
Transforming the Niche: The Emerging Role of Extracellular Vesicles in Acute Myeloid Leukaemia Progression

 , , , , and
, , , , and 
Abstract
1. Introduction
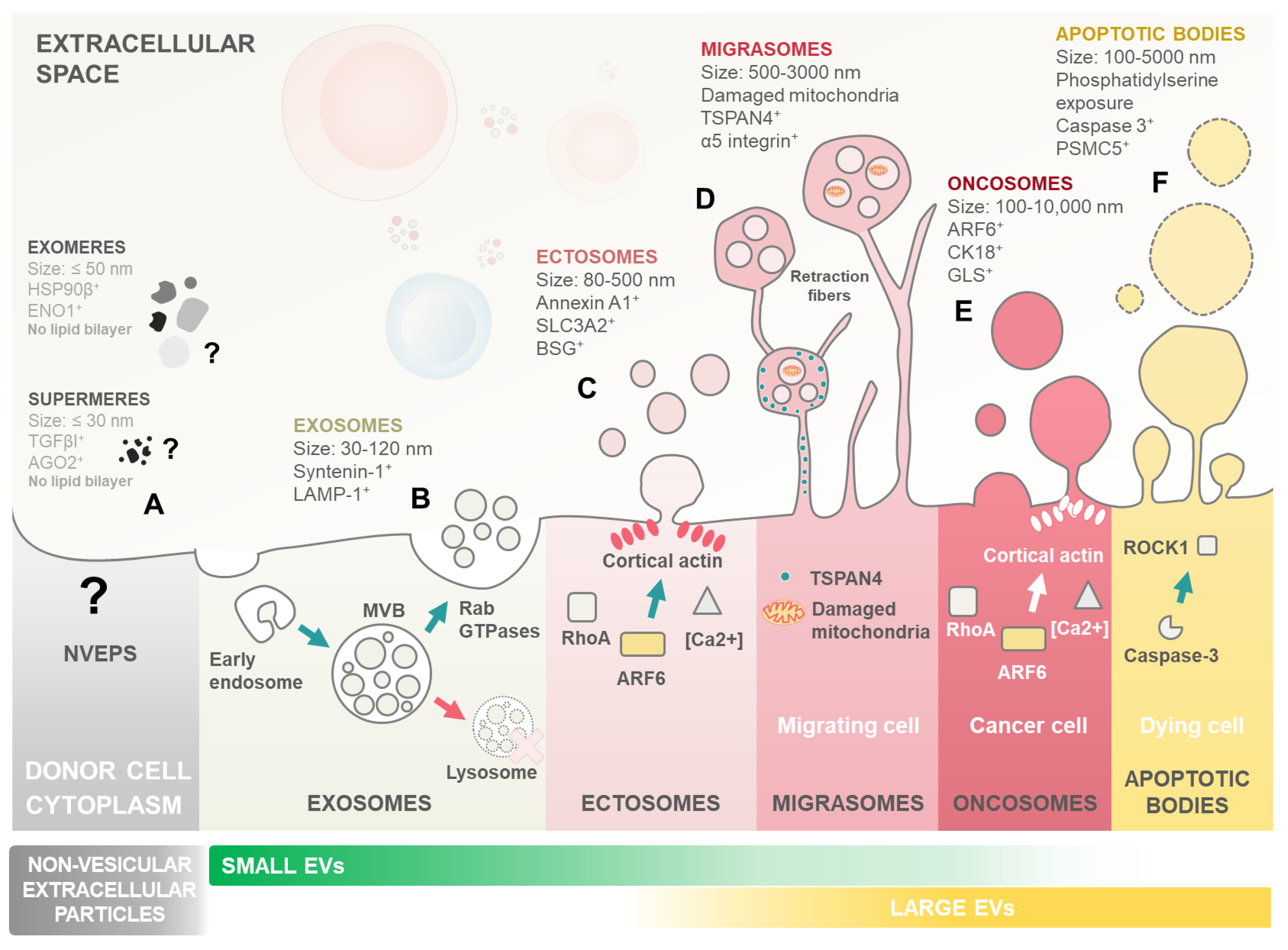
2. Extracellular Vesicles
3. The Leukaemia Heist: Niche Remodelling Favours Disease Progression
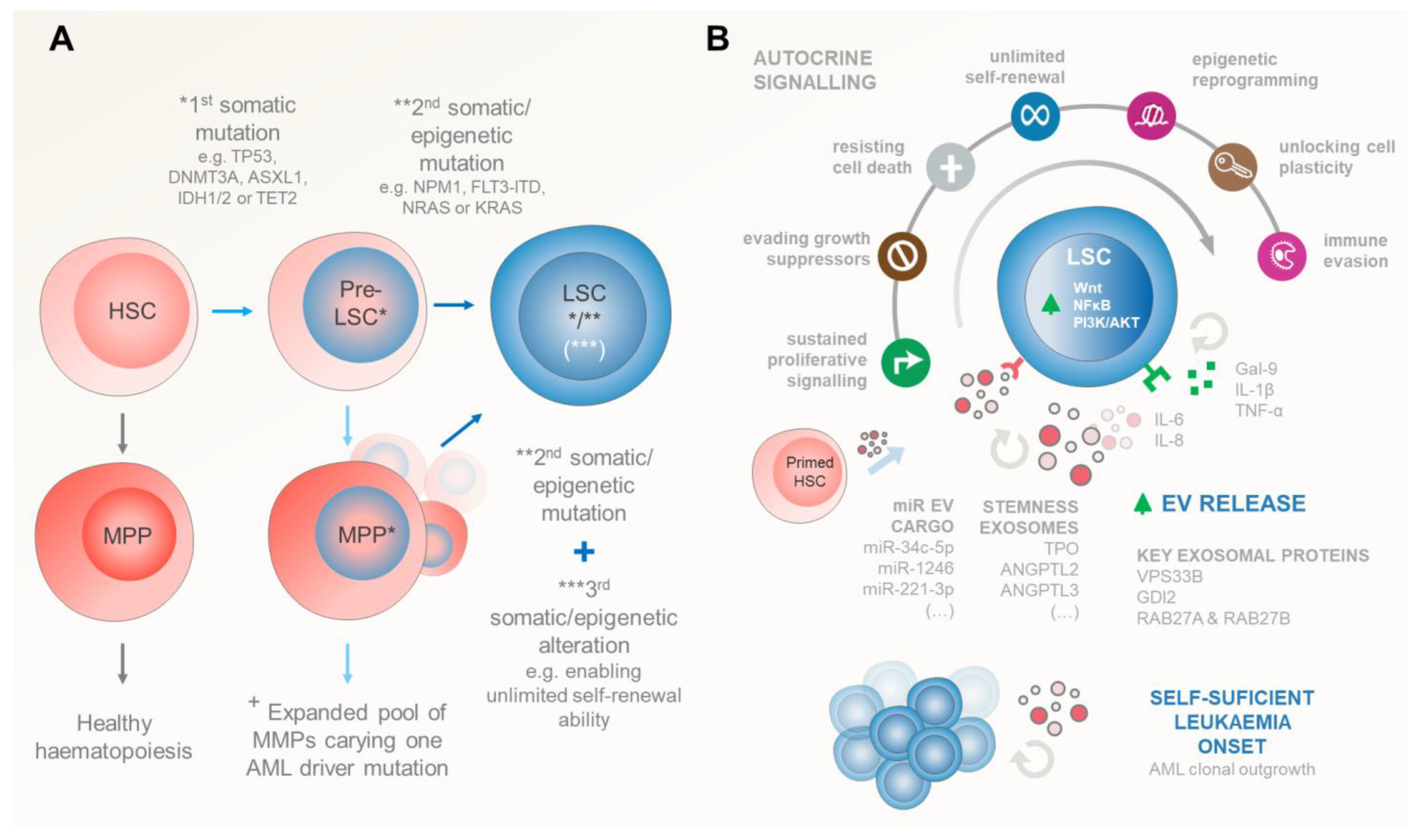
3.1. Leukaemic Stem Cell Inception
3.2. Self-Sustained Leukaemia Growth

{kind=link}
{kind=link}
{kind=link}
| Factor | Origin | Target | Signalling | Pro-Leukaemia Effect | Cancer Hallmark [20] | Ref. | Sample Origin |
|---|---|---|---|---|---|---|---|
| LEUKAEMIA SELF-SUFFICIENCY | |||||||
| Gal-9 | LSCs | LSCs | Autocrine | Promote LSC self-renewal through activation of NF-kB and Wnt pathways, by Gal-9/TIM-3 binding. | Enabling replicative immortality | [41] | Patient Sample |
| TNF-α | LSCs | LSCs | Autocrine | Promote LSC self-renewal through increase in NF-kB pathway activity in positive feedback loop. | Enabling replicative immortality | [43] | Mouse BM |
| IL1-β | LSCs | LSCs | Autocrine | Promote LSC self-renewal through increase in NF-kB pathway activity and increase in HGFs, GM-CSF, IL-6, and TNF-α production, creating a positive feedback loop of proliferative signalling. | Enabling replicative immortality | [45] | Patient Sample |
| NmU | LSC | LSC | Autocrine | Promote leukaemic cell growth and proliferation through MYB-related mechanism. | Sustaining proliferative signalling | [46] | K562 AML Line, Patient Sample |
| miR-221- 3p | AML Cells | AML Cells | Autocrine EV Cargo | Promote leukaemic cell growth and proliferation through promoting entry into cell cycle and apoptosis inhibition through downregulation of Gpb2 gene expression. | Resisting cell death | [49] | Mouse Model THP-1, HL-60, Kasumi-1, MOLM-13 (AML Cell Lines) |
| ENDOSTEAL NICHE | |||||||
| IL-8 | HSCs, BMMSCs | LSCs | Soluble Factor | Lead to induction of proliferative and oncogenic pathways and recruitment of myeloid-derived suppressor cells through binding to overexpressed CXCR1 and CXCR2 receptors. | Sustaining proliferative signalling | [50] | Patient Sample |
| MIF | AML Blasts | BMMSCs | Soluble Factor | Induce IL-8 secretion in PKCβ-regulated mechanism. | Sustaining proliferative signalling | [51] | Patient Sample |
| IL-6 | BMMSCs | AML cells | Soluble Factor | Promote chemoresistance in leukaemic cells through STAT-3 pathway activation, leading to higher OXPHOS levels. | Resisting cell death | [52] | HS-5 (BMMSC lines) HL-60, U-937,THP-1 (AML lines) |
| TPO | Niche Osteoblasts, HSCs, LSCs | HSCs and LSCs | EV Cargo | Induce HSC adhesion to osteoblastic niche. Promote SC quiescence and induce SC proliferation in endosteal niche. | Enabling replicative immortality | [53,54] | Mouse BM |
| ANGPTL3 | Endothelial cells, BMMSCs, HSCs, LSCs | HSCs and LSCs | EV Cargo | Directly bind to HSCs. Promote SC quiescence through suppression of TF Ikaros. | Enabling replicative immortality | [53,55] | Mouse BM |
| miR-34c-5p | LSCs | None, exported out of LSCs via EVs | EV Cargo | miR-34c-5p induces LSC senescence through p53/p21-dependent CDK/Cyclin or p53-independent CDK/Cyclin pathways. LSC EV-mediated export of this factor inhibits this effect, leading to worse AML prognosis. | Senescent cells evading growth suppressors | [56] | Patient Sample |
| miR-1246 | AML Cells | LSCs | EV Cargo | Activate STAT3 pathway through LRIGH1 downregulation. Increase LSC viability and proliferation. Decrease LSC differentiation and apoptosis. | Resisting cell death | [57] | KG1-A, Kasumi-1 (AML lines) |
| IFN-γ | BMMSCs, AML cells | BMMSCs, HSCs and LSCs | Soluble factor EV Cargo | Pro-inflammatory effects. Activate STAT1 signalling to induce oxidative stress, by increased ROS production, leading to decreased osteogenic differentiation of MSCs. Decrease immune response to LSCs by conditioning MSCs into anti-inflammatory activity as a response to excess IFN-γ. | Tumour promoting inflammation Avoiding immune destruction | [58,59] | Patient Sample |
| PGE2 TGF-β TSG-6 HGF HLA-G6 IL-10 IL-6 galectins | BMMSCs | Many | Soluble factor EV Cargo | Dampen immune response against LSCs through promoting anti-inflammatory environment as response to excess inflammatory factors produced by AML cells. | Avoiding immune destruction | [60,61,62] | Patient Sample [60,62] Mouse BM [61] Mouse MS-5 Stromal Line [62] |
| miR-188-5p | LSCs | BMMSCs | EV Cargo | Promote LSC proliferation through restructuring of niche MSCs, as they increase MCAM presence on their surface, increasing binding to myeloid cells, leading to ERK signalling pathway activation. | Sustaining proliferative signalling | [63] | KG1a, SKM-1 (AML Lines) HS5, HS27a (BMMSC lines) |
| miR-4532 | AML Blasts | Pre-Osteoblasts | EV Cargo | Increase DKK1 expression, which inhibits Wnt pathway signalling, leading to decrease in osteoblastic differentiation, causing disruption of endosteal niche bone formation and normal haematopoiesis. | Activating invasion | [64,65] | HL-60, Molm-14, OCI-AML3 (AML lines) |
| PRDX2 PRDX4 L-plastin | Erythroleukaemia Cells | Osteoclast precursors | EV Cargo | Promote bone resorption through induction of osteoclast differentiation. Bone resorption increases the central marrow cavity space, where AML cell growth can occur. | Activating invasion | [66,67,68] | Mouse BM Human Breast Cancer lines |
| YBX1 | AML Cells | BMMSCs | EV Cargo | Reduces osteoblastic differentiation, disrupting normal haematopoiesis. YBX1 downregulation led to impact on other EV cargo, hinting at possible cooperation between different factors. | Activating invasion | [69] | K562, MV-4–11 (AML Lines) Patient Sample (BMMSC) |
| FTO | BMMSCs | AML Blasts | EV Cargo | Increased LncRNA GLCC1 expression in AML blasts, leading to increase in LncRNA-GLCC1-IGF2BP1-c-Myc signalling pathway activation, linked with higher tumour aggressiveness. | Sustaining proliferative signalling | [70] | THP-1, Kasumi-1 (AML Lines) Patient Sample (BMMSC) |
| AML derived EVs | AML Cells | BMMSCs | EVs | Alter gene expression profile of BMMSCs, with concentration-dependent effects. Increased MSC survival, proliferation, and metabolic activity through increased Ki-67 and BCL2 expression (at lower concentrations of AML cell-derived EVs). Downregulation of ROS production. Upregulation of apoptosis (at higher AML cell-derived EV concentrations). | Resisting cell death | [71] | Patient Sample |
| VASCULAR NICHE | |||||||
| CXCL12 (SDF-1) | BMMSCs | AML Cells | Soluble Factor EV Cargo | Bind to CXCR4 expressed on AML cells to promote homing to BM niche and stromal cell–AML cell adhesion. Increase AML cell resistance to apoptosis. Promote LSC quiescence, maintenance, and proliferation. | Activating invasion Resisting cell death | [72,73,74] | Patient Sample (BMMSCs) KG-1a (AML Line) [72] Mouse BM [74] |
| ANGPL2 | Endothelial Cells | LSCs | EV Cargo | Bind to LILRB2 receptor to promote LSC maintenance and drive LSCs to localize around endothelial cells in BM niche. | Enabling replicative immortality | [75] | Mouse Model |
