
The Novel Anticancer Aryl-Ureido Fatty Acid CTU Increases Reactive Oxygen Species Production That Impairs Mitochondrial Fusion Mechanisms and Promotes MDA-MB-231 Cell Death

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
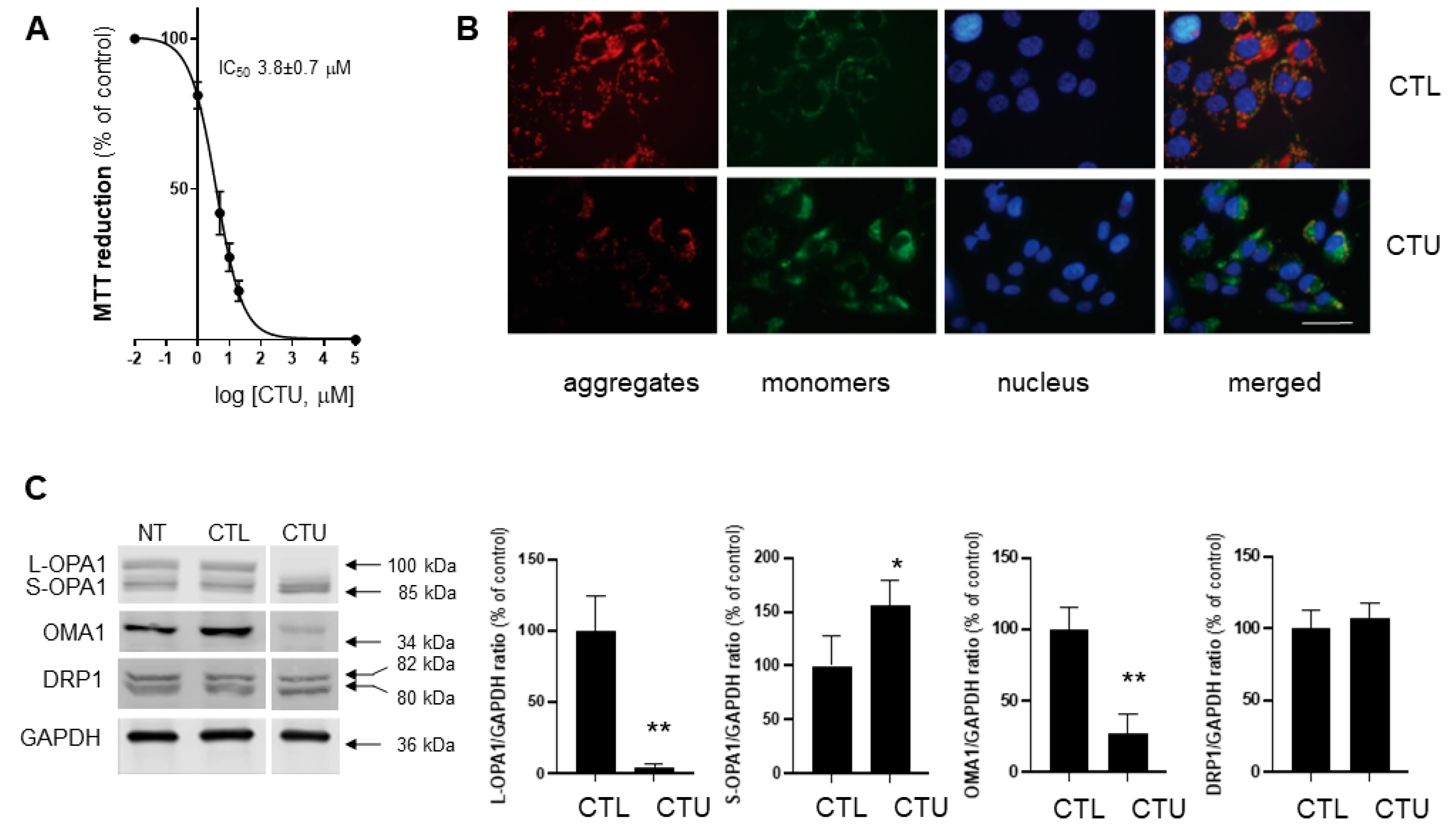
2.1. Mitochondrial Disruption, ROS Production, and Decreased Viability in CTU-Treated Cells
2.2. CTU Dysregulates the OMA1/OPA1 Pathway of IMM Fusion in Cells
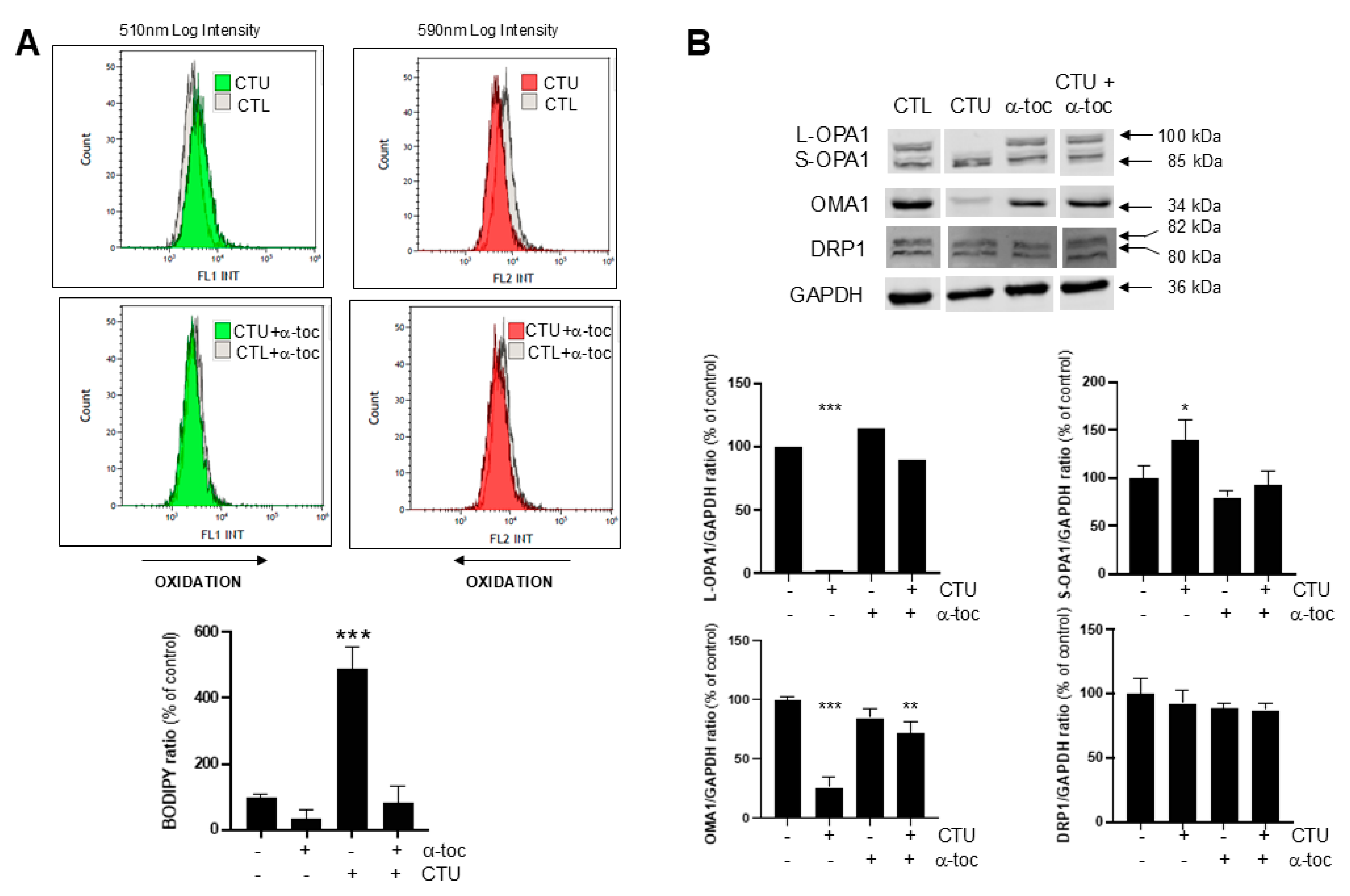
2.3. Role of CTU-Derived ROS in Dysregulation of OMA1-OPA1
3. Discussion
4. Materials and Methods
4.1. Cell Culture Reagents and Chemicals
4.2. MTT Reduction Assay of Cell Proliferation
4.3. Caspase-3/7 Assay of Cell Apoptosis
4.4. JC-1 Fluorescence Assay of the Mitochondrial Membrane Potential
4.5. BODIPY (581/591) C11 Lipid Peroxidation Assay
4.6. Preparation of Cell Lysates for Electrophoresis and Immunoblotting
4.7. Confocal Microscopy and Cell Staining for Mitochondrial Integrity and DNA
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Singh, A.P.; Kvansakul, M.; Ryan, M.T.; Osellame, L.D. Splitting up the powerhouse: Structural insights into the mechanism of mitochondrial fission. Cell. Mol. Life Sci. 2015, 72, 3695–3707. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Jabůrek, M.; Ježek, P. The mechanism of proton transport mediated by mitochondrial uncoupling proteins. FEBS Lett. 1998, 438, 10–14. [Google Scholar] [CrossRef]
- Rawling, T.; MacDermott-Opeskin, H.; Roseblade, A.; Pazderka, C.; Clarke, C.; Bourget, K.; Wu, X.; Lewis, W.; Noble, B.; Gale, P.A.; et al. Aryl urea substituted fatty acids: A new class of protonophoric mitochondrial uncoupler that utilises a synthetic anion transporter. Chem. Sci. 2020, 11, 12677–12685. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Koopman, W.J.; Nijtmans, L.G.; Dieteren, C.E.; Roestenberg, P.; Valsecchi, F.; Smeitink, J.A.; Willems, P.H. Mammalian mitochondrial complex I: Biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal. 2010, 15, 1431–1470. [Google Scholar] [CrossRef]
