
Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ER Stress Can Induce Both Autophagy and Apoptosis
3. The Characteristic of Endoplasmic Stress Response Mechanism
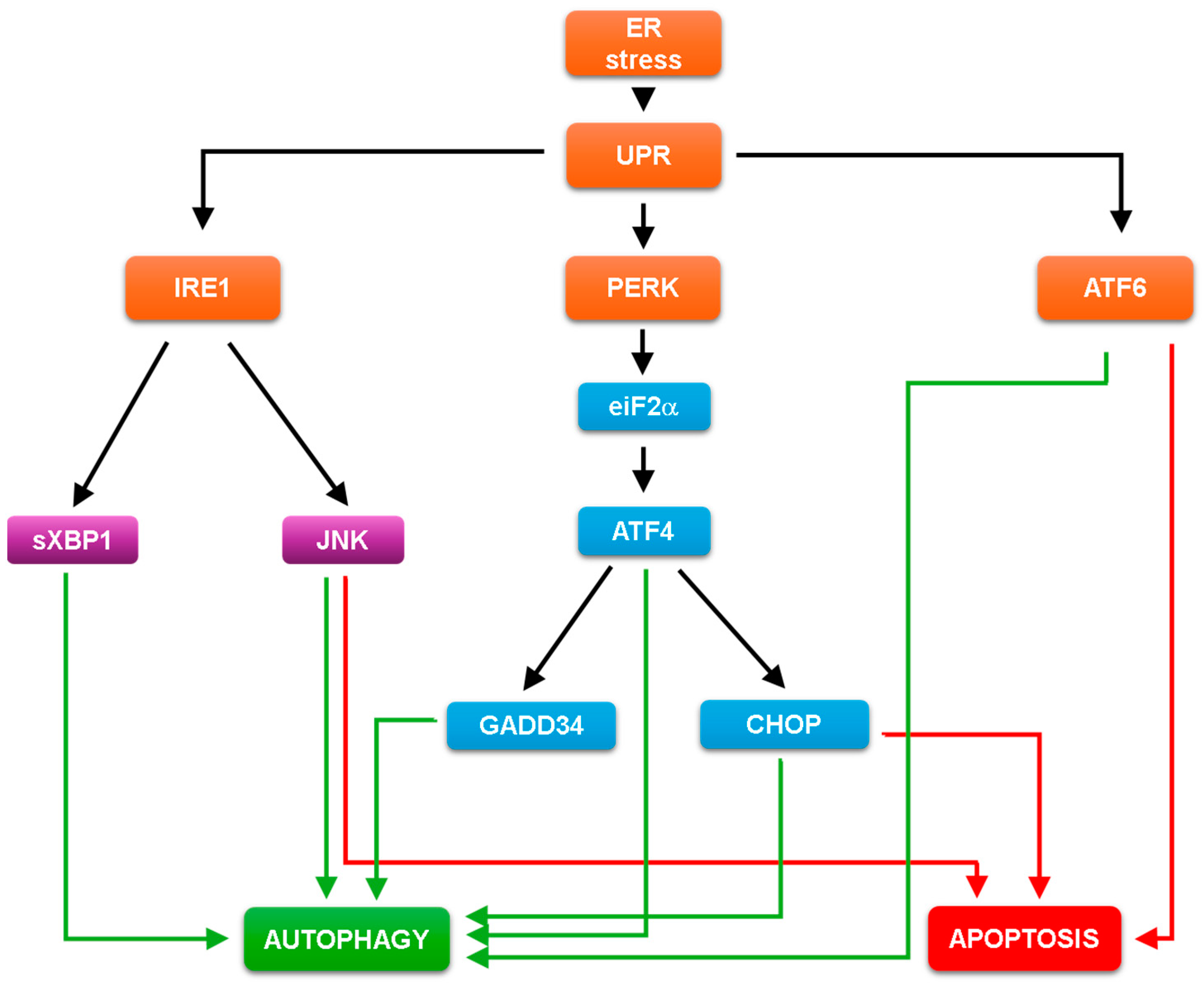
3.1. The Molecular Mechanism of Unfolded Protein Response
3.2. The UPR Dependent Regulation of Both Autophagy and Apoptosis
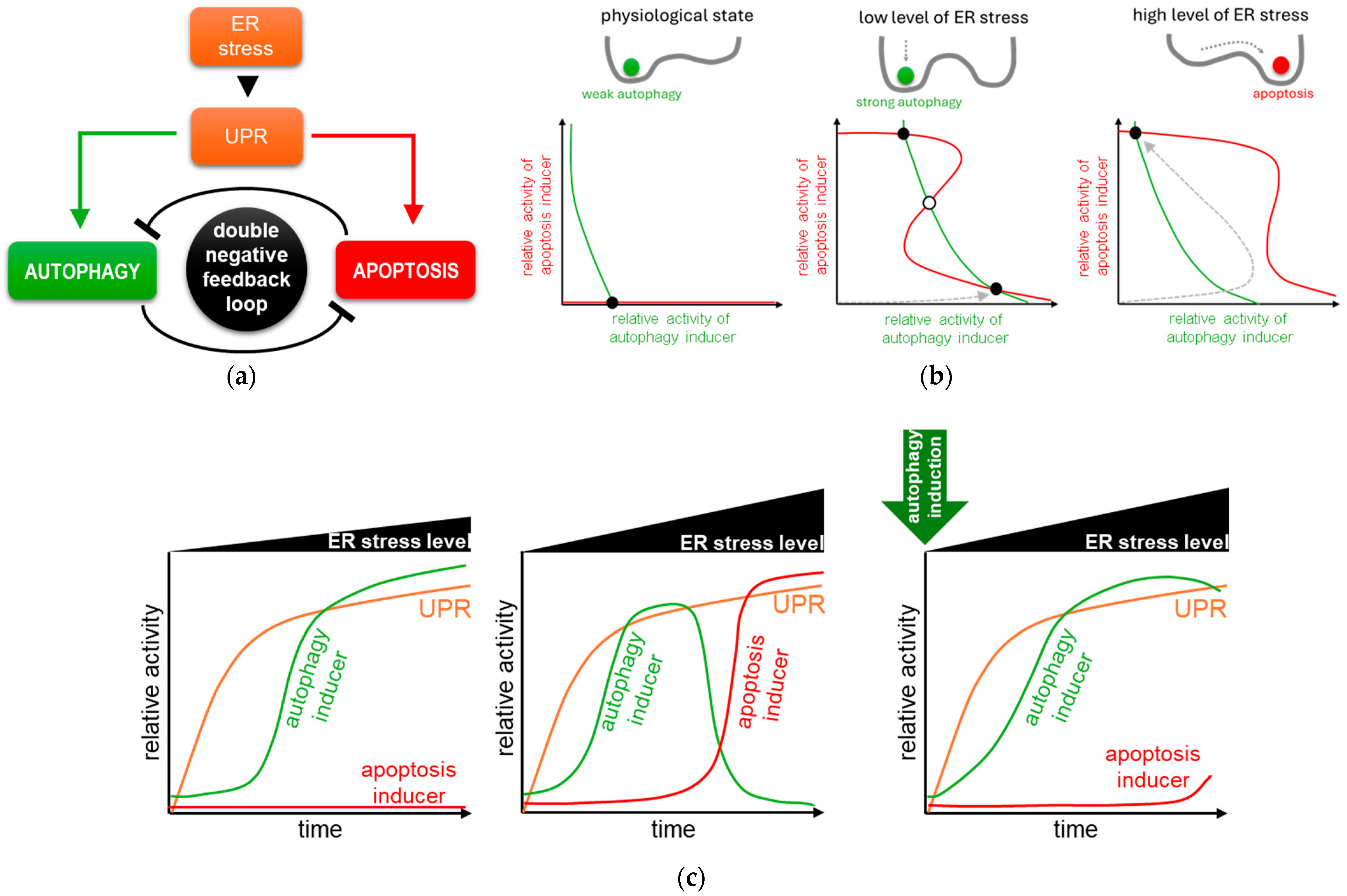
4. Systems Biological Analysis of Autophagy and Apoptosis Induction upon ER Stress
4.1. The Dynamical Analysis of Autophagy and Apoptosis Induction upon ER Stress
4.2. Autophagy Always Precedes Apoptosis upon ER Stress
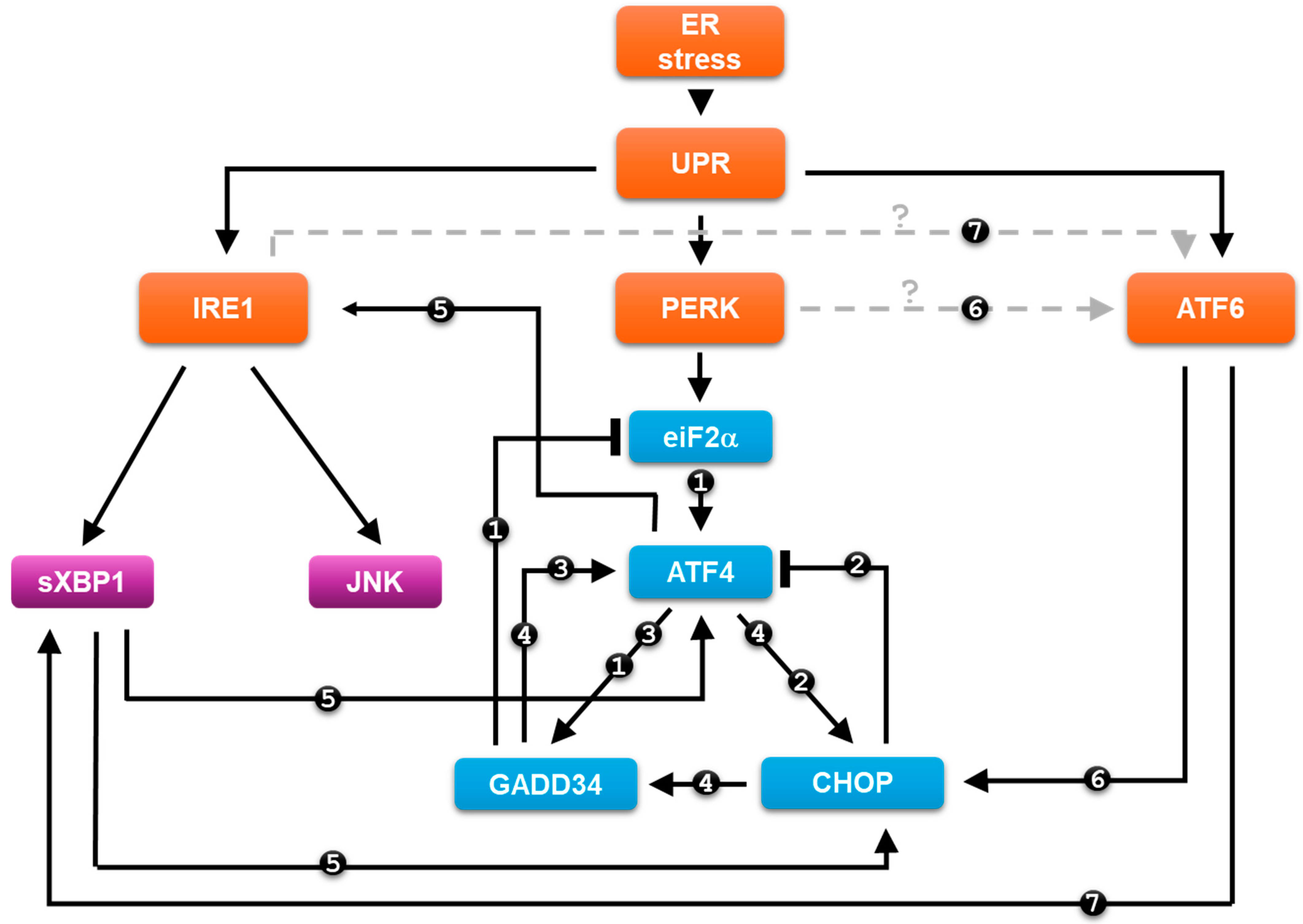
5. Crosslinks Inside and between the UPR Arms and Their Roles upon ER Stress
6. Discussion
7. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, G.; Jia, M.; Jiang, Q.; Li, S.; Wang, H.; Li, W.; Wang, Y.; Bian, X.; Zhao, Y.G.; et al. Stay in touch with the endoplasmic reticulum. Sci. China Life Sci. 2024, 67, 230–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Shi, C.R.; He, M.H.; Xiong, S.Q.; Xia, X.B. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Sanvictores, T.; Davis, D.D. Histology, Rough Endoplasmic Reticulum. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Bánhegyi, G.; Benedetti, A.; Csala, M.; Mandl, J. Stress on redox. FEBS Lett. 2007, 581, 3634–3640. [Google Scholar] [CrossRef]
- Chen, G.F.; Wei, T.Y.; Ju, F.R.; Li, H.S. Protein quality control and aggregation in the endoplasmic reticulum: From basic to bedside. Front. Cell Dev. Biol. 2023, 11, 1156152. [Google Scholar] [CrossRef]
- Wickner, W.; Schekman, R. Protein translocation across biological membranes. Science 2005, 310, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Protein folding in the endoplasmic reticulum and the unfolded protein response. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 69–91. [Google Scholar]
- Csala, M.; Marcolongo, P.; Lizak, B.; Senesi, S.; Margittai, E.; Fulceri, R.; Magyar, J.E.; Benedetti, A.; Banhegyi, G. Transport and transporters in the endoplasmic reticulum. Biochim. Biophys. Acta 2007, 1768, 1325–1341. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Ohkawa, Y.; Maeda, K.; Taniguchi, N. Glycan quality control in and out of the endoplasmic reticulum of mammalian cells. FEBS J. 2022, 289, 7147–7162. [Google Scholar] [CrossRef] [PubMed]
- van Anken, E.; Braakman, I. Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 191–228. [Google Scholar] [CrossRef]
- Tao, Y.; Yang, Y.Q.; Zhou, R.B.; Gong, T. Golgi Apparatus: An Emerging Platform for Innate Immunity. Trends Cell Biol. 2020, 30, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2022, 21, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Khanna, M.; Agrawal, N.; Chandra, R.; Dhawan, G. Targeting unfolded protein response: A new horizon for disease control. Expert Rev. Mol. Med. 2021, 23, e1. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic beta-Cell Dysfunction and Senescence in Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 4843. [Google Scholar] [CrossRef]
