Abstract
Chronic kidney disease (CKD) is a condition caused by the gradual decline of renal function that approximatively affects 10–12% of the world population, thus representing a public health priority. In CKD patients, chronic and systemic low-grade inflammation is observed, and it significantly contributes to disease development and progression, especially for patients with advanced disease. It also results in CKD-associated complications and increased mortality. The low-grade inflammation is due to different factors, such as the decline of glomerular filtration rate, increased immune system activation, reactive oxygen species release, and intestinal homeostasis. Therefore, the possibility to control chronic low-grade inflammation in CKD deserves great attention. In this review, we will examine the current possible pharmacological approaches to counteract the inflammatory state in CKD, focusing our attention both on the pro-inflammatory factors and the pro-resolving mediators involved in CKD inflammatory state.
1. Introduction
Chronic kidney disease (CKD) is a complex and multifactorial pathological condition that affects more than 800 million individuals in the world and represents a global health problem, becoming a leading cause of death worldwide with an increase in deaths over the past two decades [1,2,3]. The main causes of CKD are hypertension, diabetes, immune-mediated diseases, glomerulonephritis, tubulointerstitial disease, and inherited kidney diseases. CKD treatment aims to slow down the progression of kidney damage, primarily by managing the underlying cause. However, even with proper management of the underlying cause, kidney damage may continue to progress, leading to end-stage renal disease (ESRD), which is life-threatening without artificial filtration through dialysis or a kidney transplant. As renal replacement therapy only partially corrects the uremic state, kidney transplantation is the treatment of choice, although it requires lifelong immunosuppression [4]. The number of people receiving renal replacement therapy, which aims to replace nonendocrine kidney function in patients with renal failure using techniques such as intermittent hemodialysis, continuous hemofiltration and hemodialysis, and peritoneal dialysis, exceeds 2.5 million and is expected to double to 5.4 million by 2030 [5]. Dialysis is the most common treatment for CKD. It contributes to reduce the accumulation of high levels of metabolic end products, normally excreted by a healthy kidney, uremic toxins, the accumulation of which is clinically relevant in CKD progression and related complications [6,7,8,9,10,11]. In fact, CKD affects more than just the kidneys, as patients frequently experience various complications, including hypertension, cardiovascular diseases, anemia, metabolic acidosis [12], impaired immune response, mineral and bone disorders, and neurological complications [13].
Among these complications, cardiovascular dysfunction and infections, exacerbated by an impaired immune response, have been identified as major contributors to increased morbidity and mortality [14]. Inflammation also plays a central role in CKD-related complications [15]. While inflammation is essential for defending against infections, its dysregulation can trigger harmful effects, including the excessive production of pro-inflammatory cytokines, which sustain a persistent inflammatory response throughout the body [13]. Patients with CKD suffer from chronic and low-grade inflammation. This worsens with the degree of renal failure [11]. Over the past two decades, numerous lines of evidence point to chronic inflammation as a pivotal element of the uremic phenotype, which significantly contributes to the progression and acceleration of CKD-related complications. Moreover, in addition to the immune system’s activation, pathogens and other abnormal metabolites (e.g., uremic toxins) can trigger chronic inflammation, thus contributing to the CKD-associated inflammatory state [16,17], Despite great progress in understanding chronic inflammation, how to prevent and treat it, mostly in association with other chronic diseases, such as CKD, remains a challenging problem.
2. Materials and Methods
To identify relevant studies, an accurate literature search was performed using PubMed (MEDLINE), Scopus, and Web of Science databases. These platforms were searched using keywords such as “chronic kidney disease,” “CKD”, “inflammation in CKD”, “pro-resolving mediators in CKD”, “pro-inflammatory factors in CKD”, and “oxidative stress in CKD.” Data from relevant studies were selected based on factors such as relevance of the publication, research methodology, results, statistical significance, and publication date, up to 18 March 2025. In addition, a manual search of the reference lists of key studies was conducted to identify additional relevant information. A total of 204 publications were selected, including systematic reviews, research articles, and meta-analyses.
3. Inflammation in CKD
Inflammation is a key regulator of host defense and plays a pivotal role in tissue repair, regeneration, and maintenance of homeostasis. Several highly conserved pathways govern the initiation and progression of the inflammatory response, ensuring an effective yet controlled reaction to eliminate pathogens and remove debris from damaged tissues, thereby promoting tissue repair. However, if the process is chronic and poorly controlled, it can promote a wide range of complications. Chronic and unresolved inflammation could result in scarring, dysfunction, and organ failure. An inducing stimulus, such as tissue injury or a body, triggers the inflammatory cascade, inducing pro-inflammatory cytokines cascade, with a consequent blood flow increase, upregulation of chemical mediators, and leukocyte infiltration [18]. The discontinuation of this response is then mediated by anti-inflammatory molecules. However, in the presence of discontinuation or endurance of pro-inflammatory mediators, low-grade inflammation may persist. Several factors can contribute to the persistence of a low-grade inflammatory state, including poor diet and nutrition, gut microbiota, childhood infection, and stress [19].
Persistent and unresolved inflammation is also a typical characteristic of numerous chronic conditions, where the inflammatory response is induced by internal triggers (e.g., atherosclerosis, kidney disease, diabetes, metabolic syndrome, non-alcoholic fatty liver disease, and cognitive decline) [3].
Chronic inflammation is one of the most important features of CKD, which is mostly advanced and increasingly associated with deterioration of kidney function. Evidence suggests that C-reactive protein (CRP) is elevated in more than half of CKD patients from stage 3 onwards, with a higher incidence in patients with ESRD [20]. However, the exact timing between the onset of inflammation and CKD remains unclear. It should be noted that the kidney is at higher risk of inflammation-related damage than many other organs [21], as it is one of the most metabolically active organs and is sensitive to many damaging factors, such as oxidative damage and alteration in calcium-phosphorus metabolism and bone disease, hyperhomocysteinemia, and malnutrition with uremic sarcopenia [22]. The kidneys typically receive 25% of the total blood volume but lack the anti-inflammatory defense found in other highly vascularized organs, such as antioxidants or detoxifying agents present in the liver. Additionally, alterations in intrarenal microcirculatory regulation caused by chronic inflammation lead to kidney damage, accelerating the progression of kidney disease. Many pro-inflammatory cytokines, chemokines, and fibrosis mediators are present in the renal tubules, which play a critical role in responding to kidney insults and injuries, and these mediators are tightly regulated. The causes of chronic inflammation in chronic kidney disease (CKD) are multifactorial, influenced by several factors that arise during the disease. These include pro-inflammatory cytokine production, oxidative stress, dialysis, intestinal dysbiosis, uremic toxin retention, and altered adipose tissue metabolism, all of which contribute to inflammation in CKD [22,23,24].
Progressive and chronic renal dysfunction is characterised by an increase in oxidative stress, which can be considered a consequence of the inefficient role of the renal antioxidant defence system, reduced elimination of pro-oxidant substances and replacement therapy in dialysis patients [4]. This oxidative phenomenon also results from the recruitment of monocytes and macrophages, which are a major source of free radicals and, therefore, mediate the phenomena of autophagy, renal tubular epithelial cells apoptosis, and pro-fibrotic mechanisms. The condition of oxidative stress is closely linked to a pro-inflammatory state in the course of kidney damage, as the pro-inflammatory stimulus increases the response of circulating immune cells, and the redox imbalance simultaneously contributes to the increase in the circulating pro-inflammatory cytokines such as interleukin-6 (IL-6) or tumor necrosis factor-alpha (TNF-α) [22,23,24,25,26,27,28,29]. On the other hand, in uremic patients, mostly on maintenance hemodialysis, in addition to a reactive oxygen species (ROS) overproduction, CKD is associated with a significant impairment of antioxidants, e.g., superoxide dismutase (SOD) and glutathione peroxidase (GPx), thus generating an oxidative stress condition [22].
In patients with CKD, oxidative stress is also related to an increased incidence of anaemia, atherosclerosis, and dyslipidaemia due to alterations in protein and lipid components and reduced clearance of triglyceride-rich lipoproteins. These disorders appear to be related in particular to an alteration in the lipoproteins themselves, with a reduction in LDL and IDL catabolism and in HDL plasma concentrations, as well as an inhibition of lipoprotein lipase (LPL) function [23].
Patients with CKD typically undergo hemodialysis or continuous peritoneal dialysis while awaiting a kidney transplant, and these treatments often trigger an inflammatory response. During dialysis, pro-inflammatory cytokines, uremic toxins, and pro-fibrotic markers are upregulated. Some studies suggest there are no significant differences in kidney damage markers between the two therapies, although the use of synthetic membranes in hemodialysis seems more closely associated with the development of inflammatory processes [30]. Additionally, other factors contribute to the inflammatory state of CKD. Overhydration leads to swelling in the intestinal mucosa and increased permeability of the blood–intestinal barrier, resulting in a “leaky gut” condition, where bacterial toxins and pathogens pass from the gastrointestinal tract into the bloodstream. These factors, common in patients with impaired renal function, also heighten the production of reactive oxygen species (ROS) by respiratory burst enzymes [25]. The gut microbiota–kidney interaction is identified as the “gut–kidney axis” [26]. Many factors contribute to the alteration of the microbiota in CKD patients, such as high levels of ammonia, which lower the pH in the gastrointestinal tract; prolonged colon transit; fluid overload; dietary restrictions, which result in reduced fiber consumption; and medications (e.g., proton pump inhibitors, potassium and phosphate binders, oral iron, and antibiotics). Alterations in the healthy gut microbiota can have many consequences, including intestinal dysbiosis, intestinal barrier dysfunction, and bacterial translocation, leading to increased immune system activation. It has been observed that a reduction in the overall bacterial population and diversity in CKD leads to the generation and widespread accumulation of pro-inflammatory uremic toxins, including indoxyl sulfate (IS), p-cresyl sulfate, ammonia, amines, and trimethylamine N-oxide [22].
The “leaky gut” condition in CKD, characterized by disruption of the intestinal epithelial barrier, also promotes the systemic absorption of these toxins, which are responsible for inflammation, oxidative stress, endothelial damage, and protein energy wasting (PEW). Disruption of healthy microbiota can impair gut immune function and, in the worst case, create a state of “immunoparalysis” with systemic consequences of systemic cytokine activation, uremic toxin accumulation, and endotoxemia [27]. Therapeutic approaches aimed at restoring normal gut microbiota in CKD patients may be useful to ameliorate the inflammatory state, oxidative stress malnutrition, and several comorbidities as well [28]. Taken together, all of the above factors contribute to the onset of chronic inflammation with adverse consequences, most notably poor quality of life and increased mortality, including systemic complications, such as CVD and infectious complications. As in other chronic diseases, targeting different levels of the inflammatory cascade could improve the prognosis of patients with CKD, even in more advanced stages of the disease itself, such as ESRD [29,30,31].
4. Pro-Inflammatory Factors in CKD
Elevated levels of cytokines, involved in the innate immune system, are prevalent in CKD patients, and through acute-phase effector proteins, they can control the inflammatory response and mediate some of its downstream effects. In particular, as CKD progresses, these pro-inflammatory molecules exacerbate the signs and symptoms of the uremic syndrome [32].
In this scenario, relevant inflammatory markers in CKD patients are TNF-α, IL-6, IL-1, CRP, adipokines, and adhesion molecules, as well as the ligand CD40, which is implicated in the progression of CKD. CRP is a pentameric protein of hepatic origin that is a reliable biochemical marker of systemic inflammation, as it can also be produced by many inflammatory cells [33]. High levels of CRP are considered a predictor of cardiovascular events, which makes CRP a risk factor for CKD patients [34]. Moreover, elevated levels of CRP are a risk factor also for the common mortality causes in CKD patients in stages 3 and 4 [35]. In fact, in patients with renal diseases, kidney tubular and endothelial cells, as well as macrophages, express high levels of CRP [33]. Several causes are associated with increased CRP in CKD patients [36,37]. One cause is the release of pro-inflammatory cytokines, especially IL-6, which can stimulate the liver to produce CRP [38]. Oxidative stress, which is upregulated in CKD and related to the state of systemic inflammation, also increases CRP levels [39], the elimination of which is impaired by reduced renal clearance, thus favoring its accumulation [33]. In addition, uremia and the retention of uremic toxins due to impaired renal functions increase the inflammatory state, further contributing to the increase of CRP levels [40]. CRP can have two conformational isoforms: the native pentameric CRP (pCRP) and monomeric CRP (mCRP) form, which both exert pro- and anti-inflammatory activity [41]. These two different isoforms of CRP may have different functions under different conditions: pCRP exhibits pro-inflammatory activities on several cell types, such as kidney cells and macrophages, which can lead to renal inflammation and fibrosis. In particular, pCRP from the damaged kidney can promote the infiltration of inflammatory cells and the release of inflammatory factors, such as cytokines, chemokines, and TGF-β1. Although CRP is stable, it can dissociate into the mCRP isoform, exacerbating the progression of inflammation-based diseases through the onset of atherosclerosis and ischemia-reperfusion injury (IRI). Meanwhile, mCRP could limit the amplification of tissue injury by inhibiting renal cell-directed complement activation [33].
The role of CRP in the inflammatory state associated with CKD has been well established using a mouse model of unilateral ureteral obstructive nephropathy (UOO). In this condition, CRP can stimulate NF-κB signaling to induce the expression of monocyte chemotactic protein 1 (MCP-1) and adhesion molecule (ICAM-1) by inducing Nf-kB signaling. This process can lead to macrophage infiltration and renal inflammation, inducing the expression of IL-1 β and TNF-α [42]. This suggests that CRP exerts its pro-inflammatory role in renal injury through direct or indirect activation of NF-κB. The profibrogenic mechanism underlying macrophage infiltration at the site of injury is a key factor in the transition of AKI into CKD, as the macrophage phenotype M2 not only contributes to the repair of the injured tissue but also promotes the fibrotic process. However, it has been shown that macrophage infiltration is also mediated by M1 macrophages, whose polarization is induced by CRP itself [43]. A study involving human transgenic mice affected by CRP has revealed an additional mechanism through which CRP mediates fibrosis and renal inflammation: the activation of Smad3. In kidney damage, Smad3 is involved in TGF-β signaling. In particular, when CRP binds to CD32, Smad3 is activated by the TGF-β1 pathway, as well as the ERK/p38 MAPK-Smad pathway, regulating cell death through the mechanism of Smad3-p27-dependent G1 cell cycle arrest. The incidence of acute kidney injury (AKI) and chronic kidney disease (CKD) is reduced in mice lacking Smad3, suggesting that genetic deletion or pharmacological inhibition of Smad3 may help reduce kidney damage in mouse models of ischemia-induced kidney disease [44]. Modulating CRP could offer a promising therapeutic approach from a clinical perspective. However, the therapeutic potential of anti-CRP treatment in both acute and chronic CKD is still in its early stages, and further studies are needed to establish CRP’s role in this disease.
