

Insight into the Interplay of Gd-IgA1, HMGB1, RAGE and PCDH1 in IgA Vasculitis (IgAV)

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics of IgAV Patients
2.2. Laboratory Parameters
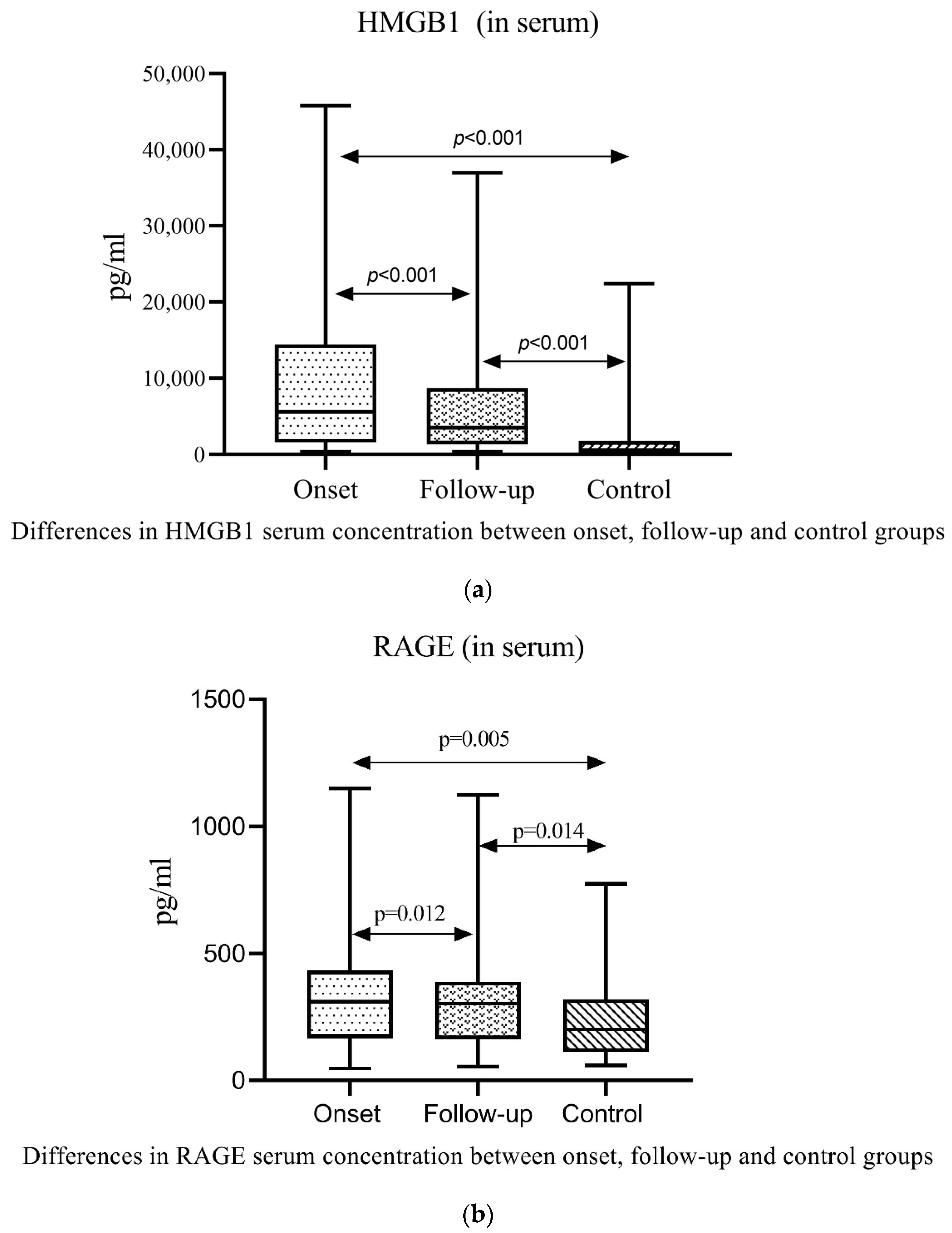
2.3. Serum and Urine Gd-IgA1, HMGB1, RAGE and PCDH1 in Patients with IgAV and Controls
2.4. Serum and Urine Gd-IgA1, HMGB1, RAGE and PCDH1 in Patients with IgAV without Nephritis and with Nephritis
2.5. ROC Analysis
2.6. Logistic Regression Analysis
2.7. Correlation of Gd-IgA1, HMGB1, RAGE and PCDH1 with Clinical Characteristics and Disease Activity
2.8. Correlation of Gd-IgA1, HMGB1, RAGE and PCDH1 with Laboratory Parameters in IgAV Patients at the Onset of the Disease and after Six-Month Follow-Up
3. Discussion
4. Materials and Methods
4.1. Participants and Study Design
4.2. Clinical Assessment
4.3. Disease Activity Assessment
4.4. Laboratory Analysis
4.5. Analysis of Gd-IgA1, HMGB1, RAGE and PCDH1
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jelusic, M.; Sestan, M.; Giani, T.; Cimaz, R. New insights and challenges associated with IgA vasculitis and IgA vasculitis with nephritis—Is it time to change the paradigm of the most common systemic vasculitis in childhood? Front. Pediatr. 2022, 10, 853724. [Google Scholar] [CrossRef]
- Ozen, S.; Pistorio, A.; Iusan, S.M.; Bakkaloglu, A.; Herlin, T.; Brik, R.; Buoncompagni, A.; Lazar, C.; Bilge, I.; Uziel, Y.; et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann. Rheum. Dis. 2010, 69, 798–806. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2012, 65, 180–186. [Google Scholar]
- Gardner-Medwin, J.M.; Dolezalova, P.; Cummins, C.; Southwood, T.R. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002, 360, 1197–1202. [Google Scholar] [CrossRef]
- Sapina, M.; Frkovic, M.; Sestan, M.; Srsen, S.; Ovuka, A.; Varga, M.B.; Kramaric, K.; Brdaric, D.; Milas, K.; Gagro, A.; et al. Geospatial clustering of childhood IgA vasculitis and IgA vasculitis-associated nephritis. Ann. Rheum. Dis. 2020, 80, 610–616. [Google Scholar] [CrossRef]
- Piram, M.; Mahr, A. Epidemiology of immunoglobulin A vasculitis (Henoch–Schönlein): Current state of knowledge. Curr. Opin. Rheumatol. 2013, 25, 171–178. [Google Scholar] [CrossRef]
- Kifer, N.; Bulimbasic, S.; Sestan, M.; Held, M.; Kifer, D.; Srsen, S.; Gudelj Gracanin, A.; Heshin-Bekenstein, M.; Giani, T.; Cimaz, R.; et al. Semiquantitative classification (SQC) and Oxford classifications predict poor renal outcome better than The International Study of Kidney Disease in Children (ISKDC) and Haas in patients with IgAV nephritis: A multicenter study. J. Nephrol. 2023, 36, 441–449. [Google Scholar] [CrossRef]
- Narchi, H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: A systematic review. Arch. Dis. Child. 2005, 90, 916–920. [Google Scholar] [CrossRef]
- Pillebout, E.; Thervet, E.; Hill, G.; Alberti, C.; Vanhille, P.; Nochy, D. Henoch-Schönlein purpura in adults: Outcome and prognostic factors. J. Am. Soc. Nephrol. 2002, 13, 1271–1278. [Google Scholar] [CrossRef]
- Heineke, M.H.; Ballering, A.V.; Jamin, A.; Ben Mkaddem, S.; Monteiro, R.C.; Van Egmond, M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schönlein purpura). Autoimmun. Rev. 2017, 16, 1246–1253. [Google Scholar] [CrossRef]
- Xu, S.; Han, S.; Dai, Y.; Wang, L.; Zhang, X.; Ding, Y. A review of the mechanism of vascular endothelial injury in immunoglobulin A vasculitis. Front. Physiol. 2022, 13, 833954. [Google Scholar] [CrossRef]
