
Analysis of the Setomimycin Biosynthetic Gene Cluster from Streptomyces nojiriensis JCM3382 and Evaluation of Its α-Glucosidase Inhibitory Activity Using Molecular Docking and Molecular Dynamics Simulations


 , ,
, , 
Abstract
:1. Introduction
2. Results
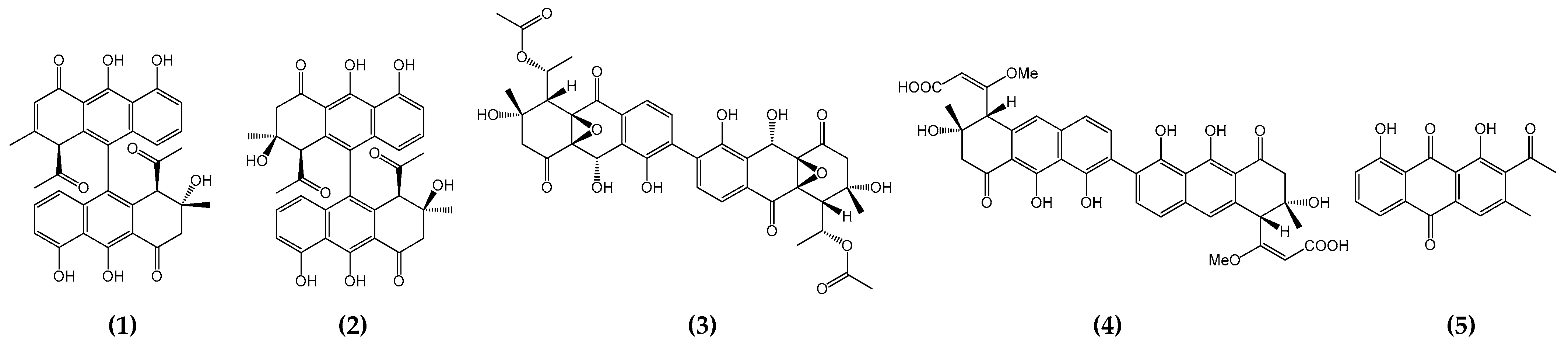
2.1. Comparative Analysis of Biosynthetic Gene Clusters between Setomimycin Producing Microorganisms
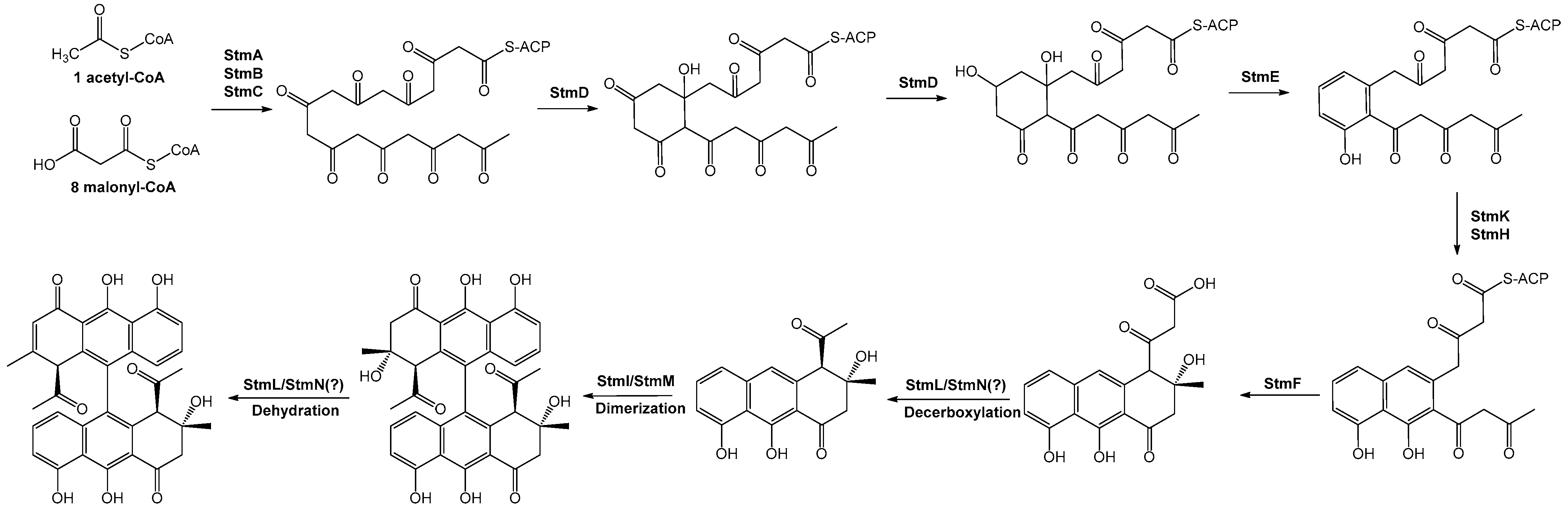
2.2. Putative Setomimycin Biosynthetic Pathway
2.2.1. Minimal PKS
2.2.2. Ketoreduction and Cyclization
2.2.3. Chain Release
2.2.4. Dimerization
2.2.5. Concluding Remarks
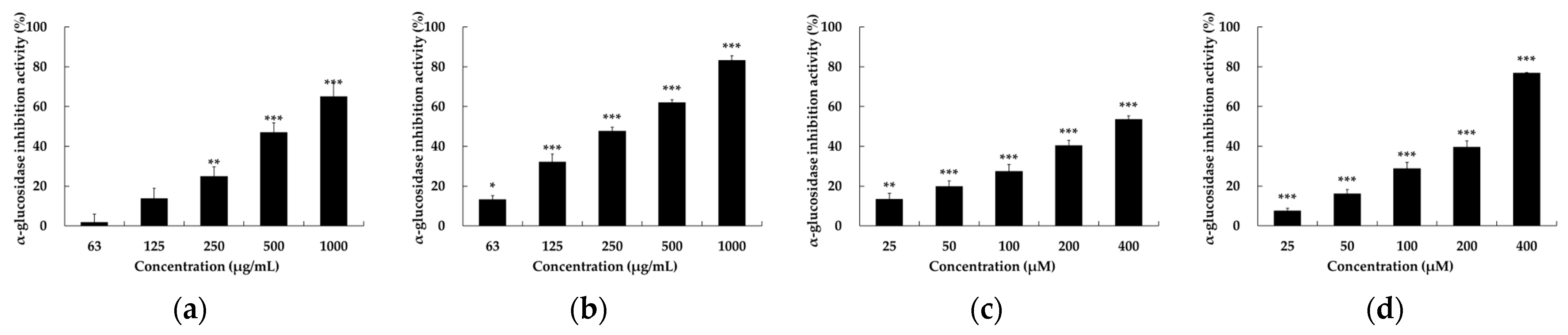
2.3. α-Glucosidase Inhibitory Activity of Setomimycin and Streptomyces nojiriensis JCM3382
2.4. Chemoinformatic Analysis
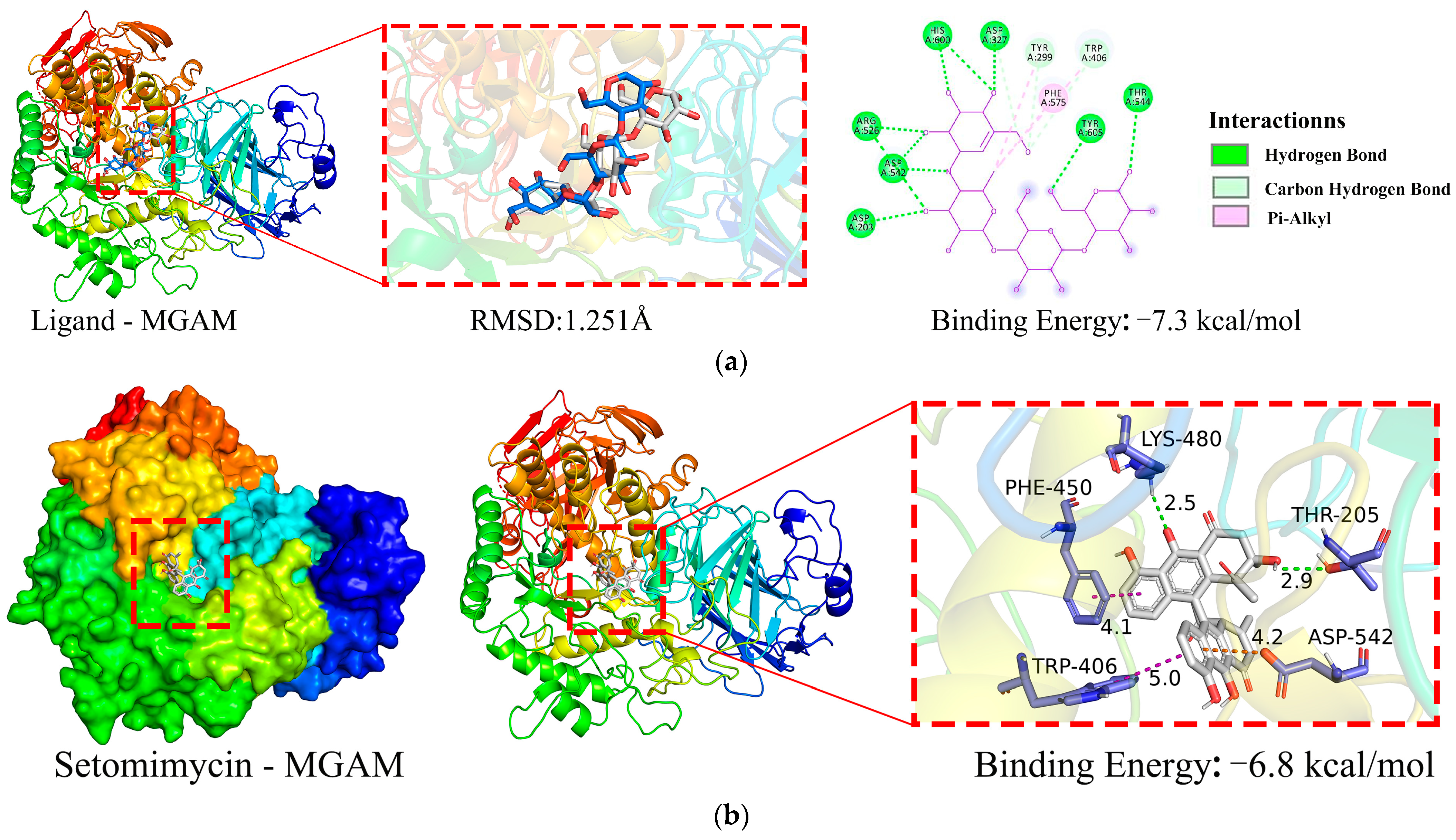
2.5. Docking and Molecular Dynamics (MD) Simulations
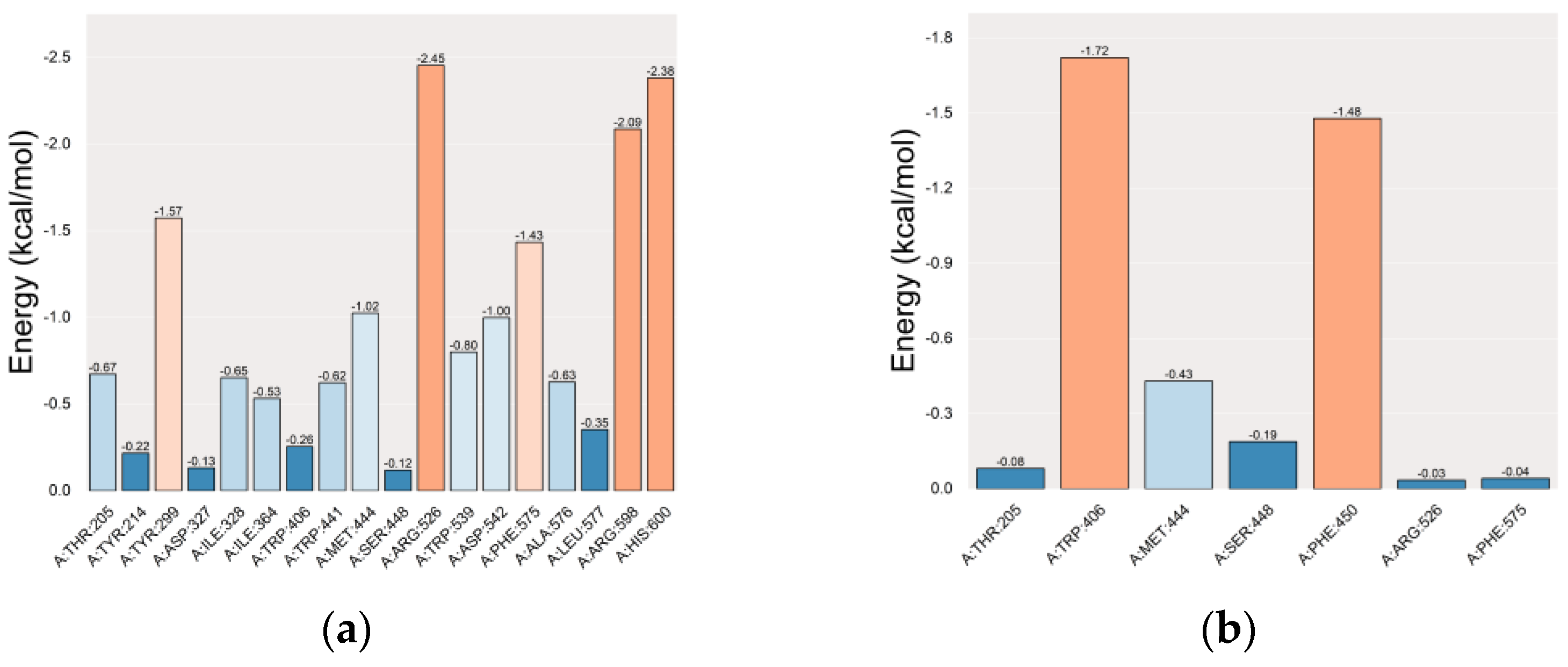
2.5.1. Molecular Docking Simulation
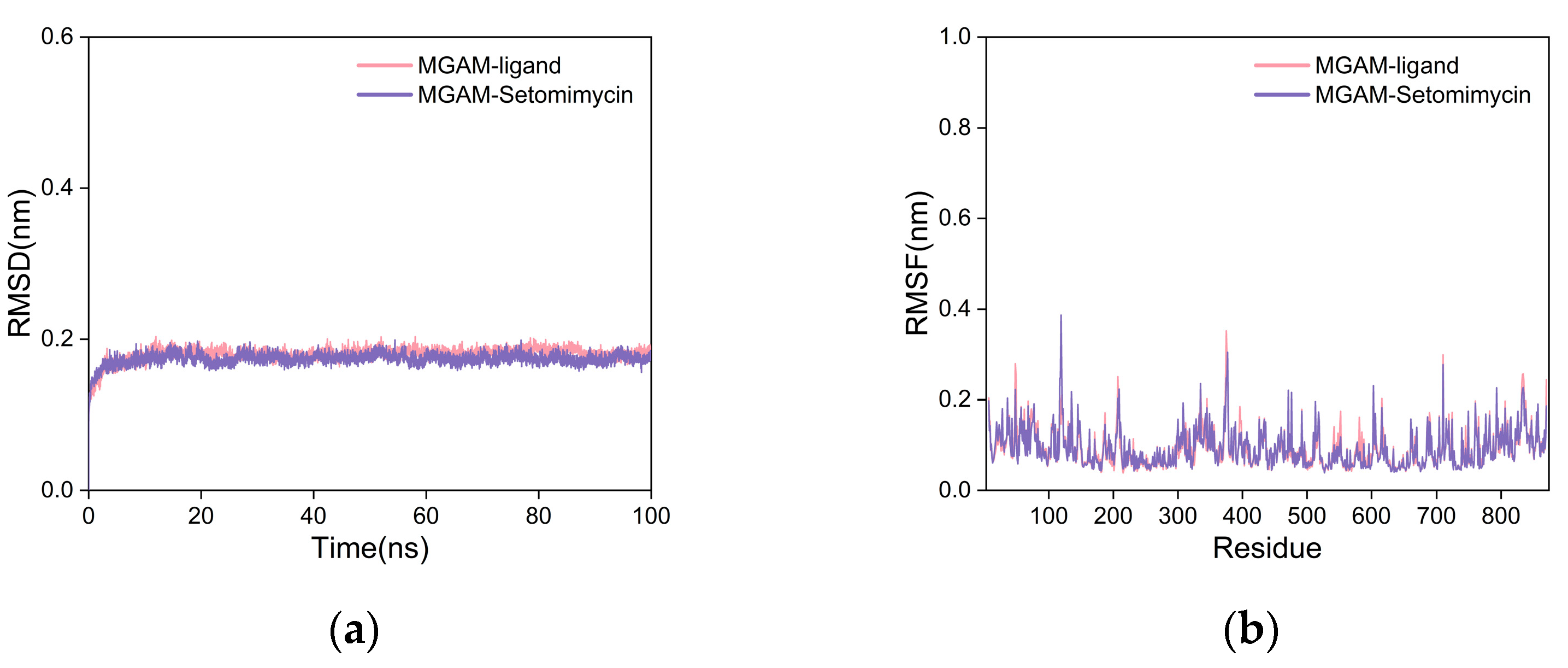
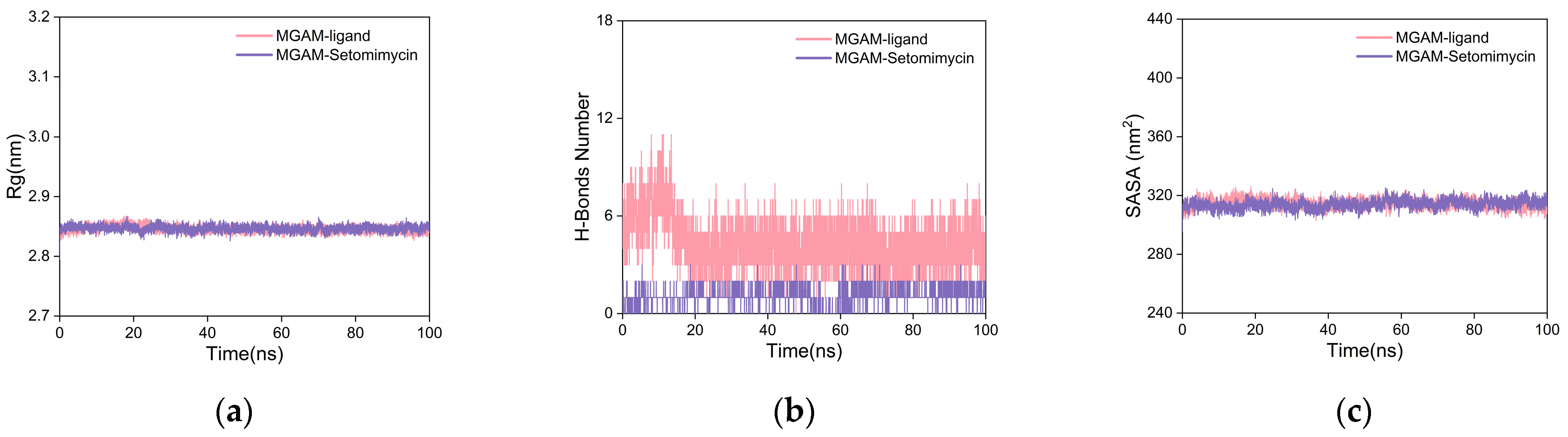
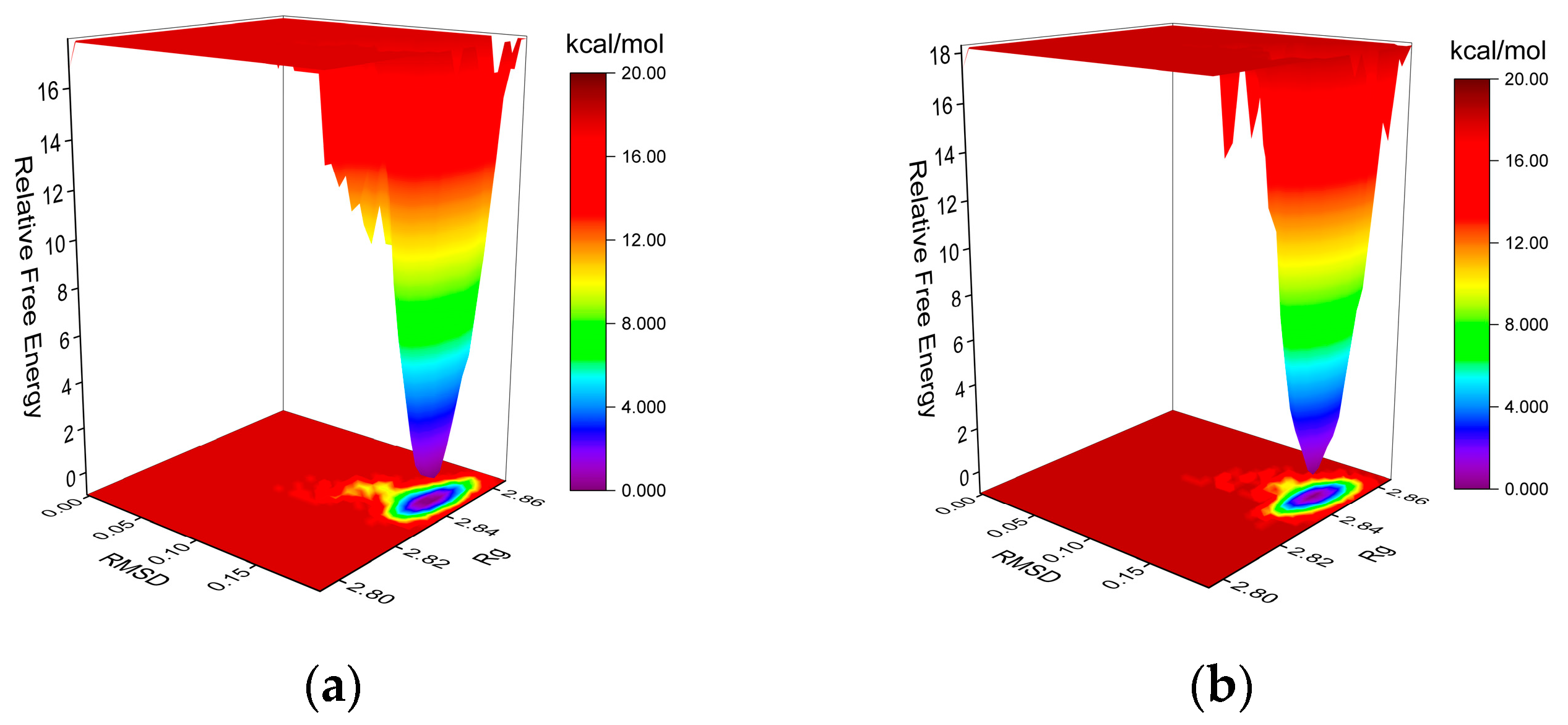
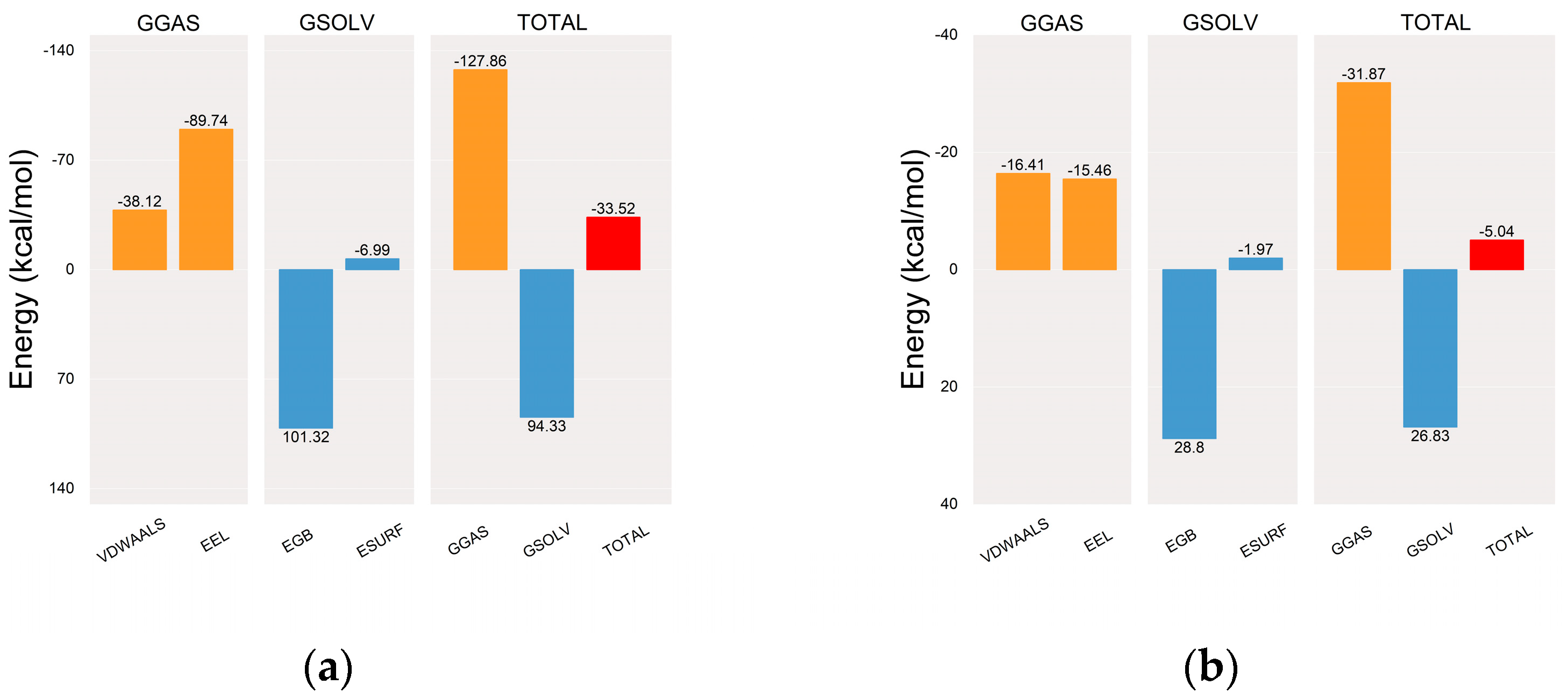
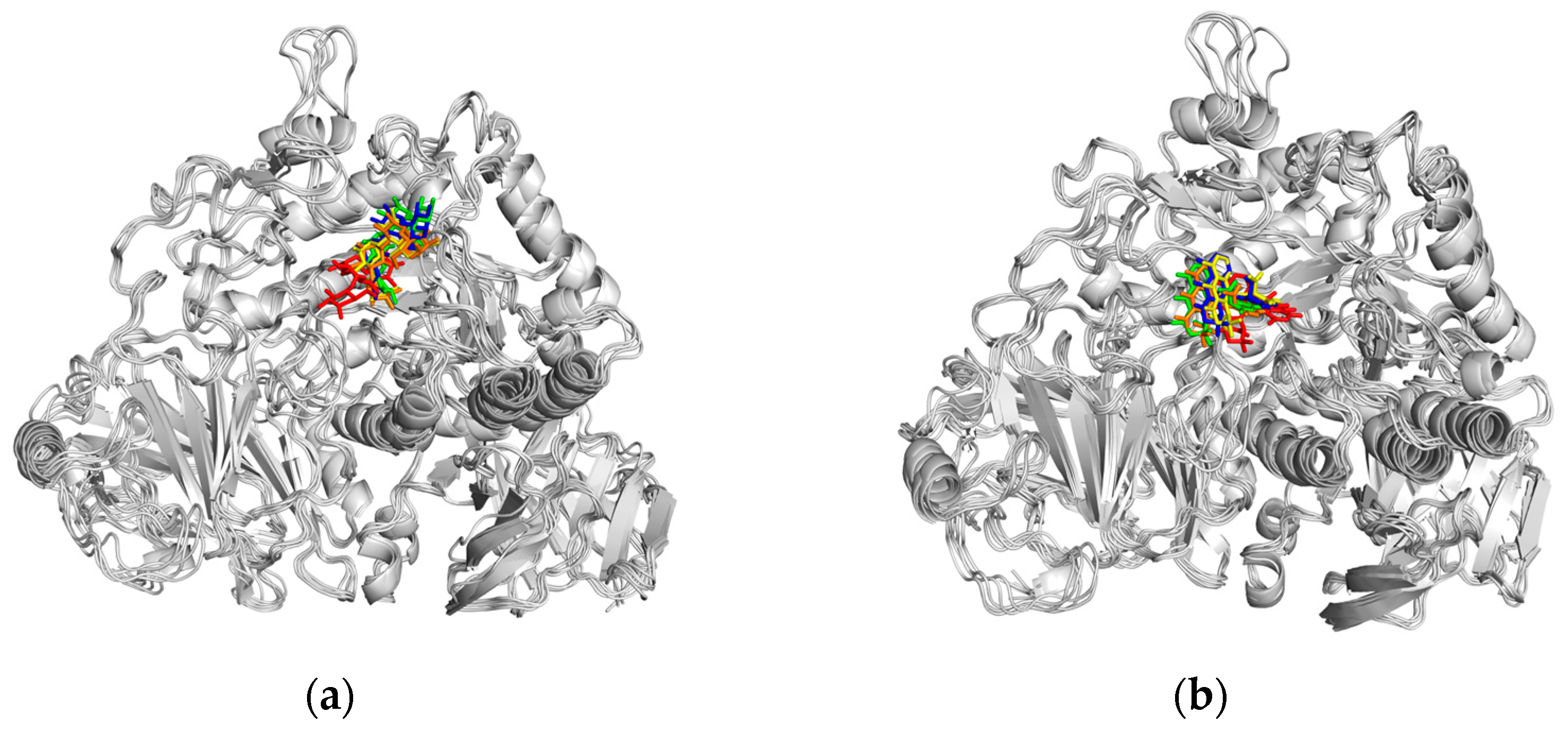
2.5.2. Molecular Dynamics (MD) Simulation
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Fermentation and Extraction
3.3. Enzymatic Analysis
3.4. Chemoinformatic Analysis
3.5. Molecular Docking Simulation Analysis
3.6. Molecular Dynamics (MD) Simulations Analysis
3.7. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McBride, C.M.; Miller, E.L.; Charkoudian, L.K. An updated catalogue of diverse type II polyketide synthase biosynthetic gene clusters captured from large-scale nucleotide databases. Microb. Genom. 2023, 9, mgen000965. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, R.; Chen, X.; Sun, X.; Yan, Y.; Shen, X.; Yuan, Q. Biosynthesis of aromatic polyketides in microorganisms using type II polyketide synthases. Microb. Cell Fact. 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Eun, H.; Prabowo, C.P.S. Metabolic Engineering and Synthetic Biology Approaches for the Heterologous Production of Aromatic Polyketides. Int. J. Mol. Sci. 2023, 24, 8923. [Google Scholar] [CrossRef] [PubMed]