Another common element in patients with CKD is the elevated plasma level of IL-6 [22,45], one of the most studied cytokines in CKD, which regulates many metabolic and neural processes; it is also well known for its modulation of acute phase proteins and B cells [46]. In CKD, IL-6 is released by renal cells (such as tubular epithelial cells, podocytes, and mesangial cells), endothelial cells, adipose tissue, and lymphocytes under the influence of stimuli such as lipopolysaccharide (LPS), TNF-α, IL-1β, and oxidative stress [47,48]. IL-6 is an interesting molecule in the pathogenesis and progression of CKD as it has both pro- and anti-inflammatory effects since IL-6 is involved in the recruitment of leukocytes, and it can also induce lymphocyte proliferation, as well as their activation and B-cell differentiation [25]. IL-6 also exerts its pro-inflammatory role through increased hypertension, alteration of erythropoiesis, anemia, and upregulation of fibroblast growth factor (FGF23), all of which contribute to increased mortality in patients [49].
In addition to its deleterious role in renal injury, IL-6 produced by renal cells may have an anti-inflammatory effect. In particular, following the signal induced by TNF-α, podocytes secrete IL-6, which induces its immunosuppressive action and, consequently, the recruitment of neutrophils into the endothelium. The pro- and anti-inflammatory action of IL-6 suggests that further studies are needed to clarify its role in acute and chronic diseases through further studies [47]. In particular, in the context of CKD, IL-6 accelerates the progression of CKD and also contributes to its complications, especially CVD [47,48]. This cytokine plays an important role in atherosclerosis, a condition found in dialysis patients and closely associated with the presence of carotid plaques and increased aortic stiffness [50]. IL-6 has been shown to induce endothelial damage also by reducing endothelial expression of nitric oxide synthase (eNOS) and adiponectin and increasing levels of ROS, which would explain the contribution to the development of atherosclerosis itself and vasoconstriction [51,52]. IL-6 can also cause malnutrition by increasing protein catabolism and eating behavior, and indeed, anorexia in patients on hemodialysis or with kidney disease is associated with higher levels of IL-6 [53]. Patients with advanced CKD who develop anemia and concomitant erythropoietin resistance may require erythropoiesis-stimulating agents, such as high-dose erythropoietin. Preclinical findings in animal models suggest a different role between IL-6, its receptor, and its anti-IL-6 receptor in kidney diseases. In fact, neutralization of IL-6 [54] and IL-6 receptor (IL-6R) reduced disease severity, while it has been highlighted that anti-IL-R6 or anti-IL-6 strategies increased the severity of nephritis in the study models [48,55].
IL-1 is a primary pro-inflammatory cytokine with a key role in both acute and chronic inflammation by enhancing inflammatory cell infiltration and increasing the expression of adhesion molecules [56,57,58,59]. Patients with CKD have elevated serum IL-1 levels [60]. Over the years, IL-1 has been hypothesized to underlie many complications in chronic dialysis patients. However, the inflammatory process associated with CKD is known to begin long before the need for chronic dialysis. The inflammasome, a protein structure that causes IL-1 activation, is active in CKD regardless of the etiology.
The primary effects of IL-1 in chronic kidney disease (CKD) are related to mineral and bone disorders [61], but IL-1 also plays a role as a potential mediator of inflammation in heart failure and the cardiovascular complications associated with CKD [62]. It has been suggested that inhibiting IL-1 could reduce cardiovascular risk in CKD patients. In a randomized, double-blind study, 42 adult patients with stage 3–4 CKD were treated with either rilonacept, an IL-1 inhibitor, or a placebo for 12 weeks. The study found that inhibiting IL-1 improved endothelial-dependent dilation (EDD), a key precursor to cardiovascular disease (CVD), in patients with moderate to severe CKD. Specifically, rilonacept treatment was associated with a reduction in systemic inflammation and vascular oxidative stress, as evidenced by lower levels of the oxidative enzyme, NADPH oxidase, and C-reactive protein (CRP), as compared to the placebo group [61].
IL-1 in CKD also induces the expression and production of ICAM-1 by glomerular cells and tubular epithelium, which facilitates the adhesion and infiltration of leukocytes and endothelium into renal tissue and promotes renal fibrosis [63]. It is currently unknown whether IL-1 inhibition in patients with CKD reduces cardiovascular risk in patients with CKD is possible. However, a double-blind, randomized, placebo-controlled, 12-week trial using an IL-1 inhibitor provided the first evidence that IL-1 inhibition improves endothelial-dependent dilation (EDD), a crucial predictor of future cardiovascular events and associated mortality in patients with moderate to severe CKD who do not require chronic dialysis [64]. A clinical trial of canakinumab (a neutralizing antibody against IL-1β) observed a reduction in the rate of cardiovascular events in patients with CKD without modifying renal function, which is clinically relevant, as few therapies have been shown to be effective in reducing the likelihood of cardiovascular events in patients with kidney disease. In the future, it may also be useful to test both the efficacy of canakinumab in patients with end-stage renal disease on dialysis and the efficacy of other anti-inflammatory agents that may be useful in CKD, such as anakinra, an IL-1 receptor antagonist, which can inhibit IL-1β and IL-1α, on which few studies have been conducted [65].
TNF-α is the main regulator of the cytokine cascade and is a potent mediator of the inflammatory response produced by various cells such as macrophages, mesangial cells, and tubular epithelial cells [66] in response to numerous stimuli such as LPS, angiotensin II, calcium-sensitive receptor (CaSR) activation, hypertension, renal failure, glomerulonephritis, diabetic nephropathy, and interstitial tubular nephritis [67]. Kidney-resident dendritic cells have been shown to be particularly involved in the secretion of TNF-α in patients with early renal ischemia-reperfusion injury [68]. In CKD, TNF-α is implicated in the progressive loss of kidney function, and its production is directly related to the severity of renal injury [69,70,71].
Caspases pathway, NF-κB pathway, and mitogen-activated protein kinase (MAPK) pathway are the primary mechanisms activated by TNF-α. The pleiotropic effects of TNF-α are mediated by two tumor necrosis factor receptors (TNFR1 and TNFR2), which activate downstream signaling pathways. Binding of TNF-α to TNFR1 enhances inflammation and tissue damage, whereas binding to TNFR2 is potentially involved in the regulation of immunity, as well as the regeneration of tissue [72]. TNF-α exerts various effects on cells and is also associated with ROS in a variety of cells, including mesangial cells [73], leading to the impairment of the barrier function of the glomerular capillary wall [74]. A member of the TNFR superfamily, commonly referred to as cluster of differentiation 40 (CD40), and its natural ligand, CD40L, are mediators involved in the inflammatory process. In fact, circulating levels of the CD40 receptor and its ligand are closely related to kidney damage. This is evidenced by the fact that CD40 is expressed on the membrane of different types of kidney cells, allowing the interaction between immune and renal cells during the progression of kidney damage. CD40 and CD40L are also expressed by activated T and B cells, but CD40L also exists in a soluble form (sCD40L) [75,76]. Elevated levels of circulating sCD40L are associated with a higher risk of cardiovascular morbidity and mortality, particularly in patients on hemodialysis (HD) [77,78]. In addition, plasma levels of soluble CD40 receptor (sCD40), an isoform of the CD40 receptor, predict the progression of renal dysfunction in patients with CKD [79]. The involvement of CD40 in immune cells has been associated with local kidney inflammatory state, and CD40/CD40L signaling in various renal cell types has been implicated in mediating glomerular permeability, interstitial inflammation, and fibrosis in kidney disease. Several factors in renal cells can modulate CD40 expression. In particular, the binding of CD40 to its ligand plays a key role in the interaction between immune cells and local renal cells in CKD [23]. Therefore, targeting CD40 may be a promising approach and a potential therapeutic method for the treatment of kidney disease. TNF-α is involved in the release of other mediators of the inflammation in CKD, such as IL-1, prostaglandins, oxygen radicals, NO, and platelet-activating factor [80,81] by activating the nuclear factor kappa B (NF-kB) and MAPK signaling pathways. The therapeutic use of anti-TNF monoclonal antibodies, such as the TNF inhibitor, etanercept, is currently approved for the treatment of autoimmune diseases, such as Crohn’s disease and rheumatoid arthritis. An in vivo study investigated the modulation of TNF levels by etanercept and the consequences of its inhibition on the progression of renal damage. In particular, this study in rats with adenosine-induced chronic renal failure showed that etanercept was able to significantly reduce serum TNF-α levels within 2 to 4 weeks of treatment. In addition, etanercept administration can reduce interstitial fibrosis, tubular atrophy, and inflammatory infiltrate induced by adenosine administration after 2 weeks. However, etanercept did not prevent renal fibrosis that occurred 4 weeks after adenosine administration [80]. Given the importance of TNF signaling pathways in the progression of CKD, further studies on the effects of TNF-targeted drugs would be useful. In addition, specific inhibition of TNFR1 or TNFR2 may help to elucidate the pro-inflammatory and immunomodulatory roles of each of the two receptors [82].
In recent years, adipokines have been studied for their role in CKD. Adipokines are proteins released by adipose tissue that play an important role in cell signaling and regulation of metabolism and inflammation, and their dysregulation contributes to the development of disorders associated with obesity, cancer, kidney damage, and cardiovascular disease. White adipocytes in adipose tissue are able to convert surplus energy in triglycerides and secrete several adipokines, including leptin and adiponectin, and TNF-α and IL-6. Brown adipocytes, on the other hand, store energy in the form of small lipid droplets and secrete IL-10, which has beneficial effects on energy expenditure, glucose homeostasis, lipid metabolism, and the inflammatory process [83]. Among the adipokines, leptin and adiponectin are associated with CKD. The pro-inflammatory effect of leptin is an increase in cytokines, such as TNF-alpha, IL-1, and IL-2, which causes an increased incidence of insulin resistance, atherosclerosis, and endothelial dysfunction. Leptin is involved not only in the release of inflammatory cytokines but also in the proliferation of CD4+ T cells and the inhibition of neutrophil chemotaxis, leading to an increased risk of persistent CKD infections [84]. Low levels of adiponectin are a strong risk factor for cardiovascular disease and even hypertension in patients with CKD; in fact, higher levels of adiponectin have been reported to be negatively associated with progression to renal failure [85]. Endothelial dysfunction may also be related to increased synthesis of intercellular adhesion molecules (ICAM)-1, which, together with adiponectin, is indicative of albuminuria. Moreover, the inhibition of ICAM-1 during renal damage is linked to the anti-inflammatory activity of adiponectin [86]. In fact, increased ICAM-1 levels are associated with the accumulation of immune cells in the renal tissue and with both loss of renal function and fibrosis development [87]. Adhesion molecules are important players that, once activated, they are released from the surface of endothelial cells [88] and are promising biomarkers reflecting endothelial activation and vascular inflammatory status [89]. The expression of cell adhesion molecules can be altered by several factors, including hypertension [90] and immunosuppressive therapy [91,92].
In pre-dialysis patients, ICAM-1 is also associated with CRP [93]. Although high levels of VCAM-1 molecules are a predictor of cardiovascular events in patients with CKD, there is little evidence to support this increase in atherosclerotic vascular tissue during CKD. However, a 2021 study on arterial smooth muscle cells showed that VCAM-1 expression in arterial smooth muscle cells was more significant in patients with advanced kidney disease than in those with normal kidney function. This suggests that VCAM-1 could be a cardiovascular prognostic biomarker in patients with CKD [90]. In addition to cytokine production and tissue damage, inflammasome activation in CKD can induce cellular stress responses, including oxidative stress and mitochondrial dysfunction [21]. ROS generated during inflammasome activation contributes to renal oxidative stress, which further exacerbates inflammation and tissue damage in the kidney. However, the relationship between the inflammasomes and chronic renal failure remains controversial. Activation of the inflammasome induces the cleavage and the subsequent activation of caspase-1. This mechanism can regulate the maturation and release of pro-inflammatory cytokines [84].
One of the most studied inflammasomes in relation to CKD is the pyrin domain-containing NOD-like receptor family 3 (NLRP3) inflammasome [83,94]. NLRP3 is a cytosolic sensor protein that forms a multiprotein complex in response to various danger signals, such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [95,96,97].
In CKD, dysregulation of the NLRP3 inflammasome has been implicated in the progression of renal injury and inflammation. Uremic toxins, such as IS, have been shown to induce NLRP3 activation and IL-1β production in renal cells [95]. Furthermore, the inflammasome is activated in dialysis patients with uremia, and this condition is associated with mitochondrial dysfunction, which is essential for NLRP3 inflammasome activation. In this context, excess ROS overstimulates the inflammasome, leading to severe tubulointerstitial damage and renal tubular cell apoptosis [98]. A 2021 study attributed poor NLPR3 activation to the presence of IS, suggesting that a lack of activation of the inflammasome itself causes a reduction in responsiveness to pathogens, with downregulation of caspase-1 and IL-1β levels. This concept is supported by the fact that CKD presents as a state of persistent low-grade inflammation, with continued exposure to pro-inflammatory agents reducing the expression of the inflammasome as well as NF-kB [99]. Macrophages also play an important role in modulating NLPR3 and maintaining renal homeostasis. In fact, they are able to activate NLRP3 through the NF-κB signaling pathway following noxious stimuli. This complex stimulates the activation of caspase-1, resulting in the production of pro-inflammatory cytokines like IL-1β and IL-18. Macrophages in this state polarise from an M1 phenotype, which causes renal inflammation, pyroptosis, and renal fibrosis, to an M2 phenotype, which is decisive in kidney repair and regeneration processes. Previous studies in murine peritoneal macrophages have shown that IS is able to induce an increase in the expression of pro-inflammatory mediators, such as cyclooxygenase (COX-2), inducible nitric oxide synthase (iNOS), as well as the pro-apoptotic protein Bax, an index of macrophage dysfunction [100]. Given the important role of macrophages in the inflammatory process in kidney disease, inhibiting macrophage recruitment and activation may be a good therapeutic strategy for kidney damage, including targeting the inflammasome itself [101].
In patients with CKD, kidney damage is linked to a decrease in the protective ability of antioxidant systems and a disruption in the nuclear translocation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Downregulation of Nrf2 in CKD leads to increased renal fibrosis, tubular damage, and worsening of the disease. In addition, reduced expression of Nrf2 in hemodialysis patients contributes to defective mitophagy, overproduction of ROS, and dysregulation of cellular metabolism, exacerbating CKD [102]. As shown in an in vitro study on intestinal epithelial cells, pro-inflammatory stimuli, such as LPS or IS, are able to influence antioxidant defense cell systems, such as HO-1, NAD(P)H quinone oxidoreductase 1 (NQO1), and SOD, causing a state of oxidative stress not only at the renal level but also at the systemic level and particularly in the gut, which is considered a primary source of inflammation in CKD [103].