- Choucair, H.; Rahman, M.K.; Umashankar, B.; Al-Zubaidi, Y.; Bourget, K.; Chen, Y.; Dunstan, C.; Rawling, T.; Murray, M. The aryl-ureido fatty acid CTU activates endoplasmic reticulum stress and PERK/NOXA-mediated apoptosis in tumor cells by a dual mitochondrial-targeting mechanism. Cancer Lett. 2022, 526, 131–141. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA. 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Gilkerson, R.; De La Torre, P.; St. Vallier, S. Mitochondrial OMA1 and OPA1 as gatekeepers of organellar structure/function and cellular stress response. Front. Cell Developm. Biol. 2021, 9, 626117. [Google Scholar] [CrossRef]
- Ježek, J.; Cooper, K.; Strich, R. Reactive oxygen species and mitochondrial dynamics: The yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants 2018, 7, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Rawling, T.; Choucair, H.; Koolaji, N.; Bourget, K.; Allison, S.E.; Chen, Y.J.; Dunstan, C.R.; Murray, M. A novel aryl-urea fatty acid that targets the mitochondrion and depletes cardiolipin to promote killing of breast cancer cells. J. Med. Chem. 2017, 60, 8661–8666. [Google Scholar] [CrossRef]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef]
- Guillery, O.; Malka, F.; Landes, T.; Guillou, E.; Blackstone, C.; Lombès, A.; Belenguer, P.; Arnoult, D.; Rojo, M. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol. Cell 2008, 100, 315–325. [Google Scholar] [CrossRef]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA 1 activation and autocatalytic turnover regulate OPA 1-dependent mitochondrial dynamics. EMBO J. 2014, 18, 578–593. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease–the long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef]