- Ridley, C.P.; Lee, H.Y.; Khosla, C. Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Y.; Tang, Y. Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 20683–20688. [Google Scholar] [CrossRef]
- Präg, A.; Grüning, B.A.; Häckh, M.; Lüdeke, S.; Wilde, M.; Luzhetskyy, A.; Richter, M.; Luzhetska, M.; Günther, S.; Müller, M. Regio- and stereoselective intermolecular oxidative phenol coupling in Streptomyces. J. Am. Chem. Soc. 2014, 136, 6195–6198. [Google Scholar] [CrossRef]
- Jiang, L.; Pu, H.; Xiang, J.; Su, M.; Yan, X.; Yang, D.; Zhu, X.; Shen, B.; Duan, Y.; Huang, Y. Huanglongmycin A-C, Cytotoxic Polyketides Biosynthesized by a Putative Type II Polyketide Synthase From Streptomyces sp. CB09001. Front. Chem. 2018, 6, 254. [Google Scholar] [CrossRef]
- Ji, X.; Dong, Y.; Ling, C.; Zhou, Z.; Li, Q.; Ju, J. Elucidation of the Tailoring Steps in Julichrome Biosynthesis by Marine Gastropod Mollusk-Associated Streptomyces sampsonii SCSIO 054. Org. Lett. 2020, 22, 6927–6931. [Google Scholar] [CrossRef]
- Jiang, L.; Xiang, J.; Zhu, S.; Tang, D.; Gong, B.; Pu, H.; Duan, Y.; Huang, Y. Undescribed benzophenone and xanthones from cave-derived Streptomyces sp. CB09001. Nat. Prod. Res. 2022, 36, 1725–1733. [Google Scholar] [CrossRef]
- Mohamed, O.G.; Khalil, Z.G.; Salim, A.A.; Cui, H.; Blumenthal, A.; Capon, R.J. Lincolnenins A-D: Isomeric Bactericidal Bianthracenes from Streptomyces lincolnensis. J. Org. Chem. 2021, 86, 11011–11018. [Google Scholar] [CrossRef]
- Shen, T.; Li, L.M.; Xu, Z.Y.; Wang, Y.D.; Xie, W.D. Julichrome derivatives and gliotoxin from a soil derived Streptomyces sp. Nat. Prod. Res. 2021, 35, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Thuaud, F.; Sekine, D.; Ito, A.; Maeda, S.; Koshino, H.; Hashizume, D.; Muranaka, A.; Cruchter, T.; Uchiyama, M.; et al. Synthesis of All Stereoisomers of Monomeric Spectomycin A1/A2 and Evaluation of Their Protein SUMOylation-Inhibitory Activity. Chemistry 2019, 25, 8387–8392. [Google Scholar] [CrossRef] [PubMed]
- Manhas, R.S.; Chander, D.; Chaubey, A. Identification and taxonomy of Streptomyces justiciae strain RA-WS2: A novel setomimycin producing actinobacterium. 3 Biotech 2023, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, D.E.; Hong, S.C.; Kim, S.Y.; Kwon, H.C.; Hyun, C.G.; Choi, J. Genome Analysis of Streptomyces nojiriensis JCM 3382 and Distribution of Gene Clusters for Three Antibiotics and an Azasugar across the Genus Streptomyces. Microorganisms 2021, 9, 1802. [Google Scholar] [CrossRef]