- Goswami, P.; Akhter, J.; Mangla, A.; Suramya, S.; Jindal, G.; Ahmad, S.; Raisuddin, S. Downregulation of ATF-4 Attenuates the Endoplasmic Reticulum Stress-Mediated Neuroinflammation and Cognitive Impairment in Experimentally Induced Alzheimer’s Disease Model. Mol. Neurobiol. 2023, 1–12. [Google Scholar] [CrossRef]
- An, Y.; Wang, X.; Guan, X.; Yuan, P.; Liu, Y.; Wei, L.; Wang, F.; Qi, X. Endoplasmic reticulum stress-mediated cell death in cardiovascular disease. Cell Stress. Chaperones 2024, 29, 158–174. [Google Scholar] [CrossRef]
- Zou, M.; Liang, Q.; Zhang, W.; Zhu, Y.; Xu, Y. Endoplasmic reticulum stress related genome-wide Mendelian randomization identifies therapeutic genes for ulcerative colitis and Crohn’s disease. Front. Genet. 2023, 14, 1270085. [Google Scholar] [CrossRef]
- Shi, Y.; Jiang, B.; Zhao, J. Induction mechanisms of autophagy and endoplasmic reticulum stress in intestinal ischemia-reperfusion injury, inflammatory bowel disease, and colorectal cancer. Biomed. Pharmacother. 2024, 170, 115984. [Google Scholar] [CrossRef]
- Wang, Z.; Kou, M.; Deng, Q.; Yu, H.; Mei, J.; Gao, J.; Fu, W.; Ning, B. Acupuncture activates IRE1/XBP1 endoplasmic reticulum stress pathway in Parkinson’s disease model rats. Behav. Brain Res. 2024, 462, 114871. [Google Scholar] [CrossRef]
- Ekundayo, B.E.; Obafemi, T.O.; Adewale, O.B.; Obafemi, B.A.; Oyinloye, B.E.; Ekundayo, S.K. Oxidative Stress, Endoplasmic Reticulum Stress and Apoptosis in the Pathology of Alzheimer’s Disease. Cell Biochem. Biophys. 2024, 1–21. [Google Scholar] [CrossRef]
- Jiang, Y.; Chadwick, S.R.; Lajoie, P. Endoplasmic reticulum stress: The cause and solution to Huntington’s disease? Brain Res. 2016, 1648, 650–657. [Google Scholar] [CrossRef]
- Maity, S.; Komal, P.; Kumar, V.; Saxena, A.; Tungekar, A.; Chandrasekar, V. Impact of ER Stress and ER-Mitochondrial Crosstalk in Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 780. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.; Majsterek, I.; Galecki, P.; Talarowska, M. The role of the endoplasmic reticulum stress in depression. Psychiatr. Pol. 2020, 54, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Hu, Y.; Ruan, L.; Ji, Y.; Lou, Z. Role of endoplasmic reticulum stress in depression (Review). Mol. Med. Rep. 2019, 20, 4774–4780. [Google Scholar] [CrossRef]
- Sreedhar, R.; Giridharan, V.V.; Arumugam, S.; Karuppagounder, V.; Palaniyandi, S.S.; Krishnamurthy, P.; Quevedo, J.; Watanabe, K.; Konishi, T.; Thandavarayan, R.A. Role of MAPK-mediated endoplasmic reticulum stress signaling in the heart during aging in senescence-accelerated prone mice. Biofactors 2016, 42, 368–375. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J.; Liu, Y.; Wang, J.; Nie, Y.; Si, B.; Liu, Y.; Wang, X.; Chen, S.; Hei, T.K.; et al. Subcellular Targets of Zinc Oxide Nanoparticles During the Aging Process: Role of Cross-talk Between Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in the Genotoxic Response. Toxicol. Sci. 2019, 171, 159–171. [Google Scholar] [CrossRef]