- Brogan, P.; Eleftheriou, D. Vasculitis update: Pathogenesis and biomarkers. Pediatr. Nephrol. 2017, 33, 187–198. [Google Scholar] [CrossRef]
- Suzuki, H.; Moldoveanu, Z.; Julian, B.A.; Wyatt, R.J.; Novak, J. Autoantibodies specific for galactose-deficient IgA1 in IgA vasculitis with nephritis. Kidney Int. Rep. 2019, 4, 1717–1724. [Google Scholar] [CrossRef]
- Tang, M.; Zhang, X.; Li, X.; Lei, L.; Zhang, H.; Ling, C.; Ni, J.; Lv, J.; Liu, X.; Chen, X. Serum levels of galactose-deficient IgA1 in Chinese children with IgA nephropathy, IgA vasculitis with nephritis, and IgA vasculitis. Clin. Exp. Nephrol. 2020, 25, 37–43. [Google Scholar] [CrossRef]
- Mizerska-Wasiak, M.; Gajewski, Ł.; Cichoń-Kawa, K.; Małdyk, J.; Dziedzic-Jankowska, K.; Leszczyńska, B.; Rybi-Szumińska, A.; Wasilewska, A.; Pukajło-Marczyk, A.; Zwolińska, D.; et al. Serum Gd-IgA1 levels in children with IgA nephropathy and Henoch-Schönlein nephritis. Cent. Eur. J. Immunol. 2018, 43, 162–167. [Google Scholar] [CrossRef]
- Lau, K.K.; Wyatt, R.J.; Moldoveanu, Z.; Tomana, M.; Julian, B.A.; Hogg, R.J.; Lee, J.Y.; Huang, W.-Q.; Mestecky, J.; Novak, J. Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch-Schönlein purpura. Pediatr. Nephrol. 2007, 22, 2067–2072. [Google Scholar] [CrossRef]
- Mizerska-Wasiak, M.; Gajewski, Ł.; Cichoń-Kawa, K.; Siejko, A.; Małdyk, J.; Spława-Neyman, A.; Zachwieja, J.; Firszt-Adamczyk, A.; Stankiewicz, R.; Drożyńska-Duklas, M.; et al. Relationship between Gd-IgA1 and TNFR1 in IgA nephropathy and IgA vasculitis nephritis in children—Multicenter study. Cent. Eur. J. Immunol. 2021, 46, 199–209. [Google Scholar] [CrossRef]
- Pillebout, E.; Jamin, A.; Ayari, H.; Housset, P.; Pierre, M.; Sauvaget, V.; Viglietti, D.; Deschenes, G.; Monteiro, R.C.; Berthelot, L.; et al. Biomarkers of IgA vasculitis nephritis in children. PLoS ONE 2017, 12, e0188718. [Google Scholar] [CrossRef]
- Neufeld, M.; Molyneux, K.; Pappelbaum, K.I.; Mayer-Hain, S.; von Hodenberg, C.; Ehrchen, J.; Barratt, J.; Suzuki, Y.; Sunderkötter, C. Galactose-deficient IgA1 in skin and serum from patients with skin-limited and systemic IgA vasculitis. J. Am. Acad. Dermatol. 2019, 81, 1078–1085. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, X.; Shi, S.; Liu, L.; Lv, J.; Zhang, H. Plasma galactose-deficient immunoglobulin A1 and loss of kidney function in patients with immunoglobulin A vasculitis nephritis. Nephrol. Dial. Transplant. 2019, 35, 2117–2123. [Google Scholar] [CrossRef]
- Berthelot, L.; Jamin, A.; Viglietti, D.; Chemouny, J.M.; Ayari, H.; Pierre, M.; Housset, P.; Sauvaget, V.; Hurtado-Nedelec, M.; Vrtovsnik, F.; et al. Value of biomarkers for predicting immunoglobulin A vasculitis nephritis outcome in an adult prospective cohort. Nephrol. Dial. Transplant. 2017, 33, 1579–1590. [Google Scholar] [CrossRef]
- Allen, A.; Willis, F.; Beattie, T.; Feehally, J. Abnormal IgA glycosylation in Henoch-Schonlein purpura restricted to patients with clinical nephritis. Nephrol. Dial. Transplant. 1998, 13, 930–934. [Google Scholar] [CrossRef]
- Zhu, B.; Zhu, Q.; Li, N.; Wu, T.; Liu, S.; Liu, S. Association of serum/plasma high mobility group box 1 with autoimmune diseases. Medicine 2018, 97, e11531. [Google Scholar] [CrossRef]
