
Hemochromatosis: Ferroptosis, ROS, Gut Microbiome, and Clinical Challenges with Alcohol as Confounding Variable


Abstract
:1. Introduction
2. Role of Iron in Human Health
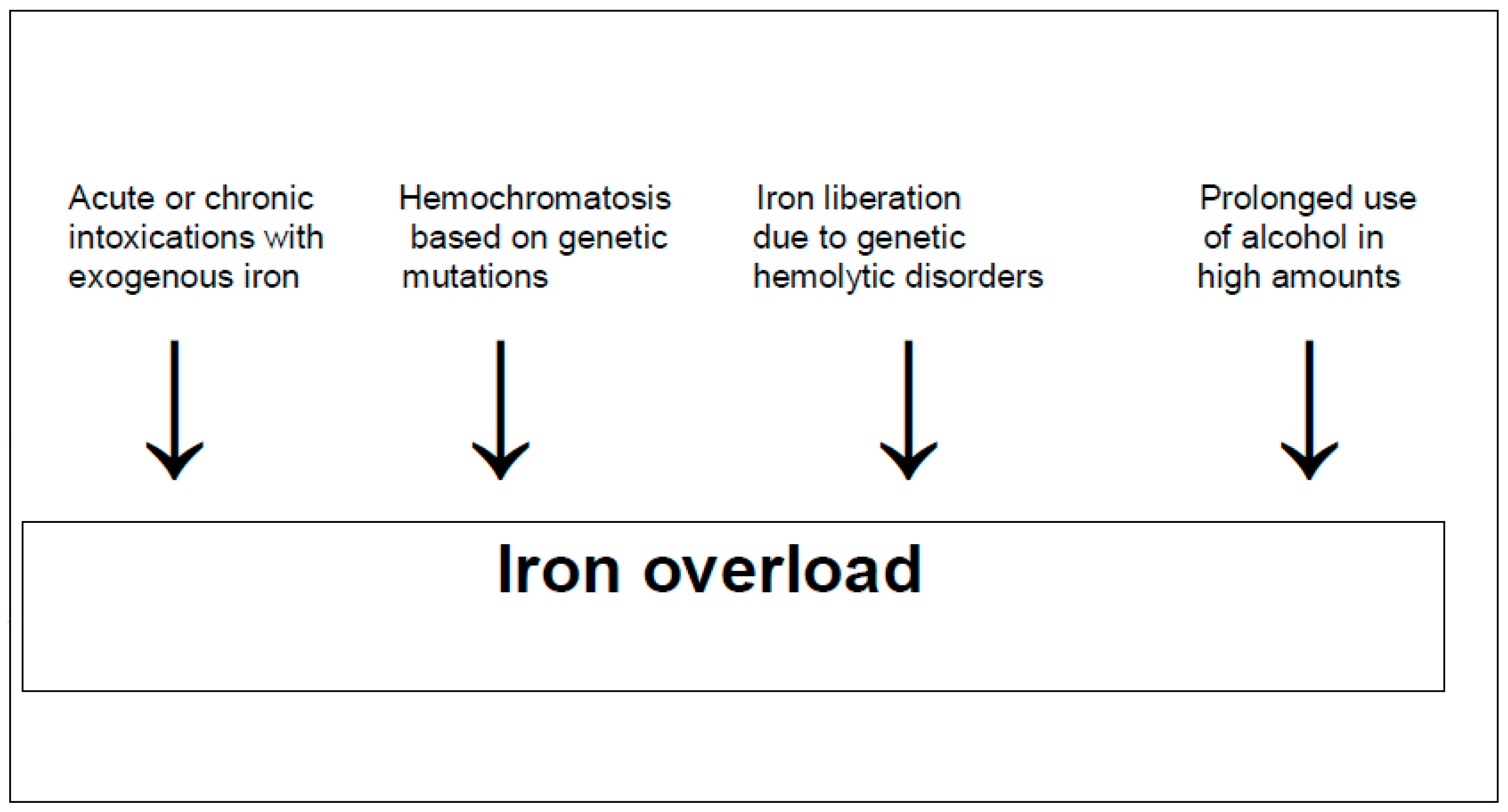
3. Iron Deficiency and Iron Overload
4. Factors Ensuring Sustained Iron Homeostasis
5. Hemochromatosis Caused by Iron as a Toxic Element
5.1. Pathophysiology
5.2. Natural Course
5.3. Prevalence
5.4. Clinical Features
5.5. Diagnosis
5.6. Therapy
5.7. Prognosis
6. Mechanistic Steps Involved in the Iron Liver Injury