| VEGF VEGFR | AML Cells | Endothelial Cells | EV Cargo | Promote vascular remodelling and angiogenesis. | Inducing or accessing vasculature | [25] | Patient Sample HUVECs |
| IGF-1R coding mRNA | AML Cells | BMMSCs | EV Cargo | Promote IGF-1R expression, which increases VEGF secretion, leading to increased angiogenesis and proliferation. | Inducing or accessing vasculature | [76] | HEL, HL-60, Molm-14, U937 (AML Lines) Patient Samples |
| miR-92a | AML Cells | Endothelial Cells | EV Cargo | Promote endothelial cell migration and tube formation, but not growth. Decrease expression of the pro-angiogenic Integrin-α5. | Inducing or accessing vasculature | [77] | K562 AML Line HUVECs |
| miR-3064-3p miR-339-5p miR-3622a-5p | AML Cells | Endothelial Cells | EV Cargo | Promote angiogenesis in HUVECs, regulated by P62 expression. | Inducing or accessing vasculature | [78] | U937 AML Line HUVECs |
| CXCL12 SCF IL-7 IL-15 M-CSF BMP-4 CCL-2 | BM Adipocytes | BMMSCs | Soluble Factors | Promote HSC proliferation and haematopoietic regeneration, upregulated and hijacked in AML. | Sustaining proliferative signalling | [79] | Mouse Model |
| GDF15 | AML Cells | BM Adipocytes | Soluble Factor | Induce lipolysis in BM adipocytes, releasing fatty acids (FAs) into the vascular environment. FAs are uptaken by AML blasts via an FABP4-dependent mechanism and used as an energy source. | Deregulating cellular metabolism | [80] | THP-1, K562, HEL, HL-60 and Kasumi AML Lines Patient Sample (BMMSCs) differentiated into Adipocytes |
| IL-8 CCL2 TIMP-1 TIMP-2 VEGF-D | ADSCs | Endothelial Cells | EV Cargo | Induce tube formation and angiogenesis. | Inducing or accessing vasculature | [81] | Canine adipose tissue sample SVEC-4 Mouse endothelial line |
| miR-155-5p miR-106a-5p miR-106b-5p miR130b-3p miR-16-5p miR-181a-5p miR-19b-3p miR-466k miR-93-5p miR-126a-5p | AML Cells | HPSCs | EV Cargo | Induce activation of inflammatory secretion profiles in HPSCs, leading to increased AML progression. | Avoiding immune destruction | [82] | C1498 Mouse AML Line Mouse Model |
| UNKNOWN MECHAMISM | |||||||
| MPIF-1 (CCL23) | Unknown | Found in blood plasma | Soluble Factor | Found at elevated levels in AML patient plasma. MPIF has reported to inhibit proliferation and differentiation of myeloid progenitors, but role in AML has not been described. | Unknown | [83] | Patient Sample |
| BMP10 CCL3 CX3CL1 OPN CD105 PTHLH CHRDL1 MMP7 | Many | Found in blood plasma | Soluble Factors | Found at elevated levels in AML patient plasma. Factors linked with bone homeostasis through multiple different pathways. Potential coordinated mechanism of action in BM niche activity in AML | Unknown | [83] | Patient Sample |
| CD31/endomucin-expressing cellular debris particles | Endothelium | Found in vascular lumen | EVs | Particles of endothelial EV origin found in the vasculature of leukaemic mice, but not in healthy control specimens, suggesting it is a possible risk factor. | Unknown | [84] | Mouse model |
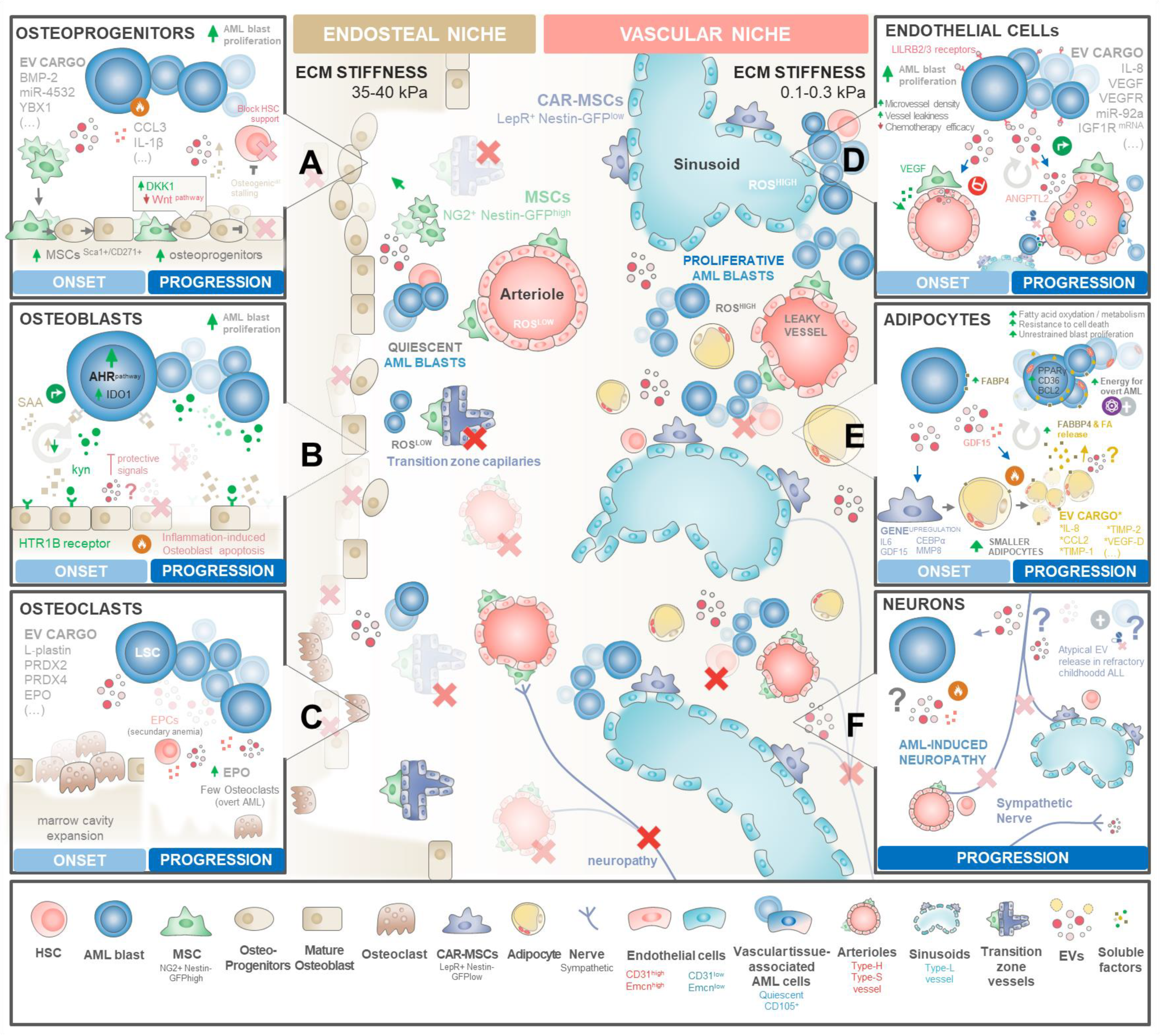
3.3. Rewiring the Haematopoietic Niches for LSC Traction
3.3.1. Endosteal Niche Remodelling
BM Mesenchymal Stromal/Stem Cells
Osteoprogenitors, Bone Lining Cells, and Osteoclasts
3.3.2. Vascular Niche Remodelling
Endothelial Cells and Progenitors
Adipocytes
Sympathetic Neurons
4. Emerging Organ-on-a-Chip Technologies to Unravel EV Signalling Networks
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of acute myeloid leukemia: Recent progress and enduring challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef] [PubMed]
- Laverdiere, I.; Boileau, M.; Neumann, A.L.; Frison, H.; Mitchell, A.; Ng, S.W.K.; Wang, J.C.Y.; Minden, M.D.; Eppert, K. Leukemic stem cell signatures identify novel therapeutics targeting acute myeloid leukemia. Blood Cancer J. 2018, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscoll, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Di Vizio, D.; Driedonks, T.A.P.; Erdbrugger, U.; et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 2024, 13, e12404. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.H.; Sharifi, L.M.A.; Kakhharov, A.J.; Opulencia, M.J.C.; Alsaikhan, F.; Bokov, D.O.; Majdi, H.S.; Jawad, M.A.; Hammid, A.T.; Shalaby, M.N.; et al. Role of Acute Myeloid Leukemia (AML)-Derived exosomes in tumor progression and survival. Biomed. Pharmacother. 2022, 150, 113009. [Google Scholar] [CrossRef] [PubMed]
- Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimaraes, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, M.H.; Caires, H.R.; Abols, A.; Xavier, C.P.R.; Line, A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drug Resist. Updates 2019, 47, 100647. [Google Scholar] [CrossRef] [PubMed]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Malkin, E.Z.; Bratman, S.V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020, 11, 584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Klein, S.; Franco, M.; Chardin, P.; Luton, F. Role of the Arf6 GDP/GTP cycle and Arf6 GTPase-activating proteins in actin remodeling and intracellular transport. J. Biol. Chem. 2006, 281, 12352–12361. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zucker, B.; Zhang, S.; Elias, S.; Zhu, Y.; Chen, H.; Ding, T.; Li, Y.; Sun, Y.; Lou, J.; et al. Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains. Nat. Cell Biol. 2019, 21, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Halicka, H.D.; Bedner, E.; Darzynkiewicz, Z. Segregation of RNA and separate packaging of DNA and RNA in apoptotic bodies during apoptosis. Exp. Cell Res. 2000, 260, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, A.; Alini, M.; Baghaban Eslaminejad, M.; Hosseini, S. Engineering strategies for customizing extracellular vesicle uptake in a therapeutic context. Stem Cell Res. Ther. 2022, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogne, J.M. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Viola, S.; Traer, E.; Huan, J.; Hornick, N.I.; Tyner, J.W.; Agarwal, A.; Loriaux, M.; Johnstone, B.; Kurre, P. Alterations in acute myeloid leukaemia bone marrow stromal cell exosome content coincide with gains in tyrosine kinase inhibitor resistance. Br. J. Haematol. 2016, 172, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Hornick, N.I.; Doron, B.; Abdelhamed, S.; Huan, J.; Harrington, C.A.; Shen, R.; Cambronne, X.A.; Verghese, S.C.; Kurre, P. AML suppresses hematopoiesis by releasing exosomes that contain microRNAs targeting c-MYB. Sci. Signal. 2016, 9, ra88. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Garnier, D.; Lee, T.H.; D’Asti, E.; Montermini, L.; Meehan, B.; Rak, J. PML-RARa modulates the vascular signature of extracellular vesicles released by acute promyelocytic leukemia cells. Angiogenesis 2016, 19, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Hou, D.; Huang, Q.; Zhan, W.; Chen, C.; Liu, J.; You, R.; Xie, J.; Chen, P.; et al. Exosomes derived from acute myeloid leukemia cells promote chemoresistance by enhancing glycolysis-mediated vascular remodeling. J. Cell. Physiol. 2019, 234, 10602–10614. [Google Scholar] [CrossRef] [PubMed]
