
Base-Labile Safety-Catch Linker: Synthesis and Applications in Solid-Phase Peptide Synthesis




Abstract
:
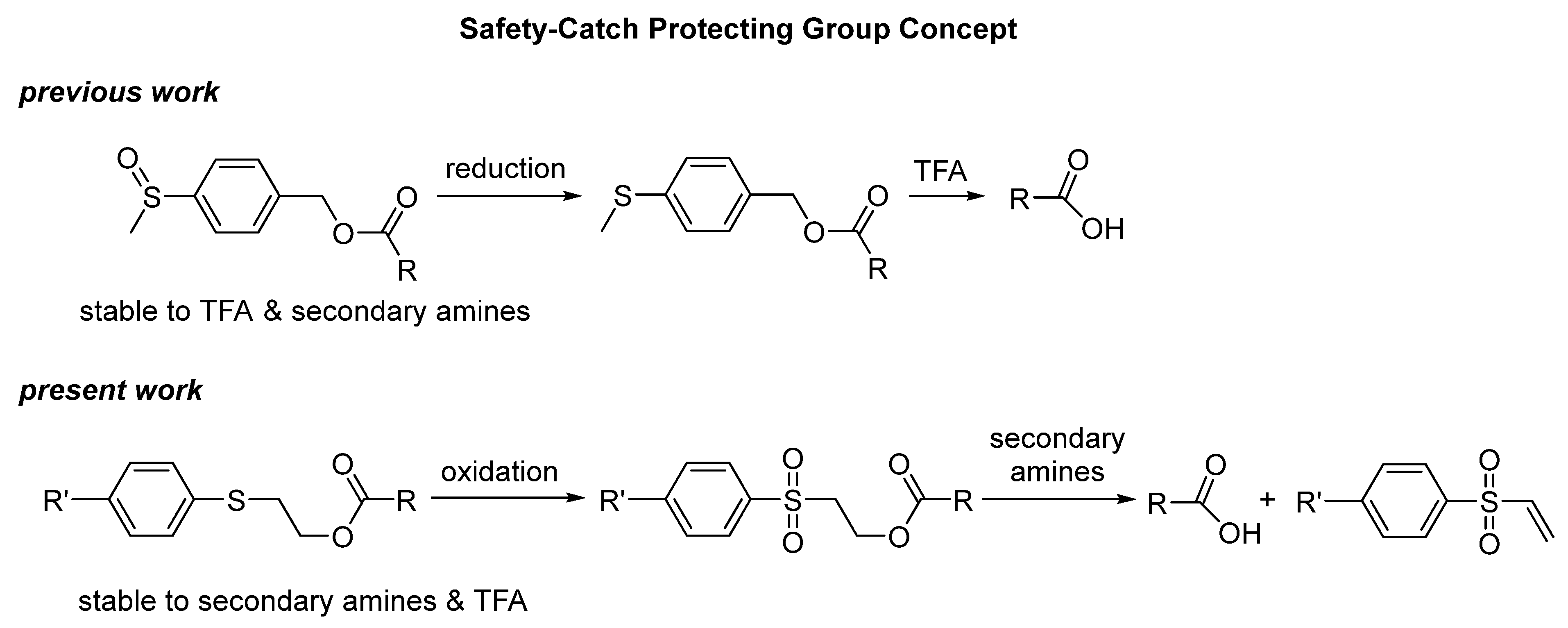
1. Introduction
2. Results and Discussion
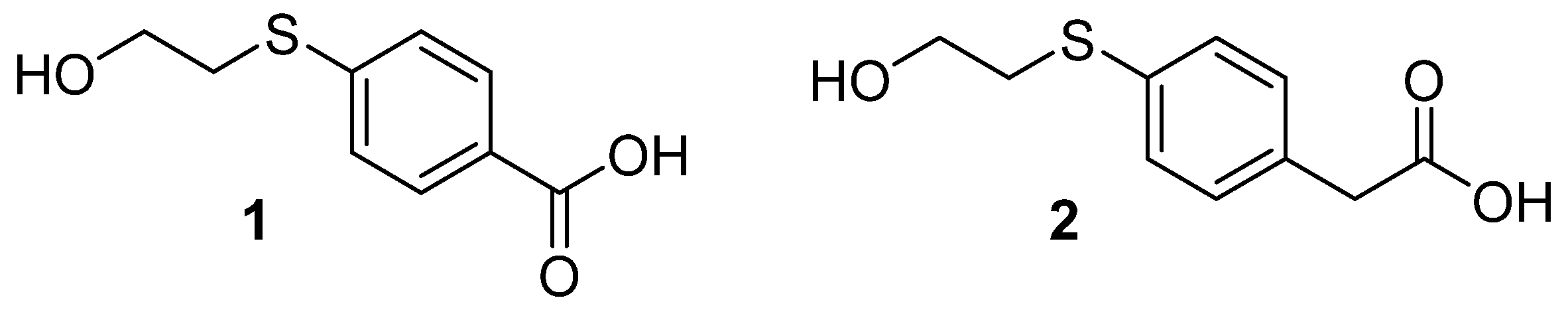
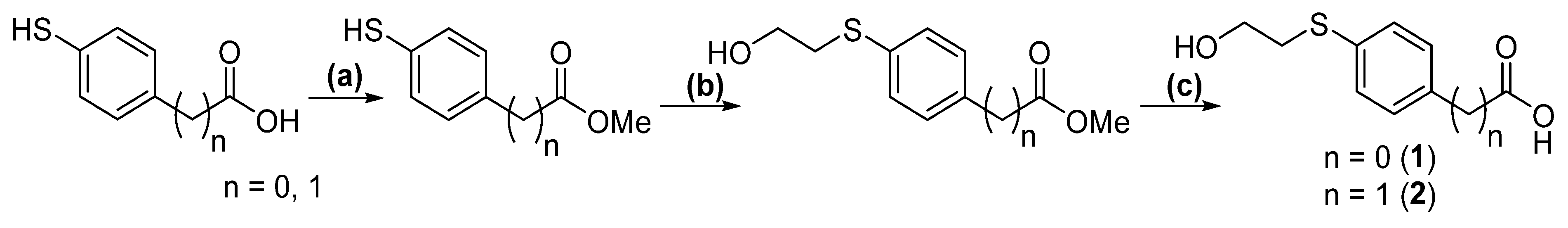
2.1. Linker Syntheses
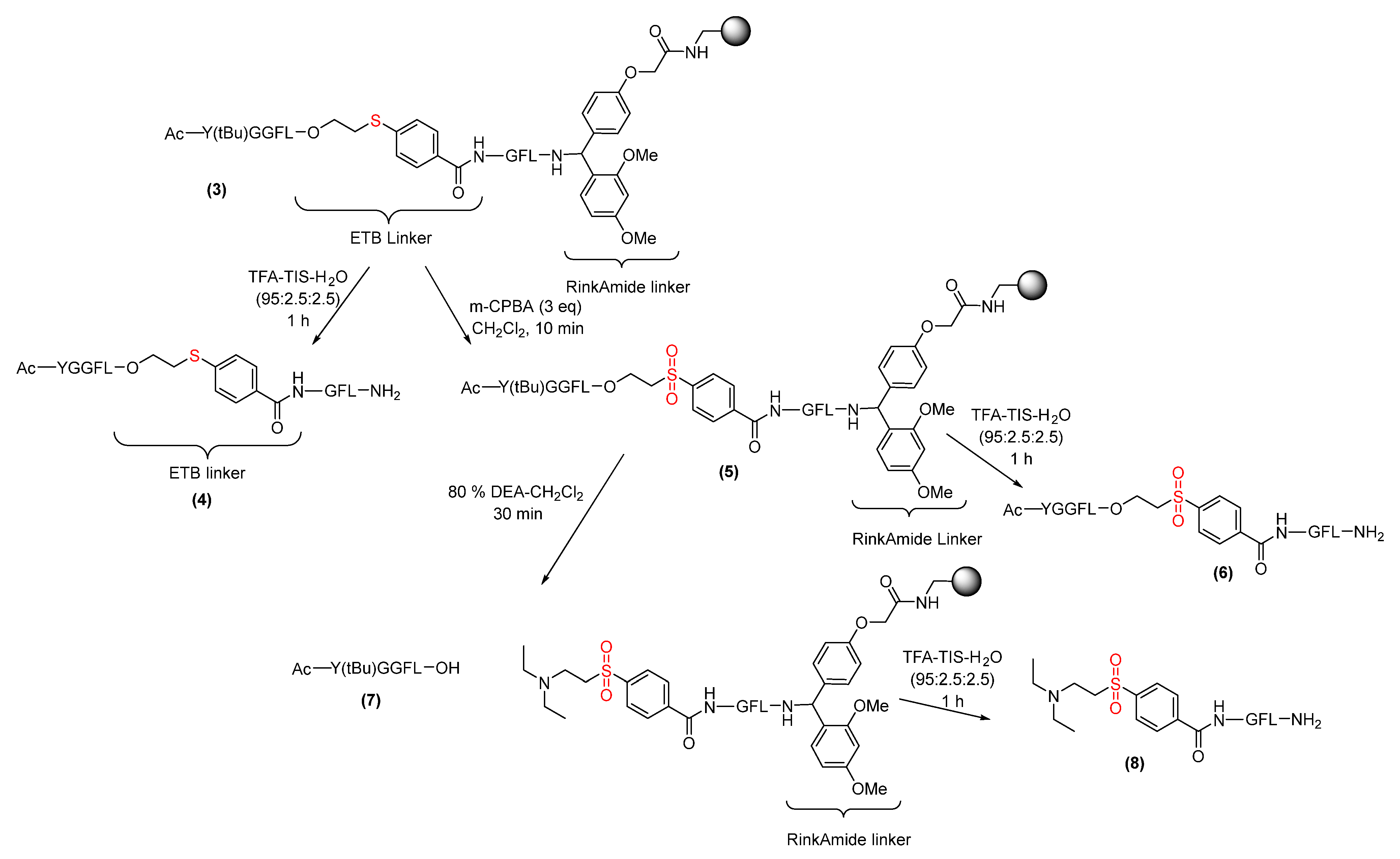
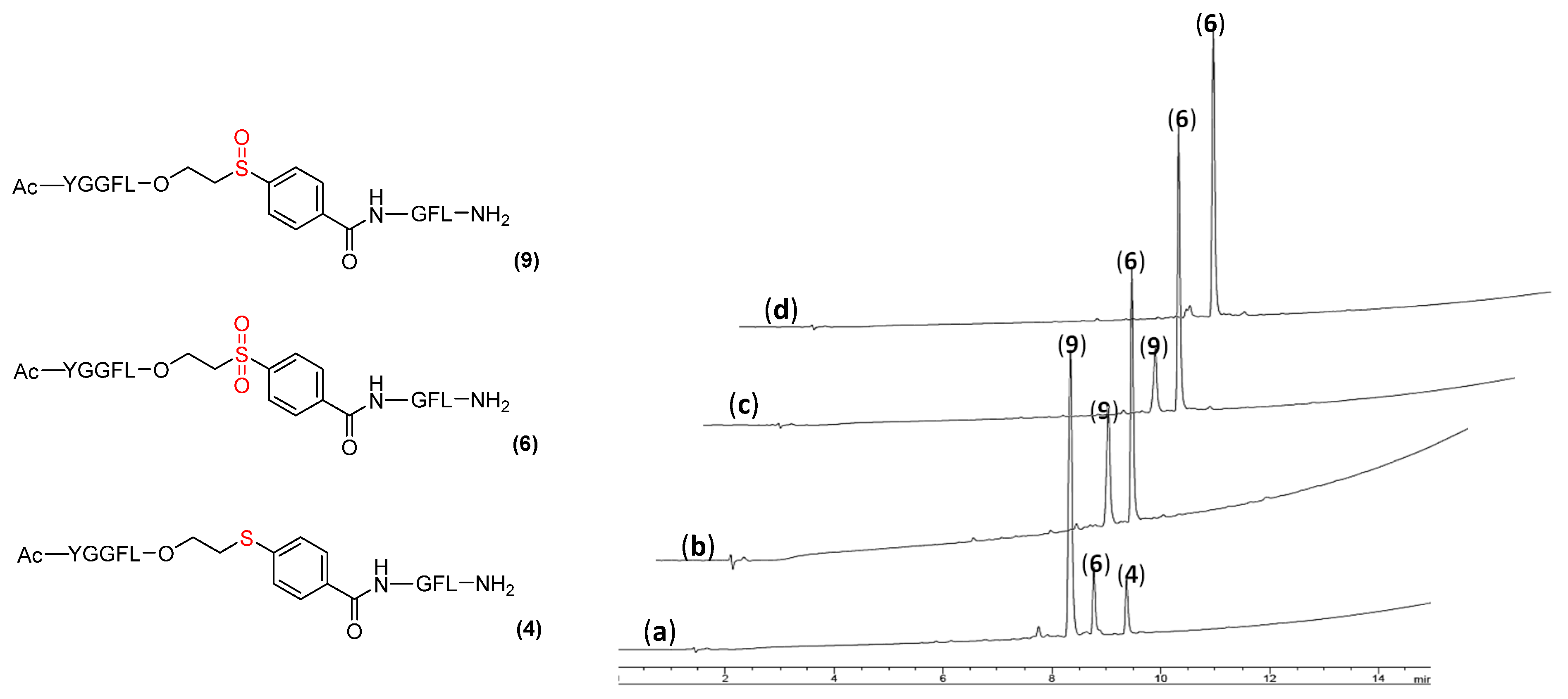
2.2. ETB Linker Activation [Oxidation of Sulfide (Thio) to Sulfone]
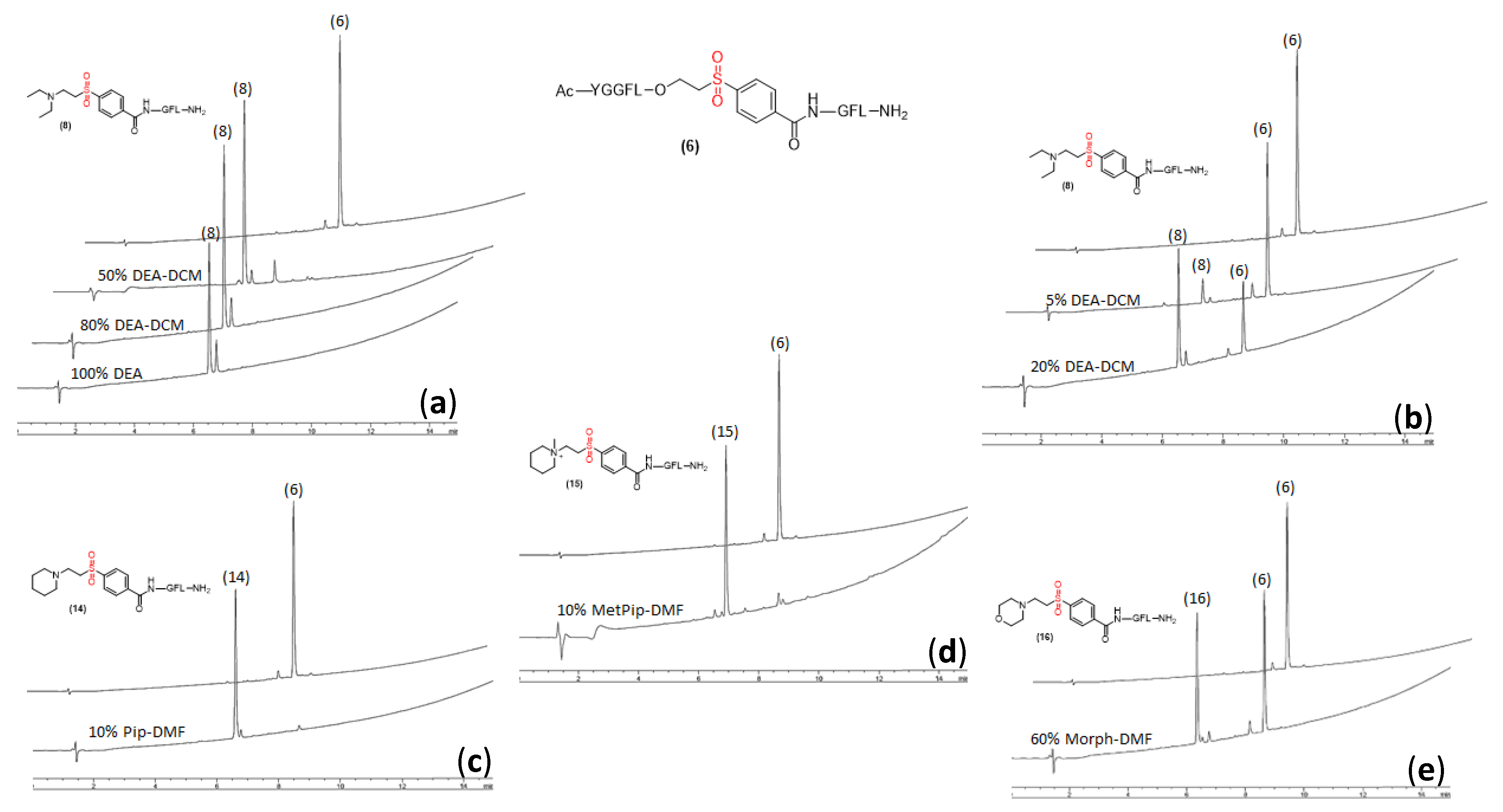
2.3. Peptide Cleavage from Activated ETB Linker