- Pilzweger, C.; Holdenrieder, S. Circulating HMGB1 and RAGE as clinical biomarkers in malignant and autoimmune diseases. Diagnostics 2015, 5, 219–253. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High mobility group box 1: Biological functions and relevance in oxidative stress related chronic diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef]
- Bobek, D.; Grčević, D.; Kovačić, N.; Lukić, K.K.; Jelušić, M. The presence of high mobility group box-1 and soluble receptor for advanced glycation end-products in juvenile idiopathic arthritis and juvenile systemic lupus erythematosus. Pediatr. Rheumatol. 2014, 12, 50. [Google Scholar] [CrossRef]
- Wang, C.; de Souza, A.W.; Westra, J.; Bijl, M.; Chen, M.; Zhao, M.H.; Kallenberg, C.G. Emerging role of high mobility group box 1 in ANCA-associated vasculitis. Autoimmun. Rev. 2015, 14, 1057–1065. [Google Scholar] [CrossRef]
- Page, T.H.; Chiappo, D.; Brunini, F.; Garnica, J.; Blackburn, J.; Dudhiya, F.; Prendecki, M.; McAdoo, S.P.; Pusey, C.D. Danger-associated molecular pattern molecules and the receptor for advanced glycation end products enhance ANCA-induced responses. Rheumatology 2021, 61, 834–845. [Google Scholar] [CrossRef]
- Chen, T.; Guo, Z.; Wang, W.; Qin, S.; Cao, N.; Li, M. Increased serum HMGB1 levels in patients with Henoch-Schönlein purpura. Exp. Dermatol. 2014, 23, 419–423. [Google Scholar] [CrossRef]
- SSato, F.; Maruyama, S.; Hayashi, H.; Sakamoto, I.; Yamada, S.; Uchimura, T.; Morita, Y.; Ito, Y.; Yuzawa, Y.; Maruyama, I.; et al. High mobility group box chromosomal protein 1 in patients with renal diseases. Nephron Clin. Pr. 2008, 108, c194–c201. [Google Scholar] [CrossRef]
- Yue, Q.; Song, Y.; Liu, Z.; Zhang, L.; Yang, L.; Li, J. Receptor for advanced glycation end products (RAGE): A pivotal hub in immune diseases. Molecules 2022, 27, 4922. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for advanced glycation endproducts), RAGE ligands and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef]
- Mahajan, N.; Dhawan, V.; Malik, S.; Jain, S. Serum levels of soluble receptor for advanced glycation end products (sRAGE) in Takayasu’s arteritis. Int. J. Cardiol. 2010, 145, 589–591. [Google Scholar] [CrossRef]
- Wittkowski, H.; Hirono, K.; Ichida, F.; Vogl, T.; Ye, F.; Yanlin, X.; Saito, K.; Uese, K.; Miyawaki, T.; Viemann, D.; et al. Acute Kawasaki disease is associated with reverse regulation of soluble receptor for advance glycation end products and its proinflammatory ligand S100A12. Arthritis Rheum. 2007, 56, 4174–4181. [Google Scholar] [CrossRef]
- Frank, M.; Kemler, R. Protocadherins. Curr. Opin. Cell Biol. 2002, 14, 557–562. [Google Scholar] [CrossRef]
- Modak, D.; Sotomayor, M. Identification of an adhesive interface for the non-clustered δ1 protocadherin-1 involved in respiratory diseases. Commun. Biol. 2019, 2, 354. [Google Scholar] [CrossRef]
- Tellez, G.F.; Vandepoele, K.; Brouwer, U.; Koning, H.; Elderman, R.M.; Hackett, T.-L.; Willemse, B.W.M.; Holloway, J.; Van Roy, F.; Koppelman, G.H.; et al. Protocadherin-1 binds to SMAD3 and suppresses TGF-1-induced gene transcription. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L725–L735. [Google Scholar] [CrossRef]
- Koning, H.; Sayers, I.; Stewart, C.E.; Jong, D.; Postma, D.S.; Oosterhout, A.J.M.; Nawijn, M.C.; Koppelman, G.H.; de Jong, D.; Hacken, N.H.T.T.; et al. Characterization of protocadherin-1 expression in primary bronchial epithelial cells: Association with epithelial cell differentiation. FASEB J. 2011, 26, 439–448. [Google Scholar] [CrossRef]