The renin-angiotensin-aldosterone system (RAAS) is another key player involved in the promotion of inflammatory processes in CKD through several mechanisms. Specifically, angiotensin II, a key component of the RAAS, exerts direct pro-inflammatory effects by stimulating the production of cytokines, such as IL-6 and TNF-alpha, along with adhesion molecules and chemokines that facilitate leukocyte recruitment and inflammation in the kidneys [104,105]. Indeed, the RAAS interacts with several inflammatory pathways, including NF-Κb and mitogen-activated protein kinase (MAPK) signaling pathways, to amplify inflammatory responses and the expression of pro-inflammatory mediators in kidneys [106,107]. In addition, angiotensin II can modulate immune cell activation and promote the differentiation and activation of immune cells, such as macrophages and T lymphocytes, which further contribute to inflammation and renal damage in CKD [75]. All these processes are schematized in Figure 1 below:
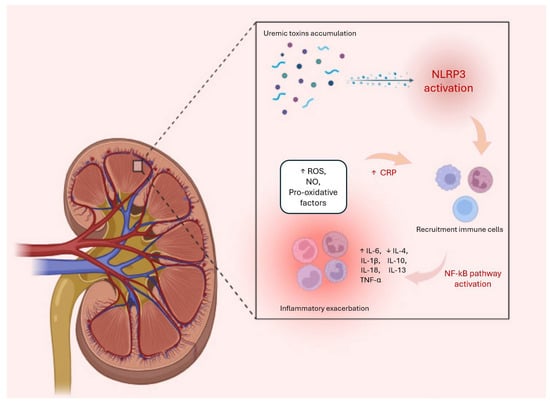
Figure 1.
During the progression of CKD, uremic toxins play a significant role in amplifying the inflammatory response by activating NLRP3, which triggers cellular stress responses. These include increases in nitric oxide (NO), reactive oxygen species (ROS), and levels of pro-inflammatory cytokines, such as IL-1, IL-1β, and TNF-α. Meanwhile, the production of pro-inflammatory cytokines and chemokines promotes the recruitment of leukocytes into the kidney. This process leads to an exacerbation of the inflammatory system, also represented by an increase in CRP levels and a decrease in anti-inflammatory cytokines, such as IL-10. The combination of these processes is responsible for the onset of various systemic complications, including fibrosis, atherosclerosis, anaemia, mineral disorders, and cardiovascular events.
5. Available Treatments
The management of CKD is mainly directed to prevent the disease progression to ESRD and other complications associated with CKD, such as cardiovascular disease (CVD), hypertension, and type 2 diabetes. Despite this, drugs employed in CKD treatment can induce severe side effects in patients. For example, administration of renin–angiotensin–aldosterone system inhibitors (RAASi) is suggested to manage hypertension, but patients on RAASi therapy have to occasionally discontinue their treatment or reduce dosage because RAASi can induce hyperkalaemia [108,109]. In CKD, treatment is fundamental to consider the management of inflammation since CKD is associated with high levels of pro-inflammatory factors. In fact, investigations have been conducted on the effectiveness of TNF-α inhibitors, such as etanercept, which showed a significant decrease in rat serum TNF-α levels, even if unsuccessful in preventing renal fibrosis [110]. Also, statin therapy is contemplated in therapies for CKD patients, especially to reduce the risk of CVD [111], addressing key points of inflammation. In fact, statins can induce inhibition of the NLPR3 inflammasome, IL-1β production; decrease serum c-reactive protein (CRP) and ROS levels; avoid NF-κB accumulation; and increase catalase and superoxide dismutase activity [112]. However, statin drugs are associated with musculoskeletal side effects, such as rhabdomyolysis, which can induce tubular injury and ischaemia [113] and increase the levels of creatine kinase, which exacerbate renal impairment with myoglobinuria and electrolyte alteration [114].
Recently, results were reported from a phase 2 trial, the RESCUE study (Reduction in Inflammation in Patients with Advanced Chronic Renal Using Antibody-Medicated IL-6 Inhibition), on patients with advanced CKD who showed high C-reactive protein levels, which investigated the anti-inflammatory activity of ziltivekimab, a human anti-IL-6 monoclonal antibody. Subjects treated with ziltivekimab were shown to have a reduced need for erythropoiesis-stimulating agents. Although these results with ziltivekimab do not provide evidence of a direct benefit of Ziltivekimab on renal function, it is believed that treatment with ziltivekimab may lead to a reduced risk of CVD in patients with CKD. Although the results of the study are not enough to affirm the direct benefit of ziltivekimab on renal function, it is believed that treatment with ziltivekimab may lead to a reduced risk of CVD in patients with CKD. Targeting IL-6 in CKD appears to be a promising avenue, as it has a key role in the progression of renal disease, as well as in associated complications. However, the IL-6 inhibitors currently approved for clinically inhibit IL-6 release in CKD therapy are not promising at this time [115].
Preclinical findings in animal models suggest a different role between IL-6, its receptor, and its anti-IL-6 receptor in kidney diseases. In fact, neutralization of IL-6 [55] and IL-6 receptor (IL-6R) reduced disease severity, while it has been highlighted that anti-IL-R6 or anti-IL-6 therapeutical approaches can worsen nephritis in the study models [116]. CKD patients are required to follow a diet low in protein and sodium in order to control renal dysfunction, hypertension, and complications associated with CKD. In fact, to prevent diabetes and cardiovascular complications, changes in patients’ eating habits are suggested, such as introducing the Mediterranean diet [117] or PUFA supplementation, which results are beneficial in decreasing oxidative stress and cytokine levels involved in inflammatory pathways in CKD [118]. A healthy diet is obviously associated with a correct lifestyle, with reduced alcohol consumption, smoking, increased physical activity, and specific dietary behaviours for each stage of the disease [119]. Very often, CKD patients are subject to polypharmacy, and concomitant use of drugs such as antibiotics can have a negative impact on the gut microbiota. For this reason, patients themselves are advised to follow a diet rich in fibre, which promotes the growth of saccharolytic SCFA-producing bacteria and concomitant reduction of intestinal-derived uremic toxins; this approach may in fact preserve the integrity of the intestinal barrier and mucus production, thereby reducing inflammation [120]. Another therapeutic strategy is the supplementation of prebiotics, probiotics, and symbiotics; however, studies are limited by small sample sizes, variations in doses, strains, outcome measures, and duration of intervention. Agents such as activated charcoal (e.g., AST-120) have also been shown to significantly reduce oxidative stress and inflammation and IS and p-CS concentrations in animal models. [121]. Despite the benefits of a healthy diet and available drug therapies, more research needs to be conducted, considering that this kind of intervention can only slow the progression of ESRD. One target of research in this context could be strategies aimed at fostering the development of a symbiotic microbiota to further investigate the effect on CKD progression. However, therapeutic choices could be influenced by an accurate and early diagnosis in order to intervene early in the progression of the disease; in this regard, genetic tests could help in identifying the aetiology and therapy to be chosen. This approach would also make it possible to have a personalised therapy for each patient that can take into account the risk factors, lifestyle, environmental factors, and co-morbidities and drug tolerance associated with that same patient. Pharmacogenomic studies can provide important information on drug selection and dosage choice to reduce side effects, an important aspect for patients with reduced renal function. Despite its potential, personalised medicine has not yet been widely adopted in the clinical setting due to high costs and problems with the accessibility of genetic testing and associated patient data [122].
6. Inflammatory Resolution Mediators
To prevent the transition from acute to chronic inflammation, it is essential to regulate the inflammatory response. Resolution of inflammation is a spatially and temporally controlled process characterised by reduction of leukocyte infiltration, apoptosis of neutrophils at the site of injury, and efferocytosis by macrophages of apoptotic cells and debris without systemic host immune suppression. The events involved in the resolution process were first presented by Robbins and Cotran [106] and complemented by Savill and collaborators [107]. They suggested that in the resolution phase, the inflammatory exudate is reorganised, with macrophages engaging in phagocytic activity to remove dead cells and debris caused by inflammation. Later, other researchers expanded on this idea, identifying a possible role for lipid mediators derived from omega-6 (n-6) and omega-3 (n-3) polyunsaturated fatty acids; in 2002, Serhan and co-workers studied inflammatory cellular exudate to elucidate the mechanism of inflammatory response in vitro and in animal models [123,124], defining the resolution of inflammation as a dynamic and hierarchical process in which lipid-derived mediators play a central role.
In particular, specialized pro-resolving lipid mediators (SPMs) are endogenous bioactive mediators that stimulate self-limited innate immune responses, kill and clear microbes, and protect organs. Compared to most anti-inflammatory agents, SPMs show potent resolving effects in various animal models of disease; therefore, these molecules, together with their receptors, represent interesting targets for the control of inflammation [125]. In the early stages of inflammation, there is localized production of pro-inflammatory cytokines (such as TNF, IL-1β, and IL-6) and eicosanoids, which facilitate the recruitment of neutrophils to the site of tissue damage. However, for the resolution process to be effective, it is crucial to halt granulocyte recruitment and simultaneously promote the recruitment and differentiation of macrophages from a pro-inflammatory to an anti-inflammatory phenotype [126]. This macrophage polarization stimulates efferocytosis, leading to the production of specialized pro-resolving mediators (SPMs), which contribute to restoring vascular integrity and regenerating damaged tissues [127]. SPMs are classified into different subgroups according to their origin and structures, as shown in the figure below (Figure 2):
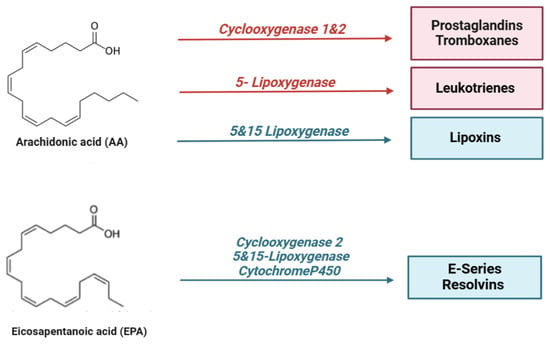
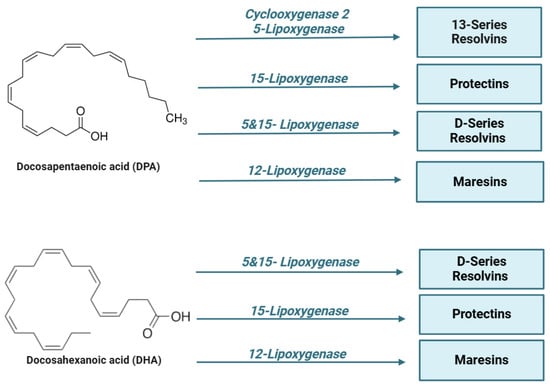
Figure 2.
Lipid mediators are derived from ω-6 and ω-3 polyunsaturated fatty acids (PUFAs). During the early initiation phase of inflammation, cyclooxygenase (COX) and lipoxygenase (LO) enzymes begin the conversion of the ω-6 polyunsaturated fatty acid, arachidonic acid, into a series of bioactive lipid molecules, such as prostaglandins (PGs), thromboxanes (TXs), and leukotrienes (LTs). During the resolution phase of inflammation, specialized pro-resolving lipid mediators (SPMs) are synthetized. Arachidonic acid produces lipoxins (LXs), while eicosapentaenoic acid (EPA), n-docosapentaenoic acid (DPA), and docosahexaenoic acid (DHA) produce lipid mediators, including resolvins (RVs), protectins (PD), and maresins (MaRs). Of note, the mouse 15-lipoxygenase carries both 12- and 15-lipoxygenase activity.
Therapeutic strategies that promote the resolution of inflammation offer a promising approach in the management of CKD. The following is a summary of preclinical and clinical studies in experimental models of kidney disease, which suggest that ω-6 and ω-3 polyunsaturated fatty acids and derivatives are useful in preserving renal function.
6.1. Arachidonic Acid Derivatives in CKD
Arachidonic acid (AA) is a ω-6 polyunsaturated fatty acid found in phospholipid form in the cell membrane [128]. In response to a cell-damaging stimulus, arachidonic acid (AA) is released from phospholipids by the action of phospholipase A2 and phospholipase C in its free form. This free AA serves as a precursor for pro-inflammatory bioactive mediators through three main metabolic routes: via the cyclooxygenase (COX) pathway, AA is converted into prostaglandins (PGs) and thromboxanes (TXs); through the lipoxygenase (LOX) pathway, leukotrienes (LTs) and lipoxins (LXs) are produced; additionally, AA generates epoxyeicosatrienoic acids (EETs) or hydroxyeicosatetraenoic acids (HETEs) via the LOX pathway. Together, these metabolites, also known as eicosanoids, play a central role in modulating the inflammatory process [129]. The metabolism of arachidonic acid (AA) is involved in various physiological and pathological processes in the body. AA affects the fluidity and permeability of cell membranes, regulates platelet function, and modulates immune system activation. Additionally, AA and its metabolites play a crucial role in the primary mechanisms contributing to chronic kidney damage by influencing glomerular and tubular function, podocyte pathophysiology, and the progression of renal fibrosis. In vitro experiments on human mesangial cell cultures incubated with arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA) demonstrated that AA could counteract the angiotensin II-induced upregulation of TGF-β, fibronectin 1 (FN1), connective tissue growth factor (CTGF), and collagen IV gene expression, thus activating the mechanisms responsible for renal damage. Treatment of the same cells with EPA or DHA suppressed the upregulation induced by Ang II and AA, suggesting that the balance between different omega-3 and omega-6 fatty acids may negatively or positively affect kidney damage [127]. Furthermore, in immortalised human podocyte cultures, AA treatment activated protein kinase A, which promotes the activation of the adaptor protein c-Abl and the phosphorylation of nephrine, which in its phosphorylated form mediates podocyte survival and function. When dephosphorylated, nephrine releases c-Abl, which accelerates actin and cytoskeletal remodeling, leading to podocyte lesion formation, a critical event in kidney injury [130].
Kidney damage is closely linked to the production of prostaglandins. A well-established connection exists between prostaglandins (PGs) and tubulointerstitial damage, as well as glomerulonephritis. PGE2, the main product of the COX-2 pathway in the kidney, is found to be elevated in conditions such as diabetic nephropathy [131] and plays a crucial role in renal hemodynamics, renin release, and renal tubular sodium/water reabsorption. In contrast, PGI2 and PGE1 help relax blood vessels and protect renal tissue from hypoxia-induced damage. Hydroxyeicosatetraenoic acids (HETEs), which are formed from arachidonic acid (AA) via the LOX pathway, can activate PPARγ in macrophages, induce apoptosis of tubulointerstitial cells, and contribute to inflammation and renal tubular fibrosis [128]. HETEs also play a role in regulating renal ion transport during renal inflammation. Specifically, 20-HETE modulates cell proliferation, angiogenesis, and serves as a key regulator of renal function [132]. However, not all metabolites derived from AA metabolism have pro-inflammatory effects, especially in renal damage; lipoxins, which are derived from the same precursor, have a pro-resolving action.