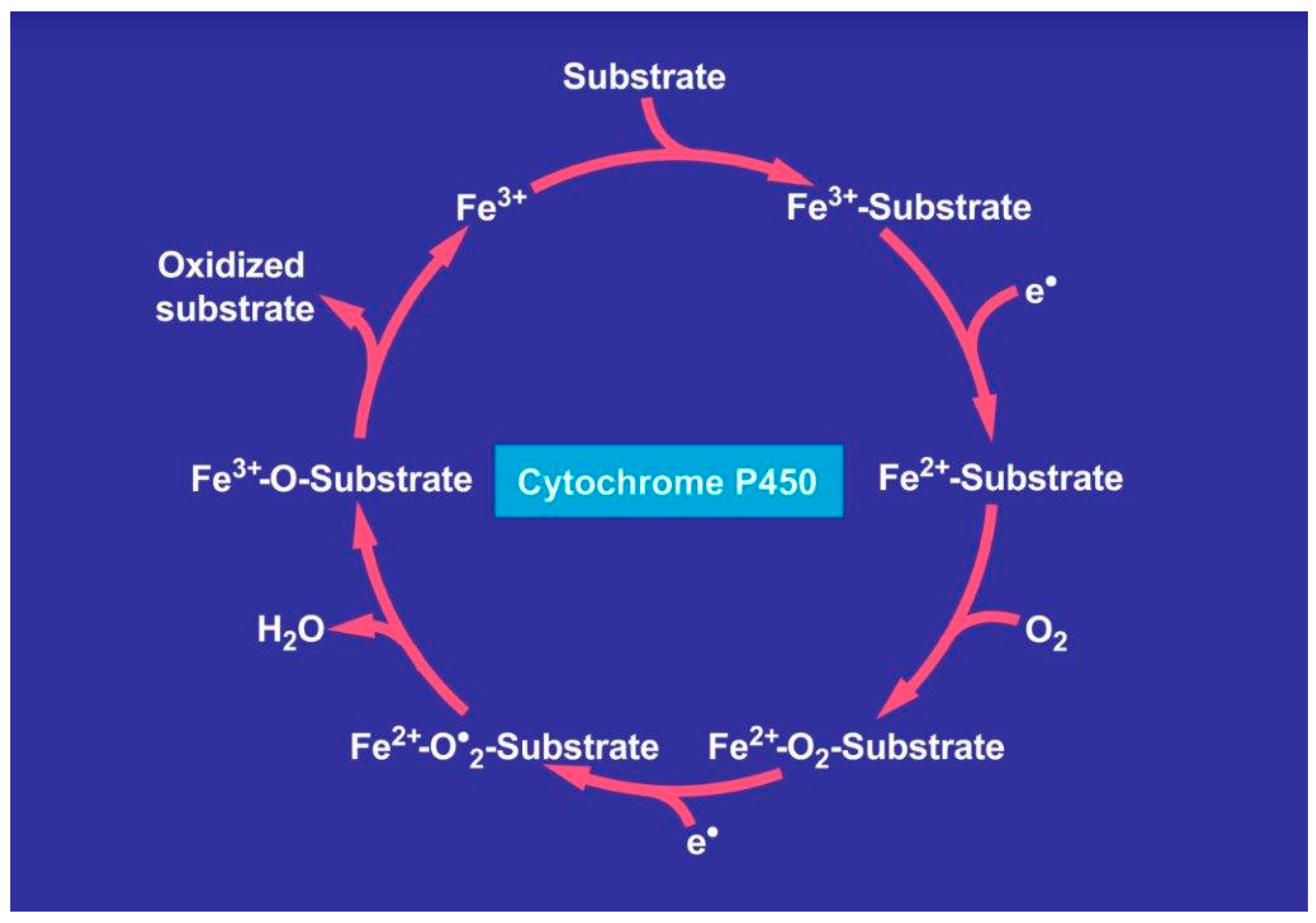
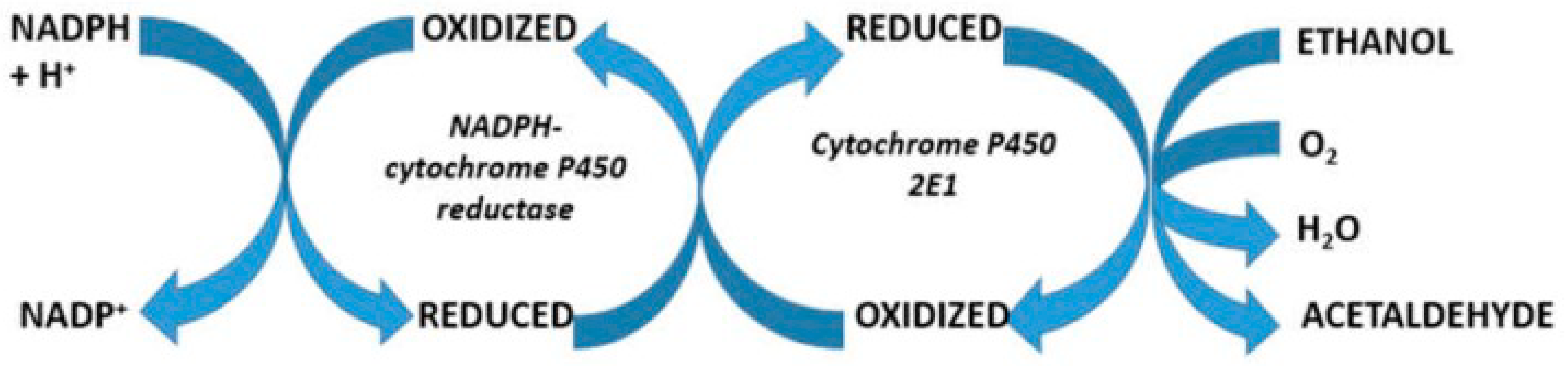
7. Alcohol
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 2012, 1820, 161–187. [Google Scholar] [CrossRef]
- Frebel, A.; Beers, T.C. The formation of the heaviest elements. Phys. Today 2018, 71, 30–37. [Google Scholar] [CrossRef]
- Clery, D. Some of the Universe’s Heavier Elements Are Created by Neutron Star Collisions. Available online: https://e.org/content/article/some-universe-s-heavier-elements-are-created-neutron-star-collisions (accessed on 19 November 2023).