2.4. Stability of Peptides with Sensitive Amino Acids (His, Trp, and Cys) in m-CPBA Oxidizing Reagent
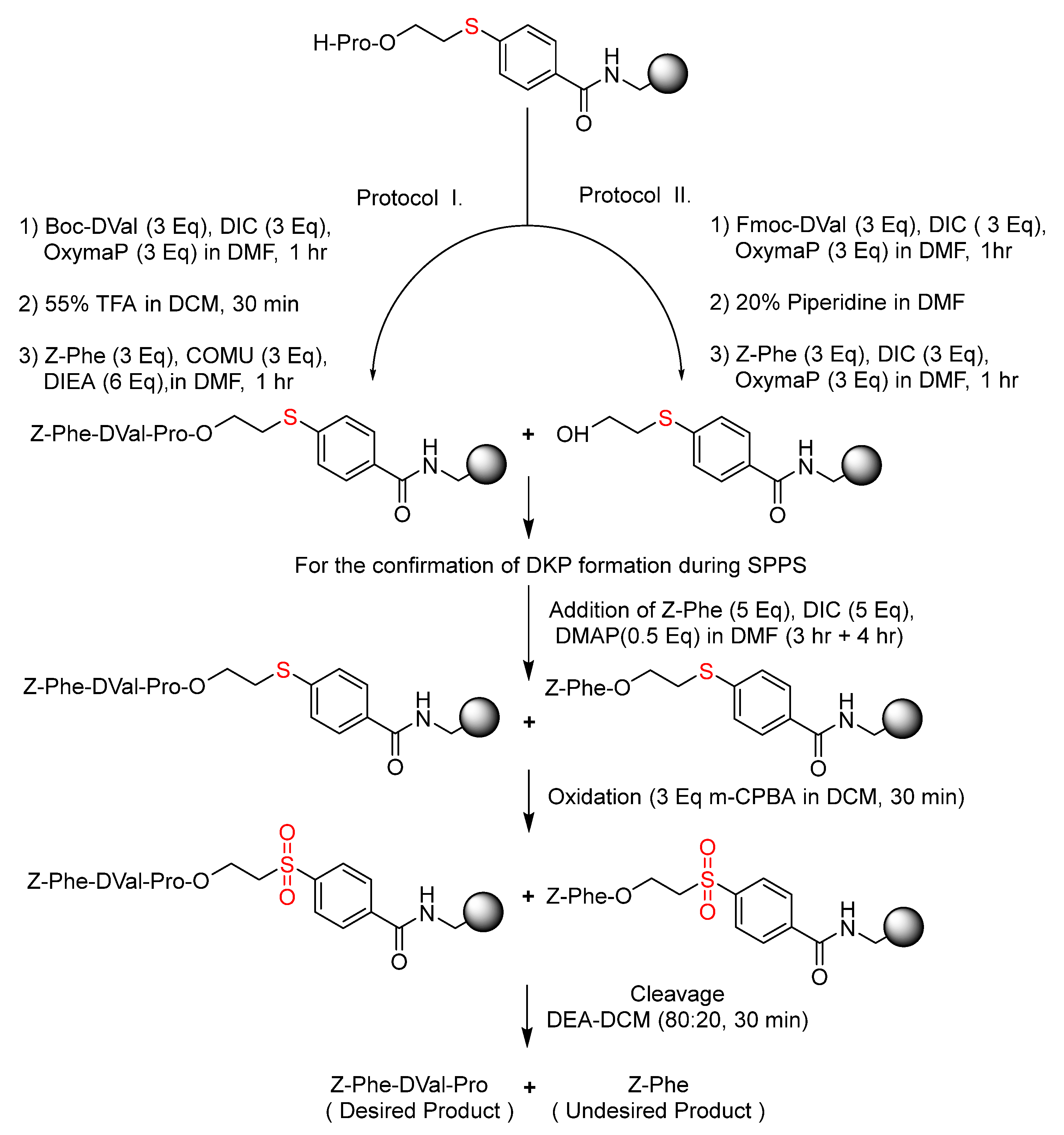
2.5. Compatibility of ETB Resin with Boc Chemistry to Minimize DKP Formation
2.6. Synthesis of Unprotected Peptides Without TFA Cleavage
3. Material and Methods
3.1. Preparation of Methyl 4-Mercaptobenzoate
 HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 8.493 min; 1H NMR (600 MHz, DMSO-d6): δ =7.81 (d, J = 7.9 Hz, 2H; ArH), 7.43 (d, J = 7.8 Hz, 2H; ArH), 3.83 (s, 3H, CH3), 3.34 (s, 1H, SH); 13C NMR (150 MHz, DMSO-d6): 166.3, 140.8, 130.5, 130.1, 128.3, 126.7, 126.3, 52.4.
HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 8.493 min; 1H NMR (600 MHz, DMSO-d6): δ =7.81 (d, J = 7.9 Hz, 2H; ArH), 7.43 (d, J = 7.8 Hz, 2H; ArH), 3.83 (s, 3H, CH3), 3.34 (s, 1H, SH); 13C NMR (150 MHz, DMSO-d6): 166.3, 140.8, 130.5, 130.1, 128.3, 126.7, 126.3, 52.4.3.2. Preparation of Methyl 4-((2-Hydroxyethyl)thio)benzoate
 HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 7.078 min; 1H NMR (600 MHz, DMSO-d6): δ =7.85 (dd, J = 6.3 Hz, 2H; ArH), 7.42 (dd, J = 6.3 Hz, 2H, ArH), 5.04 (s, 1H, OH), 3.83 (s, 3H, CH3), 3.62 (t, J = 5.1 Hz, 2H), 3.14 (m, J = 6.4 Hz, 2H);13C NMR (150 MHz, DMSO-d6): 166.4, 144.6, 130.1, 126.5, 126.3, 60.1, 52.4, 34.2. HRMS: m/z: for C10H12O3S+: calcd: 213.0580 [M + H]+; found: 213.0577.
HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 7.078 min; 1H NMR (600 MHz, DMSO-d6): δ =7.85 (dd, J = 6.3 Hz, 2H; ArH), 7.42 (dd, J = 6.3 Hz, 2H, ArH), 5.04 (s, 1H, OH), 3.83 (s, 3H, CH3), 3.62 (t, J = 5.1 Hz, 2H), 3.14 (m, J = 6.4 Hz, 2H);13C NMR (150 MHz, DMSO-d6): 166.4, 144.6, 130.1, 126.5, 126.3, 60.1, 52.4, 34.2. HRMS: m/z: for C10H12O3S+: calcd: 213.0580 [M + H]+; found: 213.0577.3.3. Preparation of 4-((2-Hydroxyethyl)thio)Benzoic Acid
 HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 5.448 min; 1H NMR (600 MHz, DMSO-d6): δ = 12.8 (s, 1H, COOH), 7.84 (dd, J = 8.3 Hz, 2H; ArH), 7.39 (dd, J = 8.4 Hz, 2H, ArH), 5.03 (s, 1H, OH), 3.61 (dd, J = 7.86 Hz, 2H, CH2), 3.13 (dd, J = 7.9 Hz, 2H, CH2);13C NMR (150 MHz, DMSO-d6): 167.5, 143.9, 130.2, 127.6, 126.5, 60.1, 34.2. HRMS: m/z: for C9H9O3S−: calcd: 197.0278 [M − H]−; found: 197.0282.