Lipoxins
Arachidonic acid (AA). Among all the SPMs that promote the resolution of inflammation, the LX lipoxins were the first to be identified [133]; LXs were isolated from human leukocytes by Serhan and co-workers, and they named these mediators “lipoxins” because they are “lipoxygenase interaction products” [134].
LX, together with their synthetic analogues, showed anti-inflammatory and pro-resolving effects on in vitro and in vivo models [135]. However, although the native lipoxins, LXA4 and LXB4, have anti-inflammatory and pro-resolving effects, they are rapidly metabolically inactivated via PG dehydrogenase-mediated oxidation and through reduction reactions, which limits their potential application in therapy development [136]. As a result, many LX analogues have been chemically synthesized to prolong the half-life of LX in vivo, and some of these analogues show better stability and more potent activity than native LXs [137]. LXs have several actions in the inflammatory process: they inhibit the migration of neutrophils across the endothelium and their interactions with epithelial cells. Moreover, LXs inhibit neutrophils to release the toxic products of their granules [138] and the formation of peroxynitrite at the site of inflammation, lowering the levels of nuclear factor κB (NFκB) and activator protein-1 (AP-1) in the nucleus, thereby reducing the production of IL-8 [139]. In addition, LXA4 production is involved in the inhibition of superoxide release and colonocyte apoptosis, which results in pro-resolving during the inflammatory process [140]. These anti-inflammatory effects are mediated by lipoxins through signals activated by binding to the high-affinity G protein-coupled lipoxin A4 receptor (ALX)/formyl peptide receptor (FPR2) [141]. Other receptors that interact with LX include G protein-coupled receptor 32 (GPR32), the Aryl hydrocarbon receptor, estrogen receptor [142], and the high-affinity cysteinyl leukotriene receptor [143]. LXs have been shown to be pro-resolving in CKD and in various associated CKD complications. Specifically, lipoxins exert their effects by modulating various signalling pathways in the kidney. For example, in the presence of lipoxin in epithelial cells, an increase in the level of let-7c miRNA is observed [144]. Fibrotic damage can also be negatively regulated by LXA4 through modulation of the transcription factor EGR-1, which regulates the transcription of the pro-inflammatory cytokines, IL-2 and TNFα, in T cells. The transcriptional repressor NAB1 downregulates EGR1 expression. In neutrophils, LXs have been shown to upregulate NAB1 expression and thereby reduce EGR1 levels [145,146]. LXs and their analogues have also proven effective in treating acute renal failure in dendritic cell mouse models. When mice with renal failure were treated with lipoxin, there was an increase in the mRNA levels of cytokine signaling suppressors (SOCS-1, 2), which bind to other cytokine receptors and inhibit their pro-inflammatory effects [147]. However, the precise impact of LXs on renal dendritic cells remains to be fully understood. In recent years, the role of LX in CKD associated with complications such as diabetes has also been investigated: in a 2018 study using mice with diabetes-associated CKD as an in vivo experimental model, LXA4 was shown to suppress diabetes-induced kidney damage, as evidenced by the reduction in albuminuria. LX also attenuated glomerular dilatation and mesangial matrix deposition [146]. The mice were fed a high-fat diet for 3 months to induce this pathological condition and were treated with LX between the fifth and twelfth week of the study. They stimulated macrophage polarization toward a resolution phenotype. In particular, LXA4 attenuated the diet-induced increase in the proportion of pro-inflammatory M1 macrophages and partially restored the population proportion of anti-inflammatory M2 macrophages. LXA4 treatment also resulted in an obesity-induced reduction in the expression of the pro-inflammatory cytokine TNF-α and partially restored adiponectin levels that had been reduced by the diet. LXA4 was also shown to significantly attenuate obesity-induced albuminuria and H2O2-mediated oxidative damage, indicating protection against kidney damage by attenuating free radical production [148]. LX may, therefore, represent a therapeutic strategy for both CKD and its associated inflammatory complications.
6.2. Eicosapentenoic Acid Derivatives in CKD
Eicosapentaenoic acid (EPA) is an omega-3 polyunsaturated fatty acid known as an anti-hyperlipidemic agent, which is known to have antioxidant or anti-inflammatory effects in cardiovascular disease and atherosclerosis; it also appears to reduce levels of pro-inflammatory cytokines and chemokines [149]. Serum EPA levels in patients with kidney disease are due to dialysis treatment, inflammation, and associated metabolic changes, and this deficiency is negatively correlated with cardiovascular events. Under normal conditions, ω-3 PUFA modulates lipid metabolism, improves cardiovascular parameters, reduces inflammation and oxidative stress, and is associated with a reduction in progression to end-stage renal disease. Interestingly, this effect has been seen to a greater extent in patients undergoing dialysis, while studies demonstrating the effect of supplementation of these compounds in patients not undergoing renal replacement therapy are still needed [150].
In this context, the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) advises incorporating food sources rich in omega-3 polyunsaturated fatty acids (PUFAs) into the diet at least twice a week.
Three trials involving more than 800 hemodialysis patients suggest that EPA therapy may improve clinical outcomes in these patients, in particular, reducing mortality related to cardiovascular events. Among these studies mentioned above, in particular, a randomized, multicenter, double-blind, controlled trial involving 206 patients carried out by Svensson and co-workers showed that the treatment of chronic hemodialysis patients with purified EPA + DHA (daily EPA dose less than 1 g/day) did not lead to a significant reduction in mortality related to cardiovascular events but did result in a significant reduction in the incidence of secondary myocardial infarction. The authors hypothesize that the reduction in myocardial infarction may be due to the anti-inflammatory antithrombotic or antioxidant effects of omega-3 PUFAs [151]. EPA has also been shown to reduce lipotoxicity at the level of renal proximal tubular cells (PTEC) in obesity-related kidney diseases. When there is lipid overload at the cellular level due to suppression of the autophagic system, lipids accumulate, resulting in renal lipotoxicity. EPA supplementation in mice fed a high-fat diet (HFD) reduced lipotoxicity in PTECs (reduction in lysosomal phospholipid accumulation, inflammation, and fibrosis). This suggests a potential use of EPA in complications associated with kidney injury [152].
6.2.1. Resolvins
Resolvins are metabolites produced from the ω-3 polyunsaturated fatty acids, DHA, EPA, and DPA through enzymatic processes catalyzed by lipoxygenases (LOXs), cytochrome P450s (CYP450s), and cyclooxygenases (COXs) [153]. Resolvins (RVs) are categorized into two primary classes: E-series resolvins, which are derived from eicosapentaenoic acid (EPA), and D-series resolvins, derived from docosahexaenoic acid (DHA). E-series resolvins (RvE1 and RvE2) are produced by 5-lipoxygenase or COX-II in vascular endothelial cells. These resolvins can also be acetylated by aspirin to form aspirin-triggered resolvins (AT-Rv) [154,155,156].
6.2.2. E-Series Resolvins
E-series resolvins are currently in clinical trials for eye, lung, kidney, skin, and bowel diseases [157]. RvE1 at nanomolar concentrations has been shown to reduce neutrophil infiltration into inflammatory sites by 50–70% in a TNF-α-induced dorsal air pouch model of inflammation. RvE1 can also be metabolized through 12-oxo-dehydrogenation, producing a biologically inactive product that may serve as a potential biomarker for resolving activity [158]. The anti-inflammatory effects of RvE1 have been demonstrated in a variety of pathological settings; RvE1 reduces IL-12 production by dendritic cells [159] and, in the skin, reduces contact hypersensitivity [160]. RvE1 and RvE2 are potent stimulators of IL-10 and phagocytosis [161].
However, a study reported that RvE1 can also exert direct antifibrotic effects in kidney disease: in a model of unilateral ureteral obstruction (UUO)-induced tissue fibrosis, RvE1 can inhibit fibroblast proliferation in vivo and in vitro. Indeed, administration of RvE1 to mice significantly reduced the accumulation of α-smooth actin myofibroblasts (SMA) and IV collagen deposition over six days. Moreover, at 2–4 days after UUO induction, resolvin administration inhibited the proliferation of myofibroblasts [162]. Therefore, further studies on the effect of RvE1 may be useful to identify the potential antifibrotic effect of this resolvin in renal injury.
6.3. Docosohexaenoic Acid Derivatives in CKD
DHA is a long-chain, highly unsaturated omega-3 (N-3) fatty acid and is also known as the precursor of maresins, protectins, and D-series resolvins, which attenuate inflammatory disorders by promoting the resolution of inflammation. DHA is crucial in communication mechanisms, membrane structure and function, synthesis of lipid mediators, cell signaling, and gene expression [163]. DHA is effective in counteracting the chronic inflammatory state found in patients with CKD. In fact, a 2018 study demonstrated the combined effects of n-3 fatty acids (EPA and DHA) and CoQ supplementation on neutrophil release in patients with CKD. In a double-blind controlled study on 85 CKD patients, supplements of n-3 FA (4 g) or CoQ (200 mg) or control (4 g olive oil) have been randomly daily administered for 8 weeks. N-3 FA results to have a potential role in limiting the inflammation since patients who received N-3 FA supplementation exhibited higher levels of LTB5 and SPMs released by neutrophils and lower plasm levels of MPO. These findings offer additional insight into the mechanisms through which omega-3 fatty acids (n-3 FAs) may help reduce low-grade inflammation in patients with CKD [164].
17-HDHA is a metabolite of DHA and a precursor of protectins (PD1), which has been shown to be clinically important in a number of immune mechanisms involved in the progression of kidney disease [165,166]. The crucial role of DHA in CKD was demonstrated using an in vivo experimental model in which rats underwent nephrectomy, which resulted in increased urinary albumin excretion, reactive oxygen species, inflammation, and tubulointerstitial fibrosis. Rats on DHA supplementation exhibited an improvement in renal dysfunction and fibrosis by showing higher levels of DHA, EPA, and their metabolites and decreased levels of pro-inflammatory cytokines and cells in the renal inflamed site. Moreover, rats on DHA supplementation exhibited lower IS levels in the kidney, which results are particularly interesting since IS is a protein-bound uremic toxin. In fact, the authors suggested a potential role of DHA as a competitor for IS binding. Although further clinical trials are needed, a potential suppression of the CKD progression and protection against tubular injury could be realized by administrating DHA in order to reduce the levels of PTECs while enhancing IS-induced oxidative stress [167].
DHA appears to slow the progression of kidney failure by reducing IS accumulation and associated oxidative stress. A DHA diet appears to suppress fibrosis in the kidney, as shown in a recent study from 2023. The authors had previously studied the effect of a DHA diet, which was demonstrated in the following study; after rats were nephrectomised for 4 weeks, a DHA diet was administered, which attenuated urinary excretion of albumin, ROS levels in the kidney, and plasma IS and TNF- α levels, which were increased 4 weeks after nephrectomy. The authors also highlighted the role of metabolites derived from DHA (protectin D) and EPA (18-HEPE) in reducing inflammation, oxidative stress, and fibrosis 4 weeks after nephrectomy [168].
In 2022, Kobayashi S. et al. conducted an initial study in CKD cats that were orally administered fish oil enriched with DHA in liquid form for 28 days [169]. Although this study has a limitation regarding the number of animals involved, it suggests a beneficial action of DHA in reducing urinary SDMA, UPC, and NAG levels, parameters of renal function. In addition, DHA appears to have reduced arachidonic acid levels in the same study, and consequently, the authors hypothesised that docosanoids, i.e., bioactive lipids converted by DHA from oxidative enzymes in vivo, may antagonise inflammatory eicosanoids, thus contributing to the kidney-protective effect.
A 2017 study had also shown that DHA can inhibit TGFβ1-stimulated fibroblast activation; these cells play an essential role in the production of extracellular matrix and the progression of renal fibrosis. In the study, this effect was demonstrated in NRK-49F cells and treated for 30 min with DHA and subsequently with TGFβ1 for 12, 24, and 48 h. TGFβ1, at a concentration of 2 ng/mL, stimulates NRK-49F cell activation, resulting in increased α-SMA, fibronectomy, and type I collagen expression. DHA significantly reduced TGFβ1-induced fibroblast activation in a time- and dose-dependent manner, and these data suggest a positive action in modulating renal fibrosis [170].
6.3.1. Maresins
Maresins (MaRs) are lipid mediators synthesized by macrophages from DHA through the action of human 12-lipoxygenase (12-LOX). These mediators have anti-inflammatory and pro-resolving properties, which may support tissue regeneration and potentially serve as therapeutic agents in chronic inflammatory diseases [171]. MARs are thought to have a protective role since they can inhibit the macrophage function [172]. In fact, MaRs can release several substances, which probably exert a pro-resolving action by enhancing phagocytosis and efferocytosis when human macrophages are incubated with MaRs. Additionally, MaRs promote tissue regeneration by downregulating pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, thus facilitating the resolution of the inflammatory process [173]. MaR1, in particular, has been shown to limit neutrophil infiltration and reduce the production and release of CXCL1, a key chemokine involved in neutrophil recruitment. Furthermore, MaR1 promotes neutrophil apoptosis, helping to resolve the inflammatory response [174].
