
Modeling PAH Mixture Interactions in a Human In Vitro Organotypic Respiratory Model

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
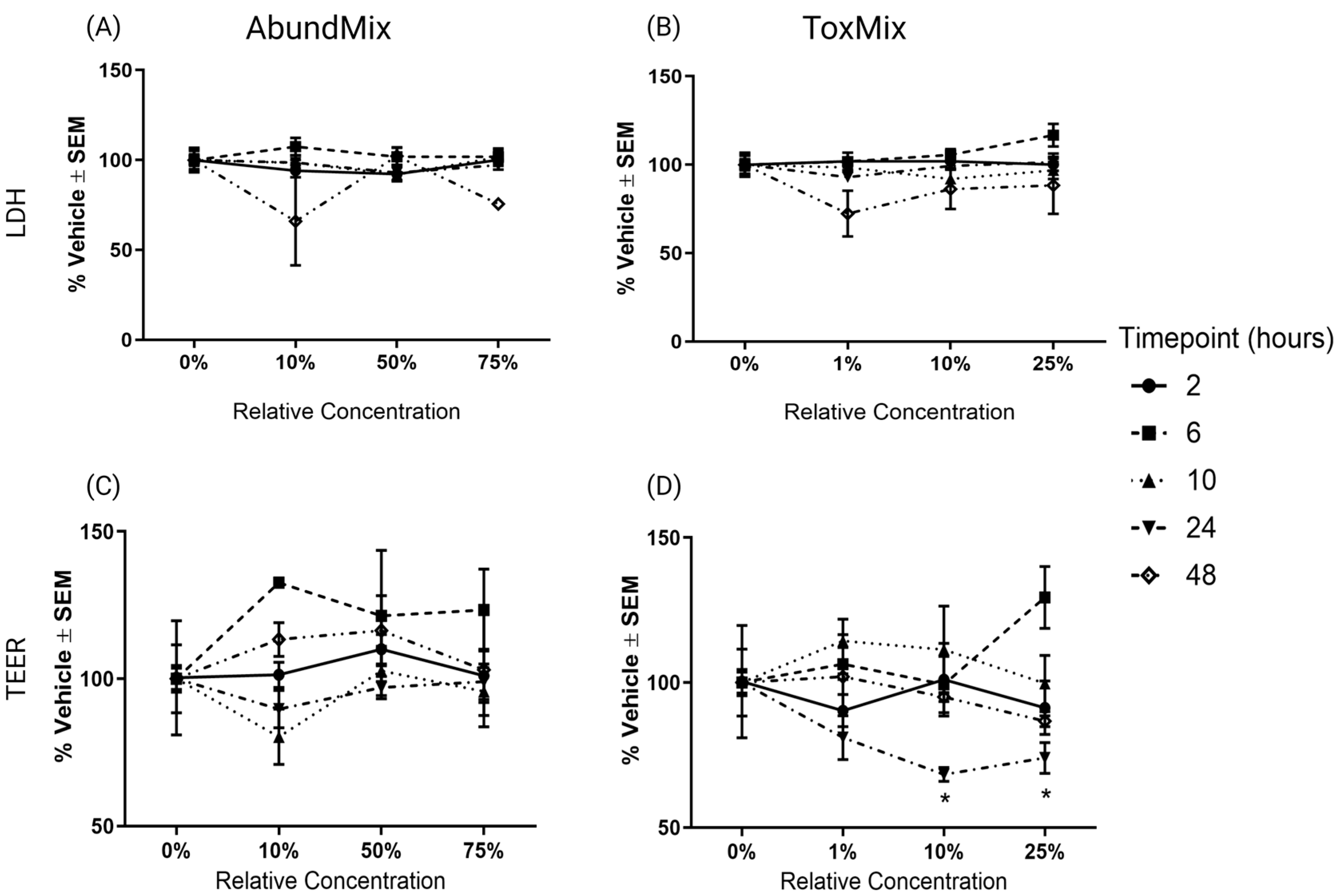
2.1. Evaluation of PAH Mixtures in ALI-HBECs
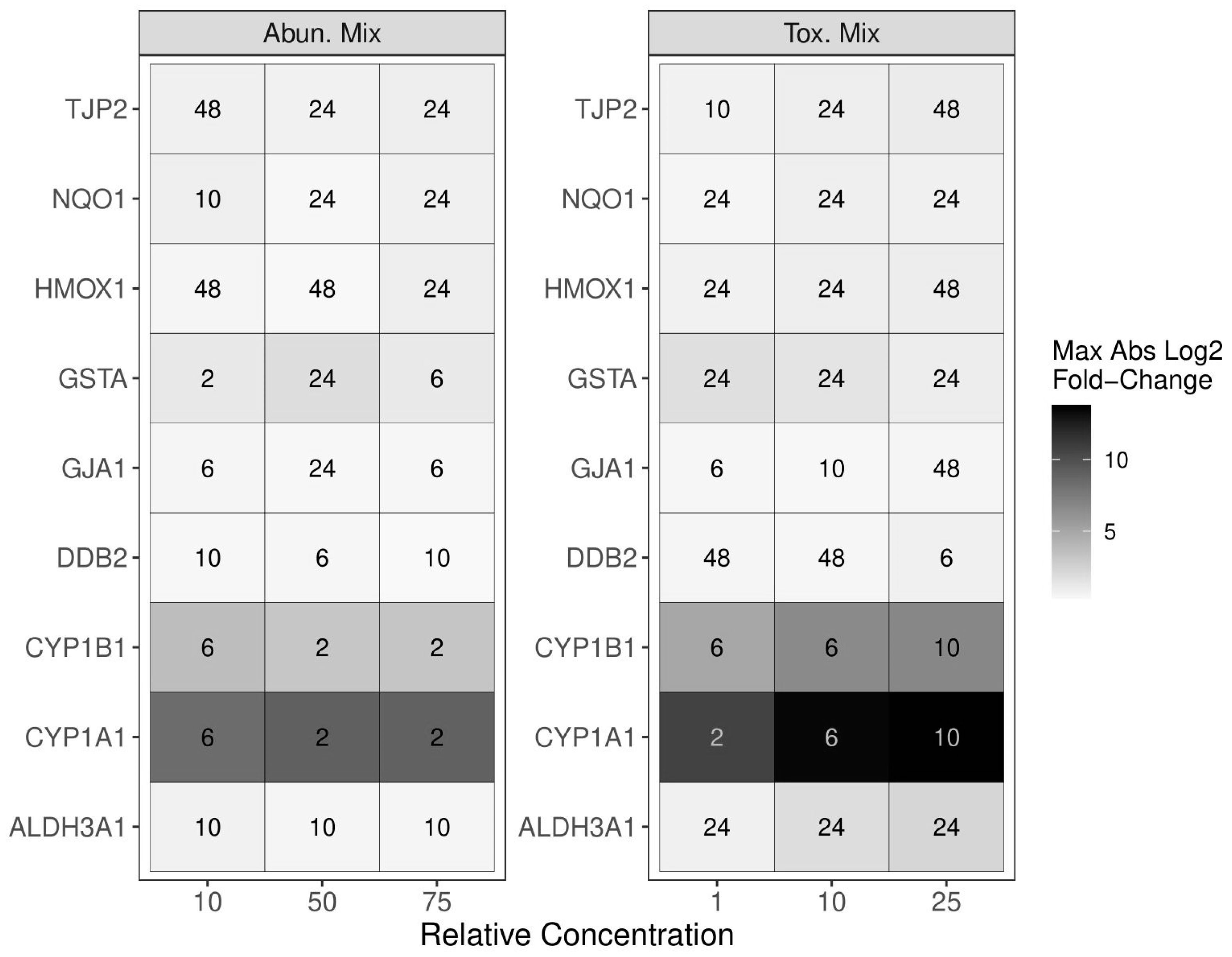
2.2. Selecting Mixture Parameters for Additivity Modeling
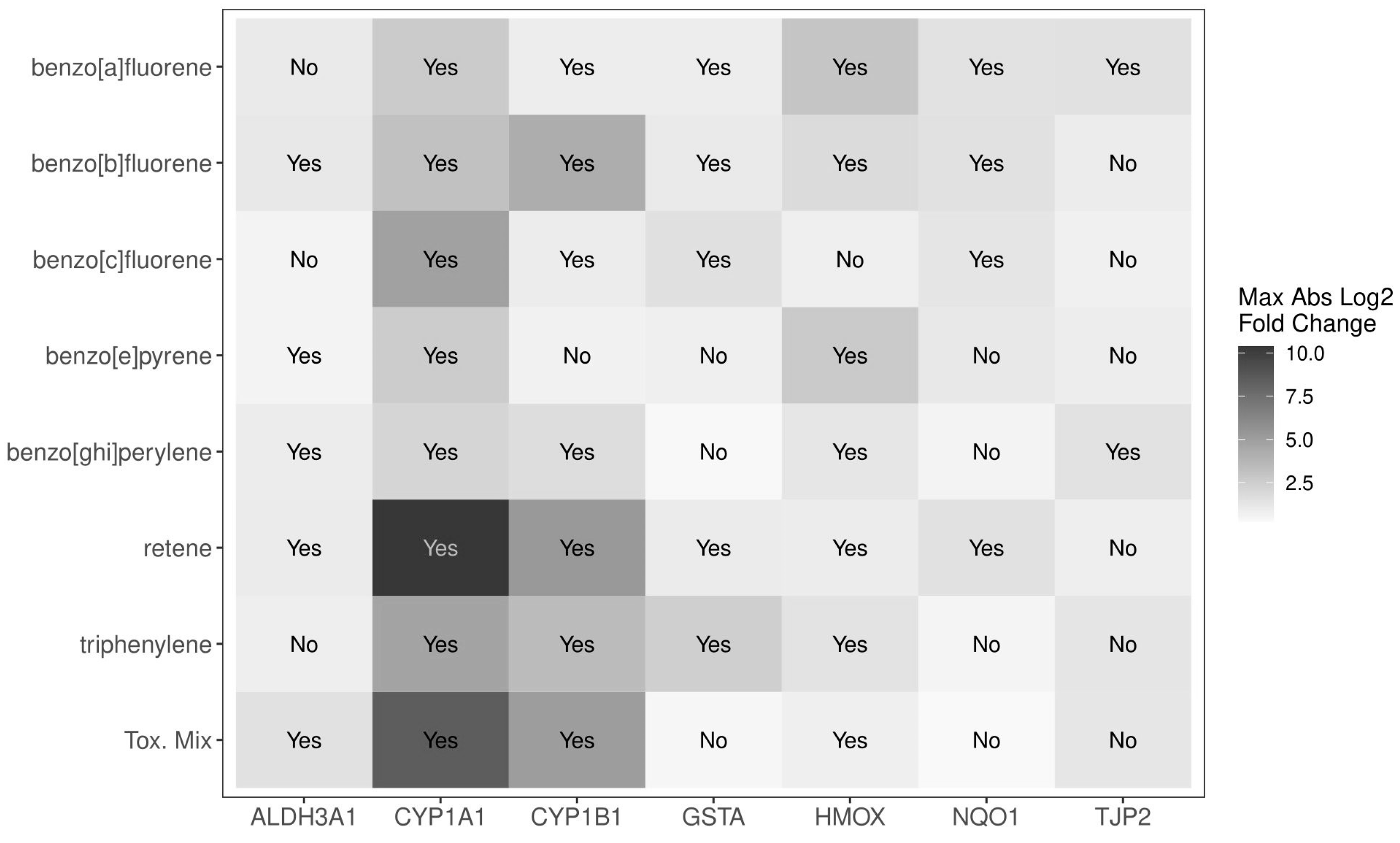
2.3. Evaluation of PAH Mixture Components in ALI-HBECs
2.4. Evaluating Mixture Additivity
3. Discussion
3.1. Differential Expression of Transcriptional Biomarkers by PAH Mixtures
3.2. Differential Expression of Transcriptional Biomarkers by Mixture Components
3.3. Modeling Mixture Response Based on Component Additivity
3.4. Best Practices for Additivity Modeling of PAH Mixtures
4. Materials and Methods
4.1. Mixture Formulation
4.2. Primary Human Bronchial Epithelial Cell Maintenance and Exposure
4.3. Transepithelial Electrical Resistance (TEER)
4.4. Cytotoxicity
4.5. Real-Time Quantitative PCR (RT-qPCR)
4.6. Statistical Analysis
4.7. Dose–Response Analysis
4.8. Independent Action Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heys, K.A.; Shore, R.F.; Pereira, M.G.; Jones, K.C.; Martin, F.L. Risk assessment of environmental mixture effects. RSC Adv. 2016, 6, 47844–47857. [Google Scholar] [CrossRef]
- Hassold, E.; Galert, W.; Schulze, J. Options for an environmental risk assessment of intentional and unintentional chemical mixtures under REACH: The status and ways forward. Environ. Sci. Eur. 2021, 33, 131. [Google Scholar] [CrossRef]
- Carlin, D.J.; Rider, C.V.; Woychik, R.; Birnbaum, L.S. Unraveling the Health Effects of Environmental Mixtures: An NIEHS Priority. Environ. Health Perspect. 2013, 121, a6–a8. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.A.; Goldsmith, J.; Kioumourtzoglou, M.-A. Complex Mixtures, Complex Analyses: An Emphasis on Interpretable Results. Curr. Environ. Health Rep. 2019, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency. Supplementary Guidance for Conducting Health Risk Assessment of Chemical Mixtures. 2000. Available online: https://cfpub.epa.gov/ncea/risk/recordisplay.cfm?deid=20533 (accessed on 3 July 2023).
- van Gestel, C.A.M.; Jonker, M.J.; Kammenga, J.E.; Laskowski, R.; Svendsen, C. Mixture Toxicity Linking Approaches from Ecological and Human Toxicology; Society of Environmental Toxicology and Chemistry (SETAC); CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Bopp, S.K.; Kienzler, A.; Richarz, A.-N.; van der Linden, S.C.; Paini, A.; Parissis, N.; Worth, A.P. Regulatory assessment and risk management of chemical mixtures: Challenges and ways forward. Crit. Rev. Toxicol. 2019, 49, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Kapraun, D.F.; Wambaugh, J.F.; Ring, C.L.; Tornero-Velez, R.; Setzer, R.W. A Method for Identifying Prevalent Chemical Combinations in the U.S. Population. Environ. Health Perspect. 2017, 125, 087017. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Gennings, C.; Teuschler, L.K.; Stork, L.G.; Tornero-Velez, R.; Crofton, K.M.; Rice, G.E. An Empirical Approach to Sufficient Similarity: Combining Exposure Data and Mixtures Toxicology Data: Testing for Sufficient Similarity. Risk Anal. 2013, 33, 1582–1595. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.N.; Ghetu, C.C.; Chang, Y.; Truong, L.; Tanguay, R.L.; Anderson, K.A.; Tilton, S.C. Leveraging Multiple Data Streams for Prioritization of Mixtures for Hazard Characterization. Toxics 2022, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Kumar, A. Identification of component-based approach for prediction of joint chemical mixture toxicity risk assessment with respect to human health: A critical review. Food Chem. Toxicol. 2020, 143, 111458. [Google Scholar] [CrossRef]
