
Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
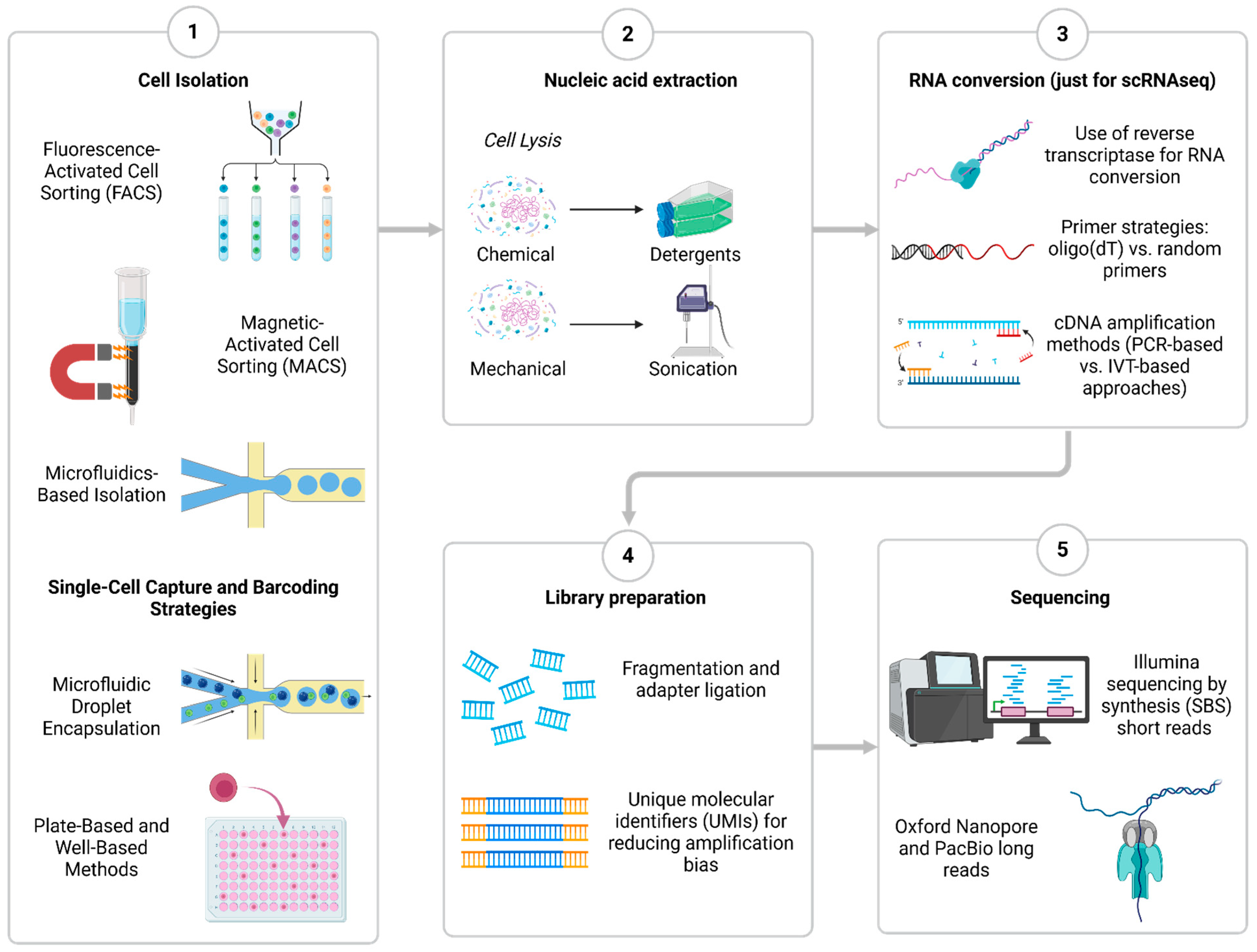
2. Overview of Core Chemistry and Methods of Single-Cell Sequencing
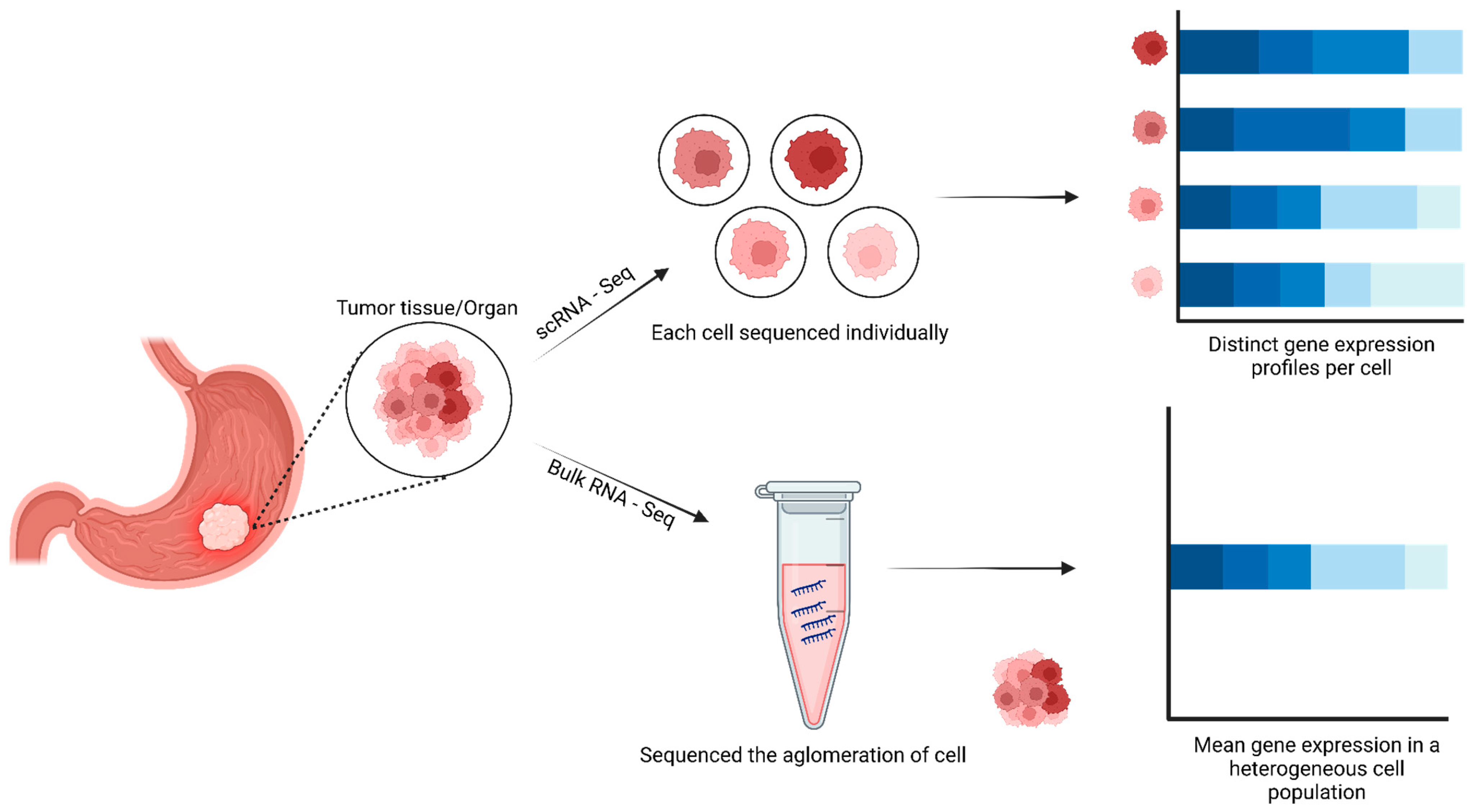
3. Cancer Research
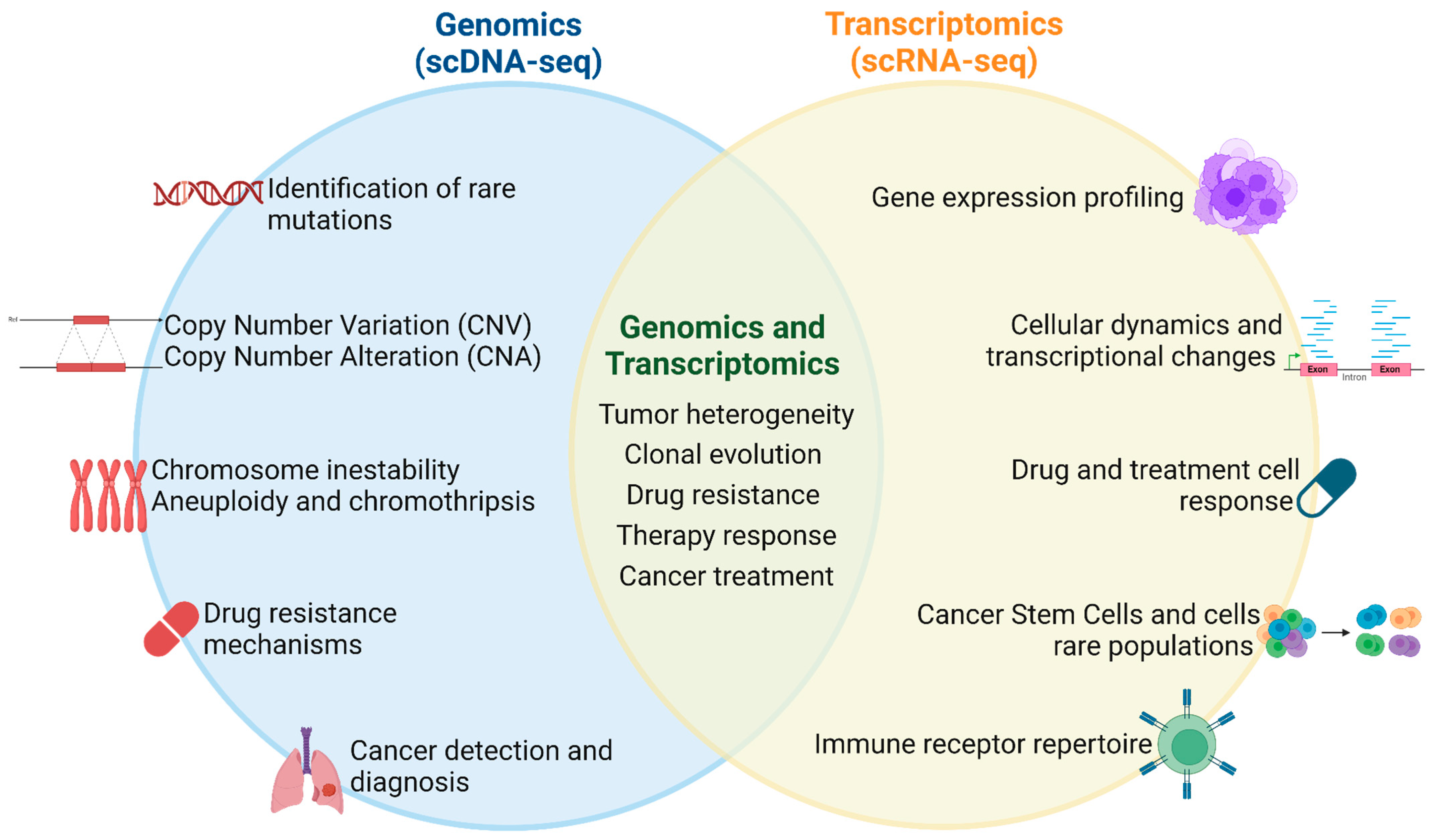
4. Applications of Single-Cell Sequencing for Genomic Profiling in Human Cancer Cells
- (a)
- Tumor Heterogeneity and Clonal Evolution
- (b)
- Identification of Rare Mutations
- (c)
- Drug Resistance and Mechanisms
- (d)
- Detection and Diagnosis of the Presence of Cancer
5. Applications of Single-Cell Sequencing for Transcriptomic Profiling in Human Cancer Cells
- (a)
- Identifying cancer stem cells and rare cell populations
- (b)
- Determining heterogeneity within a cell population
- (c)
- Tumor immunology
- (d)
- Cancer progression, drug development, and cancer treatment
6. ‘Co-Presence’ and ‘Phenotypic Association’ Capability of scSeq Technology
7. Immune Cell Response in Tumor Microenvironment Using SCS
- (a)
- Tumor microenvironment
- (b)
- Cellular Components of Tumor Microenvironment
- (c)
- Overview of tumor microenvironment at single-cell resolution
8. Role of Single-Cell Data Analysis Technologies in Cancer Therapy
- (a)
- Preprocessing and Integration
- (b)
- Clustering and downstream analysis
- (c)
- Single-Cell DNA and Whole-Genome Sequencing (scWGS)
9. Emerging Technologies and Future Directions in scWGS in Cancer Biology
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- Suvà, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef]
- Han, Y.; Wang, D.; Peng, L.; Huang, T.; He, X.; Wang, J.; Ou, C. Single-Cell Sequencing: A Promising Approach for Uncovering the Mechanisms of Tumor Metastasis. J. Hematol. Oncol. 2022, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shen, F.; Yang, X.; Han, T.; Long, Z.; Wen, J.; Huang, J.; Shen, J.; Guo, Q. Single-Cell Sequencing Technology Applied to Epigenetics for the Study of Tumor Heterogeneity. Clin. Epigenetics 2023, 15, 161. [Google Scholar] [CrossRef]
- Loo, J.F.-C.; Ho, H.P.; Kong, S.K.; Wang, T.-H.; Ho, Y.-P. Technological Advances in Multiscale Analysis of Single Cells in Biomedicine. Adv. Biosyst. 2019, 3, e1900138. [Google Scholar] [CrossRef] [PubMed]
- Rognoni, E.; Watt, F.M. Skin Cell Heterogeneity in Development, Wound Healing, and Cancer. Trends Cell Biol. 2018, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Zhang, M.; Chen, L. The Comparison of Two Single-Cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium. Curr. Genomics 2020, 21, 602–609. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S.A. Exponential Scaling of Single-Cell RNA-Seq in the Past Decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Schmitz, U. Single-Cell and Long-Read Sequencing to Enhance Modelling of Splicing and Cell-Fate Determination. Comput. Struct. Biotechnol. J. 2023, 21, 2373–2380. [Google Scholar] [CrossRef]
- Evrony, G.D.; Hinch, A.G.; Luo, C. Applications of Single-Cell DNA Sequencing. Annu. Rev. Genomics Hum. Genet. 2021, 22, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-Seq: Parallel Sequencing of Single-Cell Genomes and Transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar]
- Demaree, B.; Delley, C.L.; Vasudevan, H.N.; Peretz, C.A.C.; Ruff, D.; Smith, C.C.; Abate, A.R. Joint Profiling of DNA and Proteins in Single Cells to Dissect Genotype-Phenotype Associations in Leukemia. Nat. Commun. 2020, 12, 1583. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible Copy Number Variation Patterns among Single Circulating Tumor Cells of Lung Cancer Patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef] [PubMed]
- Dago, A.E.; Stepansky, A.; Carlsson, A.; Luttgen, M.; Kendall, J.; Baslan, T.; Kolatkar, A.; Wigler, M.; Bethel, K.; Gross, M.E.; et al. Rapid Phenotypic and Genomic Change in Response to Therapeutic Pressure in Prostate Cancer Inferred by High Content Analysis of Single Circulating Tumor Cells. PLoS ONE 2014, 9, e101777. [Google Scholar] [CrossRef]