- Tellez, G.F.; Nawijn, M.C.; Koppelman, G.H. Protocadherin-1: Epithelial barrier dysfunction in asthma and eczema. Eur. Respir. J. 2014, 43, 671–674. [Google Scholar] [CrossRef]
- Tellez, G.F.; Willemse, B.W.M.; Brouwer, U.; Nijboer-Brinksma, S.; Vandepoele, K.; Noordhoek, J.A.; Heijink, I.; de Vries, M.; Smithers, N.P.; Postma, D.S.; et al. Protocadherin-1 localization and cell adhesion function in airway epithelial cells in asthma. PLoS ONE 2016, 11, e0163967. [Google Scholar]
- Ye, Z.; Yang, Y.; Wei, Y.; Li, L.; Wang, X.; Zhang, J. PCDH1 promotes progression of pancreatic ductal adenocarcinoma via activation of NF-κB signalling by interacting with KPNB1. Cell Death Dis. 2022, 13, 633. [Google Scholar] [CrossRef]
- Rook, A. William Heberden’s cases of anaphylactoid purpura. Arch. Dis. Child. 1958, 33, 271. [Google Scholar] [CrossRef]
- Song, Y.; Huang, X.; Yu, G.; Qiao, J.; Cheng, J.; Wu, J.; Chen, J. Pathogenesis of IgA vasculitis: An up-to-date review. Front. Immunol. 2021, 12, 771619. [Google Scholar] [CrossRef]
- Guilpain, P.; Mouthon, L. Antiendothelial cells autoantibodies in vasculitis-associated systemic diseases. Clin. Rev. Allergy Immunol. 2008, 35, 59–65. [Google Scholar] [CrossRef]
- Papa, N.D.; Guidali, L.; Sala, A.; Buccellati, C.; Khamashta, M.A.; Ichikawa, K.; Koike, T.; Balestrieri, G.; Tincani, A.; Hughes, G.R.V.; et al. Endothelial cells as target for antiphospholipid antibodies. Human polyclonal and monoclonal anti-β2-glycoprotein I antibodies react in vitro with endothelial cells through adherent β2-glycoprotein I and induce endothelial activation. Arthritis Rheum. 1997, 40, 551–561. [Google Scholar] [CrossRef]
- Yang, Y.H.; Wang, S.J.; Chuang, Y.H.; Lin, Y.T.; Chiang, B.L. The level of IgA antibodies to human umbilical vein endothelial cells can be enhanced by TNF-α treatment in children with Henoch–Schönlein purpura. Clin. Exp. Immunol. 2002, 130, 352–357. [Google Scholar] [CrossRef]
- Yang, Y.; Chang, C.; Chuang, Y.; Hsu, H.; Yu, H.; Lee, J.; Wang, L.; Lin, Y.; Chiang, B. Identification and characterization of IgA antibodies against β2-glycoprotein I in childhood Henoch-Schönlein purpura. Br. J. Dermatol. 2012, 167, 874–881. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Huang, Y.-H.; Wang, L.-C.; Chuang, Y.-H.; Yu, H.-H.; Lin, Y.-T.; Chiang, B.-L. Circulating IgA from acute stage of childhood Henoch-Schönlein purpura can enhance endothelial interleukin (IL)-8 production through MEK/ERK signalling pathway. Clin. Exp. Immunol. 2006, 144, 247–253. [Google Scholar] [CrossRef]
- Gok, F.; Ugur, Y.; Ozen, S.; Dagdeviren, A. Pathogenesis-related adhesion molecules in Henoch–Schonlein vasculitis. Rheumatol. Int. 2007, 28, 313–316. [Google Scholar] [CrossRef]
- Yang, Y.H.; Tsai, I.J.; Chang, C.J.; Chuang, Y.H.; Hsu, H.Y.; Chiang, B.L. The interaction between circulating complement proteins and cutaneous microvascular endothelial cells in the development of childhood Henoch-Schönlein purpura. PLoS ONE 2015, 10, e0120411. [Google Scholar] [CrossRef]
- Yang, Y.H.; Lai, H.J.; Huang, C.M.; Wang, L.C.; Lin, Y.T.; Chiang, B.L. Sera from children with active Henoch-Schonlein purpura can enhance the production of interleukin 8 by human umbilical venous endothelial cells. Ann. Rheum. Dis. 2004, 63, 1511–1513. [Google Scholar] [CrossRef]