- Bureš, M.S.; Cvetnić, M.; Miloloža, M.; Grgić, D.K.; Markić, M.; Kušić, H.; Bolanča, T.; Rogošić, M.; Ukić, Š. Modeling the toxicity of pollutants mixtures for risk assessment: A review. Environ. Chem. Lett. 2021, 19, 1629–1655. [Google Scholar] [CrossRef]
- Hertzberg, R.C.; Mumtaz, M.M. Component-Based Risk Assessment Approaches with Additivity and Interactions. In Chemical Mixtures and Combined Chemical and Nonchemical Stressors; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Cedergreen, N.; Christensen, A.M.; Kamper, A.; Kudsk, P.; Mathiassen, S.K.; Streibig, J.C.; Sørensen, H. A review of independent action compared to concentration addition as reference models for mixtures of compounds with different molecular target sites. Environ. Toxicol. Chem. 2008, 27, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.J.; Webster, T.F. Generalized concentration addition: A method for examining mixtures containing partial agonists. J. Theor. Biol. 2009, 259, 469–477. [Google Scholar] [CrossRef] [PubMed]
- White, P.A. The genotoxicity of priority polycyclic aromatic hydrocarbons in complex mixtures. Mutat. Res. Toxicol. Environ. Mutagen. 2002, 515, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Labib, S.; Williams, A.; Kuo, B.; Yauk, C.L.; White, P.A.; Halappanavar, S. A framework for the use of single-chemical transcriptomics data in predicting the hazards associated with complex mixtures of polycyclic aromatic hydrocarbons. Arch. Toxicol. 2017, 91, 2599–2616. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, N.; Taxvig, C.; Pedersen, M.; Nellemann, C.; Hass, U.; Vinggaard, A.M. Concentration Addition, Independent Action and Generalized Concentration Addition Models for Mixture Effect Prediction of Sex Hormone Synthesis In Vitro. PLoS ONE 2013, 8, e70490. [Google Scholar] [CrossRef]
- Howard, G.J.; Schlezinger, J.J.; Hahn, M.E.; Webster, T.F. Generalized Concentration Addition Predicts Joint Effects of Aryl Hydrocarbon Receptor Agonists with Partial Agonists and Competitive Antagonists. Environ. Health Perspect. 2010, 118, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.H. Hazard and risk assessment of chemical mixtures using the toxic equivalency factor approach. Environ. Health Perspect. 1998, 106, 1051–1058. [Google Scholar] [PubMed]
- Bliss, C.I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J.C. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Lawal, A.T. Polycyclic aromatic hydrocarbons. A review. Cogent Environ. Sci. 2017, 3, 1339841. [Google Scholar] [CrossRef]
- Lee, J.H.; Gigliotti, C.L.; Offenberg, J.H.; Eisenreich, S.J.; Turpin, B.J. Sources of polycyclic aromatic hydrocarbons to the Hudson River Airshed. Atmos. Environ. 2004, 38, 5971–5981. [Google Scholar] [CrossRef]
- Masclet, P.; Mouvier, G.; Nikolaou, K. Relative decay index and sources of polycyclic aromatic hydrocarbons. Atmos. Environ. 1986, 20, 439–446. [Google Scholar] [CrossRef]
- Mitra, S.; Ray, B. Patterns and sources of polycyclic aromatic hydrocarbons and their derivatives in indoor air. Atmos. Environ. 1995, 29, 3345–3356. [Google Scholar] [CrossRef]
- Nikolaou, K.; Masclet, P.; Mouvier, G. Sources and chemical reactivity of polynuclear aromatic hydrocarbons in the atmosphere—A critical review. Sci. Total. Environ. 1984, 32, 103–132. [Google Scholar] [CrossRef]