- Wang, R.; Mishra, P.; Garbis, S.D.; Moradian, A.; Sweredoski, M.J.; Chan, D.C. Identification of new OPA1 cleavage site reveals that short isoforms regulate mitochondrial fusion. Mol. Biol. Cell 2021, 32, 157–168. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Ma, J.; Zhu, C.; Niu, T.; Chen, W.; Pang, X.; Zhai, Y.; Sun, F. Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. eLife 2020, 9, e50294. [Google Scholar] [CrossRef]
- Alavi, M.V. Recent advances in, and challenges of, designing OMA1 drug screens. Pharmacol. Res. 2022, 176, 106063. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Smith, S.B.; Sheu, S.S.; Yoon, Y. The short variant of optic atrophy 1 (OPA1) improves cell survival under oxidative stress. J. Biol. Chem. 2020, 295, 6543–6560. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Dietz, J.V.; Swenson, S.; Zahayko, N.; Khalimonchuk, O. Redox regulation of the mitochondrial quality control protease Oma1. Antioxid. Redox Signal. 2019, 31, 429–443. [Google Scholar] [CrossRef]
- Bohovych, I.; Fernandez, M.R.; Rahn, J.J.; Stackley, K.D.; Bestman, J.E.; Anandhan, A.; Franco, R.; Claypool, S.M.; Lewis, R.E.; Chan, S.S.L.; et al. Metalloprotease OMA1 fine-tunes mitochondrial bioenergetic function and respiratory supercomplex stability. Sci. Rep. 2015, 5, 13989. [Google Scholar] [CrossRef]
- Richter, U.; Lahtinen, T.; Marttinen, P.; Suomi, F.; Battersby, B.J. Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. J. Cell Biol. 2015, 211, 373–389. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef]
- He, L.; Qi, Y.; Rong, X.; Jiang, J.; Yang, Q.; Yamahara, J.; Murray, M.; Li, Y. The Ayurvedic medicine Salacia oblonga attenuates diabetic renal fibrosis in rats: Suppression of angiotensin II/AT1 signaling. Evid. Based Complement. Altern. Med. 2011, 2011, 807451. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, Z.; Wang, K.; Ali, Y.; Shu, W.; Bao, X.; Zhu, L.; Murray, M.; Zhou, F. Procyanidin B2 and rutin protect human Retinal Pigment Epithelial (RPE) cells from oxidative stress by modulating Nrf2 and Erk1/2 signalling pathways. Exp. Eye Res. 2021, 207, 108586. [Google Scholar] [CrossRef]