- Arkhypov, I.; Lasser, S.; Petrova, V.; Weber, R.; Groth, C.; Utikal, J.; Altevogt, P.; Umansky, V. Myeloid Cell Modulation by Tumor-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 6319. [Google Scholar] [CrossRef] [PubMed]
- Swatler, J.; Turos-Korgul, L.; Brewinska-Olchowik, M.; De Biasi, S.; Dudka, W.; Le, B.V.; Kominek, A.; Cyranowski, S.; Pilanc, P.; Mohammadi, E.; et al. 4-1BBL-containing leukemic extracellular vesicles promote immunosuppressive effector regulatory T cells. Blood Adv. 2022, 6, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Tien, H.F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27, 81. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Lind, K.; Heitzer, E.; Ulz, P.; Aubell, K.; Kashofer, K.; Middeke, J.M.; Thiede, C.; Schulz, E.; Rosenberger, A.; et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood 2017, 129, 2587–2591. [Google Scholar] [CrossRef] [PubMed]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Boyd, A.L.; Lu, J.; Hollands, C.G.; Alsostovar, L.; Murali, S.; Reid, J.C.; Ye, W.; Vandersluis, S.; Johnson, P.; ElRafie, A.; et al. Leukemic progenitor compartment serves as a prognostic measure of cancer stemness in patients with acute myeloid leukemia. Cell Rep. Med. 2023, 4, 101108. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Publisher Correction: Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020, 11, 5996. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef]
- Diaz de la Guardia, R.; Lopez-Millan, B.; Lavoie, J.R.; Bueno, C.; Castano, J.; Gomez-Casares, M.; Vives, S.; Palomo, L.; Juan, M.; Delgado, J.; et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Rep. 2017, 8, 1573–1586. [Google Scholar] [CrossRef]
- Hong, C.S.; Diergaarde, B.; Whiteside, T.L. Small extracellular vesicles in plasma carry luminal cytokines that remain undetectable by antibody-based assays in cancer patients and healthy donors. BJC Rep. 2024, 2, 16. [Google Scholar] [CrossRef]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, T.; Kikushige, Y.; Miyamoto, T.; Irifune, H.; Harada, T.; Hatakeyama, K.; Kunisaki, Y.; Kato, K.; Akashi, K. TIM-3 signaling hijacks the canonical Wnt/beta-catenin pathway to maintain cancer stemness in acute myeloid leukemia. Blood Adv. 2023, 7, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing leukemia stem cells to NF-kappaB inhibitor treatment in vivo by inactivation of both TNF and IL-1 signaling. Oncotarget 2017, 8, 8420–8435. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.; Edwards, D.K.T.; Eide, C.A.; Newell, L.; Traer, E.; Medeiros, B.C.; Pollyea, D.A.; Deininger, M.W.; Collins, R.H.; Tyner, J.W.; et al. Identification of Interleukin-1 by Functional Screening as a Key Mediator of Cellular Expansion and Disease Progression in Acute Myeloid Leukemia. Cell Rep. 2017, 18, 3204–3218. [Google Scholar] [CrossRef] [PubMed]
- Shetzline, S.E.; Rallapalli, R.; Dowd, K.J.; Zou, S.; Nakata, Y.; Swider, C.R.; Kalota, A.; Choi, J.K.; Gewirtz, A.M. Neuromedin U: A Myb-regulated autocrine growth factor for human myeloid leukemias. Blood 2004, 104, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, K.; Forbes, L.; Minuesa, G.; Gindin, T.; Brown, F.; Kharas, M.G.; Krivtsov, A.V.; Armstrong, S.A.; Still, E.; de Stanchina, E.; et al. Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat. Commun. 2018, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, G.; Zhao, J.; Pu, S.; Lv, Y.; Wang, Y.; Li, Y.; Zhao, X.; Wang, Y.; Yang, S.; et al. Small extracellular vesicles derived from acute myeloid leukemia cells promote leukemogenesis by transferring miR-221-3p. Haematologica 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Schinke, C.; Giricz, O.; Li, W.; Shastri, A.; Gordon, S.; Barreyro, L.; Bhagat, T.; Bhattacharyya, S.; Ramachandra, N.; Bartenstein, M.; et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 2015, 125, 3144–3152. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Shafat, M.S.; Mehta, T.K.; Di Palma, F.; Lawes, M.J.; Rushworth, S.A.; Bowles, K.M. MIF-Induced Stromal PKCbeta/IL8 Is Essential in Human Acute Myeloid Leukemia. Cancer Res. 2017, 77, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Wang, B.; You, R.; Wang, X.; Liu, J.; Zhan, W.; Chen, P.; Qin, T.; Zhang, X.; Huang, H. Stromal cells promote chemoresistance of acute myeloid leukemia cells via activation of the IL-6/STAT3/OXPHOS axis. Ann. Transl. Med. 2020, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Chen, C.; Hao, X.; Wang, C.; Zhang, X.; Li, Z.; Shao, H.; Zeng, H.; Yu, Z.; Xie, L.; et al. Sorting protein VPS33B regulates exosomal autocrine signaling to mediate hematopoiesis and leukemogenesis. J. Clin. Investig. 2016, 126, 4537–4553. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Huynh, H.; Umikawa, M.; Silvany, R.; Zhang, C.C. Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood 2011, 117, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Wang, H.; Li, L.; Ma, X.; Chen, Y.; Zhou, H.; Luo, Y.; Xiao, Y.; Liu, L. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia 2018, 32, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, Z.; Zhou, Y.; Ni, J.; Zhu, J.; Fan, X.; Chen, X.; Liu, Y.; Li, Z.; Zhou, H. microRNA-1246-containing extracellular vesicles from acute myeloid leukemia cells promote the survival of leukemia stem cells via the LRIG1-meditated STAT3 pathway. Aging 2021, 13, 13644–13662. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Yang, F.; Feng, S.; Chang, K.; Yu, X.; Guan, F.; Li, X. Clonal MDS/AML cells with enhanced TWIST1 expression reprogram the differentiation of bone marrow MSCs. Redox Biol. 2023, 67, 102900. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Pang, X.; Xie, C.; Deeg, J.H.; Wang, H.; Wan, T.; Wu, J.; Guan, F.; Li, X. Elevated TWIST1 expression in myelodysplastic syndromes/acute myeloid leukemia reduces efficacy of hypomethylating therapy with decitabine. Haematologica 2020, 105, e502. [Google Scholar] [CrossRef]
- Neo, S.H.; Her, Z.; Othman, R.; Tee, C.A.; Ong, L.C.; Wang, Y.; Tan, I.; Tan, J.; Yang, Y.; Yang, Z.; et al. Expansion of human bone marrow-derived mesenchymal stromal cells with enhanced immunomodulatory properties. Stem Cell Res. Ther. 2023, 14, 259. [Google Scholar] [CrossRef]
- English, K.; Barry, F.P.; Field-Corbett, C.P.; Mahon, B.P. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol. Lett. 2007, 110, 91–100. [Google Scholar] [CrossRef]
- Schelker, R.C.; Iberl, S.; Muller, G.; Hart, C.; Herr, W.; Grassinger, J. TGF-beta1 and CXCL12 modulate proliferation and chemotherapy sensitivity of acute myeloid leukemia cells co-cultured with multipotent mesenchymal stromal cells. Hematology 2018, 23, 337–345. [Google Scholar] [CrossRef]