- Lohr, J.G.; Adalsteinsson, V.A.; Cibulskis, K.; Choudhury, A.D.; Rosenberg, M.; Cruz-Gordillo, P.; Francis, J.M.; Zhang, C.-Z.; Shalek, A.K.; Satija, R.; et al. Whole-Exome Sequencing of Circulating Tumor Cells Provides a Window into Metastatic Prostate Cancer. Nat. Biotechnol. 2014, 32, 479–484. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Chen, C.; Zhang, J.; Xu, L.; Zhang, X.; Li, Z.; Chen, Y.; Zhou, J.; Ji, F.; Wang, L. Detection of Circulating Tumor Cells: Opportunities and Challenges. Biomark. Res. 2022, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular Profiling of Single Circulating Tumor Cells with Diagnostic Intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, A.; Lai, S.; Guo, G.; Yuan, G.-C. Single-Cell Analysis in Cancer Genomics. Trends Genet. 2015, 31, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian Approach to Single-Cell Differential Expression Analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef]
- Shekhar, K.; Brodin, P.; Davis, M.M.; Chakraborty, A.K. Automatic Classification of Cellular Expression by Nonlinear Stochastic Embedding (ACCENSE). Proc. Natl. Acad. Sci. USA 2014, 111, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef]
- Wills, Q.F.; Mead, A.J. Application of Single-Cell Genomics in Cancer: Promise and Challenges. Hum. Mol. Genet. 2015, 24, R74–R84. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, W.; Du, Y.; Yang, M.; Pan, Y.; Li, H.; Cui, M. Single-Cell Analysis Technologies for Cancer Research: From Tumor-Specific Single Cell Discovery to Cancer Therapy. Front. Genet. 2023, 14, 1276959. [Google Scholar] [CrossRef]
- Thiele, J.-A.; Pitule, P.; Hicks, J.; Kuhn, P. Single-Cell Analysis of Circulating Tumor Cells. Methods Mol. Biol. 2019, 1908, 243–264. [Google Scholar] [PubMed]
- Reza, K.K.; Dey, S.; Wuethrich, A.; Wang, J.; Behren, A.; Antaw, F.; Wang, Y.; Sina, A.A.I.; Trau, M. In Situ Single Cell Proteomics Reveals Circulating Tumor Cell Heterogeneity during Treatment. ACS Nano 2021, 15, 11231–11243. [Google Scholar] [CrossRef] [PubMed]
- Chew, V.; Toh, H.C.; Abastado, J.-P. Immune Microenvironment in Tumor Progression: Characteristics and Challenges for Therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Cui, J. Development of Immunotherapy Strategies Targeting Tumor Microenvironment Is Fiercely Ongoing. Front. Immunol. 2022, 13, 890166. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Eum, H.H.; Lee, H.-O. Clinical Perspectives of Single-Cell RNA Sequencing. Biomolecules 2021, 11, 1161. [Google Scholar] [CrossRef]
- Rajan, S.; Zaccaria, S.; Cannon, M.V.; Cam, M.; Gross, A.C.; Raphael, B.J.; Roberts, R.D. Structurally Complex Osteosarcoma Genomes Exhibit Limited Heterogeneity within Individual Tumors and across Evolutionary Time. Cancer Res. Commun. 2023, 3, 564–575. [Google Scholar] [CrossRef]
- Meyers, S.; Alberti-Servera, L.; Gielen, O.; Erard, M.; Swings, T.; De Bie, J.; Michaux, L.; Dewaele, B.; Boeckx, N.; Uyttebroeck, A.; et al. Monitoring of Leukemia Clones in B-Cell Acute Lymphoblastic Leukemia at Diagnosis and During Treatment by Single-Cell DNA Amplicon Sequencing. Hemasphere 2022, 6, e700. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Durruthy-Durruthy, R.; Eastburn, D.J.; Pellegrino, M.; Shah, O.; Meyer, E.; Zehnder, J. Clonal Evolution and Changes in Two AML Patients Detected with A Novel Single-Cell DNA Sequencing Platform. Sci. Rep. 2019, 9, 11119. [Google Scholar] [CrossRef] [PubMed]
- Borgsmüller, N.; Bonet, J.; Marass, F.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Beerenwinkel, N. BnpC: Bayesian Non-Parametric Clustering of Single-Cell Mutation Profiles. Bioinformatics 2020, 36, 4854–4859. [Google Scholar] [CrossRef]