- Chen, X.Q.; Tu, L.; Tang, Q.; Huang, L.; Qin, Y.H. An emerging role for neutrophil extracellular traps in IgA vasculitis: A mini-review. Front. Immunol. 2022, 13, 912929. [Google Scholar] [CrossRef]
- Damman, J.; Mooyaart, A.L.; van den Bosch, T.P.P.; Seelen, M.A.; van Doorn, M.B. Lectin and alternative complement pathway activation in cutaneous manifestations of IgA-vasculitis: A new target for therapy? Mol. Immunol. 2022, 143, 114–121. [Google Scholar] [CrossRef]
- Demir, S.; Yet, I.; Ekiz, M.S.; Sag, E.; Bilginer, Y.; Celikbicak, O.; Lay, I.; Ozen, S. Plasma proteomic analysis reveals the potential role of lectin and alternative complement pathways in IgA vasculitis pathogenesis. Diagnostics 2023, 13, 1729. [Google Scholar] [CrossRef]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Pullerits, R.; Brisslert, M.; Jonsson, I.; Tarkowski, A. Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via β2 integrin Mac-1. Arthritis Rheum. 2006, 54, 3898–3907. [Google Scholar] [CrossRef]
- Zhang, Q.; Geng, M.; Li, K.; Gao, H.; Jiao, X.; Ai, K.; Wei, X.; Yang, J. TGF-β1 suppresses the T-cell response in teleost fish by initiating Smad3- and Foxp3-mediated transcriptional networks. J. Biol. Chem. 2022, 299, 102843. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.; Zhu, X.; Ren, H.; Wang, Y.; Sun, R. The imbalance of Th17/Treg in Chinese children with Henoch-Schonlein purpura. Int. Immunopharmacol. 2013, 16, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ren, Q.; Ling, J.; Tao, Z.; Yang, X.; Li, Y. The change of Th17/Treg cells and IL-10/IL-17 in Chinese children with Henoch-Schonlein purpura. Medicine 2019, 98, e13991. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.; Zhu, D.; Wang, Y. The role of T cells in the development of Henoch-Schonlein purpura. Front. Biosci. 2018, 23, 837–851. [Google Scholar]
- Donadio, M.E.; Loiacono, E.; Peruzzi, L.; Amore, A.; Camilla, R.; Chiale, F.; Vergano, L.; Boido, A.; Conrieri, M.; Bianciotto, M.; et al. Toll-like receptors, immunoproteasome and regulatory T cells in children with Henoch-Schönlein purpura and primary IgA nephropathy. Pediatr. Nephrol. 2014, 29, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Dolezalova, P.; E Price-Kuehne, F.; Özen, S.; Benseler, S.M.; A Cabral, D.; Anton, J.; Brunner, J.; Cimaz, R.; O’Neil, K.M.; A Wallace, C.; et al. Disease activity assessment in childhood vasculitis: Development and preliminary validation of the Paediatric Vasculitis Activity Score (PVAS). Ann. Rheum. Dis. 2012, 72, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Demirkaya, E.; Ozen, S.; Pistorio, A.; Galasso, R.; Ravelli, A.; Hasija, R.; Baskin, E.; Dressler, F.; Fischbach, M.; Consuegra, J.G.; et al. Performance of the Birmingham Vasculitis Activity Score and disease extent index in childhood vasculitides. Clin. Exp. Rheumatol. 2012, 30 (Suppl. S70), 162–168. [Google Scholar]
- Counahan, R.; Winterborn, M.H.; White, R.H.; Heaton, J.M.; Meadow, S.R.; Bluett, N.H.; Swetschin, H.; Cameron, J.S.; Chantler, C. Prognosis of Henoch-Schonlein nephritis in children. BMJ 1977, 2, 11–14. [Google Scholar] [CrossRef]
- StatSoft, Inc. STATISTICA (Data Analysis Software System), Version 12.0. 2013. Available online: www.statsoft.com (accessed on 13 November 2023).