MaRs also play a crucial role in the early stages of kidney injury, including the acute phase, when hospitalized patients experience critical complications such as sepsis. In vivo studies have shown that treatment with MaR1 can be a therapeutic strategy in this area: in an experimental model of acute septic kidney injury in vivo on male mice, it has been shown that in the presence of a pro-inflammatory stimulation by a single intraperitoneal injection of LPS (10 mg/kg), the administration of MaR1 reduced the levels of TNF-α, IL-6, and IL-1β; downregulated the expression of BAX and caspase-3; and increased the expression of BCL-2 in damaged renal tissue. In addition, the same study highlighted the antioxidant action of MaR1: it improved the activity of superoxide dismutase in renal tissues and inhibited ROS production. In conclusion, MaR1 attenuated LPS-induced renal inflammation and oxidative stress by inhibiting the NOX4/ROS/NF-κB p65 pathway [175]. In the early stages of kidney injury, MaR1 treatment is also able to enhance antioxidant defenses through increased nuclear translocation of Nrf2 and expression of HO-1, SOD, and NQO-1, thereby attenuating oxidative stress [176]. Another study also investigated the protective role of MaR-1 in the pathogenesis of diabetes-associated chronic kidney injury and its clinical relevance, using male C57BL/6 J mice as an in vivo experimental model in which diabetes was induced by a high-fat diet combined with streptozotocin (STZ) treatment. Human renal proximal tubular epithelial cells (HK-2) were then used as an in vitro experimental model, and serum MaR1 levels were finally analyzed in a clinical study involving 104 subjects with type 2 diabetes (T2DM). In these subjects, serum MaR1 levels decreased significantly during the course of kidney disease compared to unaffected subjects. In CKD patients, LGR6 levels were significantly lower compared to healthy controls as in mouse models, but MaR1 treatment restored these levels at both protein and mRNA levels. Furthermore, in high glucose-treated cell models, MaR1 counteracted the reduction of LGR6, suggesting that the protective effects of MaR1 may be linked to the enhancement of LGR6 function. Knocking down LGR6 in cells blocked the beneficial effects of MaR1 on reducing ROS and inflammation, further confirming that LGR6 is essential for the actions of MaR1, reduced hyperglycemia, and slowed the pathological progression of kidney damage. The fact that this study showed that decreased serum MaR1 levels are closely related to the development of CKD in the presence of diabetes suggested that MaR-1 can be considered a predictor and a potential therapeutic target for this disease [177]. One of the key mechanisms through which MaR1 exerts its effects is an antioxidant pathway mediated by cAMP and SOD2. MaR1 treatment increased cAMP and SOD2 levels in both cells and kidneys of mouse models, reducing ROS accumulation and modulating inflammation. These findings support the hypothesis that MaR1 ameliorates DKD through an antioxidant mechanism mediated by the LGR6 receptor, specifically by modulating the cAMP/SOD2 pathway [178].
6.3.2. Protectins
Protectins (PDs) are lipid mediators produced by neutrophils, macrophages, and T cells from docosahexaenoic acid (DHA). They are of great biomedical interest because of their ability to limit the extent and duration of the acute inflammatory response and to regulate tissue regeneration [179]. Among several products derived from DHA metabolism, a lipidomic study in 2002 by Serhan et al. also reported biosynthetic and structural information of a product, later named protectin D1 (PD1) [178]. PD1 is a compound of interest since it has a pro-resolving effect on the inflammatory state and defends the host against bacterial and viral infections [179,180]. The loss of balance between anti-inflammatory and pro-inflammatory factors is often at the basis of the loss of kidney function in CKD, but the experimental evidence highlighted how the administration of PD1 can contribute to the reduction of kidney damage, leading to an anti-inflammatory response that is critical for the resolution of kidney damage [181]. In kidney disease, ischemic-inflammatory damage to the kidney plays an important role in regulating this balance; this has been demonstrated using a mouse model of ischemic kidney injury, in which polymorphonuclear cells produce both bioactive D-series and DHA precursor during the inflammatory response. First, the precursor DHA was shown to be present in the kidney before and after ischaemic damage. However, greater amounts of PDs and resolvins were produced in the post-ischaemic kidney, and the administration of these compounds protected the kidney from further damage by reducing leukocyte influx and the increase in serum creatinine with a reduction in post-ischaemic renal fibrosis [182]. Based on these findings, it is likely that endogenous anti-inflammatory compounds involved in resolving the inflammatory response play a crucial role in the progression of both acute and chronic kidney injury. Notably, after the onset of ischemic injury, higher doses of PD1 are necessary to achieve therapeutic levels in damaged kidneys compared to the doses of RVs required. Further research is needed, as a single dose of 10 μg of PD1 was selected for the post-injury phase of the study [183].
PD1 has also been shown to have renoprotective and antioxidant effects in CKD, in particular, acting on the heme oxygenase-1 (HO-1) pathway, which has been shown to be protective both in rodent and in human kidney disease models [184]. In fact, HO-1 is an enzyme involved in the degradation of toxic heme to bile pigment biliverdin and carbon monoxide (CO), showing antioxidant and anti-inflammatory properties, as well as to admeliorate the circulatory flow in the rat model [164]. Furthermore, a study by Hassan and Gronert on in vitro and in vivo models suggests that PD-1 induces the HO-1 pathway in mesangial cells [185].
However, the administration of PDs, which retain anti-inflammatory and antifibrotic properties, could be therapeutically valuable in treating renal injury. This approach may facilitate the early resolution of damage and help prevent the irreversible progression of lesions in chronic kidney disease (CKD).
6.3.3. D-Series Resolvins
Endothelial cells, located in the inner wall of blood vessels, are a target of RvD1 and RvD2. RvD1 is among the most extensively studied resolvins due to its demonstrated pro-resolving effects in various acute inflammatory diseases in animal models. These conditions include lung injury, kidney damage, pancreatitis, hepatitis, and peritonitis [186]. RvD1 also upregulates the expression of ZO-1, occludin, and narrow-junction proteins to protect against endothelial barrier dysfunction via the IκBα pathway [187]. RvD1 can enhance macrophage efferocytosis of apoptotic neutrophils. The same effect on M2 macrophages is also exerted by RvD3 and RvD5 [188]. RvD1 has also effects on podocytes during nephropathy that is characterized by proteinuria and glomerulosclerosis, induced by adriamycin treatment. This injury results in a progressive loss of synaptopodine, an actin-associated protein that plays a role in the shape and motility of actin-based cells in renal podocytes [189]. Synaptopodine downregulation in course of nephropathy can be modulated by early administration of RvD1. Similarly, in a podocyte cell line, resolvin D1 was able to prevent synaptodin downregulation induced by TNF-α treatment [190]. Supplementation with 4 g/day of n-3 fatty acids for 8 weeks enhances the synthesis of SPMs (specialized pro-resolving mediators) that promote inflammation resolution. Specifically, n-3 fatty acids significantly increase the levels of RvD1 and the upstream precursors of RvE and RvD, 18-HEPE, and 17-HDHA, respectively [191]. RvD1 has been shown to have a protective effect against intestinal damage mediated by certain drugs, such as NSAIDs, as demonstrated by an in vivo study conducted in 2021. Indeed, this study showed that RvD1 derived from 12/15-lipoxygenase contributes to mucoprotection against NSAID-induced small intestinal damage by exerting anti-inflammatory effects through activation of the ALX/FPR2 receptor [192]. Furthermore, exogenous supplementation of RvD1 protected the small intestine from NSAID-induced damage. In contrast, inhibiting 12/15-lipoxygenase, the enzyme responsible for producing resolvin D1 and blocking ALX/FPR2, worsened NSAID-induced small intestinal damage. These findings align with previous studies suggesting that RvD1 protects against NSAID-induced intestinal damage by suppressing the expression of TNF-α and IL-1β through the inhibition of the TLR4-NF-κB and NLRP3 inflammasome signaling pathways [193]. This mechanism on toll-like receptors, which are implicated in the progression of kidney damage, was also highlighted in another study, underlining the important role of RvD1 in counteracting disease progression [194]. RvD2 regulates NO production and adhesion molecule receptor expression in endothelial cells [195].
In a study conducted in 2014 by Katakura M. et al., it was shown that dietary supplementation of a ω-3 PUFA (EPA + DHA) formulation in SHRcp rats (used as an animal model of metabolic syndrome) was able to attenuate renal dysfunction and tissue damage [196]. Supplementation with EPA + DHA would also appear to stimulate the levels of DHA in plasma and its derivatives (PD1, RvD1, and RvD2) in the kidneys in contrast to supplementation with EPA alone. The fact that the increased production of DHA derivatives was associated with a reduction in glomerular damage suggests that these compounds could contribute to the suppression of the progression of renal dysfunction in metabolic syndrome types.
7. Conclusions
Inflammation is a central element in kidney disease, and its resolution is crucial to repristinate the physiological renal function after damage. In this context, it is important to note that SPMs play an important role in the resolution of the inflammatory response. New therapeutic strategies based on SPM aim to reduce the inflammatory response associated with kidney disease, acting on several fronts: limiting neutrophil recruitment (PMN), relieving pain, modulating fibrosis, stimulating macrophage-mediated efferocytosis, and preserving renal function through the activation of antioxidant pathways (Figure 3).
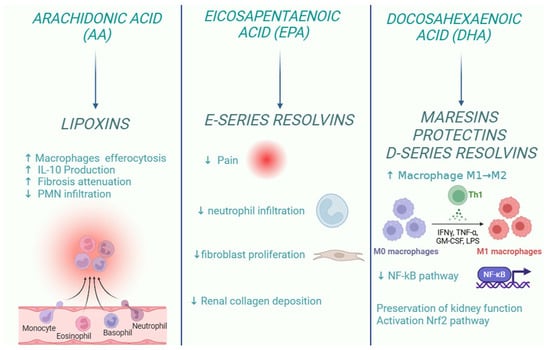
Figure 3.
Role of specialized pro-resolving lipid mediators in the resolution of kidney inflammation.
Supplementation with SPM precursors has attracted increasing interest as a therapeutic approach in acute and chronic inflammatory diseases, as evidence suggests that levels of these mediators are reduced in patients suffering from inflammatory and immune disorders. In particular, in the context of renal disease, experimental evidence suggests that SPMs, both endogenous and synthetic, are effective in preserving renal function (Table 1).

Table 1.
Summary of studies based on SPMs in various experimental models of renal disease.
CKD currently represents a serious global health challenge, with a shortage of effective treatments. Chronic dialysis is a key strategy to support many patients, especially in the advanced stages of kidney disease. However, several toxic products are not easily eliminated by this treatment. Moreover, the high costs associated with dialysis highlight the urgency of developing alternative therapies. Renal transplantation remains a solution in the most severe cases, but the increasing shortage of organs and limited access to effective immunosuppressive drugs, which could reduce side effects, emphasise the importance of early diagnosis. However, this therapy is associated with risk factors, such as tumour occurrence and fibrosis, and the long-term effects of these treatments are unknown. Early diagnosis reduces the need for complex treatments and also reduces related complications. Research is exploring new therapeutic strategies, such as the use of embryonic stem cells for renal regeneration or gene therapy. Stem cell therapies, in particular, induced pluripotent stem cells, mesenchymal stem cells, and renal stem cells, could stimulate the regeneration of damaged kidney tissue and new nephrons, thus reducing the need for dialysis and transplantation. However, this therapy is associated with risk factors such as cancer and fibrosis, and the long-term effects of these treatments are unknown. Similarly, new drugs that act on specific genes could enable personalised treatments and improve patients’ quality of life [204]. New therapeutic strategies based also on pro-resolving mediators aim to slow down the complications associated with kidney disease, providing valuable support against this global health challenge. The therapeutic approach that exploits the resolving properties of these mediators has the potential not only to stop the action of pro-inflammatory mediators but also to actively contribute to the resolution of the inflammatory process without suppressing the host’s immune response. Although preclinical studies have led to the development of compounds that are more resistant to metabolic inactivation than their endogenous homologues, further studies are certainly needed to improve the stability of these lipid compounds. Formulations based on liposomes, lipid nanoparticles, or nanotechnologies are currently under study to obtain greater stability, bioavailability of SPMs, and their targeted delivery to inflamed tissues [182]. Such research could shed light on aspects that are still unclear, such as the optimal timing and methods of administration of these mediators in the context of renal disease. Given the multiple actions of SPMs in different organs, including the kidney, the identification of metabolically stable mimetics molecules could be a significant breakthrough, increasing their potential to promote the resolution of inflammation. In this way, defects in the resolution of inflammation, an aspect that characterizes many chronic human diseases, could be addressed. Incorporating these compounds into the treatment of kidney disease could open up new therapeutic perspectives and significantly improve the management of chronic inflammatory diseases.
Author Contributions
Conceptualization, S.M.; investigation, R.M.R.; writing—original draft preparation, R.M.R., S.M. and V.D.F.; writing—review and editing, A.P., J.D., G.A. and R.d.d.V.B.; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FARB 2023/2024—ORSA232311/ORSA240949—University of Salerno (granted to S. Marzocco).
Acknowledgments
The authors are grateful to Alessia Brizzi for her support in the graphical abstract creation.
Conflicts of Interest
Only for JD: JD is an inventor on patents related to the composition of matter and/or use of pro-resolving mediators, some of which are licensed by Brigham and Women’s Hospital or Queen Mary University of London for clinical development.
