Ultrastructure of the “fringe-layer”, the innermost epidermis of cotton seed coats. Protoplasma 1988, 147, 81–90. [Google Scholar] [CrossRef]
- Ruan, Y.L.; Chourey, P.S.; Delmer, D.P.; PerezGrau, L. The differential expression of sucrose synthase in relation to diverse patterns of carbon partitioning in developing cotton seed. Plant. Physiol. 1997, 115, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Pugh, D.A.; Offler, C.E.; Talbot, M.J.; Ruan, Y.L. Evidence for the Role of Transfer Cells in the Evolutionary Increase in Seed and Fiber Biomass Yield in Cotton. Mol. Plant 2010, 3, 1075–1086. [Google Scholar] [CrossRef]
- West, M.A.L.; Harada, J.J. Embryogenesis in Higher Plants: An Overview. Plant Cell 1993, 5, 1361–1369. [Google Scholar] [CrossRef]
- Reeves, R.G.; Beasley, J.O. The development of the cotton embryo. J. Agric. Res. 1935, 51, 935–944. [Google Scholar]
- Pollock, E.G.; Jensen, W.A. Cell development during early embryogenesis in Capsella and Gossypium. Am. J. Bot. 1964, 51, 915–921. [Google Scholar] [CrossRef]
- Yeung, E.C.; Meinke, D.W. Embryogenesis in Angiosperms: Development of the Suspensor. Plant Cell 1993, 5, 1371–1381. [Google Scholar] [CrossRef]
- Turley, R.B.; Chapman, K.D. Ontogeny of Cotton Seeds: Gametogenesis, Embryogenesis, Germination, and Seedling Growth; Springer: Dordrecht, The Netherlands, 2010; pp. 332–341. [Google Scholar]
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot. 2007, 100, 1391–1401. [Google Scholar] [CrossRef]
- Yang, S.X.; Huang, L.; Song, J.K.; Liu, L.S.; Bian, Y.Y.; Jia, B.; Wu, L.Y.; Xin, Y.; Wu, M.; Zhang, J.F.; et al. Genome-Wide Analysis of DA1-Like Genes in Gossypium and Functional Characterization of GhDA1-1A Controlling Seed Size. Front. Plant Sci. 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yang, S.X.; Wu, L.Y.; Xin, Y.; Song, J.K.; Wang, L.; Pei, W.F.; Wu, M.; Yu, J.W.; Ma, X.Y.; et al. Genome-Wide Analysis of the GW2-Like Genes in Gossypium and Functional Characterization of the Seed Size Effect of GhGW2-2D. Front. Plant Sci. 2022, 13, 10. [Google Scholar] [CrossRef]
- Zhao, T.; Wu, H.Y.; Wang, X.T.; Zhao, Y.Y.; Wang, L.Y.; Pan, J.Y.; Mei, H.; Han, J.; Wang, S.Y.; Lu, K.N.; et al. Integration of eQTL and machine learning to dissect causal genes with pleiotropic effects in genetic regulation networks of seed cotton yield. Cell Rep. 2023, 42, 21. [Google Scholar] [CrossRef]
- Liu, X.Y.; Hou, J.; Chen, L.; Li, Q.Q.; Fang, X.M.; Wang, J.X.; Hao, Y.S.; Yang, P.; Wang, W.W.; Zhang, D.S.; et al. Natural variation of GhSI7 increases seed index in cotton. Theor. Appl. Genet. 2022, 135, 3661–3672. [Google Scholar] [CrossRef]
- Futschik, M.E.; Carlisle, B. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 2005, 3, 965–988. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 13. [Google Scholar] [CrossRef]