- Feng, J.; Wang, Y.; Li, B.; Yu, X.; Lei, L.; Wu, J.; Zhang, X.; Chen, Q.; Zhou, Y.; Gou, J.; et al. Loss of bisecting GlcNAcylation on MCAM of bone marrow stoma determined pro-tumoral niche in MDS/AML. Leukemia 2023, 37, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Du, F.; Zhao, Y.; Wang, S.; Qi, L. Acute myeloid leukemia cells secrete microRNA-4532-containing exosomes to mediate normal hematopoiesis in hematopoietic stem cells by activating the LDOC1-dependent STAT3 signaling pathway. Stem Cell Res. Ther. 2019, 10, 384. [Google Scholar] [CrossRef]
- Fleming, H.E.; Janzen, V.; Lo Celso, C.; Guo, J.; Leahy, K.M.; Kronenberg, H.M.; Scadden, D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell 2008, 2, 274–283. [Google Scholar] [CrossRef]
- Sadvakassova, G.; Tiedemann, K.; Steer, K.J.D.; Mikolajewicz, N.; Stavnichuk, M.; In-Kyung Lee, I.; Sabirova, Z.; Schranzhofer, M.; Komarova, S.V. Active hematopoiesis triggers exosomal release of PRDX2 that promotes osteoclast formation. Physiol. Rep. 2021, 9, e14745. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, S.; Tiedemann, K.; Tabaries, S.; Siegel, P.M.; Komarova, S.V. Peroxiredoxin 4: A novel secreted mediator of cancer induced osteoclastogenesis. Cancer Lett. 2015, 361, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, K.; Sadvakassova, G.; Mikolajewicz, N.; Juhas, M.; Sabirova, Z.; Tabaries, S.; Gettemans, J.; Siegel, P.M.; Komarova, S.V. Exosomal Release of L-Plastin by Breast Cancer Cells Facilitates Metastatic Bone Osteolysis. Transl. Oncol. 2019, 12, 462–474. [Google Scholar] [CrossRef]
- Chetty, V.K.; Ghanam, J.; Licha, K.; Brenzel, A.; Reinhardt, D.; Thakur, B.K. Y-box binding protein 1 in small extracellular vesicles reduces mesenchymal stem cell differentiation to osteoblasts-implications for acute myeloid leukaemia. J. Extracell. Vesicles 2024, 13, e12417. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.; Li, T.; Fu, C.; Jiang, D.; Wang, Y.; Meng, J.; Zhong, R.; Liang, C.; Dong, M. Exosome-shuttled FTO from BM-MSCs contributes to cancer malignancy and chemoresistance in acute myeloid leukemia by inducing m6A-demethylation: A nano-based investigation. Environ. Res. 2023, 244, 117783. [Google Scholar] [CrossRef]
- Kargar-Sichani, Y.; Mohammadi, M.H.; Amiri, V.; Barzegar, M.; Keshavarz, A.; Bashash, D.; Farsani, M.A. Effect of Acute Myeloid Leukemia-derived Extracellular Vesicles on Bone Marrow Mesenchymal Stromal Cells: Expression of Poor Prognosis Genes. Arch. Med. Res. 2023, 54, 95–104. [Google Scholar] [CrossRef]
- Deniz, I.A.; Karbanova, J.; Wobus, M.; Bornhauser, M.; Wimberger, P.; Kuhlmann, J.D.; Corbeil, D. Mesenchymal stromal cell-associated migrasomes: A new source of chemoattractant for cells of hematopoietic origin. Cell Commun. Signal. 2023, 21, 36. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Burkle, A. The CXCR4 chemokine receptor in acute and chronic leukaemia: A marrow homing receptor and potential therapeutic target. Br. J. Haematol. 2007, 137, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sun, G.; Hao, X.; He, X.; Zheng, Z.; Chen, C.; Yu, Z.; Xie, L.; Ma, S.; Liu, L.; et al. ANGPTL2-containing small extracellular vesicles from vascular endothelial cells accelerate leukemia progression. J. Clin. Investig. 2021, 131, 1. [Google Scholar] [CrossRef]
- Huan, J.; Hornick, N.I.; Shurtleff, M.J.; Skinner, A.M.; Goloviznina, N.A.; Roberts, C.T., Jr.; Kurre, P. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013, 73, 918–929. [Google Scholar] [CrossRef]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef]
- Li, C.; Long, X.; Liang, P.; Liu, Z.; Wang, C.; Hu, R. Analysis of microRNA expression profiles in exosomes derived from acute myeloid leukemia by p62 knockdown and effect on angiogenesis. PeerJ 2022, 10, e13498. [Google Scholar] [CrossRef]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Lu, W.; Wan, Y.; Li, Z.; Zhu, B.; Yin, C.; Liu, H.; Yang, S.; Zhai, Y.; Yu, Y.; Wei, Y.; et al. Growth differentiation factor 15 contributes to marrow adipocyte remodeling in response to the growth of leukemic cells. J. Exp. Clin. Cancer Res. 2018, 37, 66. [Google Scholar] [CrossRef]
- Gangadaran, P.; Rajendran, R.L.; Oh, J.M.; Oh, E.J.; Hong, C.M.; Chung, H.Y.; Lee, J.; Ahn, B.C. Identification of Angiogenic Cargo in Extracellular Vesicles Secreted from Human Adipose Tissue-Derived Stem Cells and Induction of Angiogenesis In Vitro and In Vivo. Pharmaceutics 2021, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.W.; Fan, J.M.; Schrey, J.M.; Mitchell, D.V.; Jung, S.K.; Hurwitz, S.N.; Perez, E.B.; Muraro, M.J.; Carroll, M.; Taylor, D.M.; et al. Inflammatory recruitment of healthy hematopoietic stem and progenitor cells in the acute myeloid leukemia niche. Leukemia 2024, 38, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Celik, H.; Lindblad, K.E.; Popescu, B.; Gui, G.; Goswami, M.; Valdez, J.; DeStefano, C.; Lai, C.; Thompson, J.; Ghannam, J.Y.; et al. Highly multiplexed proteomic assessment of human bone marrow in acute myeloid leukemia. Blood Adv. 2020, 4, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77.e66. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, N.; Gupta, M.; Schwartz, L.; Todorova, T.; Shastri, A.; Will, B.; Steidl, U.; Verma, A. Role of IL8 in myeloid malignancies. Leuk. Lymphoma 2023, 64, 1742–1751. [Google Scholar] [CrossRef]
- Cheng, J.; Li, Y.; Liu, S.; Jiang, Y.; Ma, J.; Wan, L.; Li, Q.; Pang, T. CXCL8 derived from mesenchymal stromal cells supports survival and proliferation of acute myeloid leukemia cells through the PI3K/AKT pathway. FASEB J. 2019, 33, 4755–4764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, H.; Zhang, Z.; Lu, W.; Zhu, J.; Shi, J. IL-6 promotes chemoresistance via upregulating CD36 mediated fatty acids uptake in acute myeloid leukemia. Exp. Cell Res. 2022, 415, 113112. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Ling, X.; Yang, T.; Zhang, J.; Gu, X.; Li, F.; Chen, H.; Wen, Y.; Li, Z.; Zou, Y.; et al. Cytokine levels in patients with non-M3 myeloid leukemia are key indicators of how well the disease responds to chemotherapy. Clin. Exp. Med. 2023, 23, 4623–4632. [Google Scholar] [CrossRef]
- Luciano, M.; Krenn, P.W.; Horejs-Hoeck, J. The cytokine network in acute myeloid leukemia. Front. Immunol. 2022, 13, 1000996. [Google Scholar] [CrossRef]