- Yuan, H.; Tao, S.; Li, B.; Lang, C.; Cao, J.; Coveney, R.M. Emission and outflow of polycyclic aromatic hydrocarbons from wildfires in China. Atmos. Environ. 2008, 42, 6828–6835. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, S. Global atmospheric emission inventory of polycyclic aromatic hydrocarbons (PAHs) for 2004. Atmos. Environ. 2009, 43, 812–819. [Google Scholar] [CrossRef]
- Chang, Y.; Siddens, L.K.; Heine, L.K.; Sampson, D.A.; Yu, Z.; Fischer, K.A.; Löhr, C.V.; Tilton, S.C. Comparative mechanisms of PAH toxicity by benzo[a]pyrene and dibenzo[def,p]chrysene in primary human bronchial epithelial cells cultured at air-liquid interface. Toxicol. Appl. Pharmacol. 2019, 379, 114644. [Google Scholar] [CrossRef]
- Hermann, M. Synergistic effects of individual polycyclic aromatic hydrocarbons on the mutagenicity of their mixtures. Mutat. Res. Toxicol. 1981, 90, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Libalova, H.; Milcova, A.; Cervena, T.; Vrbova, K.; Rossnerova, A.; Novakova, Z.; Topinka, J.; Rossner, P. Kinetics of ROS generation induced by polycyclic aromatic hydrocarbons and organic extracts from ambient air particulate matter in model human lung cell lines. Mutat. Res. Toxicol. Environ. Mutagen. 2018, 827, 50–58. [Google Scholar] [CrossRef]
- Osgood, R.S.; Upham, B.L.; Bushel, P.R.; Velmurugan, K.; Xiong, K.-N.; Bauer, A.K. Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol. Sci. 2017, 157, 156–171. [Google Scholar] [CrossRef]
- Silva, E.; Rajapakse, N.; Kortenkamp, A. Something from “Nothing”—Eight Weak Estrogenic Chemicals Combined at Concentrations below NOECs Produce Significant Mixture Effects. Environ. Sci. Technol. 2002, 36, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. (Ed.) Polycyclic Aromatic Hydrocarbons. In Who Guidelines for indoor Air Quality: Selected Pollutants; WHO: Geneva, Switzerland, 2010; pp. 289–345. [Google Scholar]
- Boffetta, P.; Jourenkova, N.; Gustavsson, P. Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Causes Control. 1997, 8, 444–472. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, S.; Hebbern, C.; Cakmak, J.D.; Dales, R.E. The influence of polycyclic aromatic hydrocarbons on lung function in a representative sample of the Canadian population. Environ. Pollut. 2017, 228, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, B.; Chu, C.; Carlin, D.J. Polycyclic Aromatic Hydrocarbons: From Metabolism to Lung Cancer. Toxicol. Sci. 2015, 145, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, A.; Tilley, A.E.; Shaykhiev, R.; Wang, R.; Crystal, R.G. Do Airway Epithelium Air–Liquid Cultures Represent the In Vivo Airway Epithelium Transcriptome? Am. J. Respir. Cell Mol. Biol. 2011, 44, 465–473. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.-G.; Crystal, R.G.; McCray, P.B., Jr.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [PubMed]
- Boei, J.J.W.A.; Vermeulen, S.; Klein, B.; Hiemstra, P.S.; Verhoosel, R.M.; Jennen, D.G.J.; Lahoz, A.; Gmuender, H.; Vrieling, H. Xenobiotic metabolism in differentiated human bronchial epithelial cells. Arch. Toxicol. 2017, 91, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Maunders, H.; Patwardhan, S.; Phillips, J.; Clack, A.; Richter, A. Human bronchial epithelial cell transcriptome: Gene expression changes following acute exposure to whole cigarette smoke in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1248–L1256. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Huynh, C.T.T.; Bastin, K.M.; Rivera, B.N.; Siddens, L.K.; Tilton, S.C. Classifying polycyclic aromatic hydrocarbons by carcinogenic potency using in vitro biosignatures. Toxicol. Vitr. 2020, 69, 104991. [Google Scholar] [CrossRef]