References
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2011, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Beyer-Westendorf, J.; Kreutz, R.; Posch, F.; Ay, C. The CHA2DS2-VASc score strongly correlates with glomerular filtration rate and predicts renal function decline over time in elderly patients with atrial fibrillation and chronic kidney disease. Int. J. Cardiol. 2018, 253, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Jarmi, T.; Agarwal, A. Heme oxygenase and renal disease. Curr. Hypertens. Rep. 2009, 11, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Neto, J.S.; Kanno, S.; Stolz, D.B.; Kimizuka, K.; Liu, F.; Bach, F.H.; Billiar, T.R.; Choi, A.M.; Otterbein, L.E.; et al. Protection Against Ischemia/Reperfusion Injury in Cardiac and Renal Transplantation with Carbon Monoxide, Biliverdin and Both. Am. J. Transplant. 2005, 5, 282–291. [Google Scholar] [CrossRef]
- Hassan, I.R.; Gronert, K. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J. Immunol. 2009, 182, 3223–3232. [Google Scholar] [CrossRef]
- Liu, C.; Fan, D.; Lei, Q.; Lu, A.; He, X. Roles of Resolvins in Chronic Inflammatory Response. Int. J. Mol. Sci. 2022, 23, 14883. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, T.; Gui, P.; Yao, C.; Sun, W.; Wang, L.; Wang, H.; Xie, W.; Yao, S.; Lin, Y.; et al. Resolvin D1 reverts lipopolysaccharide-induced TJ proteins disruption and the increase of cellular permeability by regulating IκBα signaling in human vascular endothelial cells. Oxid. Med. Cell. Longev. 2013, 2013, 185715. [Google Scholar] [CrossRef]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that de-pend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef]
- Zhang, X.; Qu, X.; Sun, Y.B.; Caruana, G.; Bertram, J.F.; Nikolic-Paterson, D.J.; Li, J. Resolvin D1 protects podocytes in adriamycin-induced nephropathy through modulation of 14-3-3β acetylation. PLoS ONE 2013, 8, e67471. [Google Scholar] [CrossRef]
- Mundel, P.; Heid, H.W.; Mundel, T.M.; Krüger, M.; Reiser, J.; Kriz, W. Synaptopodin: An actin-associated protein in telencephalic dendrites and renal podocytes. J. Cell Biol. 1997, 139, 193–204. [Google Scholar] [CrossRef]
- Barden, A.E.; Mas, E.; Mori, T.A. n-3 Fatty acid supplementation and proresolving mediators of inflammation. Curr. Opin. Lipidol. 2016, 27, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Kuzumoto, T.; Tanigawa, T.; Higashimori, A.; Kitamura, H.; Nadatani, Y.; Otani, K.; Fukunaga, S.; Hosomi, S.; Tanaka, F.; Kamata, N.; et al. Protective role of resolvin D1, a pro-resolving lipid mediator, in nonsteroidal anti-inflammatory drug-induced small intestinal damage. PLoS ONE 2021, 16, e0250862. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, A.; Watanabe, T.; Nadatani, Y.; Takeda, S.; Otani, K.; Tanigawa, T.; Yamagami, H.; Shiba, M.; Tominaga, K.; Fujiwara, Y.; et al. Mechanisms of NLRP3 inflammasome activation and its role in NSAID-induced enteropathy. Mucosal Immunol. 2016, 9, 659–668. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, X.; Cheng, Y.; Zhang, Y.L.; Wen, H.X.; Tao, Z.; Li, H.; Hao, Y.; Gao, Y.; Yang, L.M.; et al. Resolvin D1 Stimulates Alveolar Fluid Clearance through Alveolar Epithelial Sodium Channel, Na,K-ATPase via ALX/cAMP/PI3K Pathway in Lipopolysaccharide-Induced Acute Lung Injury. J. Immunol. 2014, 192, 3765–3777. [Google Scholar] [CrossRef]
- Mas, E.; Barden, A.; Burke, V.; Beilin, L.J.; Watts, G.F.; Huang, R.C.; Puddey, I.B.; Irish, A.B.; Mori, T.A. A randomized controlled trial of the effects of n-3 fatty acids on resolvins in chronic kidney disease. Clin. Nutr. 2016, 35, 331–336. [Google Scholar] [CrossRef]
- Kieran, N.E.; Doran, P.P.; Connolly, S.B.; Greenan, M.C.; Higgins, D.F.; Leonard, M.; Godson, C.; Taylor, C.T.; Henger, A.; Kretzler, M.; et al. Modification of the transcriptomic response to renal ischemia/reperfusion injury by lipoxin analog. Kidney Int. 2003, 64, 480–492. [Google Scholar] [CrossRef]
- Katakura, M.; Hashimoto, M.; Inoue, T.; Mamun, A.A.; Tanabe, Y.; Iwamoto, R.; Arita, M.; Tsuchikura, S.; Shido, O. Omega-3 Fatty Acids Protect Renal Functions by Increasing Docosahexaenoic Acid-Derived Metabolite Levels in SHR.Cg-Leprcp/NDmcr Rats, a Metabolic Syndrome Model. Molecules 2014, 19, 3247–3263. [Google Scholar] [CrossRef]
- Börgeson, E.; Docherty, N.G.; Murphy, M.; Rodgers, K.; Ryan, A.; O’Sullivan, T.P.; Guiry, P.J.; Goldschmeding, R.; Higgins, D.F.; Godson, C. A₄ and benzo-lipoxin A₄ attenuate experimental renal fibrosis. FASEB J. 2011, 25, 2967–2979. [Google Scholar] [CrossRef]
- Wu, S.-H.; Chen, X.-Q.; Lü, J.; Wang, M.-J. BML-111 Attenuates Renal Ischemia/Reperfusion Injury Via Peroxisome Proliferator-Activated Receptor-α-Regulated Heme Oxygenase-1. Inflammation 2016, 39, 611–624. [Google Scholar] [CrossRef]
- Luan, H.; Wang, C.; Sun, J.; Zhao, L.; Li, L.; Zhou, B.; Shao, S.; Shen, X.; Xu, Y. Resolvin D1 Protects Against Ischemia/Reperfusion-Induced Acute Kidney Injury by Increasing Treg Percentages via the ALX/FPR2 Pathway. Front. Physiol. 2020, 11, 285. [Google Scholar] [CrossRef]
- Duffield, J.S.; Hong, S.; Vaidya, V.S.; Lu, Y.; Fredman, G.; Serhan, C.N.; Bonventre, J.V. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006, 177, 5902–5911. [Google Scholar] [CrossRef] [PubMed]
- Elmarakby, A.A.; Ibrahim, A.S.; Katary, M.A.; Elsherbiny, N.M.; El-Shafey, M.; Abd-Elrazik, A.M.; Abdelsayed, R.A.; Maddipati, K.R.; Al-Shabrawey, M. A dual role of 12/15-lipoxygenase in LPS-induced acute renal inflammation and injury. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liang, Y.; Zhao, H.; Zhao, M. Effects of AT-RvD1 on paraquat-induced acute renal injury in mice. Int. Immunopharmacol. 2019, 67, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, Z.; Bhat, O.M.; Zhang, Q.; Abais-Battad, J.M.; Conley, S.M.; Ritter, J.K.; Li, P.L. NLRP3 inflammasome as a novel target for docosahexaenoic acid metabolites to abrogate glomerular injury. J. Lipid Res. 2017, 58, 1080–1090. [Google Scholar] [CrossRef]
- Kurokawa, K.; Nangaku, M.; Saito, A.; Inagi, R.; Miyata, T. Current issues and future perspectives of chronic renal failure. J. Am. Soc. Nephrol. 2002, 13 (Suppl. S1), S3–S6. [Google Scholar] [CrossRef]
- Wen, C.P.; Cheng, T.Y.; Tsai, M.K.; Chang, Y.C.; Chan, H.T.; Tsai, S.P.; Chiang, P.H.; Hsu, C.C.; Sung, P.K.; Hsu, Y.H.; et al. All-cause mortality attributable to chronic kidney disease: A prospective cohort study based on 462 293 adults in Taiwan. Lancet 2008, 371, 2173–2182. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef]
- Popolo, A.; Adesso, S.; Pinto, A.; Autore, G.; Marzocco, S. L-Arginine and its metabolites in kidney and cardio-vascular disease. Amino Acids 2014, 46, 2271–2286. [Google Scholar] [CrossRef]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Ribecco, M.T.; Sorrentino, R.; Domenico, B.; Genesio, M.; Pinto, A.; Autore, G.; Cuzzocrea, S. Effect of Methylguanidine in a Model of Septic Shock Induced by LPS. Free Radic. Res. 2004, 38, 1143–1153. [Google Scholar] [CrossRef]
- Marzocco, S.; Di Paola, R.; Genovese, T.; Sorrentino, R.; Britti, D.; Scollo, G.; Pinto, A.; Cuzzocrea, S.; Autore, G. Methylguanidine reduces the development of non septic shock induced by zymosan in mice. Life Sci. 2004, 75, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Di Paola, R.; Serraino, I.; Sorrentino, R.; Meli, R.; Mattaceraso, G.; Cuzzocrea, S.; Pinto, A.; Autore, G. Effect of methylguanidine in carrageenan-induced acute inflammation in the rats. Eur. J. Pharmacol. 2004, 484, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Autore, G.; Marzocco, S.; Sorrentino, R.; Mirone, V.G.; Baydoun, A.; Pinto, A. In vitro and in vivo TNFalpha synthesis modulation by methylguanidine, an uremic catabolite. Life Sci. 1999, 65, PL121–PL127. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.R.; Di Micco, L.; Marzocco, S.; De Simone, E.; De Blasio, A.; Sirico, M.L.; Nardone, L. UBI Study Group. Very Low-Protein Diet (VLPD) Reduces Metabolic Acidosis in Subjects with Chronic Kid-ney Disease: The “Nutritional Light Signal” of the Renal Acid Load. Nutrients 2017, 9, 69. [Google Scholar] [CrossRef]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl Sulfate Affects Glial Function Increasing Oxidative Stress and Neuroinflammation in Chronic Kidney Disease: Interaction between Astrocytes and Microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef]
- Masako, K.; Kentaro, K.; Yoshiki, S.; Kunitoshi, I.; Yusuke, O. Chronic kidney disease, inflammation and cardiovascular disease risk in rheumatoid arthritis. J. Cardiol. 2018, 71, 277–283. [Google Scholar] [CrossRef]
- Ikizler, T.A. Nutrition, inflammation and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2008, 17, 162–167. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diab. Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Pahwa, R.; Goyal, A.; Jialal, I. Chronic Inflammation. In StatPearls [Internet] 2023; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK493173/ (accessed on 8 January 2025).
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef]
- Lee, D.E.; Qamar, M.; Wilke, R.A. Relative Contribution of Genetic and Environmental Factors in CKD. S. D. Med. 2021, 74, 306–309. [Google Scholar] [PubMed]
- Eustace, J.A.; Astor, B.; Muntner, P.M.; Ikizler, T.A.; Coresh, J. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int. 2004, 65, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef] [PubMed]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. [Google Scholar] [CrossRef]
- Katadane, S.P.; Satariano, M.; Massey, M.; Mongan, K.; Raina, R. The Role of Inflammation in CKD. Cells 2023, 12, 1581. [Google Scholar] [CrossRef]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimbürger, O.; Bárány, P.; Lindholm, B.; Lindström, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of Cardiovascular Disease and Mortality Risk in Patients with Advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-alpha: Central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2017, 32, 2005–2014. [Google Scholar] [CrossRef]
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 9, 208. [Google Scholar] [CrossRef]
- Iorember, F.M. Malnutrition in Chronic Kidney Disease. Front. Pediatr. 2018, 6, 161. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed]
- Oncel, M.; Akbulut, S.; Toka Ozer, T.; Kiyici, A.; Keles, M.; Baltaci, B.; Turk, S. Cytokines, adipocytokines and inflammatory markers in patients on continuous ambulatory peritoneal dialysis and hemodialysis. Ren. Fail. 2016, 38, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, M.; Cobo, G.; Lindholm, B.; Stenvinkel, P. Inflammation and Protein-Energy Wasting in the Uremic Milieu. Contrib. Nephrol. 2017, 191, 58–71. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Lan, H.Y.; Tang, Y. Role of C-Reactive Protein in Kidney Diseases. Kidney Dis. 2023, 9, 73–81. [Google Scholar] [CrossRef]
- Soriano, S.; González, L.; Martín-Malo, A.; Rodríguez, M.; Aljama, P. C-reactive protein and low albumin are predictors of morbidity and cardiovascular events in chronic kidney disease (CKD) 3–5 patients. Clin. Nephrol. 2007, 67, 352–357. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Gómez-Campderá, F.; Aragoncillo, I.; Verdalles, U.; Mosse, A.; Luño, J. Serum fibrinogen levels are an independent predictor of mortality in patients with chronic kidney disease (CKD) stages 3 and 4. Kidney Int. Suppl. 2008, 74, S67–S70. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, G.; Shi, X. Advances in the Progression and Prognosis Biomarkers of Chronic Kidney Disease. Front. Pharmacol. 2021, 12, 785375. [Google Scholar] [CrossRef]
- Lousa, I.; Reis, F.; Beirão, I.; Alves, R.; Belo, L.; Santos-Silva, A. New Potential Biomarkers for Chronic Kidney Disease Management-A Review of the Literature. Int. J. Mol. Sci. 2020, 22, 43. [Google Scholar] [CrossRef]
- Hassan, M.O.; Duarte, R.; Dickens, C.; Dix-Peek, T.; Naidoo, S.; Vachiat, A.; Grinter, S.; Manga, P.; Naicker, S. Interleukin-6 gene polymorhisms and interleukin-6 levels are associated with atherosclerosis in CKD patients. Clin. Nephrol. 2020, 93, 82–86. [Google Scholar] [CrossRef]
- Annuk, M.; Soveri, I.; Zilmer, M.; Lind, L.; Hulthe, J.; Fellström, B. Endothelial function, CRP and oxidative stress in chronic kidney disease. J. Nephrol. 2005, 18, 721–726. [Google Scholar] [PubMed]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.I.; Chung, A.C.; Zhou, L.; Huang, X.R.; Liu, F.; Fu, P.; Fan, J.M.; Szalai, A.J.; Lan, H.Y. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Lab. Investig. 2011, 91, 837–851. [Google Scholar] [CrossRef]
- Devaraj, S.; Jialal, I. C-reactive protein polarizes human macrophages to an M1 phenotype and inhibits trans-formation to the M2 phenotype. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1397–1402. [Google Scholar] [CrossRef]
- Lai, W.; Tang, Y.; Huang, X.R.; Tang, P.M.; Xu, A.; Szalai, A.J.; Lou, T.Q.; Lan, H.Y. C-reactive protein promotes acute kidney injury via Smad3-dependent inhibition of CDK2/cyclin E. Kidney Int. 2016, 90, 610–626. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Heimbürger, O.; Bárány, P.; Suliman, M.; Fehrman-Ekholm, I.; Lindholm, B.; Stenvinkel, P. Associations between circulating inflammatory markers and residual renal function in CRF patients. Am. J. Kidney Dis. 2003, 41, 1212–1218. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef]