- Huang, A.Y.; Lee, E.A. Identification of Somatic Mutations From Bulk and Single-Cell Sequencing Data. Front Aging 2021, 2, 800380. [Google Scholar] [CrossRef]
- Tang, J.; Tu, K.; Lu, K.; Zhang, J.; Luo, K.; Jin, H.; Wang, L.; Yang, L.; Xiao, W.; Zhang, Q.; et al. Single-Cell Exome Sequencing Reveals Multiple Subclones in Metastatic Colorectal Carcinoma. Genome Med. 2021, 13, 148. [Google Scholar] [CrossRef]
- Jaberi, E.; Tresse, E.; Grønbæk, K.; Weischenfeldt, J.; Issazadeh-Navikas, S. Identification of Unique and Shared Mitochondrial DNA Mutations in Neurodegeneration and Cancer by Single-Cell Mitochondrial DNA Structural Variation Sequencing (MitoSV-Seq). EBioMedicine 2020, 57, 102868. [Google Scholar] [CrossRef]
- Gráf, A.; Enyedi, M.Z.; Pintér, L.; Kriston-Pál, É.; Jaksa, G.; Bálind, Á.; Ezer, É.; Horváth, P.; Sükösd, F.; Kiss, E.; et al. The Combination of Single-Cell and Next-Generation Sequencing Can Reveal Mosaicism for BRCA2 Mutations and the Fine Molecular Details of Tumorigenesis. Cancers 2021, 13, 2354. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Q.; Wang, C.; Guo, H.; Mukwaya, V.; Chen, R.; Xu, Y.; Wei, X.; Chen, X.; Zhang, S.; et al. Single-Cell Diagnosis of Cancer Drug Resistance through the Differential Endocytosis of Nanoparticles between Drug-Resistant and Drug-Sensitive Cancer Cells. ACS Nano 2023, 17, 19372–19386. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Ding, J.; Ge, Y.; Fan, J.; Fan, S.-K. Single-Cell-Derived Tumor-Sphere Formation and Drug-Resistance Assay Using an Integrated Microfluidics. Anal. Chem. 2019, 91, 8318–8325. [Google Scholar] [CrossRef]
- Lee, P.; Yim, R.; Fung, S.-H.; Miu, K.-K.; Wang, Z.; Wu, K.-C.; Au, L.; Leung, G.M.-K.; Lee, V.H.-F.; Gill, H. Single-Nucleotide Variations, Insertions/Deletions and Copy Number Variations in Myelodysplastic Syndrome during Disease Progression Revealed by a Single-Cell DNA Sequencing Platform. Int. J. Mol. Sci. 2022, 23, 4647. [Google Scholar] [CrossRef]
- Peretz, C.A.C.; McGary, L.H.F.; Kumar, T.; Jackson, H.; Jacob, J.; Durruthy-Durruthy, R.; Levis, M.J.; Perl, A.; Huang, B.J.; Smith, C.C. Single-Cell DNA Sequencing Reveals Complex Mechanisms of Resistance to Quizartinib. Blood Adv. 2021, 5, 1437–1441. [Google Scholar] [CrossRef]
- Grant, C.R.; Benjamin, D.J.; Cramer, S.; Rezazadeh Kalebasty, A. A Remnant Never Forgotten: The Utility of Circulating Tumor DNA in Treatment Guidance of Urachal Cancer. Ther. Adv. Med. Oncol. 2024, 16, 17588359241230743. [Google Scholar] [CrossRef]
- Shen, X.; Dai, J.; Guo, L.; Liu, Z.; Yang, L.; Gu, D.; Xie, Y.; Wang, Z.; Li, Z.; Xu, H.; et al. Single-Cell Low-Pass Whole Genome Sequencing Accurately Detects Circulating Tumor Cells for Liquid Biopsy-Based Multi-Cancer Diagnosis. NPJ Precis Oncol. 2024, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y.; Shen, X.; Zhao, Y.; Zhang, Z.; Yin, H.; Zhao, X.; Liu, H.; Shi, Q. Single-Cell Genomics-Based Molecular Algorithm for Early Cancer Detection. Anal. Chem. 2022, 94, 2607–2614. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, G.; Yan, Y.; Peng, J. A Pan-Cancer Analysis of Copper Homeostasis-Related Gene Lipoyltransferase 1: Its Potential Biological Functions and Prognosis Values. Front. Genet. 2022, 13, 1038174. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, Y.; Zhu, J.; Ge, X.; Wu, J.; Xu, P.; Yao, J. Advancement of Single-Cell Sequencing for Clinical Diagnosis and Treatment of Pancreatic Cancer. Front. Med. 2023, 10, 1213136. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Y.; Zhang, H.; Zhang, N.; Dai, Z.; Cheng, Q.; Li, Y. Single-Cell Sequencing: High-Resolution Analysis of Cellular Heterogeneity in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2024, 66, 376–400. [Google Scholar] [CrossRef]