- Wong, L.N.; Aung, H.; Lamé, M.; Wegesser, T.; Wilson, D. Fine particulate matter from urban ambient and wildfire sources from California’s San Joaquin Valley initiate differential inflammatory, oxidative stress, and xenobiotic responses in human bronchial epithelial cells. Toxicol. Vitr. 2011, 25, 1895–1905. [Google Scholar] [CrossRef]
- Woenckhaus, M.; Klein-Hitpass, L.; Grepmeier, U.; Merk, J.; Pfeifer, M.; Wild, P.; Bettstetter, M.; Wuensch, P.; Blaszyk, H.; Hartmann, A.; et al. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J. Pathol. 2006, 210, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lee, J.J.; Tang, H.; Fan, Y.-H.; Xiao, L.; Ren, H.; Kurie, J.; Morice, R.C.; Hong, W.K.; Mao, L. Impact of Smoking Cessation on Global Gene Expression in the Bronchial Epithelium of Chronic Smokers. Cancer Prev. Res. 2008, 1, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.K.; Velmurugan, K.; Plöttner, S.; Siegrist, K.J.; Romo, D.; Welge, P.; Brüning, T.; Xiong, K.-N.; Käfferlein, H.U. Environmentally prevalent polycyclic aromatic hydrocarbons can elicit co-carcinogenic properties in an in vitro murine lung epithelial cell model. Arch. Toxicol. 2018, 92, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Totlandsdal, A.I.; Låg, M.; Lilleaas, E.; Cassee, F.; Schwarze, P. Differential proinflammatory responses induced by diesel exhaust particles with contrasting PAH and metal content: Effects of Diesel Particles with Contrasting PAH and Metal Content. Environ. Toxicol. 2015, 30, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Šimečková, P.; Pěnčíková, K.; Kováč, O.; Slavík, J.; Pařenicová, M.; Vondráček, J.; Machala, M. In vitro profiling of toxic effects of environmental polycyclic aromatic hydrocarbons on nuclear receptor signaling, disruption of endogenous metabolism and induction of cellular stress. Sci. Total. Environ. 2022, 815, 151967. [Google Scholar] [CrossRef] [PubMed]
- Glatt, H.; Rost, K.; Frank, H.; Seidel, A.; Kollock, R. Detoxification of promutagenic aldehydes derived from methylpyrenes by human aldehyde dehydrogenases ALDH2 and ALDH3A1. Arch. Biochem. Biophys. 2008, 477, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Jernström, B.; Seidel, A.; Funk, M.; Oesch, F.; Mannervik, B. Glutathione conjugation of trans-3,4-dihydroxy 1,2-epoxy l,2,3,4-tetrahydrobenzo[c]phenanthrene isomers by human glutathione transferases. Carcinogenesis 1992, 13, 1549–1555. [Google Scholar] [CrossRef]
- Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; A Solis, W.; Yang, Y.; Dalton, T.P. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem. Pharmacol. 2000, 59, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Rybicki, B.A.; Nock, N.L.; Savera, A.T.; Tang, D.; Rundle, A. Polycyclic aromatic hydrocarbon-DNA adduct formation in prostate carcinogenesis. Cancer Lett. 2006, 239, 157–167. [Google Scholar] [CrossRef]
- Shimada, T. Xenobiotic-Metabolizing Enzymes Involved in Activation and Detoxification of Carcinogenic Polycyclic Aromatic Hydrocarbons. Drug Metab. Pharmacokinet. 2006, 21, 257–276. [Google Scholar] [CrossRef]
- Shimada, T.; Guengerich, F.P. Inhibition of Human Cytochrome P450 1A1-, 1A2-, and 1B1-Mediated Activation of Procarcinogens to Genotoxic Metabolites by Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2006, 19, 288–294. [Google Scholar] [CrossRef]