HPLC [5–95% of MeCN (0.1% TFA/H2O (0.1% TFA) over 15 min] tR = 5.448 min; 1H NMR (600 MHz, DMSO-d6): δ = 12.8 (s, 1H, COOH), 7.84 (dd, J = 8.3 Hz, 2H; ArH), 7.39 (dd, J = 8.4 Hz, 2H, ArH), 5.03 (s, 1H, OH), 3.61 (dd, J = 7.86 Hz, 2H, CH2), 3.13 (dd, J = 7.9 Hz, 2H, CH2);13C NMR (150 MHz, DMSO-d6): 167.5, 143.9, 130.2, 127.6, 126.5, 60.1, 34.2. HRMS: m/z: for C9H9O3S−: calcd: 197.0278 [M − H]−; found: 197.0282. 3.4. General Peptide Synthesis Protocol
3.5. ETB Linker Attachment to Aminomethyl Polystyrene Resin
3.6. Coupling First Amino Acid to ETB Resin
3.7. Fmoc Removal
3.8. Elongation of Peptide
3.9. Oxidation of ETB Resin (Sulfide to Sulfone)
3.10. Removal of the Side-Chain Protecting Groups
3.11. Cleavage of Unprotected Peptide from ETB Resin
3.12. Cleavage of Protected Peptide from ETB Resin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Bruckdorfer, T.; Marder, O.; Albericio, F. From Production of Peptides in Milligram Amounts for Research to Multi-Tons Quantities for Drugs of the Future. Curr. Pharm. Biotechnol. 2004, 5, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Guillier, F.; Orain, D.; Bradley, M. Linkers and Cleavage Strategies in Solid-Phase Organic Synthesis and Combinatorial Chemistry. Chem. Rev. 2000, 100, 2091–2158. [Google Scholar] [CrossRef]
- Shelton, P.T.; Jensen, K.J. Linkers, Resins, and General Procedures for Solid-Phase Peptide Synthesis. In Peptide Synthesis and Applications; Jensen, K.J., Tofteng Shelton, P., Pedersen, S.L., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 23–41. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Pátek, M.; Lebl, M. Safety-catch and multiply cleavable linkers in solid-phase synthesis. Pept. Sci. 1998, 47, 353–363. [Google Scholar] [CrossRef]
- Kenner, G.W.; McDermott, J.R.; Sheppard, R.C. The safety catch principle in solid phase peptide synthesis. J. Chem. Soc. D 1971, 12, 636–637. [Google Scholar] [CrossRef]
- Lebl, M.; Patek, M.; Kocis, P.; Krchnak, V.; Hruby, V.J.; Salmon, S.E.; Lam, K. Multiple release of equimolar amounts of peptides from a polymeric carrier using orthogonal linkage-cleavage chemistry. Int. J. Pept. Protein Res. 1993, 41, 201–203. [Google Scholar] [CrossRef]
- Noki, S.; Brasil, E.; Zhang, H.; Bruckdorfer, T.; de la Torre, B.G.; Albericio, F. Solid-Phase Peptide Synthesis Using a Four-Dimensional (Safety-Catch) Protecting Group Scheme. J. Org. Chem. 2022, 87, 9443–9453. [Google Scholar] [CrossRef] [PubMed]
- Nandhini, K.P.; Albericio, F.; de la Torre, B.G. 2-Methoxy-4-methylsulfinylbenzyl Alcohol as a Safety-Catch Linker for the Fmoc/tBu Solid-Phase Peptide Synthesis Strategy. J. Org. Chem. 2022, 87, 9433–9442. [Google Scholar] [CrossRef] [PubMed]
- Tesser, G.I.; Balvert-Geers, I.C. The methylsulfonylethyloxycarbonyl group, a new and versatile amino protective function. Int. J. Pept. Protein Res. 1975, 7, 295–305. [Google Scholar] [CrossRef]
- Tesser, G.I.; Buis, J.T.W.A.R.M.; Wolters, E.T.M.; Bothé-Helmes, E.G.A.M. Synthesis of 2-hydroxyethylsulfonyl-methyl-substituted polystyrenes and their application in solid phase peptide synthesis. Tetrahedron 1976, 32, 1069–1071. [Google Scholar] [CrossRef]
- Buis, J.T.W.A.R.M.; Tesser, G.I.; Nivard, R.J.F. Comparison of the applicability of 2-hydroxyethylsulfonylmethyl- and chloromethyl-polystyrenes in the solid-phase synthesis of protected peptides. Tetrahedron 1976, 32, 2321–2325. [Google Scholar] [CrossRef]
- Carpino, L.A.; Han, G.Y. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J. Am. Chem. Soc. 1970, 92, 5748–5749. [Google Scholar] [CrossRef]
- Carreño, C.; Méndez, M.E.; Andreu, D.; Kim, Y.D.; Kim, H.J.; Kates, S.A.; Albericio, F. Nsc and Fmoc Nα-amino protection for solid-phase peptide synthesis: A parallel study. Int. J. Pept. Res. 2000, 56, 63–69. [Google Scholar] [CrossRef]
- Katti, S.B.; Misra, P.K.; Haq, W.; Mathur, K.B. A new base-labile linker for solid-phase peptide synthesis. J. Chem. Soc. Chem. Commun. 1992, 11, 843–844. [Google Scholar] [CrossRef]