- Zheng, J.; Umikawa, M.; Cui, C.; Li, J.; Chen, X.; Zhang, C.; Huynh, H.; Kang, X.; Silvany, R.; Wan, X.; et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 2012, 485, 656–660. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wen, J.; Li, Q.; Peng, D.; Liao, C.; Ma, X.; Wang, M.; Niu, J.; Wang, D.; Li, Y.; et al. RAB27B-regulated exosomes mediate LSC maintenance via resistance to senescence and crosstalk with the microenvironment. Leukemia 2023, 38, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Fu, J.; Chen, X. A combined immune and exosome-related risk signature as prognostic biomakers in acute myeloid leukemia. Hematology 2024, 29, 2300855. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Hornick, N.I.; Goloviznina, N.A.; Kamimae-Lanning, A.N.; David, L.L.; Wilmarth, P.A.; Mori, T.; Chevillet, J.R.; Narla, A.; Roberts, C.T., Jr.; et al. Coordinate regulation of residual bone marrow function by paracrine trafficking of AML exosomes. Leukemia 2015, 29, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Aparici Herraiz, I.; Caires, H.R.; Castillo-Fernandez, O.; Sima, N.; Mendez-Mora, L.; Risueno, R.M.; Sattabongkot, J.; Roobsoong, W.; Hernandez-Machado, A.; Fernandez-Becerra, C.; et al. Advancing Key Gaps in the Knowledge of Plasmodium vivax Cryptic Infections Using Humanized Mouse Models and Organs-on-Chips. Front. Cell. Infect. Microbiol. 2022, 12, 920204. [Google Scholar] [CrossRef]
- Nelson, M.R.; Ghoshal, D.; Mejias, J.C.; Rubio, D.F.; Keith, E.; Roy, K. A multi-niche microvascularized human bone marrow (hBM) on-a-chip elucidates key roles of the endosteal niche in hBM physiology. Biomaterials 2021, 270, 120683. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e1916. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pronk, E.; van Dijk, C.; Bian, Y.; Feyen, J.; van Tienhoven, T.; Yildirim, M.; Pisterzi, P.; de Jong, M.M.E.; Bastidas, A.; et al. A Single-Cell Taxonomy Predicts Inflammatory Niche Remodeling to Drive Tissue Failure and Outcome in Human AML. Blood Cancer Discov. 2023, 4, 394–417. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.G.X.; Iacobucci, I.; Shah, S.; Mitchell, A.; Wong, G.; Bansal, S.; Gao, Q.; Kim, H.; Kennedy, J.A.; Minden, M.D.; et al. Precise single-cell transcriptomic mapping of normal and leukemic cell states reveals unconventional lineage priming in acute myeloid leukemia. bioRxiv 2023. [Google Scholar] [CrossRef]
- Crippa, S.; Bernardo, M.E. Mesenchymal Stromal Cells: Role in the BM Niche and in the Support of Hematopoietic Stem Cell Transplantation. Hemasphere 2018, 2, e151. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.; Guezguez, B. Dynamic Changes of the Bone Marrow Niche: Mesenchymal Stromal Cells and Their Progeny During Aging and Leukemia. Front. Cell Dev. Biol. 2021, 9, 714716. [Google Scholar] [CrossRef] [PubMed]
- Braga, C.L.; da Silva, L.R.; Santos, R.T.; de Carvalho, L.R.P.; Mandacaru, S.C.; de Oliveira Trugilho, M.R.; Rocco, P.R.M.; Cruz, F.F.; Silva, P.L. Proteomics profile of mesenchymal stromal cells and extracellular vesicles in normoxic and hypoxic conditions. Cytotherapy 2022, 24, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Lenzini, S.; Debnath, K.; Joshi, J.C.; Wong, S.W.; Srivastava, K.; Geng, X.; Cho, I.S.; Song, A.; Bargi, R.; Lee, J.C.; et al. Cell-Matrix Interactions Regulate Functional Extracellular Vesicle Secretion from Mesenchymal Stromal Cells. ACS Nano 2021, 15, 17439–17452. [Google Scholar] [CrossRef]
- Gomariz, A.; Helbling, P.M.; Isringhausen, S.; Suessbier, U.; Becker, A.; Boss, A.; Nagasawa, T.; Paul, G.; Goksel, O.; Szekely, G.; et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun. 2018, 9, 2532. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 2015, 16, 239–253. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Varma, S.; Montgomery, K.; Greenberg, P.L.; Gratzinger, D. Distinctive contact between CD34+ hematopoietic progenitors and CXCL12+ CD271+ mesenchymal stromal cells in benign and myelodysplastic bone marrow. Lab. Investig. 2012, 92, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef]
- Morikawa, S.; Mabuchi, Y.; Kubota, Y.; Nagai, Y.; Niibe, K.; Hiratsu, E.; Suzuki, S.; Miyauchi-Hara, C.; Nagoshi, N.; Sunabori, T.; et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 2009, 206, 2483–2496. [Google Scholar] [CrossRef]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Borella, G.; Da Ros, A.; Borile, G.; Porcu, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting the plasticity of mesenchymal stromal cells to reroute the course of acute myeloid leukemia. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, Q.; Cang, H.; Wang, Z.; Hu, X.; Pan, R.; Yang, Y.; Chen, Y. Acute Myeloid Leukemia Cells Educate Mesenchymal Stromal Cells toward an Adipogenic Differentiation Propensity with Leukemia Promotion Capabilities. Adv. Sci. 2022, 9, 2105811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.G.; Liang, Y.; Li, K.; Li, W.M.; Li, Q.B.; Chen, Z.C.; Zou, P. Phenotypic and functional comparison of mesenchymal stem cells derived from the bone marrow of normal adults and patients with hematologic malignant diseases. Stem Cells Dev. 2007, 16, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Yehudai-Resheff, S.; Attias-Turgeman, S.; Sabbah, R.; Gabay, T.; Musallam, R.; Fridman-Dror, A.; Zuckerman, T. Abnormal morphological and functional nature of bone marrow stromal cells provides preferential support for survival of acute myeloid leukemia cells. Int. J. Cancer 2019, 144, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Shipounova, I.N.; Petinati, N.A.; Bigildeev, A.E.; Sorokina, T.V.; Kuzmina, L.A.; Parovichnikova, E.N.; Savchenko, V.G. Alterations in multipotent mesenchymal stromal cells from the bone marrow of acute myeloid leukemia patients at diagnosis and during treatment. Leuk. Lymphoma 2019, 60, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yuan, Y.; Chen, T. Morphology, differentiation and adhesion molecule expression changes of bone marrow mesenchymal stem cells from acute myeloid leukemia patients. Mol. Med. Rep. 2014, 9, 293–298. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells from De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Xiao, P.; Sandhow, L.; Heshmati, Y.; Kondo, M.; Bouderlique, T.; Dolinska, M.; Johansson, A.S.; Sigvardsson, M.; Ekblom, M.; Walfridsson, J.; et al. Distinct roles of mesenchymal stem and progenitor cells during the development of acute myeloid leukemia in mice. Blood Adv. 2018, 2, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hoffmeister, L.M.; Zaun, Y.; Arnold, L.; Schmid, K.W.; Giebel, B.; Klein-Hitpass, L.; Hanenberg, H.; Squire, A.; Reinhardt, H.C.; et al. Acute myeloid leukemia-induced remodeling of the human bone marrow niche predicts clinical outcome. Blood Adv. 2020, 4, 5257–5268. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-C.; Chang, H.; Sun, C.-F.; Shih, L.-Y.; Dunn, P.; Wang, P.-N.; Wu, J.-H.; Kuo, M.-C.; Lin, T.-L.; Hung, Y.-S.; et al. Clinical Implications of Reticulin Fibrosis of Bone Marrow in De Novo Acute Myeloid Leukemia. Blood 2012, 120, 2585. [Google Scholar] [CrossRef]
- Boyd, A.L.; Reid, J.C.; Salci, K.R.; Aslostovar, L.; Benoit, Y.D.; Shapovalova, Z.; Nakanishi, M.; Porras, D.P.; Almakadi, M.; Campbell, C.J.V.; et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat. Cell Biol. 2017, 19, 1336–1347. [Google Scholar] [CrossRef]
- von der Heide, E.K.; Neumann, M.; Vosberg, S.; James, A.R.; Schroeder, M.P.; Ortiz-Tanchez, J.; Isaakidis, K.; Schlee, C.; Luther, M.; Johrens, K.; et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2017, 31, 1069–1078. [Google Scholar] [CrossRef]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef]