- Yuan, S.W.; Chen, S.H.; Guo, H.; Chen, L.T.; Shen, H.J.; Liu, L.; Gao, Z.Z. Elucidation of the Complete Biosynthetic Pathway of Phomoxanthone A and Identification of a Para-Para Selective Phenol Coupling Dimerase. Org. Lett. 2022, 24, 3069–3074. [Google Scholar] [CrossRef]
- Zetzsche, L.E.; Yazarians, J.A.; Chakrabarty, S.; Hinze, M.E.; Murray, L.A.M.; Lukowski, A.L.; Joyce, L.A.; Narayan, A.R.H. Biocatalytic oxidative cross-coupling reactions for biaryl bond formation. Nature 2022, 603, 79–85. [Google Scholar] [CrossRef]
- Hüttel, W.; Müller, M. Regio- and stereoselective intermolecular phenol coupling enzymes in secondary metabolite biosynthesis. Nat. Prod. Rep. 2021, 38, 1011–1043. [Google Scholar] [CrossRef]
- Manhas, R.S.; Kumar, A.; Chaubey, A. A biostatistical approach for augmenting rare bianthraquinone antibiotic production by Streptomyces sp. RA-WS2 using Taguchi design. AMB Express 2022, 12, 155. [Google Scholar] [CrossRef]
- Krysenko, S. Impact of Nitrogen-Containing Compounds on Secondary Metabolism in Streptomyces spp.—A Source of Metabolic Engineering Strategies. SynBio 2023, 1, 204–225. [Google Scholar] [CrossRef]
- Hirohama, M.; Kumar, A.; Fukuda, I.; Matsuoka, S.; Igarashi, Y.; Saitoh, H.; Takagi, M.; Shin-ya, K.; Honda, K.; Kondoh, Y.; et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 2013, 8, 2635–2642. [Google Scholar] [CrossRef]
- Dong, Y.; Ding, W.; Sun, C.; Ji, X.; Ling, C.; Zhou, Z.; Chen, Z.; Chen, X.; Ju, J. Julichrome Monomers from Marine Gastropod Mollusk-Associated Streptomyces and Stereochemical Revision of Julichromes Q3·5 and Q3·3. Chem. Biodivers. 2020, 17, e2000057. [Google Scholar] [CrossRef] [PubMed]