- Chen, Q.; Kovilakath, A.; Allegood, J.; Thompson, J.; Hu, Y.; Cowart, L.A.; Lesnefsky, E.J. Endoplasmic reticulum stress and mitochondrial dysfunction during aging: Role of sphingolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2023, 1868, 159366. [Google Scholar] [CrossRef]
- Parashar, S.; Ferro-Novick, S. Architecture of the endoplasmic reticulum plays a role in proteostasis. Autophagy 2022, 18, 937–938. [Google Scholar] [CrossRef]
- Wright, M.T.; Plate, L. Revealing functional insights into ER proteostasis through proteomics and interactomics. Exp. Cell Res. 2021, 399, 112417. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.F.; Yang, N.N.; Huang, Y.; Hu, T.T.; Rao, C.L. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022, 13, 1051. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Spencer, B.G.; Finnie, J.W. The Role of Endoplasmic Reticulum Stress in Cell Survival and Death. J. Comp. Pathol. 2020, 181, 86–91. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Cao, W.Y.; Li, J.H.; Yang, K.P.; Cao, D.L. An overview of autophagy: Mechanism, regulation and research progress. B Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- Kapuy, O.; Holczer, M.; Márton, M.; Korcsmáros, T. Autophagy-dependent survival is controlled with a unique regulatory network upon various cellular stress events. Cell Death Dis. 2021, 12, 309. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Krasovec, G.; Horkan, H.R.; Quéinnec, E.; Chambon, J.P. The constructive function of apoptosis: More than a dead-end job. Front. Cell Dev. Biol. 2022, 10, 1033645. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef]
- Dutreix, M. Safety control for apoptotic irreversibility. Proc. Natl. Acad. Sci. USA 2012, 109, 12844–12845. [Google Scholar] [CrossRef]
- Kapuy, O.; Vinod, P.K.; Mandl, J.; Banhegyi, G. A cellular stress-directed bistable switch controls the crosstalk between autophagy and apoptosis. Mol. Biosyst. 2013, 9, 296–306. [Google Scholar] [CrossRef]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling-from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.Z.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, A.O.; Simpson, K.E.; Oyelere, S.F.; Nur, M.; Ngule, C.M.; Owoyemi, B.C.D.; Ayarick, V.A.; Oyelami, F.F.; Obaleye, O.; Esoe, D.P.; et al. Unveiling the dark side of glucose-regulated protein 78 (GRP78) in cancers and other human pathology: A systematic review. Mol. Med. 2023, 29, 112. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, G.; Martinotti, S.; Ranzato, E. Endoplasmic Reticulum Stress and Cancer: Could Unfolded Protein Response Be a Druggable Target for Cancer Therapy? Int. J. Mol. Sci. 2023, 24, 1566. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, Z.; Cheng, W.S.; Zhao, Q.Q.; Zhang, X.Y.; Zhang, H.; Yu, M.; Xu, H.; Gao, Y.C.; Jiang, Q.R.; et al. The PERK Branch of the Unfolded Protein Response Safeguards Protein Homeostasis and Mesendoderm Specification of Human Pluripotent Stem Cells. Adv. Sci. 2023, 10, e2303799. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.Q.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the mRNA (vol 415, pg 92, 2002). Nature 2002, 420, 202. [Google Scholar] [CrossRef]