- Belford, R. The Origin of the Elements. Available online: https://chem.libretexts.org/Courses/University_of_Arkansas_Little_Rock/Chem_1403%3A_General_Chemistry_2/Text/21%3A_Nuclear_Chemistry/21.06%3A_The_Origin_of_the_Elements (accessed on 19 November 2023).
- Teschke, R.; Xuan, T.D. Heavy metals, halogenated hydrocarbons, phthalates, glyphosate, cordycepin, alcohol, drugs, and herbs, assessed for liver injury and mechanistic steps. Front. Biosci. Landmark 2022, 27, 314. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. Aluminum, Arsenic, Beryllium, Cadmium, Chromium, Cobalt, Copper, Iron, Lead, Mercury, Molybdenum, Nickel, Platinum, Thallium, Titanium, Vanadium, and Zinc: Molecular aspects in experimental liver injury. Int. J. Mol. Sci. 2022, 23, 12213. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Merle, U.; Weiskirchen, R. Clinical features of Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S61. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Meyerrose, K.W.; Niederau, C.; Hefter, H.; Kreuzpaintner, G.; Strohmeyer, G. Wilson disease: Clinical presentation, treatment, and survival. Ann. Intern. Med. 1991, 115, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Strohmeyer, G.; Niederau, C.; Stremmel, W. Survival and causes of death in hemochromatosis. Observations in 163 patients. Ann. N. Y. Acad. Sci. 1988, 526, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Sonnenberg, A.; Stremmel, W.; Trampisch, H.J.; Strohmeyer, G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985, 313, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Strohmeyer, G.; Stremmel, W. Epidemiology, clinical spectrum and prognosis of hemochromatosis. Adv. Exp. Med. Biol. 1994, 356, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Fischer, R.; Pürschel, A.; Stremmel, W.; Häussinger, D.; Strohmeyer, G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996, 110, 1107–1119. [Google Scholar] [CrossRef]
- Roemhild, K.; von Maltzahn, F.; Weiskirchen, R.; Knüchel, R.; von Stillfried, S.; Lammers, T. Iron metabolism: Pathophysiology and pharmacology. Trends Pharmacol. Sci. 2021, 42, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On iron metabolism and its regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Connorton, J.M.; Balk, J.; Rodríguez-Celma, J. Iron homeostasis in plants-a brief overview. Metallomics 2017, 9, 813–823. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Matsuo, Y.; Watanabe, R.; Miyazato, M.; Matsuo, M.; Nagahama, Y.; Tanaka, H.; Ooshio, T.; Goto, M.; Okada, Y.; et al. Hepatocyte-specific damage in acute toxicity of sodium ferrous citrate: Presentation of a human autopsy case and experimental results in mice. Toxicol. Rep. 2023, 10, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.W.; Becker, W. Iron Toxicity; Updated 26 June 2023; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459224/ (accessed on 19 November 2023).
- Salomao, M.A. Pathology of hepatic iron overload. Clin. Liver Dis. 2021, 17, 232–237. [Google Scholar] [CrossRef]
- Abou Yassine, A.; MacDougall, K.; Sasso, R.; Shammaa, Y.; Alsheikh, M.; Abureesh, M.; Dahabra, L.; Alshami, M.; Mulrooney, S. The evolution of iron-related comorbidities and hospitalization in patients with hemochromatosis: An analysis of the nationwide inpatient sample. Blood Sci. 2023, 5, 131–135. [Google Scholar] [CrossRef]
- Zhang, A.S.; Enns, C.A. Molecular mechanisms of normal iron homeostasis. Hematol. Am. Soc. Hematol. Educ. Program. 2009, 2009, 207–214. [Google Scholar] [CrossRef]
- Ems, T.; St Lucia, K.; Huecker, M.R. Biochemistry, Iron Absorption; Updated 17 April 2023; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448204/ (accessed on 19 November 2023).