- Nervi, B.; Ramirez, P.; Rettig, M.P.; Uy, G.L.; Holt, M.S.; Ritchey, J.K.; Prior, J.L.; Piwnica-Worms, D.; Bridger, G.; Ley, T.J.; et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009, 113, 6206–6214. [Google Scholar] [CrossRef]
- Abdul-Aziz, A.M.; Sun, Y.; Hellmich, C.; Marlein, C.R.; Mistry, J.; Forde, E.; Piddock, R.E.; Shafat, M.S.; Morfakis, A.; Mehta, T.; et al. Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 2019, 133, 446–456. [Google Scholar] [CrossRef]
- Lee, H.R.; Yang, S.J.; Choi, H.K.; Kim, J.A.; Oh, I.H. The Chromatin Remodeling Complex CHD1 Regulates the Primitive State of Mesenchymal Stromal Cells to Control Their Stem Cell Supporting Activity. Stem Cells Dev. 2021, 30, 363–373. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.; Murphy, J.M.; Mahon, B.P. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007, 149, 353–363. [Google Scholar] [CrossRef]
- Towers, R.; Trombello, L.; Fusenig, M.; Tunger, A.; Baumann, A.L.; Savoldelli, R.; Wehner, R.; Fasslrinner, F.; Arndt, C.; Dazzi, F.; et al. Bone marrow-derived mesenchymal stromal cells obstruct AML-targeting CD8(+) clonal effector and CAR T-cell function while promoting a senescence-associated phenotype. Cancer Immunol. Immunother. 2024, 73, 8. [Google Scholar] [CrossRef]
- Kapor, S.; Santibanez, J.F. Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies. J. Clin. Med. 2021, 10, 2788. [Google Scholar] [CrossRef]
- Kim, D.S.; Jang, I.K.; Lee, M.W.; Ko, Y.J.; Lee, D.H.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-gamma. EBioMedicine 2018, 28, 261–273. [Google Scholar] [CrossRef]
- Kerkela, E.; Laitinen, A.; Rabina, J.; Valkonen, S.; Takatalo, M.; Larjo, A.; Veijola, J.; Lampinen, M.; Siljander, P.; Lehenkari, P.; et al. Adenosinergic Immunosuppression by Human Mesenchymal Stromal Cells Requires Co-Operation with T cells. Stem Cells 2016, 34, 781–790. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, X.; Munoz, N.; Logan, T.M.; Ma, T. Commitment to Aerobic Glycolysis Sustains Immunosuppression of Human Mesenchymal Stem Cells. Stem Cells Transl. Med. 2019, 8, 93–106. [Google Scholar] [CrossRef]
- Wobma, H.M.; Tamargo, M.A.; Goeta, S.; Brown, L.M.; Duran-Struuck, R.; Vunjak-Novakovic, G. The influence of hypoxia and IFN-gamma on the proteome and metabolome of therapeutic mesenchymal stem cells. Biomaterials 2018, 167, 226–234. [Google Scholar] [CrossRef]
- Larey, A.M.; Spoerer, T.M.; Daga, K.R.; Morfin, M.G.; Hynds, H.M.; Carpenter, J.; Hines, K.M.; Marklein, R.A. High throughput screening of mesenchymal stromal cell morphological response to inflammatory signals for bioreactor-based manufacturing of extracellular vesicles that modulate microglia. bioRxiv 2023. [Google Scholar] [CrossRef]
- de Kruijf, E.F.M.; Zuijderduijn, R.; Stip, M.C.; Fibbe, W.E.; van Pel, M. Mesenchymal stromal cells induce a permissive state in the bone marrow that enhances G-CSF-induced hematopoietic stem cell mobilization in mice. Exp. Hematol. 2018, 64, 59–70.e52. [Google Scholar] [CrossRef]
- Xiao, Q.; Lei, L.; Ren, J.; Peng, M.; Jing, Y.; Jiang, X.; Huang, J.; Tao, Y.; Lin, C.; Yang, J.; et al. Mutant NPM1-Regulated FTO-Mediated m(6)A Demethylation Promotes Leukemic Cell Survival via PDGFRB/ERK Signaling Axis. Front. Oncol. 2022, 12, 817584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tomasoni, C.; Arsuffi, C.; Donsante, S.; Corsi, A.; Riminucci, M.; Biondi, A.; Pievani, A.; Serafini, M. AML alters bone marrow stromal cell osteogenic commitment via Notch signaling. Front. Immunol. 2023, 14, 1320497. [Google Scholar] [CrossRef]
- Battula, V.L.; Le, P.M.; Sun, J.C.; Nguyen, K.; Yuan, B.; Zhou, X.; Sonnylal, S.; McQueen, T.; Ruvolo, V.; Michel, K.A.; et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2017, 2, e90036. [Google Scholar] [CrossRef]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, W.; Xiao, M.; Wei, T.; Qiu, Y.; Qiu, J.; Wang, H.; Qiu, Z.; Zhang, S.; Pan, Y.; et al. TREM2 acts as a receptor for IL-34 to suppress acute myeloid leukemia in mice. Blood 2023, 141, 3184–3198. [Google Scholar] [CrossRef]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef]
- Visnjic, D.; Kalajzic, Z.; Rowe, D.W.; Katavic, V.; Lorenzo, J.; Aguila, H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004, 103, 3258–3264. [Google Scholar] [CrossRef]
- Galan-Diez, M.; Borot, F.; Ali, A.M.; Zhao, J.; Gil-Iturbe, E.; Shan, X.; Luo, N.; Liu, Y.; Huang, X.P.; Bisikirska, B.; et al. Subversion of Serotonin Receptor Signaling in Osteoblasts by Kynurenine Drives Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 1106–1127. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, A.; Zhao, H.; Lu, P.; Cheng, H.; Dong, F.; Gong, Y.; Ma, S.; Zheng, Y.; Zhang, H.; et al. Leukemia cell infiltration causes defective erythropoiesis partially through MIP-1alpha/CCL3. Leukemia 2016, 30, 1897–1908. [Google Scholar] [CrossRef]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1beta as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev. 2017, 31, 306–317. [Google Scholar] [CrossRef]
- Mao, C.Y.; Wang, Y.G.; Zhang, X.; Zheng, X.Y.; Tang, T.T.; Lu, E.Y. Double-edged-sword effect of IL-1beta on the osteogenesis of periodontal ligament stem cells via crosstalk between the NF-kappaB, MAPK and BMP/Smad signaling pathways. Cell Death Dis. 2016, 7, e2296. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 2019, 33, 918–930. [Google Scholar] [CrossRef]
- Teufel, S.; Hartmann, C. Wnt-signaling in skeletal development. Curr. Top. Dev. Biol. 2019, 133, 235–279. [Google Scholar] [CrossRef]
- Kapinas, K.; Kessler, C.; Ricks, T.; Gronowicz, G.; Delany, A.M. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J. Biol. Chem. 2010, 285, 25221–25231. [Google Scholar] [CrossRef]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Cong, D.; Bai, Y. Detection and clinical significance of serum EPO levels in patients with haematological tumours. Cell. Mol. Biol. 2022, 68, 33–36. [Google Scholar] [CrossRef]
- Miller, D.; Kerkhofs, K.; Abbas-Aghababazadeh, F.; Madahar, S.S.; Minden, M.D.; Hebert, J.; Haibe-Kains, B.; Bayfield, M.A.; Benchimol, S. Heterogeneity in leukemia cells that escape drug-induced senescence-like state. Cell Death Dis. 2023, 14, 503. [Google Scholar] [CrossRef]
- Awida, Z.; Hiram-Bab, S.; Bachar, A.; Saed, H.; Zyc, D.; Gorodov, A.; Ben-Califa, N.; Omari, S.; Omar, J.; Younis, L.; et al. Erythropoietin Receptor (EPOR) Signaling in the Osteoclast Lineage Contributes to EPO-Induced Bone Loss in Mice. Int. J. Mol. Sci. 2022, 23, 12051. [Google Scholar] [CrossRef]