- Abbas, I.; Saint-Georges, F.; Billet, S.; Verdin, A.; Mulliez, P.; Shirali, P.; Garçon, G. Air pollution particulate matter (PM2.5)-induced gene expression of volatile organic compound and/or polycyclic aromatic hydrocarbon-metabolizing enzymes in an in vitro coculture lung model. Toxicol. Vitr. 2009, 23, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Penning, T.M. Genotoxic Polycyclic Aromatic Hydrocarbon ortho-Quinones Generated by Al-do-Keto Reductases Induce CYP1A1 via Nuclear Translocation of the Aryl Hydrocarbon Receptor. Cancer Res. 2000, 60, 908–915. [Google Scholar] [PubMed]
- De Oliveira Galvão, M.F.; Sadiktsis, I.; de Medeiros, S.R.B.; Dreij, K. Genotoxicity and DNA damage signaling in response to complex mixtures of PAHs in biomass burning particulate matter from cashew nut roasting. Environ. Pollut. 2020, 256, 113381. [Google Scholar] [CrossRef]
- Elovaara, E.; Mikkola, J.; Stockmann-Juvala, H.; Luukkanen, L.; Keski-Hynnilä, H.; Kostiainen, R.; Pasanen, M.; Pelkonen, O.; Vainio, H. Polycyclic aromatic hydrocarbon (PAH) metabolizing enzyme activities in human lung, and their inducibility by exposure to naphthalene, phenanthrene, pyrene, chrysene, and benzo(a)pyrene as shown in the rat lung and liver. Arch. Toxicol. 2007, 81, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Lemm, F.; Wilhelm, M.; Roos, P.H. Occupational exposure to polycyclic aromatic hydrocarbons suppresses constitutive expression of CYP1B1 on the transcript level in human leukocytes. Int. J. Hyg. Environ. Health 2004, 207, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Till, M.; Riebniger, D.; Schmitz, H.-J.; Schrenk, D. Potency of various polycyclic aromatic hydrocarbons as inducers of CYP1A1 in rat hepatocyte cultures. Chem. Interact. 1999, 117, 135–150. [Google Scholar] [CrossRef]
- Wilson, L.B.; McClure, R.S.; Waters, K.M.; Simonich, M.T.; Tanguay, R.L. Concentration-response gene expression analysis in zebrafish reveals phenotypically-anchored transcriptional responses to retene. Front. Toxicol. 2022, 4, 950503. [Google Scholar] [CrossRef]
- Wohak, L.E.; Krais, A.M.; Kucab, J.E.; Stertmann, J.; Øvrebø, S.; Seidel, A.; Phillips, D.H.; Arlt, V.M. Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53-dependent mechanism. Arch. Toxicol. 2016, 90, 291–304. [Google Scholar] [CrossRef]
- Pushparajah, D.S.; Umachandran, M.; Plant, K.E.; Plant, N.; Ioannides, C. Up-regulation of the glutathione S-transferase system in human liver by polycyclic aromatic hydrocarbons; comparison with rat liver and lung. Mutagenesis 2008, 23, 299–308. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. NAD(P)H:Quinone Oxidoreductase 1 (NQO1, DT-Diaphorase), Functions and Pharmacogenetics. Methods Enzymol. 2004, 382, 115–144. [Google Scholar] [CrossRef] [PubMed]
- Turkseven, S.; Kruger, A.; Mingone, C.J.; Kaminski, P.; Inaba, M.; Rodella, L.F.; Ikehara, S.; Wolin, M.S.; Abraham, N.G.; Cao, J.; et al. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am. J. Physiol. Circ. Physiol. 2005, 289, H701–H707. [Google Scholar] [CrossRef] [PubMed]