- Teichmann, R.; Stremmel, W. Iron uptake by human upper small intestine microvillous membrane vesicles. Indication for a facilitated transport mechanism mediated by a membrane iron-binding protein. J. Clin. Investig. 1990, 86, 2145–2153. [Google Scholar] [CrossRef]
- McLaren, G.D.; Nathanson, M.H.; Jacobs, A.; Trevett, D.; Thomson, W. Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. J. Lab. Clin. Med. 1991, 117, 390–401. [Google Scholar]
- Stremmel, W.; Schneider, M.; Lotz, G.; Niederau, C.; Teschke, R.; Strohmeyer, G. Iron uptake by rat duodenal microvillous membrane vesicles. Gastroenterology 1985, 88, A1602. [Google Scholar]
- Stremmel, W.; Schneider, M.; Lotz, G.; Niederau, C.; Teschke, R.; Strohmeyer, G. Iron uptake by rat duodenal microvillous membrane vesicles: Evidence for a carrier-mediated process. Eur. J. Clin. Investig. 1987, 17, 136–145. [Google Scholar] [CrossRef]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Sharp, P.; Srai, S.K. Molecular mechanisms involved in intestinal iron absorption. World J. Gastroenterol. 2007, 13, 4716–4724. [Google Scholar] [CrossRef]
- McLaren, G.D.; Gordeuk, V.R. Hereditary hemochromatosis: Insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematol. Am. Soc. Hematol. Educ. Program. 2009, 2009, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.C.; Edwards, C.Q.; Acton, R.T. HFE gene: Structure, function, mutations, and associated iron abnormalities. Gene 2015, 574, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Grønlien, H.K.; Christoffersen, T.E.; Nystrand, C.F.; Garabet, L.; Syvertsen, T.; Moe, M.K.; Olstad, O.K.; Jonassen, C.M. Cytokine and gene expression profiling in patients with HFE-associated hereditary hemochromatosis according to genetic profile. Acta Haematol. 2021, 144, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Cancado, R.D.; Alvarenga, A.M.; Santos, P.C.J. HFE hemochromatosis: An overview about therapeutic recommendations. Hematol. Transfus. Cell Ther. 2022, 44, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Girelli, D.; Busti, F.; Brissot, P.; Cabantchik, I.; Muckenthaler, M.U.; Porto, G.; on behalf of the Nomenclature Committee of the International Society for the Study of Iron in Biology and Medicine (BIOIRON Society). Hemochromatosis classification: Update and recommendations by the BIOIRON Society. Blood 2022, 139, 3018–3029. [Google Scholar] [CrossRef]
- Porter, J.L.; Rawla, P. Hemochromatosis; Updated 31 March 2023; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430862/ (accessed on 19 November 2023).
- Milman, N.T. Managing genetic hemochromatosis: An overview of dietary measures, which may reduce intestinal iron absorption in persons with iron overload. Gastroenterol. Res. 2021, 14, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Prim. 2018, 4, 18016. [Google Scholar] [CrossRef]
- Byrnes, V.; Ryan, E.; Barrett, S.; Kenny, P.; Mayne, P.; Crowe, J. Genetic hemochromatosis, a Celtic disease: Is it now time for population screening? Genet. Test. 2001, 5, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Lucotte, G.; Mercier, G. Celtic origin of the C282Y mutation of hemochromatosis. Genet. Test. 2000, 4, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Distante, S.; Robson, K.J.; Graham-Campbell, J.; Arnaiz-Villena, A.; Brissot, P.; Worwood, M. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum. Genet. 2004, 115, 269–279. [Google Scholar] [CrossRef]
- Kiely, P.D. Haemochromatosis arthropathy-a conundrum of the Celtic curse. J. R. Coll. Physicians Edinb. 2018, 48, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Lucotte, G. Frequency analysis and allele map in favor of the celtic origin of the C282Y mutation of hemochromatosis. Blood Cells Mol. Dis. 2001, 27, 549–556. [Google Scholar] [CrossRef]
- Lucotte, G.; Dieterlen, F. A European allele map of the C282Y mutation of hemochromatosis: Celtic versus Viking origin of the mutation? Blood Cells Mol. Dis. 2003, 31, 262–726. [Google Scholar] [CrossRef] [PubMed]
- Olynyk, J.K.; Cullen, D.J.; Aquilia, S.; Rossi, E.; Summerville, L.; Powell, L.W. A population-based study of the clinical expression of the hemochromatosis gene. N. Engl. J. Med. 1999, 3411, 718–724. [Google Scholar] [CrossRef]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Bardou-Jacquet, E.; Brissot, P. Diagnostic evaluation of hereditary hemochromatosis (HFE and non-HFE). Hematol. Oncol. Clin. N. A. 2014, 28, 625–635. [Google Scholar] [CrossRef]
- Yun, S.; Vincelette, N.D. Update on iron metabolism and molecular perspective of common genetic and acquired disorder, hemochromatosis. Crit. Rev. Oncol. Hematol. 2015, 95, 12–25. [Google Scholar] [CrossRef]
- Joshi, R.; Shvartsman, M.; Morán, E.; Lois, S.; Aranda, J.; Barqué, A.; de la Cruz, X.; Bruguera, M.; Vagace, J.M.; Gervasini, G.; et al. Functional consequences of transferrin receptor-2 mutations causing hereditary hemochromatosis type 3. Mol. Genet. Genom. Med. 2015, 3, 221–232. [Google Scholar] [CrossRef]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, P1811–P1821. [Google Scholar] [CrossRef]
- Barton, J.C.; McLaren, C.E.; Chen, W.P.; Ramm, G.A.; Anderson, G.J.; Powell, L.W.; Subramaniam, V.N.; Adams, P.C.; Phatak, P.D.; Gurrin, L.C.; et al. Cirrhosis in hemochromatosis: Independent risk factors in 368 HFE p.C282Y homozygotes. Ann. Hepatol. 2018, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.L.; Filho, R.; Cruz, S.; Franco, R.; Tavella, M.; Secaf, M.; Ramalho, L.; Zucoloto, S.; Rodrigues, S.; Zago, M. Hereditary hemochromatosis in a Brazilian university hospital in São Paulo State (1990–2000). Genet. Mol. Res. 2005, 4, 31–38. [Google Scholar] [PubMed]