- Komoda, T.; Saeki, N.; Koseki, Y.; Kiyota, H. 12T061A and 12T061C, two new julichrome family compounds, as radical scavengers from Streptomyces sp. J. Gen. Appl. Microbiol. 2016, 62, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Yang, W.; Peng, Z.; He, Y.; Wang, G. Chromone-based benzohydrazide derivatives as potential α-glucosidase inhibitor: Synthesis, biological evaluation and molecular docking study. Bioorg. Chem. 2023, 131, 106276. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Li, Y.J.; Shao, J.; Li, Y.S.; Cui, Z.N. Synthesis and biological evaluation of 2,5-disubstituted furan derivatives containing 1,3-thiazole moiety as potential α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2023, 83, 129173. [Google Scholar] [CrossRef]
- Kim, H.M.; Hyun, C.G. Miglitol, an Oral Antidiabetic Drug, Downregulates Melanogenesis in B16F10 Melanoma Cells through the PKA, MAPK, and GSK3β/β-Catenin Signaling Pathways. Molecules 2022, 28, 115. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Martinet, L.; Naômé, A.; Deflandre, B.; Maciejewska, M.; Tellatin, D.; Tenconi, E.; Smargiasso, N.; de Pauw, E.; van Wezel, G.P.; Rigali, S. A Single Biosynthetic Gene Cluster Is Responsible for the Production of Bagremycin Antibiotics and Ferroverdin Iron Chelators. mBio 2019, 10, e01230-19. [Google Scholar] [CrossRef]
- Rivers, M.A.J.; Lowell, A.N. Expanding the Biosynthetic Toolbox: The Potential and Challenges of In Vitro Type II Polyketide Synthase Research. SynBio 2024, 2, 85–111. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Korman, T.P.; Tan, Y.H.; Wong, J.; Luo, R.; Tsai, S.C. Inhibition kinetics and emodin cocrystal structure of a type II polyketide ketoreductase. Biochemistry 2008, 47, 1837–1847. [Google Scholar] [CrossRef]
- Javidpour, P.; Das, A.; Khosla, C.; Tsai, S.C. Structural and biochemical studies of the hedamycin type II polyketide ketoreductase (HedKR): Molecular basis of stereo- and regiospecificities. Biochemistry 2011, 50, 7426–7439. [Google Scholar] [CrossRef] [PubMed]
- Bililign, T.; Hyun, C.G.; Williams, J.S.; Czisny, A.M.; Thorson, J.S. The hedamycin locus implicates a novel aromatic PKS priming mechanism. Chem. Biol. 2004, 11, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Javidpour, P.; Korman, T.P.; Shakya, G.; Tsai, S.C. Structural and biochemical analyses of regio- and stereospecificities observed in a type II polyketide ketoreductase. Biochemistry 2011, 50, 4638–4649. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.D.; Lee, M.Y.; Moody, C.; Zhang, W.; Tang, Y.; Tsai, S.C. Structural and biochemical characterization of ZhuI aromatase/cyclase from the R1128 polyketide pathway. Biochemistry 2011, 50, 8392–8406. [Google Scholar] [CrossRef]