- DeMarini, D.M.; Warren, S.H.; Lavrich, K.; Flen, A.; Aurell, J.; Mitchell, W.; Greenwell, D.; Preston, W.; Schmid, J.E.; Linak, W.P.; et al. Mutagenicity and oxidative damage induced by an organic extract of the particulate emissions from a simulation of the deepwater horizon surface oil burns: Mutagenicity and Oxidative Damage Induced by Oil-Burn Emissions. Environ. Mol. Mutagen. 2017, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Abraham, V.; Chou, M.L.; George, P.; Pooler, P.; Zaman, A.; Savani, R.C.; Koval, M.; Pelletier, R.-M.; Akpovi, C.D.; Chen, L.; et al. Heterocellular gap junctional communication between alveolar epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L1085–L1093. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct Binding of Three Tight Junction-Associated Maguks, Zo-1, Zo-2, and Zo-3, with the Cooh Termini of Claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Terada, M.; Sugimoto, H. The zonula occludens protein family regulates the hepatic barrier system in the murine liver. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 165994. [Google Scholar] [CrossRef] [PubMed]
- Swartzendruber, J.A.; Nicholson, B.J.; Murthy, A.K. The Role of Connexin 43 in Lung Disease. Life 2020, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- Yang, L.; Wang, W.-C.; Lung, S.-C.C.; Sun, Z.; Chen, C.; Chen, J.-K.; Zou, Q.; Lin, Y.-H.; Lin, C.-H. Polycyclic aromatic hydrocarbons are associated with increased risk of chronic obstructive pulmonary disease during haze events in China. Sci. Total. Environ. 2017, 574, 1649–1658. [Google Scholar] [CrossRef]
- Straif, K.; Baan, R.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Cogliano, V. Carcinogenicity of polycyclic aromatic hydrocarbons. Lancet Oncol. 2005, 6, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Bagchi, S.; Raychaudhuri, P. DDB2 decides cell fate following DNA damage. Proc. Natl. Acad. Sci. USA 2009, 106, 10690–10695. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Raychaudhuri, P.; Bagchi, S. DDB2 (Damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle 2009, 8, 4067–4071. [Google Scholar] [CrossRef] [PubMed]
- Hockley, S.L.; Arlt, V.M.; Jahnke, G.; Hartwig, A.; Giddings, I.; Phillips, D.H. Identification through microarray gene expression analysis of cellular responses to benzo(a)pyrene and its diol-epoxide that are dependent or independent of p53. Carcinogenesis 2008, 29, 202–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hawliczek, A.; Nota, B.; Cenijn, P.; Kamstra, J.; Pieterse, B.; Winter, R.; Winkens, K.; Hollert, H.; Segner, H.; Legler, J. Developmental toxicity and endocrine disrupting potency of 4-azapyrene, benzo[b]fluorene and retene in the zebrafish Danio rerio. Reprod. Toxicol. 2012, 33, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Houser, W.H.; Raha, A.; Vickers, M. Induction of CYP1A1 gene expression in H4-II-E rat hepatoma cells by benzo[e]pyrene. Mol. Carcinog. 1992, 5, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Kamps, C.; Safe, S. Binding of polynuclear aromatic hydrocarbons to the rat 4S cytosolic binding protein: Structure-activity relationships. Cancer Lett. 1987, 34, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Sterling, K.; Raha, A.; Bresnick, E. Induction of CYP1A1 Gene Expression in Mouse Hepatoma Cells by Benzo(e)Pyrene, a Ligand of the 4S Polycyclic Hydrocarbon-Binding Protein. Toxicol. Appl. Pharmacol. 1994, 128, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Cherng, S.-H.; Lin, P.; Yang, J.-L.; Hsu, S.-L.; Lee, H. Benzo[g,h,i]perylene Synergistically Transactivates Benzo[a]pyrene-Induced CYP1A1 Gene Expression by Aryl Hydrocarbon Receptor Pathway. Toxicol. Appl. Pharmacol. 2001, 170, 63–68. [Google Scholar] [CrossRef]
- Delistraty, D. Toxic equivalency factor approach for risk assessment of polycyclic aromatic hydrocarbons. Toxicol. Environ. Chem. 1997, 64, 81–108. [Google Scholar] [CrossRef]