- Stål, P.; Glaumann, H.; Hultcrantz, R. Liver cell damage and lysosomal iron storage in patients with idiopathic hemochromatosis. A light and electron microscopic study. J. Hepatol. 1990, 11, 172–180. [Google Scholar] [CrossRef]
- Golfeyz, S.; Lewis, S.; Weisberg, I.S. Hemochromatosis: Pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Daru, J.; Colman, K.; Stanworth, S.J.; De La Salle, B.; Wood, E.M.; Pasricha, S.R. Serum ferritin as an indicator of iron status: What do we need to know? Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1634S–1639S. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.M.; Looker, A.C. Laboratory methodologies for indicators of iron status: Strengths, limitations, and analytical challenges. Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1606S–1614S. [Google Scholar] [CrossRef] [PubMed]
- Kotla, N.K.; Dutta, P.; Parimi, S.; Das, N.K. The role of ferritin in health and disease: Recent advances and understandings. Metabolites 2022, 12, 609. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Namaste, S.M.; Serdula, M.; Suchdev, P.S.; Rohner, F.; Flores-Ayala, R.; Addo, O.Y.; Raiten, D.J. Adjusting total body iron for inflammation: Biomarkers Reflecting Inflammation and Nutritional Determinants of Anemia (BRINDA) project. Am. J. Clin. Nutr. 2017, 106 (Suppl 1), 383S–389S. [Google Scholar] [PubMed]
- Koperdanova, M.; Cullis, J.O. Interpreting raised serum ferritin levels. BMJ 2015, 351, h3692. [Google Scholar] [CrossRef] [PubMed]
- Sandnes, M.; Ulvik, R.J.; Vorland, M.; Reikvam, H. Hyperferritinemia-a clinical overview. J. Clin. Med. 2021, 10, 2008. [Google Scholar] [CrossRef]
- DePalma, R.G.; Hayes, V.W.; O’Leary, T.J. Optimal serum ferritin level range: Iron status measure and inflammatory biomarker. Metallomics 2021, 13, mfab030. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Ferrao, K.; Mehta, K.J. Liver iron loading in alcohol-associated liver disease. Am. J. Pathol. 2023, 193, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Li, L.X.; Guo, F.F.; Liu, H.; Zeng, T. Iron overload in alcoholic liver disease: Underlying mechanisms, detrimental effects, and potential therapeutic targets. Cell. Mol. Life Sci. 2022, 79, 201. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef]
- EASL. EASL clinical practice guidelines on haemochromatosis. J. Hepatol. 2022, 77, 479–502. [Google Scholar] [CrossRef]
- Srole, D.N.; Ganz, T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J. Cell. Physiol. 2021, 236, 4888–4901. [Google Scholar] [CrossRef]
- Bacon, B.R.; Adams, P.C.; Kowdley, K.V.; Powell, L.W.; Tavill, A.S.; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011, 54, 328–343. [Google Scholar] [CrossRef]
- Tavill, A.S.; Adams, P.C. A diagnostic approach to hemochromatosis. Can. J. Gastroenterol. 2006, 20, 535–540. [Google Scholar] [CrossRef]
- Lyon, E.; Frank, E.L. Hereditary hemochromatosis since discovery of the HFE gene. Clin. Chem. 2001, 47, 1147–1156. [Google Scholar] [CrossRef]
- Sood, R.; Bakashi, R.; Vinod SHegade, V.S.; Kelly, S.M. Diagnosis and management of hereditary haemochromatosis. Br. J. Gen. Pract. 2013, 63, 331–332. [Google Scholar] [CrossRef]
- Palmer, W.C.; Vishnu, P.; Sanchez, W.; Aqel, B.; Riegert-Johnson, D.; Seaman, L.A.K.; Bowman, A.W.; Rivera, C.E. Diagnosis and management of genetic iron overload disorders. J. Gen. Intern. Med. 2018, 33, 2230–2236. [Google Scholar] [CrossRef]
- Daniłowicz-Szymanowicz, L.; Świątczak, M.; Sikorska, K.; Starzyński, R.R.; Raczak, A.; Lipiński, P. Pathogenesis, diagnosis, and clinical implications of hereditary hemochromatosis-The cardiological point of view. Diagnostics 2021, 11, 1279. [Google Scholar] [CrossRef]
- Adams, P.C. Epidemiology and diagnostic testing for hemochromatosis and iron overload. Int. J. Lab. Hematol. 2015, 37, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Teschke, R. DILI, HILI, RUCAM algorithm, and AI, the Artificial Intelligence: Provocative issues, progress, and proposals. Arch. Gastroenterol. Res. 2020, 1, 4–11. [Google Scholar]
- Teschke, R.; Danan, G. Idiosyncratic drug-induced liver injury (DILI) and herb-induced liver injury (HILI): Diagnostic algorithm based on the quantitative Roussel Uclaf Causality Assessment Method (RUCAM). Diagnostics 2021, 11, 458. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Bénichou, C. Causality assessment of adverse reactions to drugs–I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, C.; Danan, G.; Flahault, A. Causality assessment of adverse reactions of drugs–II. An original model for validation of drug causality assessment methods: Case reports with positive rechallenge. J. Clin. Epidemiol. 1993, 46, 1331–1336. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. Roussel Uclaf Causality Assessment Method for drug-induced liver injury: Present and future. Front. Pharmacol. 2019, 10, 853. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 2016, 17, 14. [Google Scholar] [CrossRef]
- European Commission. White Paper on Artificial Intelligence–A European Approach to Excellence and Trust, Released 19 February 2020. Available online: https://ec.europa.eu/info/sites/info/files/commission-white-paper-artificial-intelligence-feb2020_en.pdf (accessed on 17 December 2023).