- Yuan, P.; Yan, L.-Y.; Qiao, J. Application of Single-Cell Sequencing Technologies in Reproductive Medicine. Reprod. Dev. Med. 2017, 1, 30–35. [Google Scholar] [CrossRef]
- Yadav, S.; Mehta, P.; Soni, J.; Chattopadhyay, P.; Devi, P.; Habyarimana, T.; Tardalkar, K.; Joshi, M.; Pandey, R. Single-Cell RNA-Seq Reveals Intracellular Microbial Diversity within Immune Cells during SARS-CoV-2 Infection and Recovery. iScience 2023, 26, 108357. [Google Scholar] [CrossRef] [PubMed]
- Kuksin, M.; Morel, D.; Aglave, M.; Danlos, F.-X.; Marabelle, A.; Zinovyev, A.; Gautheret, D.; Verlingue, L. Applications of Single-Cell and Bulk RNA Sequencing in Onco-Immunology. Eur. J. Cancer 2021, 149, 193–210. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhou, C.; Lu, Y.; Ma, F.; Fan, Y.; Wang, C. Single-Cell RNA-Seq Reveals Invasive Trajectory and Determines Cancer Stem Cell-Related Prognostic Genes in Pancreatic Cancer. Bioengineered 2021, 12, 5056–5068. [Google Scholar] [CrossRef]
- Boesch, M.; Sopper, S.; Zeimet, A.G.; Reimer, D.; Gastl, G.; Ludewig, B.; Wolf, D. Heterogeneity of Cancer Stem Cells: Rationale for Targeting the Stem Cell Niche. Biochim. Biophys. Acta 2016, 1866, 276–289. [Google Scholar] [CrossRef]
- Pan, X.-W.; Zhang, H.; Xu, D.; Chen, J.-X.; Chen, W.-J.; Gan, S.-S.; Qu, F.-J.; Chu, C.-M.; Cao, J.-W.; Fan, Y.-H.; et al. Identification of a Novel Cancer Stem Cell Subpopulation That Promotes Progression of Human Fatal Renal Cell Carcinoma by Single-Cell RNA-Seq Analysis. Int. J. Biol. Sci. 2020, 16, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Segerstolpe, Å.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Rosas, P.C.; Nagaraja, G.M.; Kaur, P.; Panossian, A.; Wickman, G.; Garcia, L.R.; Al-Khamis, F.A.; Asea, A.A.A. Hsp72 (HSPA1A) Prevents Human Islet Amyloid Polypeptide Aggregation and Toxicity: A New Approach for Type 2 Diabetes Treatment. PLoS ONE 2016, 11, e0149409. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef]
- Lenz, G.; Onzi, G.R.; Lenz, L.S.; Buss, J.H.; Dos Santos, J.A.; Begnini, K.R. The Origins of Phenotypic Heterogeneity in Cancer. Cancer Res. 2022, 82, 3–11. [Google Scholar] [CrossRef]
- Wen, L.; Li, G.; Huang, T.; Geng, W.; Pei, H.; Yang, J.; Zhu, M.; Zhang, P.; Hou, R.; Tian, G.; et al. Single-Cell Technologies: From Research to Application. Innovation (Camb) 2022, 3, 100342. [Google Scholar] [CrossRef]
- Chen, H.; Ye, F.; Guo, G. Revolutionizing Immunology with Single-Cell RNA Sequencing. Cell. Mol. Immunol. 2019, 16, 242–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA Sequencing in Cancer Research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; Bravo González-Blas, C.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust Gene Expression Programs Underlie Recurrent Cell States and Phenotype Switching in Melanoma. Nat. Cell Biol. 2020, 22, 986–998. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Hui, Z.; Ren, Y.; Zhang, D.; Chen, Y.; Yu, W.; Cao, J.; Liu, L.; Wang, T.; Xiao, S.; Zheng, L.; et al. PD-1 Blockade Potentiates Neoadjuvant Chemotherapy in NSCLC via Increasing CD127+ and KLRG1+ CD8 T Cells. NPJ Precis. Oncol. 2023, 7, 48. [Google Scholar] [CrossRef]
- Rajewsky, N.; Almouzni, G.; Gorski, S.A.; Aerts, S.; Amit, I.; Bertero, M.G.; Bock, C.; Bredenoord, A.L.; Cavalli, G.; Chiocca, S.; et al. LifeTime and Improving European Healthcare through Cell-Based Interceptive Medicine. Nature 2020, 587, 377–386. [Google Scholar] [CrossRef]
- Stein, C.M.; Weiskirchen, R.; Damm, F.; Strzelecka, P.M. Single-Cell Omics: Overview, Analysis, and Application in Biomedical Science. J. Cell. Biochem. 2021, 122, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef] [PubMed]