- García-Echeverría, C. A base labile handle for solid phase organic chemistry. Tetrahedron Lett. 1997, 38, 8933–8934. [Google Scholar] [CrossRef]
- Wade, W.S.; Yang, F.; Sowin, T.J. Application of Base Cleavable Safety Catch Linkers to Solid Phase Library Production. J. Comb. Chem. 2000, 2, 266–275. [Google Scholar] [CrossRef]
- Tumelty, D.; Cao, K.; Holmes, C.P. Traceless Solid-Phase Synthesis of Substituted Benzimidazoles via a Base-Cleavable Linker. Org. Lett. 2001, 3, 83–86. [Google Scholar] [CrossRef] [PubMed]
- van Maarseveen, J.H.; Meester, W.J.N.; Veerman, J.J.N.; Kruse, C.G.; Hermkens, P.H.H.; Rutjes, F.P.J.T.; Hiemstra, H. Development and application of allyl, 2-sulfonylethyl and 2-thioethyl carbamate linkers for solid phase N-acyliminium ion chemistry. J. Chem. Soc. 2001, 9, 994–1001. [Google Scholar] [CrossRef]
- Veerman, J.J.N.; Rutjes, F.P.J.T.; van Maarseveen, J.H.; Hiemstra, H. A novel acid stable/base labile carbamate linker for N-acyliminium ion reactions on solid support. Tetrahedron Lett. 1999, 40, 6079–6082. [Google Scholar] [CrossRef]
- Kates, S.A.; Solé, N.A.; Johnson, C.R.; Hudson, D.; Barany, G.; Albericio, F. A novel, convenient, three-dimensional orthogonal strategy for solid-phase synthesis of cyclic peptides. Tetrahedron Lett. 1993, 34, 1549–1552. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Guasch-Camell, J.; Álvarez, M.; Albericio, F. p-Nitrobenzyloxycarbonyl (pNZ) as a Temporary Nα-Protecting Group in Orthogonal Solid-Phase Peptide Synthesis—Avoiding Diketopiperazine and Aspartimide Formation. EurJOC 2005, 2005, 3031–3039. [Google Scholar] [CrossRef]
- Tam, J.P.; Dimarchi, R.D.; Merrifield, R.B. Photolabile multi-detachable p-alkoxybenzyl alcohol resin supports for peptide fragment or semi-synthesis. Int. J. Pept. Protein Res. 1980, 16, 412–425. [Google Scholar] [CrossRef]
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef]
- Tonkyn, R.G.; Wynstra, J. The reactions of piperidine and diethylamine with epichlorohydrin. J. Appl. Polym. Sci. 1967, 11, 145–148. [Google Scholar] [CrossRef]
- Mthembu, S.N.; Chakraborty, A.; Schönleber, R.; Albericio, F.; de la Torre, B.G. Morpholine, a strong contender for Fmoc removal in solid-phase peptide synthesis. J. Pept. Sci. 2024, 30, e3538. [Google Scholar] [CrossRef]
- Yang, Y. Side Reactions in Peptide Synthesis; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Jadhav, S.; Martin, V.; Egelund, P.H.G.; Johansson Castro, H.; Krüger, T.; Richner, F.; Thordal Le Quement, S.; Albericio, F.; Dettner, F.; Lechner, C.; et al. Replacing DMF in solid-phase peptide synthesis: Varying the composition of green binary solvent mixtures as a tool to mitigate common side-reactions. Green Chem. 2021, 23, 3312–3321. [Google Scholar] [CrossRef]
- Wang, J.; Berglund, M.R.; Braden, T.; Embry, M.C.; Johnson, M.D.; Groskreutz, S.R.; Sayyed, F.B.; Tsukanov, S.V.; White, T.D.; Jalan, A.; et al. Mechanistic Study of Diketopiperazine Formation during Solid-Phase Peptide Synthesis of Tirzepatide. ACS Omega 2022, 7, 46809–46824. [Google Scholar] [CrossRef] [PubMed]
- Rovero, P.; Viganò, S.; Pegoraro, S.; Quartara, L. Synthesis of the bradykinin B1 antagonist [desArg10]HOE 140 on 2-chlorotrityl resin. Lett. Pept. Sci. 1996, 2, 319–323. [Google Scholar] [CrossRef]
- Alcaro, M.C.; Sabatino, G.; Uziel, J.; Chelli, M.; Ginanneschi, M.; Rovero, P.; Papini, A.M. On-resin head-to-tail cyclization of cyclotetrapeptides: Optimization of crucial parameters. J. Pept. Sci. 2004, 10, 218–228. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Bayó-Puxan, N.; Moreno, J.A.; Francesch, A.M.; Cuevas, C.; Álvarez, M.; Albericio, F. Solid-Phase Synthesis of Oxathiocoraline by a Key Intermolecular Disulfide Dimer. J. Am. Chem. Soc. 2007, 129, 5322–5323. [Google Scholar] [CrossRef]