- McCarthy, J.; Minsky, M.L.; Rochester, N.; Shannon, C.E. A Proposal for the Dartmouth Summer Research Project on Artificial Intelligence. Available online: http://www-formal.stanford.edu/jmc/history/dartmouth/dartmouth.html (accessed on 17 December 2023).
- Amato, F.; López, A.; Peña-Méndez, E.M.; Vaňhara, P.; Hampl, A.; Havel, J. Artificial neural networks in medical diagnosis. J. Appl. Biomed. 2013, 11, 47–58. [Google Scholar] [CrossRef]
- Parmanand, B.; Watson, M.; Boland, K.J.; Ramamurthy, N.; Wharton, V.; Morovat, A.; Lund, E.K.; Collier, J.; Le Gall, G.; Kellingray, L.; et al. Systemic iron reduction by venesection alters the gut microbiome in patients with haemochromatosis. JHEP Rep. 2020, 2, 100154. [Google Scholar] [CrossRef]
- Rombout-Sestrienkova, E.; Winkens, B.; Essers, B.A.; Nieman, F.H.; Noord, P.A.; Janssen, M.C.; van Deursen, C.T.; Boonen, A.; Reuser-Kaasenbrood, E.P.; Heeremans, J.; et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: Results from a randomized crossover trial. Transfusion 2016, 56, 261–270. [Google Scholar] [CrossRef]
- Cançado, R.; Melo, M.R.; de Moraves Bastos, R.; Santos, P.C.; Guerra-Shinohara, E.M.; Chiattone, C.; Ballas, S.K. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: Results of a 1-yr Phase 2 study. Eur. J. Haematol. 2015, 95, 545–550. [Google Scholar] [CrossRef]
- Phatak, P.; Brissot, P.; Wurster, M.; Adams, P.C.; Bonkovsky, H.L.; Gross, J.; Malfertheiner, P.; McLaren, G.D.; Niederau, C.; Piperno, A.; et al. A phase 1/2 dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary hemochromatosis. Hepatology 2010, 52, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Brissot, E. What’s important and new in hemochromatosis? Clin. Hematol. Int. 2020, 2, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lymberopoulos, P.; Prakash, S.; Shaikh, A.; Bhatnagar, A.; Allam, A.K.; Goli, K.; Goss, J.A.; Kanwal, F.; Rana, A.; Kowdley, K.V.; et al. Long-term outcomes and trends in liver transplantation for hereditary hemochromatosis in the United States. Liver Transpl. 2023, 29, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Brandhagen, D.J.; Alvarez, W.; Therneau, T.M.; Kruckeberg, K.E.; Thibodeau, S.N.; Ludwig, J.; Porayko, M.K. Iron overload in cirrhosis-HFE genotypes and outcome after liver transplantation. Hepatology 2000, 31, 456–460. [Google Scholar] [CrossRef]
- Tung, B.Y.; Farrell, F.J.; McCashland, T.M.; Gish, R.G.; Bacon, B.R.; Keeffe, E.B.; Kowdley, K.V. Long-term follow-up after liver transplantation in patients with hepatic iron overload. Liver Transpl. Surg. 1999, 5, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Dobrindt, E.M.; Keshi, E.; Neulichedl, J.; Schöning, W.; Öllinger, R.; Pratschke, J.; Eurich, D. Long-term outcome of orthotopic liver transplantation in patients with hemochromatosis: A summary of a 30-year transplant program. Transpl. Direct 2020, 6, e560. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Philip, J.; Lorho, R.; Ropert, M.; Latournerie, M.; Houssel-Debry, P.; Guyader, D.; Loréal, O.; Boudjema, K.; Brissot, P. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology 2014, 59, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. The molecular pathogenesis of hereditary hemochromatosis. Semin. Liver Dis. 2011, 31, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.; Mukkamalla, S.K.R. Iron Binding Capacity; Updated 2 January 2023; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Yilmaz, B.; Li, H. Gut microbiota and iron: The crucial factors in health and disease. Pharmaceuticals 2018, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Inherited disorders of iron overload. Front. Nutr. 2018, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, J. Twenty-Five years of contemplating genotype-based hereditary hemochromatosis population screening. Genes 2022, 13, 1622. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Ramm, G.A.; Crawford, D.H.; Witztum, J.L.; Powell, L.W.; Chojkier, M. Excess iron induces hepatic oxidative stress and transforming growth factor beta1 in genetic hemochromatosis. Hepatology 1997, 26, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Shizukuda, Y.; Bolan, C.D.; Nguyen, T.T.; Botello, G.; Tripodi, D.J.; Yau, Y.Y.; Waclawiw, M.A.; Leitman, S.F.; Rosing, D.R. Oxidative stress in asymptomatic subjects with hereditary hemochromatosis. Am. J. Hematol. 2007, 82, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Shizukuda, Y.; Tripodi, D.J.; Rosing, D.R. Iron overload or oxidative stress? Insight into a mechanism of early cardiac manifestations of asymptomatic hereditary hemochromatosis subjects with C282Y homozygosity. J. Cardiovasc. Transl. Res. 2016, 9, 400–401. [Google Scholar] [CrossRef]
- Niemelä, O.; Parkkila, S.; Britton, R.S.; Brunt, E.; Janney, C.; Bacon, B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J. Lab. Clin. Med. 1999, 133, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Hepatic microsomal ethanol-oxidizing system. In vitro characteristics and adaptive properties in vivo. J. Biol. Chem. 1970, 245, 2505–2512. [Google Scholar] [CrossRef]