- Suresh, S.; Lee, J.; Noguchi, C.T. Erythropoietin signaling in osteoblasts is required for normal bone formation and for bone loss during erythropoietin-stimulated erythropoiesis. FASEB J. 2020, 34, 11685–11697. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Iga, T.; Kobayashi, H.; Kusumoto, D.; Sanosaka, T.; Fujita, N.; Tai-Nagara, I.; Ando, T.; Takahashi, T.; Matsuo, K.; Hozumi, K.; et al. Spatial heterogeneity of bone marrow endothelial cells unveils a distinct subtype in the epiphysis. Nat. Cell Biol. 2023, 25, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Comazzetto, S.; Shen, B.; Morrison, S.J. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev. Cell 2021, 56, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, S.; Li, Y.; Crane, J.L. Type H blood vessels in bone modeling and remodeling. Theranostics 2020, 10, 426–436. [Google Scholar] [CrossRef]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef]
- Mosteo, L.; Storer, J.; Batta, K.; Searle, E.J.; Duarte, D.; Wiseman, D.H. The Dynamic Interface Between the Bone Marrow Vascular Niche and Hematopoietic Stem Cells in Myeloid Malignancy. Front. Cell Dev. Biol. 2021, 9, 635189. [Google Scholar] [CrossRef]
- Langen, U.H.; Pitulescu, M.E.; Kim, J.M.; Enriquez-Gasca, R.; Sivaraj, K.K.; Kusumbe, A.P.; Singh, A.; Di Russo, J.; Bixel, M.G.; Zhou, B.; et al. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat. Cell Biol. 2017, 19, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Nombela-Arrieta, C.; Pivarnik, G.; Winkel, B.; Canty, K.J.; Harley, B.; Mahoney, J.E.; Park, S.Y.; Lu, J.; Protopopov, A.; Silberstein, L.E. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat. Cell Biol. 2013, 15, 533–543. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Hussong, J.W.; Rodgers, G.M.; Shami, P.J. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 2000, 95, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Padro, T.; Ruiz, S.; Bieker, R.; Burger, H.; Steins, M.; Kienast, J.; Buchner, T.; Berdel, W.E.; Mesters, R.M. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 2000, 95, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.J.; Stussi, G.; Maertens, J.; van Montfort, K.; Biemond, B.J.; Breems, D.; Ferrant, A.; Graux, C.; de Greef, G.E.; Halkes, C.J.; et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: A randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood 2012, 120, 4706–4711. [Google Scholar] [CrossRef] [PubMed]
- Zahiragic, L.; Schliemann, C.; Bieker, R.; Thoennissen, N.H.; Burow, K.; Kramer, C.; Zuhlsdorf, M.; Berdel, W.E.; Mesters, R.M. Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia 2007, 21, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e326. [Google Scholar] [CrossRef]
- Vijay, V.; Miller, R.; Vue, G.S.; Pezeshkian, M.B.; Maywood, M.; Ast, A.M.; Drusbosky, L.M.; Pompeu, Y.; Salgado, A.D.; Lipten, S.D.; et al. Interleukin-8 blockade prevents activated endothelial cell mediated proliferation and chemoresistance of acute myeloid leukemia. Leuk. Res. 2019, 84, 106180. [Google Scholar] [CrossRef]
- Hatfield, K.; Oyan, A.M.; Ersvaer, E.; Kalland, K.H.; Lassalle, P.; Gjertsen, B.T.; Bruserud, O. Primary human acute myeloid leukaemia cells increase the proliferation of microvascular endothelial cells through the release of soluble mediators. Br. J. Haematol. 2009, 144, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Ottersbach, K. Endothelial-to-haematopoietic transition: An update on the process of making blood. Biochem. Soc. Trans. 2019, 47, 591–601. [Google Scholar] [CrossRef]
- Padro, T.; Bieker, R.; Ruiz, S.; Steins, M.; Retzlaff, S.; Burger, H.; Buchner, T.; Kessler, T.; Herrera, F.; Kienast, J.; et al. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002, 16, 1302–1310. [Google Scholar] [CrossRef]
- Kampen, K.R.; Ter Elst, A.; de Bont, E.S. Vascular endothelial growth factor signaling in acute myeloid leukemia. Cell. Mol. Life Sci. 2013, 70, 1307–1317. [Google Scholar] [CrossRef]
- Pievani, A.; Biondi, M.; Tomasoni, C.; Biondi, A.; Serafini, M. Location First: Targeting Acute Myeloid Leukemia Within Its Niche. J. Clin. Med. 2020, 9, 1513. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.P.; Testut, C.; Fauriat, C.; Mancini, S.J.C.; Aurrand-Lions, M. Adhesion Molecules Involved in Stem Cell Niche Retention During Normal Haematopoiesis and in Acute Myeloid Leukaemia. Front. Immunol. 2021, 12, 756231. [Google Scholar] [CrossRef] [PubMed]
- Cogle, C.R.; Goldman, D.C.; Madlambayan, G.J.; Leon, R.P.; Masri, A.A.; Clark, H.A.; Asbaghi, S.A.; Tyner, J.W.; Dunlap, J.; Fan, G.; et al. Functional integration of acute myeloid leukemia into the vascular niche. Leukemia 2014, 28, 1978–1987. [Google Scholar] [CrossRef]
- Skinner, A.M.; Grompe, M.; Kurre, P. Intra-hematopoietic cell fusion as a source of somatic variation in the hematopoietic system. J. Cell Sci. 2012, 125, 2837–2843. [Google Scholar] [CrossRef]
- Xu, C.; Lu, T.; Lv, X.; Cheng, T.; Cheng, H. Role of the bone marrow vascular niche in chemotherapy for MLL-AF9-induced acute myeloid leukemia. Blood Sci. 2023, 5, 92–100. [Google Scholar] [CrossRef]
- Kellaway, S.G.; Potluri, S.; Keane, P.; Blair, H.J.; Ames, L.; Worker, A.; Chin, P.S.; Ptasinska, A.; Derevyanko, P.K.; Adamo, A.; et al. Leukemic stem cells activate lineage inappropriate signalling pathways to promote their growth. Nat. Commun. 2024, 15, 1359. [Google Scholar] [CrossRef]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Baccin, C.; Al-Sabah, J.; Velten, L.; Helbling, P.M.; Grunschlager, F.; Hernandez-Malmierca, P.; Nombela-Arrieta, C.; Steinmetz, L.M.; Trumpp, A.; Haas, S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 2020, 22, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, K.; Tarnowski, M. Bone Marrow Adipocytes-Role in Physiology and Various Nutritional Conditions in Human and Animal Models. Nutrients 2021, 13, 1412. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Doucette, C.R.; Learman, B.S.; Cawthorn, W.P.; Khandaker, S.; Schell, B.; Wu, B.; Ding, S.Y.; Bredella, M.A.; Fazeli, P.K.; et al. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat. Commun. 2015, 6, 7808. [Google Scholar] [CrossRef] [PubMed]
- Miggitsch, C.; Meryk, A.; Naismith, E.; Pangrazzi, L.; Ejaz, A.; Jenewein, B.; Wagner, S.; Nagele, F.; Fenkart, G.; Trieb, K.; et al. Human bone marrow adipocytes display distinct immune regulatory properties. EBioMedicine 2019, 46, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Haren, N.; Ghali, O.; Clabaut, A.; Chauveau, C.; Hardouin, P.; Broux, O. Adipogenic RNAs are transferred in osteoblasts via bone marrow adipocytes-derived extracellular vesicles (EVs). BMC Cell Biol. 2015, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef]
- Labella, R.; Vujacic, M.; Trivanovic, D. Bone Marrow Adipose Tissue: Regulation of Osteoblastic Niche, Hematopoiesis and Hematological Malignancies. Stem Cell Rev. Rep. 2023, 19, 1135–1151. [Google Scholar] [CrossRef] [PubMed]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Fu, H.; Schiffrin, M.; Winkler, C.; Koufany, M.; Jouzeau, J.Y.; Bonnet, N.; Gilardi, F.; Renevey, F.; Luther, S.A.; et al. Lack of Adipocytes Alters Hematopoiesis in Lipodystrophic Mice. Front. Immunol. 2018, 9, 2573. [Google Scholar] [CrossRef]