- Hyrskyluoto, A.; Reijonen, S.; Kivinen, J.; Lindholm, D.; Korhonen, L. GADD34 mediates cytoprotective autophagy in mutant huntingtin expressing cells via the mTOR pathway. Exp. Cell Res. 2012, 318, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Banhegyi, G.; Kapuy, O. GADD34 Keeps the mTOR Pathway Inactivated in Endoplasmic Reticulum Stress Related Autophagy. PLoS ONE 2016, 11, e0168359. [Google Scholar] [CrossRef]
- Uddin, M.N.; Ito, S.; Nishio, N.; Suganya, T.; Isobe, K. Gadd34 induces autophagy through the suppression of the mTOR pathway during starvation. Biochem. Biophys. Res. Commun. 2011, 407, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Tambe, Y.; Okuyama, N.; Nakagawa, T.; Muramoto, A.; Hasebe, M.; Chano, T.; Inoue, H. Suppression of viral replication by tumor suppressor via mTOR dependent pathway. Cancer Lett. 2012, 314, 82–91. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- B’chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal 2014, 26, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.X.; Ding, C.; Yu, S.Q. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Adler, H.T.; Chinery, R.; Wu, D.Y.; Kussick, S.J.; Payne, J.M.; Fornace, A.J., Jr.; Tkachuk, D.C. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol. Cell Biol. 1999, 19, 7050–7060. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Kawiak, A.; Kostecka, A. Regulation of Bcl-2 Family Proteins in Estrogen Receptor-Positive Breast Cancer and Their Implications in Endocrine Therapy. Cancers 2022, 14, 279. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.J.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes. Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, C.; Adajar, R.C.; DeZwaan-McCabe, D.; Rutkowski, D.T. A beneficial adaptive role for CHOP in driving cell fate selection during ER stress. EMBO Rep. 2024, 25, 228–253. [Google Scholar] [CrossRef]
- Margariti, A.; Li, H.L.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.Z.; Zampetaki, A.; Zhang, Z.Y.; et al. mRNA Splicing Triggers an Autophagic Response in Endothelial Cells through-Transcriptional Activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef]
- Deegan, S.; Koryga, I.; Glynn, S.A.; Gupta, S.; Gorman, A.M.; Samali, A. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem. Biophys. Res. Commun. 2015, 456, 305–311. [Google Scholar] [CrossRef]
- Tian, P.G.; Jiang, Z.X.; Li, J.H.; Zhou, Z.; Zhang, Q.H. Spliced XBP1 promotes macrophage survival and autophagy by interacting with Beclin-1. Biochem. Biophys. Res. Commun. 2015, 463, 518–523. [Google Scholar] [CrossRef]
- Allagnat, F.; Christulia, F.; Ortis, F.; Pirot, P.; Lortz, S.; Lenzen, S.; Eizirik, D.L.; Cardozo, A.K. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010, 53, 1120–1130. [Google Scholar] [CrossRef]