- Caldara-Festin, G.; Jackson, D.R.; Barajas, J.F.; Valentic, T.R.; Patel, A.B.; Aguilar, S.; Nguyen, M.; Vo, M.; Khanna, A.; Sasaki, E.; et al. Structural and functional analysis of two di-domain aromatase/cyclases from type II polyketide synthases. Proc. Natl. Acad. Sci. USA 2015, 112, E6844–E6851. [Google Scholar] [CrossRef]
- Jiang, K.; Chen, X.; Yan, X.; Li, G.; Lin, Z.; Deng, Z.; Luo, S.; Qu, X. An unusual aromatase/cyclase programs the formation of the phenyldimethylanthrone framework in anthrabenzoxocinones and fasamycin. Proc. Natl. Acad. Sci. USA 2024, 121, e2321722121. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Little, R.F.; Hertweck, C. Chain release mechanisms in polyketide and non-ribosomal peptide biosynthesis. Nat. Prod. Rep. 2022, 39, 163–205. [Google Scholar] [CrossRef]
- Hua, K.; Liu, X.; Zhao, Y.; Gao, Y.; Pan, L.; Zhang, H.; Deng, Z.; Jiang, M. Offloading Role of a Discrete Thioesterase in Type II Polyketide Biosynthesis. mBio 2020, 11, e01334-20. [Google Scholar] [CrossRef]
- Bräuer, A.; Zhou, Q.; Grammbitter, G.L.C.; Schmalhofer, M.; Rühl, M.; Kaila, V.R.I.; Bode, H.B.; Groll, M. Structural snapshots of the minimal PKS system responsible for octaketide biosynthesis. Nat. Chem. 2020, 12, 755–763. [Google Scholar] [CrossRef]
- Wang, W.G.; Wang, H.; Du, L.Q.; Li, M.; Chen, L.; Yu, J.; Cheng, G.G.; Zhan, M.T.; Hu, Q.F.; Zhang, L.; et al. Molecular Basis for the Biosynthesis of an Unusual Chain-Fused Polyketide, Gregatin A. J. Am. Chem. Soc. 2020, 142, 8464–8472. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.O.; Joyce, S.A.; Jenke-Kodama, H.; Schwär, G.; Clarke, D.J.; Bode, H.B. A type II polyketide synthase is responsible for anthraquinone biosynthesis in Photorhabdus luminescens. Chembiochemistry 2007, 8, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Cheng, F.; Li, S. Cytochrome P450 enzymes in fungal natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1072–1099. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, S.M. Two Cytochrome P450 Enzymes from Streptomyces sp. NRRL S-1868 Catalyze Distinct Dimerization of Tryptophan-Containing Cyclodipeptides. Org. Lett. 2019, 21, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Chen, S.; Zhang, L. A Streptomyces P450 enzyme dimerizes isoflavones from plants. Beilstein J. Org. Chem. 2022, 18, 1107–1115. [Google Scholar] [CrossRef]
- He, J.; Liu, X.; Li, C. Engineering Electron Transfer Pathway of Cytochrome P450s. Molecules 2024, 29, 2480. [Google Scholar] [CrossRef]
- Greule, A.; Stok, J.E.; De Voss, J.J.; Cryle, M.J. Unrivalled diversity: The many roles and reactions of bacterial cytochromes P450 in secondary metabolism. Nat. Prod. Rep. 2018, 35, 757–791. [Google Scholar] [CrossRef]
- Zerbe, K.; Pylypenko, O.; Vitali, F.; Zhang, W.; Rouset, S.; Heck, M.; Vrijbloed, J.W.; Bischoff, D.; Bister, B.; Süssmuth, R.D.; et al. Crystal structure of OxyB, a cytochrome P450 implicated in an oxidative phenol coupling reaction during vancomycin biosynthesis. J. Biol. Chem. 2002, 277, 47476–47485. [Google Scholar] [CrossRef]
- Harken, L.; Li, S.M. Modifications of diketopiperazines assembled by cyclodipeptide synthases with cytochrome P450 enzymes. Appl. Microbiol. Biotechnol. 2021, 105, 2277–2285. [Google Scholar] [CrossRef]
- Brieke, C.; Tarnawski, M.; Greule, A.; Cryle, M.J. Investigating Cytochrome P450 specificity during glycopeptide antibiotic biosynthesis through a homologue hybridization approach. J. Inorg. Biochem. 2018, 185, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Shende, V.V.; Khatri, Y.; Newmister, S.A.; Sanders, J.N.; Lindovska, P.; Yu, F.; Doyon, T.J.; Kim, J.; Houk, K.N.; Movassaghi, M.; et al. Structure and Function of NzeB, a Versatile C-C and C-N Bond-Forming Diketopiperazine Dimerase. J. Am. Chem. Soc. 2020, 142, 17413–17424. [Google Scholar] [CrossRef] [PubMed]
- Macedo-Ribeiro, S.; Darimont, B.; Sterner, R.; Huber, R. Small structural changes account for the high thermostability of 1[4Fe-4S] ferredoxin from the hyperthermophilic bacterium Thermotoga maritima. Structure 1996, 4, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Krauss, N.; Schubert, W.D.; Klukas, O.; Fromme, P.; Witt, H.T.; Saenger, W. Photosystem