- Ebert, B.; Rautengarten, C.; Guo, X.Y.; Xiong, G.Y.; Stonebloom, S.; Smith-Moritz, A.M.; Herter, T.; Chan, L.J.G.; Adams, P.D.; Petzold, C.J.; et al. Identification and Characterization of a Golgi-Localized UDP-Xylose Transporter Family from Arabidopsis. Plant Cell 2015, 27, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Pan, Y.X.; Zhang, Y.; Li, Z.K.; Wu, L.Q.; Liu, H.W.; Zhang, G.Y.; Wang, X.F.; Ma, Z.Y. Antisense expression of Gossypium hirsutum UDP-glucuronate decarboxylase in Arabidopsis leads to changes in cell wall components. Genet. Mol. Res. 2016, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, M.; Griffith, C.L.; Doering, T.L. Functional cloning and characterization of a UDP-glucuronic acid decarboxylase:: The pathogenic fungus Cryptococcus neoformans elucidates UDP-xylose synthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 12003–12008. [Google Scholar] [CrossRef]
- Bindschedler, L.V.; Tuerck, J.; Maunders, M.; Ruel, K.; Petit-Conil, M.; Danoun, S.; Boudet, A.M.; Joseleau, J.P.; Bolwell, G.P. Modification of hemicellulose content by antisense down-regulation of UDP-glucuronate decarboxylase in tobacco and its consequences for cellulose extractability. Phytochemistry 2007, 68, 2635–2648. [Google Scholar] [CrossRef]
- Kuang, B.Q.; Zhao, X.H.; Zhou, C.; Zeng, W.; Ren, J.L.; Ebert, B.; Beahan, C.T.; Deng, X.M.; Zeng, Q.Y.; Zhou, G.K.; et al. Role of UDP-Glucuronic Acid Decarboxylase in Xylan Biosynthesis in Arabidopsis. Mol. Plant. 2016, 9, 1119–1131. [Google Scholar] [CrossRef]
- Zhong, R.Q.; Teng, Q.C.; Haghighat, M.; Yuan, Y.X.; Furey, S.T.; Dasher, R.L.; Ye, Z.H. Cytosol-Localized UDP-Xylose Synthases Provide the Major Source of UDP-Xylose for the Biosynthesis of Xylan and Xyloglucan. Plant Cell Physiol. 2017, 58, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Ruan, N.; Dang, Z.J.; Wang, M.H.; Cao, L.Y.; Wang, Y.; Liu, S.T.; Tang, Y.J.; Huang, Y.W.; Zhang, Q.; Xu, Q.; et al. FRAGILE CULM 18 encodes a UDP-glucuronic acid decarboxylase required for xylan biosynthesis and plant growth in rice. J. Exp. Bot. 2022, 73, 2320–2335. [Google Scholar] [CrossRef]
- Song, J.; Jia, B.; Feng, P.; Xi, H.; Zhao, W.; Xi, H.; Dong, Y.; Pei, W.; Ma, J.; Zhang, B.; et al. Transcriptome analysis reveals potential of down-regulated genes in cotton fiber improvement. Ind. Crops Prod. 2024, 217, 118737. [Google Scholar] [CrossRef]
- Yang, Z.E.; Ge, X.Y.; Yang, Z.R.; Qin, W.Q.; Sun, G.F.; Wang, Z.; Li, Z.; Liu, J.; Wu, J.; Wang, Y.; et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat. Commun. 2019, 10, 2989. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Location | HSW | SA | SG | SL | SW | SD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | ||
| 2017 | Alaer | 12.71 | 7.37 | 44.21 | 28.17 | 27.91 | 21.99 | 10.32 | 8.12 | 5.75 | 4.69 | 7.49 | 5.98 |