- Durlacher-Betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-Many, T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef] [PubMed]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Yudkin, J.S.; Kumari, M.; Humphries, S.E.; Mohamed-Ali, V. Inflammation, obesity, stress and coronary heart disease: Is interleukin-6 the link? Atherosclerosis 2000, 148, 209–214. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Block, G.; McAllister, C.J.; Humphreys, M.H.; Kopple, J.D. Appetite and inflammation, nutrition, anemia, and clinical outcome in hemodialysis patients. Am. J. Clin. Nutr. 2004, 80, 299–307. [Google Scholar] [CrossRef]
- Liang, B.; Gardner, D.B.; Griswold, D.E.; Bugelski, P.J.; Song, X.Y.R. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef]
- Kiberd, B.A. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J. Am. Soc. Nephrol. 1993, 4, 58–61. [Google Scholar] [CrossRef]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014, 5, 114. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef]
- Hu, H.; Wu, A.; Mu, X.; Zhou, H. Role of Interleukin 1 Receptor 2 in Kidney Disease. J. Interferon Cytokine Res. 2024, 44, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Afsar, B.; Covic, A.; Ortiz, A.; Afsar, R.E.; Kanbay, M. The Future of IL-1 Targeting in Kidney Disease. Drugs 2018, 78, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; Hung, A.; Ikizler, T.A.; Farmer-Bailey, H.; Salas-Cruz, N.; Sarkar, S.; Hoofnagle, A.; You, Z.; Chonchol, M. Interleukin-1 inhibition, chronic kidney disease-mineral and bone disorder, and physical function. Clin. Nephrol. 2017, 88, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Dharmadhikari, G.; Schumann, D.M.; Størling, J. Interleukin-1 beta targeted therapy for type 2 diabetes. Expert. Opin. Biol. Ther. 2009, 9, 1177–1188. [Google Scholar] [CrossRef]
- Park, C.W.; Kim, J.H.; Lee, J.H.; Kim, Y.S.; Ahn, H.J.; Shin, Y.S.; Kim, S.Y.; Choi, E.J.; Chang, Y.S.; Bang, B.K. High glucose-induced intercellular adhesion molecule-1 (ICAM-1) expression through an osmotic effect in rat mesangial cells is PKC-NF-kappa B-dependent. Diabetologia 2000, 43, 1544–1553. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1β by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef]
- Baud, L.; Fouqueray, B.; Philippe, C.; Amrani, A. Tumor necrosis factor alpha and mesangial cells. Kidney Int. 1992, 41, 600–603. [Google Scholar] [CrossRef]
- Dong, X.; Swaminathan, S.; Bachman, L.-A.; Croatt, A.-J.; Nath, K.-A.; Griffin, M.-D. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia–reperfusion injury. Kidney Int. 2007, 71, 619–628. [Google Scholar] [CrossRef]
- Ramseyer, V.D.; Garvin, J.L. Tumor necrosis factor-α: Regulation of renal function and blood pressure. Am. J. Physiol. Ren. Physiol. 2013, 304, F1231–F1242. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Cannon, J.G.; Wolff, S.M.; Bernheim, H.A.; Beutler, B.; Cerami, A.; Figari, I.S.; Palladino, M.A., Jr.; O’Connor, J.V. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J. Exp. Med. 1986, 163, 1433–1450. [Google Scholar] [CrossRef]
- Therrien, F.J.; Agharazii, M.; Lebel, M.; Larivière, R. Neutralization of tumor necrosis factor-alpha reduces renal fibrosis and hypertension in rats with renal failure. Am. J. Nephrol. 2012, 36, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Sonkar, G.K.; Usha; Singh, R.G. Evaluation of serum tumor necrosis factor alpha and its correlation with histology in chronic kidney disease, stable renal transplant and rejection cases. Saudi J. Kidney Dis. Transpl. 2009, 20, 1000–1004. [Google Scholar]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Radeke, H.H.; Meier, B.; Topley, N.; Flöge, J.; Habermehl, G.G.; Resch, K. Interleukin 1-alpha and tumor necro-sis factor-alpha induce oxygen radical production in mesangial cells. Kidney Int. 1990, 37, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Azushima, K.; Yamaji, T.; Urate, S.; Suzuki, T.; Abe, E.; Tanaka, S.; Tsukamoto, S.; Kamimura, D.; Kinguchi, S.; et al. Effects of tumor necrosis factor-α inhibition on kidney fibrosis and inflammation in a mouse model of aristolochic acid nephropathy. Sci. Rep. 2021, 11, 23587. [Google Scholar] [CrossRef]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef]
- André, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-derived CD40L: The switch-hitting player of cardiovascular disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef]
- Bontekoe, J.; Bansal, V.; Lee, J.; Syed, M.; Hoppensteadt, D.; Maia, P.; Walborn, A.; Liles, J.; Vasaiwala, S.; Fareed, J. Procalcitonin as a Marker of Comorbid Atrial Fibrillation in Chronic Kidney Disease and History of Sepsis. Clin. Appl. Thromb. Hemost. 2020, 26. [Google Scholar] [CrossRef]
- Rusu, C.; Racasan, S.; Moldovan, D.; Kacso, I.M.; Potra, A.; Bondor, C.I.; Patiu, I.M.; Vladutiu, D.; Caprioara, M.G. Soluble CD40 ligand in haemodialysis patients: Survival impact and cardiovascular prognostic role. Biomarkers 2017, 22, 232–238. [Google Scholar] [CrossRef]
- Xie, J.X.; Alderson, H.; Ritchie, J.; Kalra, P.A.; Xie, Y.; Ren, K.; Nguyen, H.; Chen, T.; Brewster, P.; Gupta, R.; et al. Circulating CD40 and sCD40L Predict Changes in Renal Function in Subjects with Chronic Kidney Disease. Sci. Rep. 2017, 7, 7942. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, T.; Chen, M.; Zhou, Y.; Yi, D.; Guo, R. CD40/CD40L Signaling as a Promising Therapeutic Target for the Treatment of Renal Disease. J. Clin. Med. 2020, 9, 3653. [Google Scholar] [CrossRef] [PubMed]
- Lousa, I.; Reis, F.; Santos-Silva, A.; Belo, L. The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD. Int. J. Mol. Sci. 2022, 23, 3284. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Martín-Rodríguez, A.; Martínez-Guardado, I.; Navarro-Jiménez, E.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. The Role of Adipokines in Health and Disease. Biomedicines 2023, 11, 1290. [Google Scholar] [CrossRef]
- Huang, G.; Zhang, Y.; Zhang, Y.; Ma, Y. Chronic kidney disease and NLRP3 inflammasome: Pathogenesis, development and targeted therapeutic strategies. Biochem. Biophys. Rep. 2023, 33, 101417. [Google Scholar] [CrossRef]
- Kishida, K.; Funahashi, T.; Shimomura, I. Adiponectin as a routine clinical biomarker. Best. Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 119–130. [Google Scholar] [CrossRef]
- Jia, T.; Carrero, J.J.; Lindholm, B.; Stenvinkel, P. The complex role of adiponectin in chronic kidney disease. Biochimie 2012, 94, 2150–2156. [Google Scholar] [CrossRef]
- Lenghel, A.R.; Kacso, I.M.; Bondor, C.I.; Rusu, C.; Rahaian, R.; Gherman Caprioara, M. Intercellular adhesion molecule, plasma adiponectin and albuminuria in type 2 diabetic patients. Diabetes Res. Clin. Pract. 2012, 95, 55–61. [Google Scholar] [CrossRef]
- Stenvinkel, P. Endothelial dysfunction and inflammation-is there a link? Nephrol. Dial. Transplant. 2001, 16, 1968–1971. [Google Scholar] [CrossRef]
- Qureshi, A.R.; Alvestrand, A.; Danielsson, A.; Divino-Filho, J.C.; Gutierrez, A.; Lindholm, B.; Bergström, J. Factors predicting malnutrition in hemodialysis patients: A cross-sectional study. Kidney Int. 1998, 53, 773–782. [Google Scholar] [CrossRef]
- Lee, P.-T.; Liao, I.-C.; Lee, C.-H.; Hsu, L.-W.; Liu, P.-Y. Expression of Vascular Cell Adhesion Molecule-1 in Peripheral Artery Disease is Enriched in Patients with Advanced Kidney Disease. Acta Cardiol. Sin. 2021, 37, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Hauser, I.A.; Riess, R.; Hausknecht, B.; Thüringer, H.; Sterzel, R.B. Expression of cell adhesion molecules in primary renal disease and renal allograft rejection. Nephrol. Dial. Transplant. 1997, 12, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, U.; Scheuer, R.; Landgraf, R.; König, A.; Feucht, H.E. Assessment of soluble adhesion molecules (sICAM-1, sVCAM-1, sELAM-1) and complement cleavage products (sC4d, sC5b-9) in urine. Clinical monitoring of renal allograft recipients. Transplantation 1994, 58, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Li, L.; Okusa, M.D. Inflammation in Acute Kidney Injury. Nephron Exp. Nephrol. 2008, 109, e102–e107. [Google Scholar] [CrossRef]
- Zheng, Z.; Xu, K.; Li, C.; Qi, C.; Fang, Y.; Zhu, N.; Bao, J.; Zhao, Z.; Yu, Q.; Wu, H.; et al. NLRP3 associated with chronic kidney disease progression after ischemia/reperfusion-induced acute kidney injury. Cell Death Discov. 2021, 7, 324. [Google Scholar] [CrossRef]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef]
- Li, L.; Tang, W.; Yi, F. Role of Inflammasome in Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 407–421. [Google Scholar] [CrossRef]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, e0122272. [Google Scholar] [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef]
- Ho, L.-C.; Wu, T.-Y.; Lin, T.-M.; Liou, H.-H.; Hung, S.-Y. Indoxyl Sulfate Mediates the Low Inducibility of the NLRP3 Inflammasome in Hemodialysis Patients. Toxins 2021, 13, 38. [Google Scholar] [CrossRef]
- Rapa, S.F.; Prisco, F.; Popolo, A.; Iovane, V.; Autore, G.; Di Iorio, B.R.; Dal Piaz, F.; Paciello, O.; Nishijima, F.; Marzocco, S. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. Int. J. Mol. Sci. 2021, 22, 1135. [Google Scholar] [CrossRef] [PubMed]
- Islamuddin, M.; Qin, X. Renal macrophages and NLRP3 inflammasomes in kidney diseases and therapeutics. Cell Death Discov. 2024, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Pedraza-Chaverri, J.; Scholze, A. Nrf2 Activation in Chronic Kidney Disease: Promises and Pitfalls. Antioxidants 2022, 11, 1112. [Google Scholar] [CrossRef]
- Adesso, S.; Ruocco, M.; Rapa, S.F.; Piaz, F.D.; Di Iorio, R.B.; Popolo, A.; Autore, G.; Nishijima, F.; Pinto, A.; Marzocco, S. Effect of Indoxyl Sulfate on the Repair and Intactness of Intestinal Epithelial Cells: Role of Reactive Oxygen Species’ Release. Int. J. Mol. Sci. 2019, 20, 2280. [Google Scholar] [CrossRef]
- Fogo, A.B. Mechanisms of progression of chronic kidney disease. Pediatr. Nephrol. 2007, 22, 2011–2022. [Google Scholar] [CrossRef]
- Siragy, H.M.; Carey, R.M. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am. J. Nephrol. 2010, 31, 541–550. [Google Scholar] [CrossRef]
- Maranduca, M.A.; Clim, A.; Pinzariu, A.C.; Statescu, C.; Sascau, R.A.; Tanase, D.M.; Serban, D.N.; Branisteanu, D.C.; Branisteanu, D.E.; Huzum, B.; et al. Role of arterial hypertension and angiotensin II in chronic kidney disease (Review). Exp. Ther. Med. 2023, 25, 153. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins & Cotran Pathologic Basis of Disease, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Savill, J.S.; Henson, P.M.; Haslett, C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J. Clin. Investig. 1989, 84, 1518–1527. [Google Scholar] [CrossRef]
- National Collaborating Centre for Chronic Conditions (Great Britain). Chronic Kidney Disease: National Clinical Guideline for Early Identification and Management in Adults in Primary and Secondary Care; Royal College of Physicians: London, UK, 2008. [Google Scholar]
- Colbert, G.B.; Patel, D.; Lerma, E.V. Patiromer for the treatment of hyperkalemia. Expert. Rev. Clin. Pharmacol. 2020, 13, 563–570. [Google Scholar] [CrossRef]
- Mendieta-Condado, E.; Villaseñor-Tapia, E.C.; Gálvez-Gastelum, F.J.; Yáñez-Sánchez, I.; Pizano-Martínez, O.; Canales-Aguirre, A.; Márquez-Aguirre, A.L. Effects of Etanercept on TNF-α Inhibition in Rats with Adenine-Induced Chronic Kidney Disease. Biomed. Res. Int. 2022, 19, 4970753. [Google Scholar] [CrossRef]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Asmus, H.G.; Krämer, W.; Kühn, K.W.; Kütemeyer, H.; Mann, J.F.; Ruf, G.; et al. Randomized controlled trial on the efficacy and safety of atorvastatin in patients with type 2 diabetes on hemodialysis (4D study): Demographic and baseline characteristics. Kidney Blood Press. Res. 2004, 27, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Z.; Qiu, Y.; Wu, L.; Wang, H.; Wu, L.; Zhao, L.; Xie, D. Statins Have an Anti-Inflammation in CKD Patients: A Meta-Analysis of Randomized Trials. Biomed. Res. Int. 2022, 22, 4842699. [Google Scholar] [CrossRef]
- Brown, W.V. Safety of statins. Curr. Opin. Lipidol. 2008, 19, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Pedro-Botet, J.; Climent, E.; Chillarón, J.J.; Toro, R.; Benaiges, D.; Flores-Le Roux, J.A. Statins for primary cardiovascular prevention in the elderly. J. Geriatr. Cardiol. 2015, 12, 431–438. [Google Scholar] [CrossRef]
- Kreiner, F.F.; Kraaijenhof, J.M.; von Herrath, M.; Hovingh, G.K.K.; von Scholten, B.J. Interleukin 6 in diabetes, chronic kidney disease, and cardiovascular disease: Mechanisms and therapeutic perspectives. Expert. Rev. Clin. Immunol. 2022, 18, 377–389. [Google Scholar] [CrossRef]
- Maraj, M.; Kuśnierz-Cabala, B.; Dumnicka, P.; Gala-Błądzińska, A.; Gawlik, K.; Pawlica-Gosiewska, D.; Ząbek-Adamska, A.; Mazur-Laskowska, M.; Ceranowicz, P.; Kuźniewski, M. Malnutrition, Inflammation, Atherosclerosis Syndrome (MIA) and Diet Recommendations among End-Stage Renal Disease Patients Treated with Maintenance Hemodialysis. Nutrients 2018, 10, 69. [Google Scholar] [CrossRef]
- Chauveau, P.; Aparicio, M.; Bellizzi, V.; Campbell, K.; Hong, X.; Johansson, L.; Kolko, A.; Molina, P.; Sezer, S.; Wanner, C.; et al. European Renal Nutrition (ERN) Working Group of the European Renal Association–European Dialysis Transplant Association (ERA-EDTA). Mediterranean diet as the diet of choice for patients with chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Baggio, B.; Musacchio, E.; Priante, G. Polyunsaturated fatty acids and renal fibrosis: Pathophysiologic link and potential clinical implications. J. Nephrol. 2005, 18, 362. [Google Scholar]