- Van de Sande, B.; Lee, J.S.; Mutasa-Gottgens, E.; Naughton, B.; Bacon, W.; Manning, J.; Wang, Y.; Pollard, J.; Mendez, M.; Hill, J.; et al. Applications of Single-Cell RNA Sequencing in Drug Discovery and Development. Nat. Rev. Drug Discov. 2023, 22, 496–520. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Li, Y.; Mitra, A.K.; Bi, L.; Abyzov, A.; van Wijnen, A.J.; Baughn, L.B.; Van Ness, B.; Rajkumar, V.; Kumar, S.; et al. Molecular Signatures of Multiple Myeloma Progression through Single Cell RNA-Seq. Blood Cancer J. 2019, 9, 2. [Google Scholar] [CrossRef]
- Tanaka, N.; Katayama, S.; Reddy, A.; Nishimura, K.; Niwa, N.; Hongo, H.; Ogihara, K.; Kosaka, T.; Mizuno, R.; Kikuchi, E.; et al. Single-Cell RNA-Seq Analysis Reveals the Platinum Resistance Gene COX7B and the Surrogate Marker CD63. Cancer Med. 2018, 7, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Zada, M.; Wang, S.-Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of Resistance Pathways and Therapeutic Targets in Relapsed Multiple Myeloma Patients through Single-Cell Sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and Applications for Single-Cell and Spatial Multi-Omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef]
- Bekaert, B.; Boel, A.; Cosemans, G.; De Witte, L.; Menten, B.; Heindryckx, B. CRISPR/Cas Gene Editing in the Human Germline. Semin. Cell Dev. Biol. 2022, 131, 93–107. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Alajez, N.M. Single-Cell Transcriptome Analysis Revealed Heterogeneity and Identified Novel Therapeutic Targets for Breast Cancer Subtypes. Cells 2023, 12, 1182. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR-Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e19. [Google Scholar] [CrossRef] [PubMed]
- Petti, A.A.; Williams, S.R.; Miller, C.A.; Fiddes, I.T.; Srivatsan, S.N.; Chen, D.Y.; Fronick, C.C.; Fulton, R.S.; Church, D.M.; Ley, T.J. A General Approach for Detecting Expressed Mutations in AML Cells Using Single Cell RNA-Sequencing. Nat. Commun. 2019, 10, 3660. [Google Scholar] [CrossRef]
- Xu, X.; Hou, Y.; Yin, X.; Bao, L.; Tang, A.; Song, L.; Li, F.; Tsang, S.; Wu, K.; Wu, H.; et al. Single-Cell Exome Sequencing Reveals Single-Nucleotide Mutation Characteristics of a Kidney Tumor. Cell 2012, 148, 886–895. [Google Scholar] [CrossRef]
- Gao, Y.; Ni, X.; Guo, H.; Su, Z.; Ba, Y.; Tong, Z.; Guo, Z.; Yao, X.; Chen, X.; Yin, J.; et al. Single-Cell Sequencing Deciphers a Convergent Evolution of Copy Number Alterations from Primary to Circulating Tumor Cells. Genome Res. 2017, 27, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Lau, B.T.; Kim, H.S.; Sathe, A.; Grimes, S.M.; Ji, H.P.; Zhang, N.R. Integrative Single-Cell Analysis of Allele-Specific Copy Number Alterations and Chromatin Accessibility in Cancer. Nat. Biotechnol. 2021, 39, 1259–1269. [Google Scholar] [CrossRef]
- Aganezov, S.; Goodwin, S.; Sherman, R.M.; Sedlazeck, F.J.; Arun, G.; Bhatia, S.; Lee, I.; Kirsche, M.; Wappel, R.; Kramer, M.; et al. Comprehensive Analysis of Structural Variants in Breast Cancer Genomes Using Single-Molecule Sequencing. Genome Res. 2020, 30, 1258–1273. [Google Scholar] [CrossRef]
- Funnell, T.; O’Flanagan, C.H.; Williams, M.J.; McPherson, A.; McKinney, S.; Kabeer, F.; Lee, H.; Salehi, S.; Vázquez-García, I.; Shi, H.; et al. Single-Cell Genomic Variation Induced by Mutational Processes in Cancer. Nature 2022, 612, 106–115. [Google Scholar] [CrossRef]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-Cell and Single-Molecule Epigenomics to Uncover Genome Regulation at Unprecedented Resolution. Nat. Genet. 2019, 51, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Schuster, L.C. Dissecting Heterogeneity, Clonal Evolution, and Epigenetic Changes in Distinct and Molecularly Defined AML Subsets by Multi Omics Single-Cell Sequencing. Heidelberg. 2022. Available online: https://archiv.ub.uni-heidelberg.de/ (accessed on 30 October 2024).