- Allison, S.E.; Chen, Y.; Petrovic, N.; Zhang, J.; Bourget, K.; Mackenzie, P.I.; Murray, M. Activation of ALDH1A1 in MDA-MB-468 breast cancer cells that over-express CYP2J2 protects against paclitaxel-dependent cell death mediated by reactive oxygen species. Biochem. Pharmacol. 2017, 143, 79–89. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, S.; Umashankar, B.; Rahman, M.K.; Choucair, H.; Rawling, T.; Murray, M. The Novel Anticancer Aryl-Ureido Fatty Acid CTU Increases Reactive Oxygen Species Production That Impairs Mitochondrial Fusion Mechanisms and Promotes MDA-MB-231 Cell Death. Int. J. Mol. Sci. 2024, 25, 10577. https://doi.org/10.3390/ijms251910577
Tam S, Umashankar B, Rahman MK, Choucair H, Rawling T, Murray M. The Novel Anticancer Aryl-Ureido Fatty Acid CTU Increases Reactive Oxygen Species Production That Impairs Mitochondrial Fusion Mechanisms and Promotes MDA-MB-231 Cell Death. International Journal of Molecular Sciences. 2024; 25(19):10577. https://doi.org/10.3390/ijms251910577
Chicago/Turabian StyleTam, Stanton, Balasubrahmanyam Umashankar, Md Khalilur Rahman, Hassan Choucair, Tristan Rawling, and Michael Murray. 2024. "The Novel Anticancer Aryl-Ureido Fatty Acid CTU Increases Reactive Oxygen Species Production That Impairs Mitochondrial Fusion Mechanisms and Promotes MDA-MB-231 Cell Death" International Journal of Molecular Sciences 25, no. 19: 10577. https://doi.org/10.3390/ijms251910577
APA StyleTam, S., Umashankar, B., Rahman, M. K., Choucair, H., Rawling, T., & Murray, M. (2024). The Novel Anticancer Aryl-Ureido Fatty Acid CTU Increases Reactive Oxygen Species Production That Impairs Mitochondrial Fusion Mechanisms and Promotes MDA-MB-231 Cell Death. International Journal of Molecular Sciences, 25(19), 10577. https://doi.org/10.3390/ijms251910577