- Labib, S.; Williams, A.; Guo, C.H.; Leingartner, K.; Arlt, V.M.; Schmeiser, H.H.; Yauk, C.L.; White, P.A.; Halappanavar, S. Comparative transcriptomic analyses to scrutinize the assumption that genotoxic PAHs exert effects via a common mode of action. Arch. Toxicol. 2016, 90, 2461–2480. [Google Scholar] [CrossRef] [PubMed]
- Streibig, J.C.; Dayan, F.E.; Rimando, A.M.; Duke, S.O. Joint action of natural and synthetic photosystem II inhibitors. Pestic. Sci. 1999, 55, 137–146. [Google Scholar] [CrossRef]
- Arrhenius, Å.; Grönvall, F.; Scholze, M.; Backhaus, T.; Blanck, H. Predictability of the mixture toxicity of 12 similarly acting congeneric inhibitors of photosystem II in marine periphyton and epipsammon communities. Aquat. Toxicol. 2004, 68, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Cedergreen, N.; Kudsk, P.; Mathiassen, S.K.; Streibig, J.C. Combination effects of herbicides on plants and algae: Do species and test systems matter? Pest Manag. Sci. 2007, 63, 282–295. [Google Scholar] [CrossRef] [PubMed]
- U.S. Environmental Protection Agency. Guidelines for the Health Risk Assessment of Chemical Mixtures. 1986. Available online: https://cfpub.epa.gov/ncea/risk/recordisplay.cfm?deid=22567 (accessed on 3 July 2023).
- Petry, T.; Schmid, P.; Schlatter, C. The use of toxic equivalency factors in assessing occupational and environmental health risk associated with exposure to airborne mixtures of polycyclic aromatic hydrocarbons (PAHs). Chemosphere 1996, 32, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Altenburger, R.; Backhaus, T.; Boedeker, W.; Faust, M.; Scholze, M.; Grimme, L.H. Predictability of the toxicity of multiple chemical mixtures to Vibrio fischeri: Mixtures composed of similarly acting chemicals. Environ. Toxicol. Chem. 2000, 19, 2341–2347. [Google Scholar] [CrossRef]
- Altenburger, R.; Walter, H.; Grote, M. What Contributes to the Combined Effect of a Complex Mixture? Environ. Sci. Technol. 2004, 38, 6353–6362. [Google Scholar] [CrossRef]
- Backhaus, T.; Altenburger, R.; Boedeker, W.; Faust, M.; Scholze, M.; Grimme, L.H. Predictability of the toxicity of a multiple mixture of dissimilarly acting chemicals to Vibrio fischeri. Environ. Toxicol. Chem. 2000, 19, 2348–2356. [Google Scholar] [CrossRef]
- Backhaus, T.; Arrhenius, Å.; Blanck, H. Toxicity of a Mixture of Dissimilarly Acting Substances to Natural Algal Communities: Predictive Power and Limitations of Independent Action and Concentration Addition. Environ. Sci. Technol. 2004, 38, 6363–6370. [Google Scholar] [CrossRef]
- Faust, M.; Altenburger, R.; Backhaus, T.; Blanck, H.; Boedeker, W.; Gramatica, P.; Hamer, V.; Scholze, M.; Vighi, M.; Grimme, L. Joint algal toxicity of 16 dissimilarly acting chemicals is predictable by the concept of independent action. Aquat. Toxicol. 2003, 63, 43–63. [Google Scholar] [CrossRef]
- Altenburger, R.; Boedeker, W.; Faust, M.; Grimme, L. Regulations for combined effects of pollutants: Consequences from risk assessment in aquatic toxicology. Food Chem. Toxicol. 1996, 34, 1155–1157. [Google Scholar] [CrossRef]
- Junghans, M.; Backhaus, T.; Faust, M.; Scholze, M.; Grimme, L. Application and validation of approaches for the predictive hazard assessment of realistic pesticide mixtures. Aquat. Toxicol. 2006, 76, 93–110. [Google Scholar] [CrossRef]