- Misra, P.; Jadhav, A.R.; Bapat, S.A. Single-Cell Sequencing: A Cutting Edge Tool in Molecular Medical Research. Armed Forces Med. J. India 2022, 78, S7–S13. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Dang, N.; Tang, G.; Li, Z.; Li, X.; Shi, B.; Xu, Z.; Li, L.; Yang, X.; Xu, C.; et al. Integrating Bulk and Single-Cell RNA Sequencing Reveals Cellular Heterogeneity and Immune Infiltration in Hepatocellular Carcinoma. Mol. Oncol. 2022, 16, 2195–2213. [Google Scholar] [CrossRef] [PubMed]
- Forcato, M.; Romano, O.; Bicciato, S. Computational Methods for the Integrative Analysis of Single-Cell Data. Brief. Bioinform. 2021, 22, 20–29. [Google Scholar] [CrossRef]
- Huang, H.; Wu, F.; Yu, Y.; Xu, B.; Chen, D.; Huo, Y.; Li, S. Multi-Transcriptomics Analysis of Microvascular Invasion-Related Malignant Cells and Development of a Machine Learning-Based Prognostic Model in Hepatocellular Carcinoma. Front. Immunol. 2024, 15, 1436131. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M. The Precision Oncology Approach to Molecular Cancer Therapeutics Targeting Oncogenic Signaling Pathways Is a Means to an End. arXiv 2023, arXiv:2304.05411. [Google Scholar] [CrossRef]
- Sierant, M.C.; Choi, J. Single-Cell Ssequencing in Cancer: Recent Applications to Immunogenomics and Multi-Omics Tools. Genomics Inform. 2018, 16, e17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; Campbell, K.R. Computational Modelling in Single-Cell Cancer Genomics: Methods and Future Directions. Phys. Biol. 2020, 17, 061001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lei, L.; Yang, X.; Ma, K.; Zheng, H.; Su, Y.; Jiao, A.; Wang, X.; Liu, H.; Zou, Y.; et al. Single-Cell Sequencing Reveals Antitumor Characteristics of Intratumoral Immune Cells in Old Mice. J. Immunother. Cancer 2021, 9, e002809. [Google Scholar] [CrossRef]
- Chen, Y.-Z.; Meng, Z.-S.; Xiang, Z.-L. HMGB2 Drives Tumor Progression and Shapes the Immunosuppressive Microenvironment in Hepatocellular Carcinoma: Insights from Multi-Omics Analysis. Front. Immunol. 2024, 15, 1415435. [Google Scholar] [CrossRef]
- Wolfe, C.; Feng, Y.; Chen, D.; Purcell, E.; Talkington, A.; Dolatshahi, S.; Shakeri, H. GeoTyper: Automated Pipeline from Raw scRNA-Seq Data to Cell Type Identification. In Proceedings of the 2022 Systems and Information Engineering Design Symposium (SIEDS), Charlottesville, VI, USA, 28–29 April 2022. [Google Scholar]
- Wang, S.; Sun, S.-T.; Zhang, X.-Y.; Ding, H.-R.; Yuan, Y.; He, J.-J.; Wang, M.-S.; Yang, B.; Li, Y.-B. The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives. Int. J. Mol. Sci. 2023, 24, 2943. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.; Greenshields-Watson, A.; Jones, E.; Smart, K.; Lauder, S.N.; Somerville, M.; Milutinovic, S.; Kendrick, H.; Hindley, J.P.; French, R.; et al. Immune Remodeling of the Extracellular Matrix Drives Loss of Cancer Stem Cells and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Alcantara, M.B.; Tang, W.S.; Wang, D.; Kaniowski, D.; Kang, E.; Dizman, N.; Chehrazi-Raffle, A.; Meza, L.; Zengin, Z.; Hall, J.; et al. Targeting STAT3 in Tumor-Associated Antigen-Presenting Cells as a Strategy for Kidney and Bladder Cancer Immunotherapy. Front. Immunol. 2023, 14, 1274781. [Google Scholar] [CrossRef]
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of Immune Cells and Cytokines on Inflammation and Immunosuppression in the Tumor Microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ramnarayanan, K.; Sundar, R.; Padmanabhan, N.; Srivastava, S.; Koiwa, M.; Yasuda, T.; Koh, V.; Huang, K.K.; Tay, S.T.; et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. 2022, 12, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Nofech-Mozes, I.; Soave, D.; Awadalla, P.; Abelson, S. Pan-Cancer Classification of Single Cells in the Tumour Microenvironment. Nat. Commun. 2023, 14, 1615. [Google Scholar] [CrossRef]
- Lee, J.J.; Bernard, V.; Semaan, A.; Monberg, M.E.; Huang, J.; Stephens, B.M.; Lin, D.; Rajapakshe, K.I.; Weston, B.R.; Bhutani, M.S.; et al. Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-World Pancreatic Cancer Biopsies. Clin. Cancer Res. 2021, 27, 5912–5921. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2021, 12, 802080. [Google Scholar] [CrossRef]
- Gao, Y.; Li, H.; Li, Z.; Xie, L.; Liu, X.; Huang, Z.; Chen, B.; Lin, X.; Wang, X.; Zheng, Y.; et al. Single-Cell Analysis Reveals the Heterogeneity of Monocyte-Derived and Peripheral Type-2 Conventional Dendritic Cells. J. Immunol. 2021, 207, 837–848. [Google Scholar] [CrossRef]
- Sun, D.; Guan, X.; Moran, A.E.; Wu, L.-Y.; Qian, D.Z.; Schedin, P.; Dai, M.-S.; Danilov, A.V.; Alumkal, J.J.; Adey, A.C.; et al. Identifying Phenotype-Associated Subpopulations by Integrating Bulk and Single-Cell Sequencing Data. Nat. Biotechnol. 2022, 40, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, T.; Jin, Y.; Huang, B.; Zhang, Y. Progress and Clinical Application of Single-Cell Transcriptional Sequencing Technology in Cancer Research. Front. Oncol. 2020, 10, 593085. [Google Scholar] [CrossRef]
- Ke, M.; Elshenawy, B.; Sheldon, H.; Arora, A.; Buffa, F.M. Single Cell RNA-Sequencing: A Powerful yet Still Challenging Technology to Study Cellular Heterogeneity. Bioessays 2022, 44, e2200084. [Google Scholar] [CrossRef] [PubMed]
- Zappia, L.; Theis, F.J. Over 1000 Tools Reveal Trends in the Single-Cell RNA-Seq Analysis Landscape. Genome Biol. 2021, 22, 301. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef]
- Wolfien, M.; David, R.; Galow, A.-M. Single-Cell RNA Sequencing Procedures and Data Analysis. In Bioinformatics; Exon Publications: Brisbane, Australia, 2021; pp. 19–35. ISBN 9780645001716. [Google Scholar]
- Vieth, B.; Parekh, S.; Ziegenhain, C.; Enard, W.; Hellmann, I. A Systematic Evaluation of Single Cell RNA-Seq Analysis Pipelines. Nat. Commun. 2019, 10, 4667. [Google Scholar] [CrossRef]
- Tjoonk, N. What Is CellRanger and How Do You Use It? Available online: https://www.scdiscoveries.com/blog/knowledge/cellranger/ (accessed on 23 August 2024).