- Lu, W.; Weng, W.; Zhu, Q.; Zhai, Y.; Wan, Y.; Liu, H.; Yang, S.; Yu, Y.; Wei, Y.; Shi, J. Small bone marrow adipocytes predict poor prognosis in acute myeloid leukemia. Haematologica 2018, 103, e21–e24. [Google Scholar] [CrossRef]
- Iversen, P.O.; Wiig, H. Tumor necrosis factor alpha and adiponectin in bone marrow interstitial fluid from patients with acute myeloid leukemia inhibit normal hematopoiesis. Clin. Cancer Res. 2005, 11, 6793–6799. [Google Scholar] [CrossRef] [PubMed]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Maryanovich, M.; Arnal-Estape, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gil, Z. The Role of Extracellular Vesicles in Cancer-Nerve Crosstalk of the Peripheral Nervous System. Cells 2022, 11, 1294. [Google Scholar] [CrossRef]
- Hunt, P.J.; Amit, M. Head and neck cancer exosomes drive microRNA-mediated reprogramming of local neurons. Extracell. Vesicles Circ. Nucleic Acid 2020, 1, 57–62. [Google Scholar] [CrossRef]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, P.D. Exosomal Induction of Tumor Innervation. Cancer Res. 2019, 79, 3529–3535. [Google Scholar] [CrossRef] [PubMed]
- Egyed, B.; Kutszegi, N.; Sagi, J.C.; Gezsi, A.; Rzepiel, A.; Visnovitz, T.; Lorincz, P.; Muller, J.; Zombori, M.; Szalai, C.; et al. MicroRNA-181a as novel liquid biopsy marker of central nervous system involvement in pediatric acute lymphoblastic leukemia. J. Transl. Med. 2020, 18, 250. [Google Scholar] [CrossRef]
- Mason, A.J.; Keeler, A.B.; Kabir, F.; Winckler, B.; Deppmann, C. Sympathetic neurons secrete retrogradely transported TrkA on extracellular vesicles. Sci. Rep. 2023, 13, 3657. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, J.; Buettner, H.; Seedhom, M.O.; Greiner, D.L.; Keck, J.G.; Ishikawa, F.; Shultz, L.D.; Brehm, M.A. Humanized mouse models for immuno-oncology research. Nat. Rev. Clin. Oncol. 2023, 20, 192–206. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2021, 20, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Glaser, D.E.; Curtis, M.B.; Sariano, P.A.; Rollins, Z.A.; Shergill, B.S.; Anand, A.; Deely, A.M.; Shirure, V.S.; Anderson, L.; Lowen, J.M.; et al. Organ-on-a-chip model of vascularized human bone marrow niches. Biomaterials 2022, 280, 121245. [Google Scholar] [CrossRef]
- Singh, D.; Mathur, A.; Arora, S.; Roy, S.; Mahindroo, N. Journey of organ on a chip technology and its role in future healthcare scenario. Appl. Surf. Sci. Adv. 2022, 9, 100246. [Google Scholar] [CrossRef]
- Santos Rosalem, G.; Gonzales Torres, L.A.; de Las Casas, E.B.; Mathias, F.A.S.; Ruiz, J.C.; Carvalho, M.G.R. Microfluidics and organ-on-a-chip technologies: A systematic review of the methods used to mimic bone marrow. PLoS ONE 2020, 15, e0243840. [Google Scholar] [CrossRef]
- Carrion, B.; Huang, C.P.; Ghajar, C.M.; Kachgal, S.; Kniazeva, E.; Jeon, N.L.; Putnam, A.J. Recreating the perivascular niche ex vivo using a microfluidic approach. Biotechnol. Bioeng. 2010, 107, 1020–1028. [Google Scholar] [CrossRef]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.P.; Lippens, E.; Duda, G.N.; et al. Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 2016, 15, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Torisawa, Y.S.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingber, D.E. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 2014, 11, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.H.; Khoneisser, J.; Huang, P.C.; Xu, X. Hydrogel as a bioactive material to regulate stem cell fate. Bioact. Mater. 2016, 1, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci. Adv. 2020, 6, eaba5536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.N.; Zhang, Q.; Jeppesen, D.K.; Scott, A.M.; Manning, H.C.; Ochieng, J.; Franklin, J.L.; Coffey, R.J. Identification and characterization of EGF receptor in individual exosomes by fluorescence-activated vesicle sorting. J. Extracell. Vesicles 2016, 5, 29254. [Google Scholar] [CrossRef]
- Lee, K.; Shao, H.; Weissleder, R.; Lee, H. Acoustic purification of extracellular microvesicles. ACS Nano 2015, 9, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Rupert, D.L.; Lasser, C.; Eldh, M.; Block, S.; Zhdanov, V.P.; Lotvall, J.O.; Bally, M.; Hook, F. Determination of exosome concentration in solution using surface plasmon resonance spectroscopy. Anal. Chem. 2014, 86, 5929–5936. [Google Scholar] [CrossRef]
- Gualerzi, A.; Niada, S.; Giannasi, C.; Picciolini, S.; Morasso, C.; Vanna, R.; Rossella, V.; Masserini, M.; Bedoni, M.; Ciceri, F.; et al. Raman spectroscopy uncovers biochemical tissue-related features of extracellular vesicles from mesenchymal stromal cells. Sci. Rep. 2017, 7, 9820. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef]
- Jorgensen, M.; Baek, R.; Pedersen, S.; Sondergaard, E.K.; Kristensen, S.R.; Varming, K. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. J. Extracell. Vesicles 2013, 2, 20920. [Google Scholar] [CrossRef]
- Lee, K.; Fraser, K.; Ghaddar, B.; Yang, K.; Kim, E.; Balaj, L.; Chiocca, E.A.; Breakefield, X.O.; Lee, H.; Weissleder, R. Multiplexed Profiling of Single Extracellular Vesicles. ACS Nano 2018, 12, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, M.; van de Nes, A.; Nieuwland, R.; Varga, Z.; van der Pol, E. Reliable measurements of extracellular vesicles by clinical flow cytometry. Am. J. Reprod. Immunol. 2021, 85, e13350. [Google Scholar] [CrossRef]
- McNamara, R.P.; Zhou, Y.; Eason, A.B.; Landis, J.T.; Chambers, M.G.; Willcox, S.; Peterson, T.A.; Schouest, B.; Maness, N.J.; MacLean, A.G.; et al. Imaging of surface microdomains on individual extracellular vesicles in 3-D. J. Extracell. Vesicles 2022, 11, e12191. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, M.; Monteiro, A.C.; Neto, E.; Barrias, C.C.; Sobrinho-Simões, M.A.; Duarte, D.; Caires, H.R. Transforming the Niche: The Emerging Role of Extracellular Vesicles in Acute Myeloid Leukaemia Progression. Int. J. Mol. Sci. 2024, 25, 4430. https://doi.org/10.3390/ijms25084430
Mendes M, Monteiro AC, Neto E, Barrias CC, Sobrinho-Simões MA, Duarte D, Caires HR. Transforming the Niche: The Emerging Role of Extracellular Vesicles in Acute Myeloid Leukaemia Progression. International Journal of Molecular Sciences. 2024; 25(8):4430. https://doi.org/10.3390/ijms25084430
Chicago/Turabian StyleMendes, Manuel, Ana C. Monteiro, Estrela Neto, Cristina C. Barrias, Manuel A. Sobrinho-Simões, Delfim Duarte, and Hugo R. Caires. 2024. "Transforming the Niche: The Emerging Role of Extracellular Vesicles in Acute Myeloid Leukaemia Progression" International Journal of Molecular Sciences 25, no. 8: 4430. https://doi.org/10.3390/ijms25084430
APA StyleMendes, M., Monteiro, A. C., Neto, E., Barrias, C. C., Sobrinho-Simões, M. A., Duarte, D., & Caires, H. R. (2024). Transforming the Niche: The Emerging Role of Extracellular Vesicles in Acute Myeloid Leukaemia Progression. International Journal of Molecular Sciences, 25(8), 4430. https://doi.org/10.3390/ijms25084430