I at 4 A resolution represents the first structural model of a joint photosynthetic reaction centre and core antenna system. Nat. Struct. Biol. 1996, 3, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, C.R.; Sieker, L.C.; Adman, E.T.; Jensen, L.H. Refined crystal structure of ferredoxin II from Desulfovibrio gigas at 1.7 A. J. Mol. Biol. 1991, 219, 693–715. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, A.; Bell, S.G.; Wong, L.L.; Zhou, W. The structure of a novel electron-transfer ferredoxin from Rhodopseudomonas palustris HaA2 which contains a histidine residue in its iron-sulfur cluster-binding motif. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1453–1464. [Google Scholar] [CrossRef]
- Gilep, A.; Varaksa, T.; Bukhdruker, S.; Kavaleuski, A.; Ryzhykau, Y.; Smolskaya, S.; Sushko, T.; Tsumoto, K.; Grabovec, I.; Kapranov, I.; et al. Structural insights into 3Fe-4S ferredoxins diversity in M. tuberculosis highlighted by a first redox complex with P450. Front. Mol. Biosci. 2023, 9, 1100032. [Google Scholar] [CrossRef]
- Nzuza, N.; Padayachee, T.; Chen, W.; Gront, D.; Nelson, D.R.; Syed, K. Diversification of Ferredoxins across Living Organisms. Curr. Issues Mol. Biol. 2021, 43, 1374–1390. [Google Scholar] [CrossRef]
- Ngcobo, P.E.; Nkosi, B.V.Z.; Chen, W.; Nelson, D.R.; Syed, K. Evolution of Cytochrome P450 Enzymes and Their Redox Partners in Archaea. Int. J. Mol. Sci. 2023, 24, 4161. [Google Scholar] [CrossRef]
- Kuatsjah, E.; Zahn, M.; Chen, X.; Kato, R.; Hinchen, D.J.; Konev, M.O.; Katahira, R.; Orr, C.; Wagner, A.; Zou, Y.; et al. Biochemical and structural characterization of a sphingomonad diarylpropane lyase for cofactorless deformylation. Proc. Natl. Acad. Sci. USA 2023, 120, e2212246120. [Google Scholar] [CrossRef]
- Vuksanovic, N.; Zhu, X.; Serrano, D.A.; Siitonen, V.; Metsä-Ketelä, M.; Melançon, C.E., 3rd; Silvaggi, N.R. Structural characterization of three noncanonical NTF2-like superfamily proteins: Implications for polyketide biosynthesis. Acta Crystallogr. F Struct. Biol. Commun. 2020, 76, 372–383. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Reviron. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, X.; Hyun, C.G. Isolation, Characterization, Genome Annotation, and Evaluation of Tyrosinase Inhibitory Activity in Secondary Metabolites of Paenibacillus sp. JNUCC32: A Comprehensive Analysis through Molecular Docking and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 2213. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liang, X.; Hyun, C.G. Isolation, Characterization, Genome Annotation, and Evaluation of Hyaluronidase Inhibitory Activity in Secondary Metabolites of Brevibacillus sp. JNUCC 41: A Comprehensive Analysis through Molecular Docking and Molecular Dynamics Simulation. Int. J. Mol. Sci. 2024, 25, 4611. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MW | HBA | HBD | RB | TPSA | Log p | MR | Lipinski’s Rule |
|---|---|---|---|---|---|---|---|---|
| Setomimycin | 580.58 | 9 | 5 | 3 | 169.43 | 3.476 | 160.66 | Yes; 1 violation: MW > 500 |
| Acarbose | 645.60 | 5 | 3 | 9 | 90.90 | 3.307 | 136.69 | No; 3 violations: MW > 500, N or O > 10, NH or OH > 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyun, K.-A.; Liang, X.; Xu, Y.; Kim, S.-Y.; Boo, K.-H.; Park, J.-S.; Chi, W.-J.; Hyun, C.-G. Analysis of the Setomimycin Biosynthetic Gene Cluster from Streptomyces nojiriensis JCM3382 and Evaluation of Its α-Glucosidase Inhibitory Activity Using Molecular Docking and Molecular Dynamics Simulations. Int. J. Mol. Sci. 2024, 25, 10758. https://doi.org/10.3390/ijms251910758
Hyun K-A, Liang X, Xu Y, Kim S-Y, Boo K-H, Park J-S, Chi W-J, Hyun C-G. Analysis of the Setomimycin Biosynthetic Gene Cluster from Streptomyces nojiriensis JCM3382 and Evaluation of Its α-Glucosidase Inhibitory Activity Using Molecular Docking and Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2024; 25(19):10758. https://doi.org/10.3390/ijms251910758
Chicago/Turabian StyleHyun, Kyung-A, Xuhui Liang, Yang Xu, Seung-Young Kim, Kyung-Hwan Boo, Jin-Soo Park, Won-Jae Chi, and Chang-Gu Hyun. 2024. "Analysis of the Setomimycin Biosynthetic Gene Cluster from Streptomyces nojiriensis JCM3382 and Evaluation of Its α-Glucosidase Inhibitory Activity Using Molecular Docking and Molecular Dynamics Simulations" International Journal of Molecular Sciences 25, no. 19: 10758. https://doi.org/10.3390/ijms251910758