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal and Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Risso, D.; Purvis, L.; Fletcher, R.B.; Das, D.; Ngai, J.; Dudoit, S.; Purdom, E. clusterExperiment and RSEC: A Bioconductor Package and Framework for Clustering of Single-Cell and Other Large Gene Expression Datasets. PLoS Comput. Biol. 2018, 14, e1006378. [Google Scholar] [CrossRef]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating Single-Cell Analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Yiu, A.; Hemberg, M. Scmap—A Tool for Unsupervised Projection of Single Cell RNA-Seq Data. bioRxiv 2017. [Google Scholar]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-Omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887.e17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ghazanfar, S.; Wang, K.Y.X.; Gagnon-Bartsch, J.A.; Lo, K.K.; Su, X.; Han, Z.-G.; Ormerod, J.T.; Speed, T.P.; Yang, P.; et al. scMerge Leverages Factor Analysis, Stable Expression, and Pseudoreplication to Merge Multiple Single-Cell RNA-Seq Datasets. Proc. Natl. Acad. Sci. USA 2019, 116, 9775–9784. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, D.; Huang, X.; Wan, C.; Li, Z.; Han, Y.; Qin, Q.; Fan, J.; Qiu, X.; Xie, Y.; et al. Integrative Analyses of Single-Cell Transcriptome and Regulome Using MAESTRO. Genome Biol. 2020, 21, 198. [Google Scholar] [CrossRef]
- Lun, A.; Griffiths, J.; McCarthy, D. DropletUtils: Utilities for Handling Single-Cell Droplet Data; Bioconductor Version 3.13; 2021.
- Zappia L, L.A. zellkonverter: Conversion Between scRNA-Seq Objects; R Foundation Team: Viennam, Austria, 2024. [Google Scholar]
- Yip, S.H.; Wang, P.; Kocher, J.-P.A.; Sham, P.C.; Wang, J. Linnorm: Improved Statistical Analysis for Single Cell RNA-Seq Expression Data. Nucleic Acids Res. 2017, 45, e179. [Google Scholar] [CrossRef]
- Ruan, X.; Lai, C.; Li, L.; Wang, B.; Lu, X.; Zhang, D.; Fang, J.; Lai, M.; Yan, F. Integrative Analysis of Single-Cell and Bulk Multi-Omics Data to Reveal Subtype-Specific Characteristics and Therapeutic Strategies in Clear Cell Renal Cell Carcinoma Patients. J. Cancer 2024, 15, 6420–6433. [Google Scholar] [CrossRef]
- Xing, Z.; Lin, D.; Hong, Y.; Ma, Z.; Jiang, H.; Lu, Y.; Sun, J.; Song, J.; Xie, L.; Yang, M.; et al. Construction of a Prognostic 6-Gene Signature for Breast Cancer Based on Multi-Omics and Single-Cell Data. Front. Oncol. 2023, 13, 1186858. [Google Scholar] [CrossRef]
- Warfvinge, R.; Geironson Ulfsson, L.; Dhapola, P.; Safi, F.; Sommarin, M.N.E.; Soneji, S.; Hjorth-Hansen, H.; Mustjoki, S.; Richter, J.; Krishna Thakur, R.; et al. Single Cell Multi-Omics Analysis of Chronic Myeloid Leukemia Links Cellular Heterogeneity to Therapy Response. bioRxiv 2023. [Google Scholar]
- Argelaguet, R.; Arnol, D.; Bredikhin, D.; Deloro, Y.; Velten, B.; Marioni, J.C.; Stegle, O. MOFA+: A Statistical Framework for Comprehensive Integration of Multi-Modal Single-Cell Data. Genome Biol. 2020, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Naghipourfar, M.; Luecken, M.D.; Khajavi, M.; Büttner, M.; Wagenstetter, M.; Avsec, Ž.; Gayoso, A.; Yosef, N.; Interlandi, M.; et al. Mapping Single-Cell Data to Reference Atlases by Transfer Learning. Nat. Biotechnol. 2022, 40, 121–130. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Gongol, B.; Hayes, J.; Borowsky, A.T.; Bailey-Serres, J.; Girke, T. spatialHeatmap: Visualizing Spatial Bulk and Single-Cell Assays in Anatomical Images. NAR Genom. Bioinform. 2024, 6, lqae006. [Google Scholar] [CrossRef] [PubMed]
- Canete, N.P.; Iyengar, S.S.; Wilmott, J.S.; Ormerod, J.T.; Harman, A.N.; Patrick, E. spicyR: Spatial Analysis of in Situ Cytometry Data in R. bioRxiv 2021. [Google Scholar]
- Righelli, D.; Weber, L.M.; Crowell, H.L.; Pardo, B.; Collado-Torres, L.; Ghazanfar, S.; Lun, A.T.L.; Hicks, S.C.; Risso, D. SpatialExperiment: Infrastructure for Spatially-Resolved Transcriptomics Data in R Using Bioconductor. Bioinformatics 2022, 38, 3128–3131. [Google Scholar] [CrossRef]
- Lun, A. bluster: Clustering Algorithms for Bioconductor; R Foundation Team: Viennam, Austria, 2024. [Google Scholar]
- Campbell, J.; Corbett, S.; Koga, Y.; Yang, S.; Reed, E.; Wang, Z. Celda: CEllular Latent Dirichlet Allocation, R package version; R Foundation Team: Viennam, Austria, 2021; Volume 181. [Google Scholar]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. SC3: Consensus Clustering of Single-Cell RNA-Seq Data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef]
- Miao, Z.; Deng, K.; Wang, X.; Zhang, X. DEsingle for Detecting Three Types of Differential Expression in Single-Cell RNA-Seq Data. Bioinformatics 2018, 34, 3223–3224. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; Crowell, H.L.; Samartsidis, P.; Weber, L.M.; Robinson, M.D. distinct: A Novel Approach to Differential Distribution Analyses. bioRxiv 2020. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating Copy Number and Clonal Substructure in Human Tumors from Single-Cell Transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Khayatan, D.; Hussain, A.; Tebyaniyan, H. Exploring Animal Models in Oral Cancer Research and Clinical Intervention: A Critical Review. Vet. Med. Sci. 2023, 9, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Wang, P.; Huang, J.; Qi, X.; Liang, Y.; Zhao, W.; Wang, H.; Lyu, J.; Zhu, H. Metabolomics, Transcriptome and Single-Cell RNA Sequencing Analysis of the Metabolic Heterogeneity between Oral Cancer Stem Cells and Differentiated Cancer Cells. Cancers 2024, 16, 237. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Garvin, T.; Aboukhalil, R.; Kendall, J.; Baslan, T.; Atwal, G.S.; Hicks, J.; Wigler, M.; Schatz, M.C. Interactive Analysis and Assessment of Single-Cell Copy-Number Variations. Nat. Methods 2015, 12, 1058–1060. [Google Scholar] [CrossRef]