- Graph-Pad Software, San Diego, CA, USA. Available online: http://www.graphpad.com (accessed on 13 November 2023).
- Unal, I. Defining an optimal cut-point value in ROC analysis: An alternative approach. Comput. Math. Methods Med. 2017, 2017, 3762651. [Google Scholar] [CrossRef]


{kind=link}
| N = 86 | % of the Cohort | |
|---|---|---|
| Demographic data | ||
| mean age (years) | 6.4 (4.5–7.8) | |
| female | 43 | 57% |
| male | 37 | 43% |
| Clinical features | ||
| skin changes | 86 | 100% |
| joint involvement | 76 | 88.4% |
| gastrointestinal involvement | 39 | 45.3% |
| nephritis | 26 | 30.2% |
| scrotal involvement | 6 | 16.2% # |
| Treatment | ||
| NSAIDs | 75 | 87.2% |
| Glucocorticoids | 36 | 41.8% |
| ACE inhibitors | 15 | 17.4% |
| immunosuppressants | 11 | 12.8% |
| PVAS | 4 (2–6) | |
| outcome of nephritis | ||
| A | 18 | 69.3% ## |
| B | 8 | 30.7% ## |
| C | 0 | 0% |
| D | 0 | 0% |
| Parameter | IgAV Group | Control Group | p * | p ** | p *** | |
|---|---|---|---|---|---|---|
| Onset | Follow Up | |||||
| ESR (mm/h) | 18 (8–30) | 9 (6–14) | 7 (4.75–11) | <0.001 | 0.012 | <0.001 |
| CRP (mg/L) | 6.8 (2.1–18.3) | 1 (0.6–1.7) | 1 (1–1) | <0.001 | 0.406 | <0.001 |
| leukocytes (109/L) | 10.1 (8.2–12.8) | 7.24 (6.1–8.03) | 6.15 (5.0–7.0) | <0.001 | <0.001 | <0.001 |
| neutrophils | 5.5 (4.2–8.1) | 3.15 (2.41–4.04) | 2.7 (2.1–3.7) | <0.001 | 0.889 | <0.001 |
| erythrocytes (1012/L) | 4.62 ± 0.42 | 4.75 ± 0.35 | 4.75 ± 0.38 | 0.063 | 0.078 | 0.04 |
| hemoglobin (g/L) | 124.3 ± 11.3 | 127.2 ± 8.9 | 130.8 ± 11.1 | <0.001 | 0.015 | 0.068 |
| platelets (109/L) | 355.3 ± 82.1 | 334.1 ± 68.5 | 291.8 ± 71.6 | <0.001 | <0.001 | 0.072 |
| creatinine (µmol/L) | 35 (28–40.25) | 36 (31.5–42) | 44 (36.25–50.75) | <0.001 | <0.001 | 0.197 |
| urea (mmol/L) | 4.5 ± 1.15 | 4.12 ± 1.01 | 4.4 ± 1.08 | 0.647 | 0.462 | 0.005 |
| fibrinogen (g/L) | 3.5 (2.8–4.2) | 2.7 (2.4–3.1) | 2.4 (2.2–2.8) | <0.001 | 0.118 | <0.001 |
| D-dimer (µg/L) | 2.9 (1.1–7.3) | 0.27 (0.19–0.36) | 0.2 (0.19–0.33) | <0.001 | 0.865 | <0.001 |
| PT | 1.01 ± 0.15 | 1.03 ± 0.16 | 0.99 ± 0.13 | 0.653 | 0.153 | 0.624 |
| aPTT (s) | 25.4 ± 3.8 | 27.4 ± 2.8 | 25.65 ± 2.09 | 0.703 | 0.425 | <0.001 |
| ferritin (ng/mL) | 58.45 (34.6–86.1) | 24.85 (16.2–41.7) | - | - | - | <0.001 |
| uACR (mg/mmol) | 5.1 (1.3–14) | 3.7 (1–12) | 0.85 (0.6–1.3) | <0.001 | <0.001 | 0.154 |
| 24-h proteinuria (g/dU) | 0.07 (0.05–0.15) | 0.07 (0.05–0.14) | - | - | - | 0.625 |
| E/mm3 (urine spot test) (%) | 22 (25.6%) | 13 (15.5%) | - | - | - | 0.103 |
| proteinuria (urine spot test) (%) | 14 (16.3%) | 8 (9.5%) | - | - | - | 0.189 |
| eGFR (mL/min/1.73 m2) | 139.2 ± 29.7 | 148.2 ± 29.4 | - | - | - | 0.111 |
| total proteins (g/L) | 68.5 (65–72) | 72 (67–74) | 72 (69.5–74) | <0.001 | 0.289 | <0.001 |