- Wu, P.B.; Tian, T.; Zhao, J.B.; Song, Q.; Wu, X.M.; Guo, Y.T.; Yu, Y.J.; Tan, S.Y.; Xia, H.M. IRE1α-JNK pathway-mediated autophagy promotes cell survival in response to endoplasmic reticulum stress during the initial phase of hepatic steatosis. Life Sci. 2021, 264, 118668. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yao, S.K. JNK-Bcl-2/Bcl-xL-Bax/Bak Pathway Mediates the Crosstalk between Matrine-Induced Autophagy and Apoptosis via Interplay with Beclin 1. Int. J. Mol. Sci. 2015, 16, 25744–25758. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Fan, Y.; Gui, Y.N.; Yang, X.L.; Ye, X.; Cao, Y.P.; Zhang, Z.L. Endoplasmic reticulum stress triggered autophagy and regulated the phenotype transformation of rheumatoid arthritis synovial fibroblasts via the IRE1/JNK pathway. Ann. Transl. Med. 2022, 10, 725. [Google Scholar] [CrossRef]
- Sharma, R.B.; Snyder, J.T.; Alonso, L.C. Atf6α impacts cell number by influencing survival, death and proliferation. Mol. Metab. 2019, 27, S69–S80. [Google Scholar] [CrossRef]
- Gade, P.; Manjegowda, S.B.; Nallar, S.C.; Maachani, U.B.; Cross, A.S.; Kalvakolanu, D.V. Regulation of the Death-Associated Protein Kinase 1 Expression and Autophagy via ATF6 Requires Apoptosis Signal-Regulating Kinase 1. Mol. Cell Biol. 2014, 34, 4033–4048. [Google Scholar] [CrossRef]
- Nakanishi, K.; Sudo, T.; Morishima, N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J. Cell Biol. 2005, 169, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Nakano, A. Activating Transcription Factor-6 (ATF6) Mediates Apoptosis with Reduction of Myeloid Cell Leukemia Sequence 1 (Mcl-1) Protein via Induction of WW Domain Binding Protein 1. J. Biol. Chem. 2011, 286, 35227–35235. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, J.; Zhu, Y.H.; Wang, L.; Jiang, X.; Liu, B.; He, G. Targeting autophagy and beyond: Deconvoluting the complexity of Beclin-1 from biological function to cancer therapy. Acta Pharm. Sin. B 2023, 13, 4688–4714. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Tyson, J.J.; Novak, B. Functional motifs in biochemical reaction networks. Annu. Rev. Phys. Chem. 2010, 61, 219–240. [Google Scholar] [CrossRef]
- Xie, Q.Q.; Liu, Y.; Li, X.F. The interaction mechanism between autophagy and apoptosis in colon cancer. Transl. Oncol. 2020, 13, 100871. [Google Scholar] [CrossRef]
- Holczer, M.; Marton, M.; Kurucz, A.; Banhegyi, G.; Kapuy, O. A Comprehensive Systems Biological Study of Autophagy-Apoptosis Crosstalk during Endoplasmic Reticulum Stress. Biomed. Res. Int. 2015, 2015, 319589. [Google Scholar] [CrossRef]
- Kapuy, O.; Márton, M.; Bánhegyi, G.; Vinod, P.K. Multiple system-level feedback loops control life-and-death decisions in endoplasmic reticulum stress. FEBS Lett. 2020, 594, 1112–1123. [Google Scholar] [CrossRef]
- Kapuy, O.; Vinod, P.K.; Banhegyi, G. mTOR inhibition increases cell viability via autophagy induction during endoplasmic reticulum stress-An experimental and modeling study. FEBS Open Bio 2014, 4, 704–713. [Google Scholar] [CrossRef]