| Shihezi | 12.51 | 7.42 | 45.91 | 28.71 | 28.31 | 22.15 | 10.59 | 8.32 | 5.75 | 4.63 | 7.63 | 6.04 | |
| Anyang | 12.82 | 7.48 | 44.89 | 29.41 | 28.19 | 22.61 | 10.51 | 8.39 | 5.66 | 4.78 | 7.55 | 6.10 | |
| Xingtai | 14.28 | 8.16 | 50.40 | 30.38 | 29.98 | 23.15 | 11.06 | 8.62 | 6.06 | 4.82 | 8.00 | 6.21 | |
| 2018 | Alaer | 11.38 | 8.05 | 41.95 | 29.92 | 27.09 | 22.33 | 10.32 | 8.53 | 5.40 | 4.74 | 7.26 | 6.17 |
| Shihezi | 10.68 | 6.91 | 43.00 | 25.67 | 27.12 | 20.63 | 10.33 | 7.82 | 5.57 | 4.39 | 7.39 | 5.71 | |
| Anyang | 11.41 | 8.21 | 43.80 | 30.69 | 27.66 | 22.77 | 10.39 | 8.58 | 5.61 | 4.86 | 7.46 | 6.24 | |
| Xingtai | 13.83 | 7.63 | 49.90 | 28.94 | 29.59 | 22.23 | 11.04 | 8.33 | 5.97 | 4.70 | 7.96 | 6.06 | |
| Year | Location | HKW | KA | KG | KL | KW | KD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | N10 | N12 | ||
| 2017 | Alaer | 7.50 | 4.42 | 28.38 | 17.86 | 22.81 | 17.55 | 8.80 | 6.78 | 4.21 | 3.49 | 6.00 | 4.76 |
| Shihezi | 7.41 | 4.64 | 28.59 | 18.92 | 23.22 | 18.64 | 9.01 | 7.18 | 4.19 | 3.50 | 6.02 | 4.90 | |
| Anyang | 8.28 | 5.28 | 30.34 | 20.39 | 23.85 | 18.72 | 9.24 | 7.23 | 4.34 | 3.78 | 6.21 | 5.09 | |
| Xingtai | 9.02 | 5.28 | 32.00 | 19.96 | 24.59 | 18.78 | 9.50 | 7.17 | 4.46 | 3.74 | 6.38 | 5.04 | |
| 2018 | Alaer | 7.33 | 5.02 | 27.04 | 19.18 | 22.12 | 18.13 | 8.81 | 7.16 | 4.02 | 3.54 | 5.86 | 4.94 |
| Shihezi | 6.17 | 4.33 | 26.10 | 17.52 | 22.72 | 17.45 | 9.00 | 6.73 | 3.86 | 3.46 | 5.76 | 4.72 | |
| Anyang | 6.99 | 5.29 | 26.39 | 20.22 | 22.33 | 18.67 | 8.97 | 7.30 | 3.85 | 3.71 | 5.79 | 5.07 | |
| Xingtai | 9.92 | 5.00 | 34.37 | 19.71 | 24.82 | 18.61 | 9.56 | 7.19 | 4.76 | 3.69 | 6.61 | 5.00 | |
| Primer Name | Sequence |
|---|---|
| GhUXS5-F | GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGGCAGCAAATTCATCAAATG |
| GhUXS5-R | GGGGACCACTTTGTACAAGAAAGCTGGGTAGTTCTCTTTGGAGACTCCGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, B.; Feng, P.; Song, J.; Zhou, C.; Wang, Y.; Zhang, B.; Wu, M.; Zhang, J.; Chen, Q.; Yu, J. Transcriptome Analysis and Identification of Genes Associated with Cotton Seed Size. Int. J. Mol. Sci. 2024, 25, 9812. https://doi.org/10.3390/ijms25189812
Jia B, Feng P, Song J, Zhou C, Wang Y, Zhang B, Wu M, Zhang J, Chen Q, Yu J. Transcriptome Analysis and Identification of Genes Associated with Cotton Seed Size. International Journal of Molecular Sciences. 2024; 25(18):9812. https://doi.org/10.3390/ijms25189812
Chicago/Turabian StyleJia, Bing, Pan Feng, Jikun Song, Caoyi Zhou, Yajie Wang, Bingbing Zhang, Man Wu, Jinfa Zhang, Quanjia Chen, and Jiwen Yu. 2024. "Transcriptome Analysis and Identification of Genes Associated with Cotton Seed Size" International Journal of Molecular Sciences 25, no. 18: 9812. https://doi.org/10.3390/ijms25189812