| serum albumin (g/L) | 39.44 ± 5.13 | 43.95 ± 4.84 | 45.16 ± 3.05 | <0.001 | 0.298 | <0.001 |
| IgA (g/L) | 1.7 (1.25–2.42) | 1.49 (1–1.9) | 1.24 (0.98–1.56) | <0.001 | 0.472 | <0.001 |
| IgG (g/L) | 10.92 ± 2.92 | 10.1 ± 2.16 | 10.68 ± 2.09 | 0.612 | 0.392 | <0.001 |
| IgM (g/L) | 0.94 (0.7–1.2) | 1.03 (0.76–1.26) | 1.1 (0.8–1.3) | 0.164 | 0.968 | <0.001 |
| C3 (g/L) | 1.32 ± 0.24 | 1.22 ± 0.21 | - | - | - | |
| C4 (g/L) | 0.27 (0.21–0.33) | 0.23 (0.17–0.26) | - | - | - | <0.001 |
| CH50 (%) | 91 (79–109) | 81 (76–97) | - | - | - | |
| positive FOBT (%) | 24 (27.9%) | 1 (1.2%) | - | - | - | <0.001 |
| fecal calprotectin (µg/g) | 37 (20–83) | 28 (20–34) | ||||
| Parameter | IgAV Group | Control Group (N = 70) | p * | p ** | p *** | ||
|---|---|---|---|---|---|---|---|
| Onset (N = 86) | Follow-Up (N = 83) | ||||||
| Gd-IgA1 (µg/mL) | serum | 52.4 (25.3–96.1) | 49.9 (22.1–87.8) | 54.1 (19.4–90.3) | 0.299 | 0.668 | 0.231 |
| urine | 10.8 (6.3–21.2) | 11.7 (5.9–18.9) | 5.4 (3.2–9.3) | <0.001 | <0.001 | 0.944 | |
| HMGB1 (pg/mL) | serum | 5573 (2274–13,829) | 3477 (1308–6445) | 1088 (574.3–2942) | <0.001 | <0.001 | <0.001 |
| urine | 178.4 (96.7–596.9) | 173.1 (94.2–380.2) | 57.1 (36.9–168) | <0.001 | <0.001 | 0.755 | |
| RAGE (pg/mL) | serum | 309.9 (166.6–432.1) | 302.4 (163.2–388) | 201.3 (112.7–319.6) | 0.005 | 0.014 | 0.012 |
| urine | 12.2 (7.3–21.2) | 10.1 (5.9–18.7) | 5.9 (4.1–10.4) | <0.001 | <0.001 | 0.077 | |
| PCDH1 (ng/mL) | serum | 44.5 (28.2–61.5) | 46.9 (31.8–59.6) | 18.8 (1.2–45.4) | <0.001 | <0.001 | 0.746 |
| urine | 174.6 (72.8–327.5) | 226.4 (102.1–414.9) | 71.1 (2.1–138.1) | <0.001 | <0.001 | 0.109 | |
| Parameter | IgAV Group | IgAVN Group | p * | p ** | |||
|---|---|---|---|---|---|---|---|
| Onset (N = 60) | Follow-Up (N = 58) | Onset (N = 26) | Follow-Up (N = 25) | ||||
| Gd-IgA1 (µg/mL) | serum | 55.7 (23.5–96.9) | 51.09 (21.52–88.75) | 50 (31.2–91.6) | 40.24 (27.93–90.1) | 0.783 | 0.863 |
| urine | 10.8 (6.3–18.9) | 12.04 (6.86–19.36) | 10.8 (6.4–25.7) | 11.17 (4.26–19.98) | 0.657 | 0.618 | |
| HMGB1 (pg/mL) | serum | 5477.5 (2266.6–14,531.6) | 3245 (1367–5552) | 6303.9 (2349–11,042.9) | 3519 (1241–9295) | 0.966 | 0.832 |
| urine | 143.1 (88.9–568.3) | 133.2 (85.9–318.6) | 340.9 (134.7–644.1) | 270.9 (146.7–542.7) | 0.083 | 0.049 | |
| RAGE (pg/mL) | serum | 294.1 (145.3–434.2) | 275.9 (140.8–369) | 314.1 (251.7–425.2) | 331.3 (236.4–416.6) | 0.492 | 0.073 |
| urine | 12.2 (8.2–17.6) | 8.6 (5.9–19.1) | 13.8 (6.9–29.1) | 11.99 (6.04–18.52) | 0.540 | 0.697 | |
| PCDH1 (ng/mL) | serum | 46.5 (32.4–62.4) | 49.74 (38.35–59.07) | 41.0 (2.6–59) | 41.27 (10.8–65.1) | 0.309 | 0.206 |