- Teschke, R.; Hasumura, Y.; Joly, J.G.; Ishii, H.; Lieber, C.S. Microsomal ethanol-oxidizing system (MEOS): Purification and properties of a rat liver system free of catalase and alcohol dehydrogenase. Biochem. Biophys. Res. Commun. 1972, 49, 1187–1193. [Google Scholar] [CrossRef]
- Teschke, R. Alcoholic liver disease: Alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef]
- Teschke, R. Alcoholic liver disease: Current mechanistic aspects with focus on their clinical relevance. Biomedicines 2019, 7, 68. [Google Scholar] [CrossRef]
- Teschke, R.; Neuman, M.G.; Liangpunsakul, S.; Seitz, H.K. Alcoholic liver disease and the co-triggering role of MEOS with its CYP 2E1 catalytic cycle and ROS. Arch. Gastroenterol. Res. 2021, 2, 9–25. [Google Scholar]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Oxygen radical generation by microsomes: Role of iron and implications for alcohol metabolism and toxicity. Free Radic. Biol. Med. 1989, 7, 559–567. [Google Scholar] [CrossRef]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in alcoholic and non-alcoholic liver injury. Roles of ROS, reactive intermediates and lipid overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Stål, P.; Johansson, I.; Ingelman-Sundberg, M.; Hagen, K.; Hultcrantz, R. Hepatotoxicity induced by iron overload and alcohol. Studies on the role of chelatable iron, cytochrome P450 2E1 and lipid peroxidation. J. Hepatol. 1996, 25, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C.; Bailey, S.M. Ethanol consumption and liver mitochondria function. Biol. Signals Recept. 2001, 10, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol. Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Fu, Y.; Chung, F.L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying hepatic cell death during liver damage: Ferroptosis—A novel form of non-apoptotic cell death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The emerging role of ferroptosis in non-cancer liver diseases: Hype or increasing hope? Cell Death Dis. 2020, 11, 518. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell. Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Luo, J.; Song, G.; Chen, N.; Xie, M.; Niu, X.; Zhou, S.; Ji, Y.; Zhu, X.; Ma, W.; Zhang, Q.; et al. Ferroptosis contributes to ethanol-induced hepatic cell death via labile iron accumulation and GPx4 inactivation. Cell Death Discov. 2023, 9, 311. [Google Scholar] [CrossRef]
- Lawless, M.W.; Mankan, A.K.; White, M.; O’Dwyer, M.J.; Norris, S. Expression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responses. BMC Cell Biol. 2007, 8, 30. [Google Scholar] [CrossRef]
- Hedges, J.C.; Singer, C.A.; Gerthoffer, W.T. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am. J. Respir. Cell Mol. Biol. 2000, 23, 86–94. [Google Scholar] [CrossRef]
- Wolff, B.; Burns, A.R.; Middleton, J.; Rot, A. Endothelial cell “memory” of inflammatory stimulation: Human venular endothelial cells store interleukin 8 in Weibel-Palade bodies. J. Exp. Med. 1998, 188, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Utgaard, J.O.; Jahnsen, F.L.; Bakka, A.; Brandtzaeg, P.; Haraldsen, G. Rapid secretion of prestored interleukin 8 from Weibel-Palade bodies of microvascular endothelial cells. J. Exp. Med. 1998, 188, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Gao, L.; Mäkitie, A.A.; Florek, E.; Czarnywojtek, A.; Saba, N.F.; Ferlito, A. Iron, Ferroptosis, and Head and Neck Cancer. Int. J. Mol. Sci. 2023, 24, 15127. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, L.M.; Powell, L.W. Hemochromatosis and alcoholic liver disease. Alcohol 2003, 30, 131–136. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Contribution of Iron to Human Health | References |
|---|---|---|
| Hemoglobin | Around 80% of the total iron in body stores are found in the hemoglobin of erythrocytes. Iron is required for hemoglobin synthesis in the context of erythropoiesis, whereby erythroblasts in the bone marrow form erythrocytes responsible for oxygen transport. Iron is recycled from senescent erythrocytes and thus conserved by the body and stored by macrophages in spleen, liver, and bone marrow. | Roemhild et al., 2021 [13], Vogt et al., 2021 [14], Abbaspour et al., 2014 [15] |
| Myoglobin | Fe2+ is bound to a heme group of myoglobin, which helps bind oxygen reversibly. Myoglobin is a protein primarily found in the striated muscles and supplies the muscle oxygen to myocytes. | Abbaspour et al., 2014 [15] |
| DNA synthesis, nucleic acid repair | Iron is involved in DNA biosynthesis and is a known indispensable functional cofactor of helicases, nucleases, glycosylases, demethylases, and ribonucleotide reductase. | Roemhild et al., 2021 [13], Vogt et al., 2021 [14] |
| Cell growth | Iron is an essential element for the growth of all cells, whereby the rapid proliferation of tumor cells is usually more dependent on iron than normal cells are. | Roemhild et al., 2021 [13], Vogt et al., 2021 [14] |