- Holczer, M.; Besze, B.; Lehel, A.; Kapuy, O. The Dual Role of Sulforaphane-Induced Cellular Stress-A Systems Biological Study. Int. J. Mol. Sci. 2024, 25, 1220. [Google Scholar] [CrossRef]
- Wang, L.; Ye, X.J.; Zhao, T.B. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. 2019, 94, 503–516. [Google Scholar] [CrossRef]
- Ferrell, J.E. Bistability, Bifurcations, and Waddington’s Epigenetic Landscape. Curr. Biol. 2012, 22, R458–R466. [Google Scholar] [CrossRef]
- Rombouts, J.; Gelens, L. Dynamic bistable switches enhance robustness and accuracy of cell cycle transitions. PLoS Comput. Biol. 2021, 17, e1008231. [Google Scholar] [CrossRef]
- Quan, H.Y.; Fan, Q.Q.; Li, C.; Wang, Y.Y.; Wang, L. The transcriptional profiles and functional implications of long non-coding RNAs in the unfolded protein response. Sci. Rep. 2018, 8, 4981. [Google Scholar] [CrossRef]
- Damiano, F.; Rochira, A.; Tocci, R.; Alemanno, S.; Gnoni, A.; Siculella, L. hnRNP A1 mediates the activation of the IRES-dependent SREBP-1a mRNA translation in response to endoplasmic reticulum stress. Biochem. J. 2013, 449, 543–553. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.J.; Spiller, D.G.; White, M.R.H.; Prior, I.A.; Paraoan, L. ER stress-linked autophagy stabilizes apoptosis effector PERP and triggers its co-localization with SERCA2b at ER-plasma membrane junctions. Cell Death Discov. 2019, 5, 132. [Google Scholar] [CrossRef] [PubMed]
- Alsamri, H.; Alneyadi, A.; Muhammad, K.; Ayoub, M.A.; Eid, A.; Iratni, R. Carnosol Induces p38-Mediated ER Stress Response and Autophagy in Human Breast Cancer Cells. Front. Oncol. 2022, 12, 911615. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Lin, T.; Gong, Z.L.; Zhu, Y. Relationship between autophagy, apoptosis and endoplasmic reticulum stress induced by melatonin in osteoblasts by septin7 expression. Mol. Med. Rep. 2020, 21, 2427–2434. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Yuan, J.Y.; Lipinski, M.M. Live imaging and single-cell analysis reveal differential dynamics of autophagy and apoptosis. Autophagy 2013, 9, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Xu, X.; Tang, J.; Sun, Z.Y.; Fu, Y.J.; Zhao, X.J.; Ma, X.M.; Ye, Q. Apatinib induces endoplasmic reticulum stress-mediated apoptosis and autophagy and potentiates cell sensitivity to paclitaxel via the IRE-1α-AKT-mTOR pathway in esophageal squamous cell carcinoma. Cell Biosci. 2021, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, A.; Guillen, C.; Benito, M. Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic β cell death. Autophagy 2012, 8, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic Cell Death and Cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef]
- Kumari, M.; Kamat, S.; Singh, S.K.; Kumar, A.; Jayabaskaran, C. Inhibition of Autophagy Increases Cell Death in HeLa Cells through Usnic Acid Isolated from Lichens. Plants-Basel 2023, 12, 519. [Google Scholar] [CrossRef]