| urine | 174.4 (86.3–302.7) | 209.1 (103–409.5) | 186.4 (6.2–364.1) | 257.1 (77.6–459.2) | 0.733 | 0.699 | |
| Parameter | AUC | 95% CI | p * | Youden Index | Cut-Off Value | Sensitivity | Specificity | |
|---|---|---|---|---|---|---|---|---|
| Gd-IgA1 (µg/mL) | serum | 0.549 | 0.467–0.628 | 0.3041 | 0.1575 | >31.716 | 68.60 | 47.14 |
| urine | 0.736 | 0.659–0.803 | <0.0001 | 0.4130 | >6.383 | 75.58 | 65.71 | |
| HMGB1 (pg/mL) | serum | 0.784 | 0.711–0.846 | <0.0001 | 0.5030 | >3926.95 | 67.44 | 82.86 |
| urine | 0.782 | 0.709–0.844 | <0.0001 | 0.4944 | >81.481 | 83.72 | 65.71 | |
| RAGE (pg/mL) | serum | 0.631 | 0.550–0.707 | 0.0037 | 0.2508 | >329.02 | 46.51 | 78.57 |
| urine | 0.720 | 0.642–0.789 | <0.0001 | 0.4146 | >9.254 | 68.60 | 72.86 | |
| PCDH1 (ng/mL) | serum | 0.705 | 0.625–0.776 | <0.0001 | 0.3496 | >29.03 | 75.58 | 59.38 |
| urine | 0.728 | 0.650–0.798 | <0.0001 | 0.3590 | >158.18 | 54.65 | 81.25 | |
| Parameter | SE | Z-Value | OR | 95% CI | p * | |
|---|---|---|---|---|---|---|
| Gd-IgA1 (µg/mL) | serum | 0.107 | 0.555 | 1.001 | 0.997–1.006 | 0.579 |
| urine | 0.341 | 1.159 | 1.029 | 0.981–1.087 | 0.246 | |
| HMGB1 (pg/mL) | serum | 0.036 | 3.923 | 1.000 | 1.0001–1.0003 | <0.001 |
| urine | 0.083 | −0.122 | 0.999 | 0.999–1.001 | 0.903 | |
| RAGE (pg/mL) | serum | 0.074 | 2.712 | 1.004 | 1.001–1.007 | 0.007 |
| urine | 0.224 | 1.968 | 1.033 | 1.002–1.070 | 0.049 | |
| PCDH1 (ng/mL) | serum | 0.487 | 1.849 | 1.018 | 0.999–1.038 | 0.064 |
| urine | 0.098 | 0.495 | 1.001 | 0.997–1.005 | 0.621 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Held, M.; Kozmar, A.; Sestan, M.; Turudic, D.; Kifer, N.; Srsen, S.; Gagro, A.; Frkovic, M.; Jelusic, M. Insight into the Interplay of Gd-IgA1, HMGB1, RAGE and PCDH1 in IgA Vasculitis (IgAV). Int. J. Mol. Sci. 2024, 25, 4383. https://doi.org/10.3390/ijms25084383
Held M, Kozmar A, Sestan M, Turudic D, Kifer N, Srsen S, Gagro A, Frkovic M, Jelusic M. Insight into the Interplay of Gd-IgA1, HMGB1, RAGE and PCDH1 in IgA Vasculitis (IgAV). International Journal of Molecular Sciences. 2024; 25(8):4383. https://doi.org/10.3390/ijms25084383
Chicago/Turabian StyleHeld, Martina, Ana Kozmar, Mario Sestan, Daniel Turudic, Nastasia Kifer, Sasa Srsen, Alenka Gagro, Marijan Frkovic, and Marija Jelusic. 2024. "Insight into the Interplay of Gd-IgA1, HMGB1, RAGE and PCDH1 in IgA Vasculitis (IgAV)" International Journal of Molecular Sciences 25, no. 8: 4383. https://doi.org/10.3390/ijms25084383
APA StyleHeld, M., Kozmar, A., Sestan, M., Turudic, D., Kifer, N., Srsen, S., Gagro, A., Frkovic, M., & Jelusic, M. (2024). Insight into the Interplay of Gd-IgA1, HMGB1, RAGE and PCDH1 in IgA Vasculitis (IgAV). International Journal of Molecular Sciences, 25(8), 4383. https://doi.org/10.3390/ijms25084383