| Host defense, cell signaling | Iron is essential for the host and pathogens in managing cellular and metabolic processes. Free iron, Fe2+, is involved in the Haber–Weiss reaction and the Fenton reaction that generate reactive oxygen species (ROS), supporting the host defense processes. Iron modulates immune cell function as well as the host-and-microbe interplay. | Vogt et al., 2021 [13] |
| Iron transporter proteins, heme enzymes, iron-containing enzymes | Iron is an essential part of iron transporter enzymes, heme enzymes, and other iron-containing enzymes involved in electron transfer and oxidation–reductions like cytochrome P450. | Vogt et al., 2021 [13], Abbaspour et al., 2014 [15] |
| Hemochromatosis Type | Details | First Author |
|---|---|---|
| Type 1 HFE-related | This is the classic form of hemochromatosis that is inherited in an autosomal-recessive fashion with a worldwide prevalence. | Bardou-Jacquet et al., 2014 [45], Yun et al., 2015 [46] |
| Type 2a Mutations in the hemojuvelin gene | Autosomal-recessive disorder that is seen both in whites and non-whites. Its onset is usually at 15–20 years. | Porter, 2023 [34] |
| Type 2b Mutations in the hepcidin gene | Autosomal-recessive disorder that is seen both in whites and non-whites. Its onset is usually at 15–20 years. | Porter, 2023 [34] |
| Type 3 Mutations in the transferrin receptor-2 gene | Autosomal-recessive disorder that is seen both in whites and non-whites. Its onset is at 30–40 years. | Joshi et al., 2015 [47] |
| Type 4 Mutations in the ferroportin gene | Autosomal-dominant disease seen both in whites and non-whites. Its onset is at 10–80 years. | Porter, 2023 [34] |
| Laboratory Test | Normal Range | Test Details in Patients with Hemochromatosis | References with First Author |
|---|---|---|---|
| Serum ferritin | <15 µg/L | >1000 µg/L | Porter et al., 2023 [34], Daru et al., 2017 [53] |
| Fasting transferrin saturation index | <45% | >45% | EASL, 2022 [63] |
| Genetic screening | NA | HFE gene mutations | Porter et al., 2023 [34] |
| Serum ALT | <40 U/L | Usually < 80 U/L | Porter et al., 2023 [34] |
| Serum iron | Variable | Not suitable as a diagnostic marker | Grønlien et al., 2021 [31] |
| Serum juvelin | NA | Under discussion as a hepcidin regulator in hemochromatosis | Porter et al., 2023 [34], EASL, 2022 [63], Srole et al., 2021 [64] |
| Serum erythroferrone | NA | Under discussion as a suppressor of induced hepcidin in hemochromatosis | Srole et al., 2021 [64] |
| Cascade of Events | Short Description | References with First Author |
|---|---|---|
| 1. Excessive intestinal uptake of iron | Mutations in the HFE gene lead the downregulation of hepcidin synthesis to excess iron absorption and iron overload in hemochromatosis | Golfeyz et al., 2018 [52] |
| 2. Uptake of high iron amounts by the liver cells from the blood | Intracellular iron initiates liver injury because the function of antioxidants is impaired due to low hepatic levels | Vogt et al., 2021 [14], Faruqi et al., 2023 [93] |
| 3. Intracellular Fe2+ reacts with ROS and facilitates ferroptosis, an iron-dependent regulated cell death, causing liver injury through phospholipid peroxidation | ROS are generated via the Haber–Weiss and Fenton reactions and attack structural proteins, lipids, nucleic acids, and carbohydrates This leads, among others, to membranous phospholipid peroxidation. Liver injury is aggravated by alcohol abuse that increases hepatic iron levels and enhances ROS production via hepatic cytochrome P450 induction | Ali et al., 2023 [60], Li et al., 2022 [61], Louvet et al., 2015 [62] |
| 4. Cytokines | Among the many mediators, including the interleukines (IL) tested, serum IL8 was elevated in patients with hemochromatosis. This indicated a close relationship of iron with hepatic macrophages, which retain IL8 in their storage vesicles | Grønlien et al., 2021 [31] |
| 5. Gut microbiome | Systemic iron reduction by phlebotomy modifies the gut microbiome through the improvement in colonic inflammation and oxidative stress | Parmanand et al., 2020 [82], Yilmaz et al., 2018 [94] |
| Details of the Haber–Weiss and Fenton Rection |
|---|
| Fe3+ + •O2− → Fe2+ + O2 (Haber–Weiss reaction) Fe2+ + H2O2 → Fe3+ + OH− + •OH (Fenton reaction) •O2− + H2O2 → OH− + •OH + O2 (Net reaction) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teschke, R. Hemochromatosis: Ferroptosis, ROS, Gut Microbiome, and Clinical Challenges with Alcohol as Confounding Variable. Int. J. Mol. Sci. 2024, 25, 2668. https://doi.org/10.3390/ijms25052668
Teschke R. Hemochromatosis: Ferroptosis, ROS, Gut Microbiome, and Clinical Challenges with Alcohol as Confounding Variable. International Journal of Molecular Sciences. 2024; 25(5):2668. https://doi.org/10.3390/ijms25052668
Chicago/Turabian StyleTeschke, Rolf. 2024. "Hemochromatosis: Ferroptosis, ROS, Gut Microbiome, and Clinical Challenges with Alcohol as Confounding Variable" International Journal of Molecular Sciences 25, no. 5: 2668. https://doi.org/10.3390/ijms25052668