- Mu, W.; Rezek, V.; Martin, H.; Carrillo, M.A.; Tomer, S.; Hamid, P.; Lizarraga, M.A.; Tibbe, T.D.; Yang, O.O.; Jamieson, B.D.; et al. Autophagy inducer rapamycin treatment reduces IFN-I-mediated Inflammation and improves anti-HIV-1 T cell response in vivo. J. Clin. Investig. 2022, 7, e159136. [Google Scholar] [CrossRef]
- Holczer, M.; Besze, B.; Zambo, V.; Csala, M.; Banhegyi, G.; Kapuy, O. Epigallocatechin-3-Gallate (EGCG) Promotes Autophagy-Dependent Survival via Influencing the Balance of mTOR-AMPK Pathways upon Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2018, 2018, 6721530. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Jin, K.H.; Luo, J.C.; Xu, W.L.; Wu, Y.J.; Zhou, J.; Wang, Y.L.; Xu, R.; Jiao, L.Q.; Wang, T.; et al. NF-κB and its crosstalk with endoplasmic reticulum stress in atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 988266. [Google Scholar] [CrossRef]
- Márton, M.; Bánhegyi, G.; Gyöngyösi, N.; Kálmán, E.É.; Pettkó-Szandtner, A.; Káldi, K.; Kapuy, O. A systems biological analysis of the ATF4-GADD34-CHOP regulatory triangle upon endoplasmic reticulum stress. FEBS Open Bio 2022, 12, 2065–2082. [Google Scholar] [CrossRef]
- Choy, M.S.; Yusoff, P.; Lee, I.C.; Newton, J.C.; Goh, C.W.; Page, R.; Shenolikar, S.; Peti, W. Structural and Functional Analysis of the GADD34:PP1 eIF2α Phosphatase. Cell Rep. 2015, 11, 1885–1891. [Google Scholar] [CrossRef]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2α. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef]
- Trusina, A.; Papa, F.R.; Tang, C. Rationalizing translation attenuation in the network architecture of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2008, 105, 20280–20285. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Alejandro, E.U. Imeglimin to the Rescue: Enhanced CHOP/GADD34/eIF2alpha Signaling Axis Promotes beta-Cell Survival. Diabetes 2022, 71, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.X.; Zhang, H.; Sun, W.; Duan, L.; Li, W.C.; Wang, D.P.; Alahdal, M. IRE1 signaling regulates chondrocyte apoptosis and death fate in the osteoarthritis. J. Cell Physiol. 2022, 237, 118–127. [Google Scholar] [CrossRef]
- Márton, M.; Kurucz, A.; Lizák, B.; Margittai, É.; Bánhegyi, G.; Kapuy, O. A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress-The Role of PERK Pathway. Int. J. Mol. Sci. 2017, 18, 58. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpedek-Kaminska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Zhang, D.; De Veirman, K.; Fan, R.; Jian, Q.; Zhang, Y.C.; Lei, L.; Evans, H.; Wang, Y.M.; Lei, L.; Wang, B.Y.; et al. ER stress arm XBP1s plays a pivotal role in proteasome inhibition-induced bone formation. Stem Cell Res. Ther. 2020, 11, 516. [Google Scholar] [CrossRef]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.P.; Mirek, E.T.; Al Baghdadi, R.J.T.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. Iscience 2020, 23, 100860. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring Er Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Olivares, S.; Henkel, A.S. Hepatic Gene Deletion Promotes Endoplasmic Reticulum Stress-induced Liver Injury and Apoptosis. J. Biol. Chem. 2015, 290, 30142–30151. [Google Scholar] [CrossRef] [PubMed]