- Geier, M.C.; Minick, D.J.; Truong, L.; Tilton, S.; Pande, P.; Anderson, K.A.; Teeguardan, J.; Tanguay, R.L. Systematic developmental neurotoxicity assessment of a representative PAH Superfund mixture using zebrafish. Toxicol. Appl. Pharmacol. 2018, 354, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, I.W.; Dreij, K.; Mattsson, Å.; Jernström, B.; Stenius, U. Interactions between polycyclic aromatic hydrocarbons in complex mixtures and implications for cancer risk assessment. Toxicology 2014, 321, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Barata, C.; Calbet, A.; Saiz, E.; Ortiz, L.; Bayona, J.M. Predicting single and mixture toxicity of petrogenic polycyclic aromatic hydrocarbons to the copepod Oithona davisae. Environ. Toxicol. Chem. 2005, 24, 2992–2999. [Google Scholar] [CrossRef]
- Belden, J.B.; Gilliom, R.J.; Lydy, M.J. How well can we predict the toxicity of pesticide mixtures to aquatic life? Integr. Environ. Assess. Manag. 2007, 3, 364–372. [Google Scholar] [CrossRef]
- Raha, A.; Joyce, T.; Gusky, S.; Bresnick, E. Glycine N-Methyltransferase Is a Mediator of Cytochrome P4501A1 Gene Expression. Arch. Biochem. Biophys. 1995, 322, 395–404. [Google Scholar] [CrossRef]
- Schüttler, A.; Jakobs, G.; Fix, J.; Krauss, M.; Krüger, J.; Leuthold, D.; Altenburger, R.; Busch, W. Transcriptome-Wide Prediction and Measurement of Combined Effects Induced by Chemical Mixture Exposure in Zebrafish Embryos. Environ. Health Perspect. 2021, 129, 47006. [Google Scholar] [CrossRef] [PubMed]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef]
- Burnham, K.P.; Anderson, D.R. Multimodel Inference: Understanding AIC and BIC in Model Selection. Sociol. Methods Res. 2004, 33, 261–304. [Google Scholar] [CrossRef]
- Messier, K.P.; Tidwell, L.G.; Ghetu, C.C.; Rohlman, D.; Scott, R.P.; Bramer, L.M.; Dixon, H.M.; Waters, K.M.; Anderson, K.A. Indoor versus Outdoor Air Quality during Wildfires. Environ. Sci. Technol. Lett. 2019, 6, 696–701. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AbundMix | ToxMix | ||
|---|---|---|---|
| Component | Percent of Mixture | Component | Percent of Mixture |
| Naphthalene | 44% | Retene | 68.6% |
| Acenaphthene | 18.3% | Benzo[a]fluorene | 16.8% |
| 2-methylnaphthalene | 15.9% | Benzo[b]fluorene | 8.42% |
| 1-methylnaphthalene | 11.9% | Benzo[c]fluorene | 4.21% |
| Fluorene | 6.7% | Triphenylene | 1.68% |
| Phenanthrene | 3.16% | Benzo[e]pyrene | 0.168% |
| Benzo[ghi]perylene | 0.0842% | ||
| Compound | TEF |
|---|---|
| Retene | 0.001 |
| Benzo[a]fluorene | 0.001 |
| Benzo[b]fluorene | 0.001 |
| Benzo[c]fluorene | 0.001 |
| Triphenylene | 0.001 |
| Benzo[e]pyrene | 1 |
| Benzo[ghi]perylene | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colvin, V.C.; Bramer, L.M.; Rivera, B.N.; Pennington, J.M.; Waters, K.M.; Tilton, S.C. Modeling PAH Mixture Interactions in a Human In Vitro Organotypic Respiratory Model. Int. J. Mol. Sci. 2024, 25, 4326. https://doi.org/10.3390/ijms25084326
Colvin VC, Bramer LM, Rivera BN, Pennington JM, Waters KM, Tilton SC. Modeling PAH Mixture Interactions in a Human In Vitro Organotypic Respiratory Model. International Journal of Molecular Sciences. 2024; 25(8):4326. https://doi.org/10.3390/ijms25084326
Chicago/Turabian StyleColvin, Victoria C., Lisa M. Bramer, Brianna N. Rivera, Jamie M. Pennington, Katrina M. Waters, and Susan C. Tilton. 2024. "Modeling PAH Mixture Interactions in a Human In Vitro Organotypic Respiratory Model" International Journal of Molecular Sciences 25, no. 8: 4326. https://doi.org/10.3390/ijms25084326
APA StyleColvin, V. C., Bramer, L. M., Rivera, B. N., Pennington, J. M., Waters, K. M., & Tilton, S. C. (2024). Modeling PAH Mixture Interactions in a Human In Vitro Organotypic Respiratory Model. International Journal of Molecular Sciences, 25(8), 4326. https://doi.org/10.3390/ijms25084326