- Pedroso, E.; Grandas, A.; de las Heras, X.; Eritja, R.; Giralt, E. Diketopiperazine formation in solid phase peptide synthesis using p-alkoxybenzyl ester resins and Fmoc-amino acids. Tetrahedron Lett. 1986, 27, 743–746. [Google Scholar] [CrossRef]
- Khosla, M.C.; Smeby, R.R.; Bumpus, F.M. Failure sequence in solid-phase peptide synthesis due to the presence of an N-alkylamino acid. J. Am. Chem. Soc. 1972, 94, 4721–4724. [Google Scholar] [CrossRef]
- Gisin, B.F.; Merrifield, R.B. Carboxyl-catalyzed intramolecular aminolysis. Side reaction in solid-phase peptide synthesis. J. Am. Chem. Soc. 1972, 94, 3102–3106. [Google Scholar] [CrossRef]
- Alsina, J.; Giralt, E.; Albericio, F. Use of N-tritylamino acids and PyAOP1 for the suppression of diketopiperazine formation in Fmoc/tBu solid-phase peptide synthesis using alkoxybenzyl ester anchoring linkages. Tetrahedron Lett. 1996, 37, 4195–4198. [Google Scholar] [CrossRef]
- Gairí, M.; Lloyd-Williams, P.; Albericio, F.; Giralt, E. Use of BOP reagent for the suppression of diketopiperazine formation in boc/bzl solid-phase peptide synthesis. Tetrahedron Lett. 1990, 31, 7363–7366. [Google Scholar] [CrossRef]
- Albericio, F.; del Fresno, M.; Frieden, A.; Royo, M.; Alsina, J.; Jensen, K.J.; Kates, S.A.; Barany, G. Backbone amide linker (BAL) for solid-phase synthesis of 2,5-piperazinediones (DKP), useful scaffolds for combinatorial chemistry. In Peptides Frontiers of Peptide Science: Proceedings of the Fifteenth American Peptide Symposium, Nashville, TN, USA, 14–19 June 1997; Tam, J.P., Kaumaya, P.T.P., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 37–39. [Google Scholar] [CrossRef]
- ECHA. European Chemical Agency. Trifluoroacetic Acid. Available online: https://echa.europa.eu/substance-information/-/substanceinfo/100.000.846 (accessed on 13 October 2024).
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef]
- Lenoir, D.; Schramm, K.-W.; Lalah, J.O. Green Chemistry: Some important forerunners and current issues. Sustain. Chem. Pharm. 2020, 18, 100313. [Google Scholar] [CrossRef]
- Kim, H.M.; An, H.S.; Bae, J.-S.; Kim, J.Y.; Choi, C.H.; Kim, J.Y.; Lim, J.H.; Choi, J.-h.; Song, H.; Moon, S.H.; et al. Effects of palmitoyl-KVK-L-ascorbic acid on skin wrinkles and pigmentation. Arch. Dermatol. Res. 2017, 309, 397–402. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basic Cleavage Conditions rt, 20 min | Yield % * |
|---|---|
| 100% DEA, | 95.1 |
| 80% DEA—DCM | 93.5 |
| 50% DEA—DCM | 91.6 |
| Basic Cleavage Conditions, rt, 30 min | Percentage of Cleaved Protected Peptide (7) (Ac-Y(tBu)GGFL-OH) * (%) | Percentage of Uncleaved Peptide (Ac-YGGFL-ETB-Linker(O=S=O)-GFL-NH2 (%) |
|---|---|---|
| 100% DEA | 100 | 0 |
| 80% DEA-DCM | >99 | <1% |
| 50% DEA-DCM | 98 | 2 |
| 20% DEA-DCM | 63 | 37 |
| 5% DEA-DCM | 15 | 85 |
| 10% Piperidine-DCM | 96 | 4 |
| 10% 4-methylpiperidine–DMF | 93 | 7 |
| 60% morpholine–DMF | 47 | 53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noki, S.; Saneii, H.; de la Torre, B.G.; Albericio, F. Base-Labile Safety-Catch Linker: Synthesis and Applications in Solid-Phase Peptide Synthesis. Int. J. Mol. Sci. 2025, 26, 2210. https://doi.org/10.3390/ijms26052210
Noki S, Saneii H, de la Torre BG, Albericio F. Base-Labile Safety-Catch Linker: Synthesis and Applications in Solid-Phase Peptide Synthesis. International Journal of Molecular Sciences. 2025; 26(5):2210. https://doi.org/10.3390/ijms26052210
Chicago/Turabian StyleNoki, Sikabwe, Hossain Saneii, Beatriz G. de la Torre, and Fernando Albericio. 2025. "Base-Labile Safety-Catch Linker: Synthesis and Applications in Solid-Phase Peptide Synthesis" International Journal of Molecular Sciences 26, no. 5: 2210. https://doi.org/10.3390/ijms26052210
APA StyleNoki, S., Saneii, H., de la Torre, B. G., & Albericio, F. (2025). Base-Labile Safety-Catch Linker: Synthesis and Applications in Solid-Phase Peptide Synthesis. International Journal of Molecular Sciences, 26(5), 2210. https://doi.org/10.3390/ijms26052210