- Zaccaria, S.; Raphael, B.J. Characterizing Allele- and Haplotype-Specific Copy Numbers in Single Cells with CHISEL. Nat. Biotechnol. 2021, 39, 207–214. [Google Scholar] [CrossRef]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical Inference of Clonal Population Structure in Cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, L.; Hao, X.; Wang, T.; Vijg, J. SCCNV: A Software Tool for Identifying Copy Number Variation from Single-Cell Whole-Genome Sequencing. Front. Genet. 2020, 11, 505441. [Google Scholar] [CrossRef]
- Dong, X.; Zhang, L.; Milholland, B.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Accurate Identification of Single-Nucleotide Variants in Whole-Genome-Amplified Single Cells. Nat. Methods 2017, 14, 491–493. [Google Scholar] [CrossRef]
- Jahn, K.; Kuipers, J.; Beerenwinkel, N. Tree Inference for Single-Cell Data. Genome Biol. 2016, 17, 86. [Google Scholar] [CrossRef]
- Erfanian, N.; Heydari, A.A.; Feriz, A.M.; Iañez, P.; Derakhshani, A.; Ghasemigol, M.; Farahpour, M.; Razavi, S.M.; Nasseri, S.; Safarpour, H.; et al. Deep Learning Applications in Single-Cell Genomics and Transcriptomics Data Analysis. Biomed. Pharmacother. 2023, 165, 115077. [Google Scholar] [CrossRef] [PubMed]
- Molho, D.; Ding, J.; Tang, W.; Li, Z.; Wen, H.; Wang, Y.; Venegas, J.; Jin, W.; Liu, R.; Su, R.; et al. Deep Learning in Single-Cell Analysis. ACM Trans. Intell. Syst. Technol. 2024, 15, 1–62. [Google Scholar] [CrossRef]
- Premkumar, R.; Srinivasan, A.; Harini Devi, K.G.; M, D.; E, G.; Jadhav, P.; Futane, A.; Narayanamurthy, V. Single-Cell Classification, Analysis, and Its Application Using Deep Learning Techniques. Biosystems 2024, 237, 105142. [Google Scholar] [CrossRef]
- Zhu, Y.; Bai, L.; Ning, Z.; Fu, W.; Liu, J.; Jiang, L.; Fei, S.; Gong, S.; Lu, L.; Deng, M.; et al. Deep Learning for Clustering Single-Cell RNA-Seq Data. Curr. Bioinform. 2024, 19, 193–210. [Google Scholar] [CrossRef]
- Halawani, R.; Buchert, M.; Chen, Y.-P.P. Deep Learning Exploration of Single-Cell and Spatially Resolved Cancer Transcriptomics to Unravel Tumour Heterogeneity. Comput. Biol. Med. 2023, 164, 107274. [Google Scholar] [CrossRef]
- Qi, R.; Zou, Q. Trends and Potential of Machine Learning and Deep Learning in Drug Study at Single-Cell Level. Research 2023, 6, 0050. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, P.; Yang, F.; Jiang, K.; Sun, S.; Xia, Z.; Yao, G.; Tang, J. Integrating Single-Cell Analysis and Machine Learning to Create Glycosylation-Based Gene Signature for Prognostic Prediction of Uveal Melanoma. Front. Endocrinol. (Lausanne) 2023, 14, 1163046. [Google Scholar] [CrossRef]
- Mou, L.; Pu, Z.; Luo, Y.; Quan, R.; So, Y.; Jiang, H. Construction of a Lipid Metabolism-Related Risk Model for Hepatocellular Carcinoma by Single Cell and Machine Learning Analysis. Front. Immunol. 2023, 14, 1036562. [Google Scholar] [CrossRef]
- Chen, H.; Yang, W.; Ma, L.; Li, Y.; Ji, Z. Machine-Learning Based Integrating Bulk and Single-Cell RNA Sequencing Reveals the SLC38A5-CCL5 Signaling as a Promising Target for Clear Cell Renal Cell Carcinoma Treatment. Transl. Oncol. 2023, 38, 101790. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Zhao, Y.-X.; Qiu, R.S.Q.; Chen, D.-N.; Zheng, Q.-S.; Xue, X.-Y.; Xu, N.; Wei, Y. Identification Macrophage Signatures in Prostate Cancer by Single-Cell Sequencing and Machine Learning. Cancer Immunol. Immunother. 2024, 73, 41. [Google Scholar] [CrossRef]
- He, X.; Liu, X.; Zuo, F.; Shi, H.; Jing, J. Artificial Intelligence-Based Multi-Omics Analysis Fuels Cancer Precision Medicine. Semin. Cancer Biol. 2023, 88, 187–200. [Google Scholar] [CrossRef]
- Li, J.; Li, L.; You, P.; Wei, Y.; Xu, B. Towards Artificial Intelligence to Multi-Omics Characterization of Tumor Heterogeneity in Esophageal Cancer. Semin. Cancer Biol. 2023, 91, 35–49. [Google Scholar] [CrossRef]
- Danishuddin; Khan, S.; Kim, J.J. From Cancer Big Data to Treatment: Artificial Intelligence in Cancer Research. J. Gene Med. 2024, 26, e3629. [Google Scholar] [CrossRef]
- Luo, J.; Pan, M.; Mo, K.; Mao, Y.; Zou, D. Emerging Role of Artificial Intelligence in Diagnosis, Classification and Clinical Management of Glioma. Semin. Cancer Biol. 2023, 91, 110–123. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega-Batista, A.; Jaén-Alvarado, Y.; Moreno-Labrador, D.; Gómez, N.; García, G.; Guerrero, E.N. Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology. Int. J. Mol. Sci. 2025, 26, 2074. https://doi.org/10.3390/ijms26052074
Ortega-Batista A, Jaén-Alvarado Y, Moreno-Labrador D, Gómez N, García G, Guerrero EN. Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology. International Journal of Molecular Sciences. 2025; 26(5):2074. https://doi.org/10.3390/ijms26052074
Chicago/Turabian StyleOrtega-Batista, Ana, Yanelys Jaén-Alvarado, Dilan Moreno-Labrador, Natasha Gómez, Gabriela García, and Erika N. Guerrero. 2025. "Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology" International Journal of Molecular Sciences 26, no. 5: 2074. https://doi.org/10.3390/ijms26052074
APA StyleOrtega-Batista, A., Jaén-Alvarado, Y., Moreno-Labrador, D., Gómez, N., García, G., & Guerrero, E. N. (2025). Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology. International Journal of Molecular Sciences, 26(5), 2074. https://doi.org/10.3390/ijms26052074