- Duwaerts, C.C.; Siao, K.; Soon, R.K.; Her, C.; Iwawaki, T.; Kohno, K.; Mattis, A.N.; Maher, J.J. Hepatocyte-specific deletion of XBP1 sensitizes mice to liver injury through hyperactivation of IRE1α. Cell Death Differ. 2021, 28, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Zhang, X.; Ye, Y.Z.; Xiong, X.X.; Zhang, S.D.; Gu, L.J.; Jian, Z.H.; Wang, H.F. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cerebral Ischemia/Reperfusion Injury. Front. Cell Neurosci. 2022, 16, 864426. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J.; Dagda, R.K.; Chu, C.T. Review: Autophagy and neurodegeneration: Survival at a cost? Neuropath Appl. Neuro 2010, 36, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.G.; Zhai, W.Q.; Lu, X.J.; Wang, G.H. The Cross-Links of Endoplasmic Reticulum Stress, Autophagy, and Neurodegeneration in Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 691881. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Yong, J.; Parekh, V.S.; Reilly, S.M.; Nayak, J.; Chen, Z.J.; Lebeaupin, C.; Jang, I.; Zhang, J.W.; Prakash, T.P.; Sun, H.; et al. Chop/Ddit3 depletion in β cells alleviates ER stress and corrects hepatic steatosis in mice. Sci. Transl. Med. 2021, 13, eaba9796. [Google Scholar] [CrossRef]
- Ma, Y.M.; Peng, Y.M.; Zhu, Q.H.; Gao, A.H.; Chao, B.; He, Q.J.; Li, J.; Hu, Y.H.; Zhou, Y.B. Novel CHOP activator LGH00168 induces necroptosis in A549 human lung cancer cells via ROS-mediated ER stress and NF-κB inhibition. Acta Pharmacol. Sin. 2016, 37, 1381–1390. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Hajdú, B.; Lorincz, T.; Szarka, A.; Bánhegyi, G.; Kapuy, O. Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation. Sci. Rep. 2020, 10, 17803. [Google Scholar] [CrossRef] [PubMed]
- Hajdú, B.; Csabai, L.; Márton, M.; Holczer, M.; Korcsmáros, T.; Kapuy, O. Oscillation of Autophagy Induction under Cellular Stress and What Lies behind It, a Systems Biology Study. Int. J. Mol. Sci. 2023, 24, 7671. [Google Scholar] [CrossRef] [PubMed]
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst. Biol. 2013, 7, 16. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapuy, O. Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2024, 25, 4368. https://doi.org/10.3390/ijms25084368
Kapuy O. Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2024; 25(8):4368. https://doi.org/10.3390/ijms25084368
Chicago/Turabian StyleKapuy, Orsolya. 2024. "Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 25, no. 8: 4368. https://doi.org/10.3390/ijms25084368
APA StyleKapuy, O. (2024). Mechanism of Decision Making between Autophagy and Apoptosis Induction upon Endoplasmic Reticulum Stress. International Journal of Molecular Sciences, 25(8), 4368. https://doi.org/10.3390/ijms25084368