- Dąbek, B.; Dybiec, J.; Frąk, W.; Fularski, P.; Lisińska, W.; Radzioch, E.; Młynarska, E.; Rysz, J.; Franczyk, B. Novel Therapeutic Approaches in the Management of Chronic Kidney Disease. Biomedicines 2023, 11, 2746. [Google Scholar] [CrossRef]
- Lambert, K.; Rinninella, E.; Biruete, A.; Sumida, K.; Stanford, J.; Raoul, P.; Mele, M.C.; Wang, A.Y.; Mafra, D. Targeting the Gut Microbiota in Kidney Disease: The Future in Renal Nutrition and Metabolism. J. Ren. Nutr. 2023, 33, S30–S39. [Google Scholar] [CrossRef]
- Adesso, S.; Paterniti, I.; Cuzzocrea, S.; Fujioka, M.; Autore, G.; Magnus, T.; Pinto, A.; Marzocco, S. AST-120 Reduces Neuroinflammation Induced by Indoxyl Sulfate in Glial Cells. J. Clin. Med. 2018, 7, 365. [Google Scholar] [CrossRef] [PubMed]
- Gembillo, G.; Siligato, R.; Santoro, D. Personalized Medicine in Kidney Disease. J. Pers. Med. 2023, 13, 1501. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef]
- Serhan, C.N. Discovery of specialized pro-resolving mediators marks the dawn of resolution physiology and pharmacology. Mol. Aspects Med. 2017, 58, 1–11. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef]
- Freire-de-Lima, C.G.; Xiao, Y.Q.; Gardai, S.J.; Bratton, D.L.; Schiemann, W.P.; Henson, P.M. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 2006, 281, 38376–38384. [Google Scholar] [CrossRef]
- Yang, L.; Pan, Y.; Wu, Y.; Lin, S.; Dai, B.; Chen, H.; Wan, J. Excessive arachidonic acid induced actin bunching remodeling and podocyte injury via a PKA-c-Abl dependent pathway. Exp. Cell Res. 2020, 388, 111808. [Google Scholar] [CrossRef]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef]
- Priante, G.; Musacchio, E.; Valvason, C.; Baggio, B. EPA and DHA suppress AngII- and arachidonic ac-id-induced expression of profibrotic genes in human mesangial cells. J. Nephrol. 2009, 22, 137–143. [Google Scholar]
- Nasrallah, R.; Hassouneh, R.; Hébert, R.L. Chronic kidney disease: Targeting prostaglandin E2 receptors. Am. J. Physiol. Renal Physiol. 2014, 307, F243–F250. [Google Scholar] [CrossRef]
- Fan, F.; Muroya, Y.; Roman, R.J. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.T.; Ahmed, F.A.; Hamm, L.L.; Teran, F.J.; Chen, C.S.; Liu, Y.; Shah, K.; Rifai, N.; Batuman, V.; Simon, E.E.; et al. Association of C-reactive protein, tumor necrosis factor-alpha, and interleukin-6 with chronic kidney disease. BMC Nephrol. 2015, 16, 77. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [PubMed]
- O’Meara, S.J.; Rodgers, K.; Godson, C. Lipoxins: Update and impact of endogenous pro-resolution lipid mediators. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 47–70. [Google Scholar] [CrossRef]
- O’Sullivan, T.P.; Vallin, K.S.; Shah, S.T.; Fakhry, J.; Maderna, P.; Scannell, M.; Sampaio, A.L.; Perretti, M.; Godson, C.; Guiry, P.J. Aromatic lipoxin A4 and lipoxin B4 analogues display potent biological activities. J. Med. Chem. 2007, 50, 5894–5902. [Google Scholar] [CrossRef]
- De Gaetano, M. Development of synthetic lipoxin-A4 mimetics (sLXms): New avenues in the treatment of cardio-metabolic diseases. Semin. Immunol. 2023, 65, 101699. [Google Scholar] [CrossRef]
- Papayianni, A.; Serhan, C.N.; Brady, H.R. Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells. J. Immunol. 1996, 156, 2264–2272. [Google Scholar]
- József, L.; Zouki, C.; Petasis, N.A.; Serhan, C.N.; Filep, J.G. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-κB and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 13266–13271. [Google Scholar] [CrossRef]
- Goh, J.; Baird, A.W.; O’Keane, C.; Watson, R.W.; Cottell, D.; Bernasconi, G.; Petasis, N.A.; Godson, C.; Brady, H.R.; MacMathuna, P. Lipoxin A(4) and aspirin-triggered 15-epi-lipoxin A(4) antagonize TNF-alpha-stimulated neutrophil-enterocyte interactions in vitro and attenuate TNF-alpha-induced chemokine release and colonocyte apoptosis in human intestinal mucosa ex vivo. J. Immunol. 2001, 167, 2772–2780. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D.; Clish, C.B.; Gronert, K.; Chiang, N. Lipoxins, aspirin-triggered 15-epi-lipoxin stable analogs and their receptors in anti-inflammation: A window for therapeutic opportunity. In Ernst Schering Res Found Workshop; Springer: Berlin/Heidelberg, Germany, 2000; pp. 143–185. [Google Scholar] [CrossRef]
- Russell, R.; Gori, I.; Pellegrini, C.; Kumar, R.; Achtari, C.; Canny, G. Lipoxin A(4) is a novel estrogen receptor modulator. FASEB J. 2011, 25, 4326–4337. [Google Scholar] [CrossRef]
- Schaldach, C.M.; Riby, J.; Bjeldanes, L.F. Lipoxin A4: A new class of ligand for the Ah receptor. Biochemistry 1999, 38, 7594–7600. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.P.; Nolan, K.A.; Börgeson, E.; Gough, O.S.; McEvoy, C.M.; Docherty, N.G.; Higgins, D.F.; Murphy, M.; Sadlier, D.M.; Ali-Shah, S.T.; et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J. Am. Soc. Nephrol. 2013, 24, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.H.; Devchand, P.R.; Wada, K.; Serhan, C.N. Aspirin-triggered lipoxin A4 and lipoxin A4 up-regulate transcriptional corepressor NAB1 in human neutrophils. FASEB J. 2001, 15, 2736–2738. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.P.; Mohan, M.; McClelland, A.; Tikellis, C.; Ziemann, M.; Kaspi, A.; Gray, S.P.; Pickering, R.; Tan, S.M.; Ali-Shah, S.T.; et al. Lipoxins Regulate the Early Growth Response-1 Network and Reverse Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 1437–1448. [Google Scholar] [CrossRef]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef]
- Börgeson, E.; Johnson, A.M.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef]
- Nelson, J.R.; Wani, O.; May, H.T.; Budoff, M. Potential benefits of eicosapentaenoic acid on atherosclerotic plaques. Vascul Pharmacol. 2017, 91, 1–9. [Google Scholar] [CrossRef]
- Svensson, M.; Schmidt, E.B.; Jørgensen, K.A.; Christensen, J.H.; OPACH Study Group. N-3 fatty acids as secondary prevention against cardiovascular events in patients who undergo chronic hemodialysis: A randomized, placebo-controlled intervention trial. Clin. J. Am. Soc. Nephrol. 2006, 1, 780–786. [Google Scholar] [CrossRef]
- Saglimbene, V.M.; Wong, G.; van Zwieten, A.; Palmer, S.C.; Ruospo, M.; Natale, P.; Campbell, K.; Teixeira-Pinto, A.; Craig, J.C.; Strippoli, G.F.M. Effects of omega-3 polyunsaturated fatty acid intake in patients with chronic kidney disease: Systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2020, 39, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Minami, S.; Sakai, S.; Fujimura, R.; Takahashi, A.; Namba-Hamano, T.; Matsuda, J.; Kimura, T.; Matsui, I.; et al. Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy 2021, 17, 1700–1713. [Google Scholar] [CrossRef]
- Serhan, C.N.; Libreros, S.; Nshimiyimana, R. E-series resolvin metabolome, biosynthesis and critical role of stereochemistry of specialized pro-resolving mediators (SPMs) in inflammation-resolution: Preparing SPMs for long COVID-19, human clinical trials, and targeted precision nutrition. Semin. Immunol. 2022, 59, 101597. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Z.; Zhang, L.; Liu, T.; Park, J.Y.; Berta, T.; Yang, R.; Serhan, C.N.; Ji, R.R. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010, 16, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 immunoresolvents: Structures and actions. Sci. Rep. 2013, 3, 1940. [Google Scholar] [CrossRef]
- Colas, R.A.; Souza, P.R.; Walker, M.E.; Burton, M.; Zasłona, Z.; Curtis, A.M.; Marques, R.M.; Dalli, J. Impaired Production and Diurnal Regulation of Vascular RvDn-3 DPA Increase Systemic Inflammation and Cardiovascular Disease. Circ. Res. 2018, 122, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Pistorius, K.; Souza, P.R.; De Matteis, R.; Austin-Williams, S.; Primdahl, K.G.; Vik, A.; Mazzacuva, F.; Colas, R.A.; Marques, R.M.; Hansen, T.V.; et al. PDn-3 DPA Pathway Regulates Human Monocyte Differentiation and Macrophage Function. Cell Chem. Biol. 2018, 25, 749–760.e9. [Google Scholar] [CrossRef]
- Clish, C.B.; O’Brien, J.A.; Gronert, K.; Stahl, G.L.; Petasis, N.A.; Serhan, C.N. Local and systemic delivery of a stable aspirin-triggered lipoxin prevents neutrophil recruitment in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 8247–8252. [Google Scholar] [CrossRef]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Sawada, Y.; Honda, T.; Hanakawa, S.; Nakamizo, S.; Murata, T.; Ueharaguchi-Tanada, Y.; Ono, S.; Amano, W.; Nakajima, S.; Egawa, G.; et al. Resolvin E1 inhibits dendritic cell migration in the skin and attenuates contact hyersensitivity responses. J. Exp. Med. 2015, 212, 1921–1930. [Google Scholar] [CrossRef]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef]
- Qu, X.; Zhang, X.; Yao, J.; Song, J.; Nikolic-Paterson, D.J.; Li, J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 2012, 228, 506–519. [Google Scholar] [CrossRef]
- Barden, A.E.; Shinde, S.; Burke, V.; Puddey, I.B.; Beilin, L.J.; Irish, A.B.; Watts, G.F.; Mori, T.A. The effect of n-3 fatty acids and coenzyme Q10 supplementation on neutrophil leukotrienes, mediators of inflammation resolution and myeloperoxidase in chronic kidney disease. Prostaglandins Other Lipid Mediat. 2018, 136, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hei, A.; Sanahanbi, L. DHA (Docosahexaenoic Acid): A Biomolecule with Diverse Roles and Health Benefits; IntechOpen: Rijeka, Croatia, 2023. [Google Scholar] [CrossRef]
- Yamamoto, T.; Isaka, Y. Dietary Omega-3 Polyunsaturated Fatty Acids and Amelioration of CKD: Possible Cellular Mechanisms. Kidney360 2023, 4, 1661–1662. [Google Scholar] [CrossRef] [PubMed]
- González-Périz, A.; Planagumà, A.; Gronert, K.; Miquel, R.; López-Parra, M.; Titos, E.; Horrillo, R.; Ferré, N.; Deulofeu, R.; Arroyo, V.; et al. Docosahexaenoic acid (DHA) blunts liver injury by conversion to protective lipid mediators: Protectin D1 and 17S-hydroxy-DHA. FASEB J. 2006, 20, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Ramon, S.; Gao, F.; Serhan, C.N.; Phipps, R.P. Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J. Immunol. 2012, 189, 1036e42. [Google Scholar]
- Liu, W.-C.; Yang, Y.-H.; Wang, Y.-C.; Chang, W.-M.; Wang, C.-W. Maresin: Macrophage Mediator for Resolving Inflammation and Bridging Tissue Regeneration-A System-Based Preclinical Systematic Review. Int. J. Mol. Sci. 2023, 24, 11012. [Google Scholar] [CrossRef]
- Muramatsu, H.; Akimoto, N.; Yajima, K.; Hashimoto, M.; Katakura, M. Suppressing Effects of Docosahexaenoic Acid–Containing Diets on Oxidative Stress and Fibrosis in 5/6 Nephrectomized Rats. Kidney360 2023, 4, 1690–1701. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kawarasaki, M.; Aono, A.; Cho, J.; Hashimoto, T.; Sato, R. Renoprotective effects of docosahexaenoic acid in cats with early chronic kidney disease due to polycystic kidney disease: A pilot study. J. Feline Med. Surgery 2022, 24, e505–e512. [Google Scholar] [CrossRef]
- Dalli, J.; Vlasakov, I.; Riley, I.R.; Rodriguez, A.R.; Spur, B.W.; Petasis, N.A.; Chiang, N.; Serhan, C.N. Maresin conjugates in tissue regeneration biosynthesis enzymes in human macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 12232–12237. [Google Scholar] [CrossRef]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef]
- Gong, J.; Liu, H.; Wu, J.; Qi, H.; Wu, Z.Y.; Shu, H.Q.; Li, H.B.; Chen, L.; Wang, Y.X.; Li, B.; et al. Maresin 1 prevents lipopolysaccharide-induced neutrophil survival and accelerates resolution of acute lung injury. Shock 2015, 44, 371–380. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Wang, L.; Jiang, L.; Qin, Z.; Zhao, Y.; Su, B. Maresin 1 Attenuates Lipopolysaccharide-Induced Acute Kidney Injury via Inhibiting NOX4/ROS/NF-κB Pathway. Front. Pharmacol. 2021, 12, 782660. [Google Scholar] [CrossRef]
- Qiu, Y.; Wu, Y.; Zhao, H.; Sun, H.; Gao, S. Maresin 1 mitigates renal ischemia/reperfusion injury in mice via inhibition of the TLR4/MAPK/NF-κB pathways and activation of the Nrf2 pathway. Drug Des. Devel Ther. 2019, 13, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Gao, C.; Long, Y.; Huang, W.; Chen, J.; Fan, F.; Jiang, C.; Xu, Y. Maresin 1 Mitigates High Glucose-Induced Mouse Glomerular Mesangial Cell Injury by Inhibiting Inflammation and Fibrosis. Mediat. Inflamm. 2017, 2017, 2438247. [Google Scholar] [CrossRef]
- Li, X.; Xu, B.; Wu, J.; Pu, Y.; Wan, S.; Zeng, Y.; Wang, M.; Luo, L.; Zhang, F.; Jiang, Z.; et al. Maresin 1 Alleviates Diabetic Kidney Disease via LGR6-Mediated cAMP-SOD2-ROS Pathway. Oxid. Med. Cell Longev. 2022, 2022, 7177889. [Google Scholar] [CrossRef]
- Ariel, A.; Li, P.L.; Wang, W.; Tang, W.X.; Fredman, G.; Hong, S.; Gotlinger, K.H.; Serhan, C.N. The docosatriene protectin D1 is produced by TH2 skewing promotes human T cell via lipid raft clustering. J. Biol. Chem. 2005, 280, 43079–43086. [Google Scholar] [CrossRef]
- Ariel, A.; Serhan, C.N. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007, 28, 176–183. [Google Scholar] [CrossRef]
- Stenvik Haatveit, Å.; Hansen, T.V. The biosynthetic pathways of the protectins. Prostaglandins Other Lipid Mediat. 2023, 169, 106787. [Google Scholar] [CrossRef]
- Jaworska, K.; Ratajczak, J.; Huang, L.; Whalen, K.; Yang, M.; Stevens, B.K.; Kinsey, G.R. Both PD-1 ligands protect the kidney from ischemia reperfusion injury. J. Immunol. 2015, 194, 325–333. [Google Scholar] [CrossRef]
- Giardini, E.; Moore, D.; Sadlier, D.; Godson, C.; Brennan, E. The dual role of lipids in chronic kidney disease: Pathogenic culprits and therapeutic allies. Atherosclerosis 2024, 398, 118615. [Google Scholar] [CrossRef]
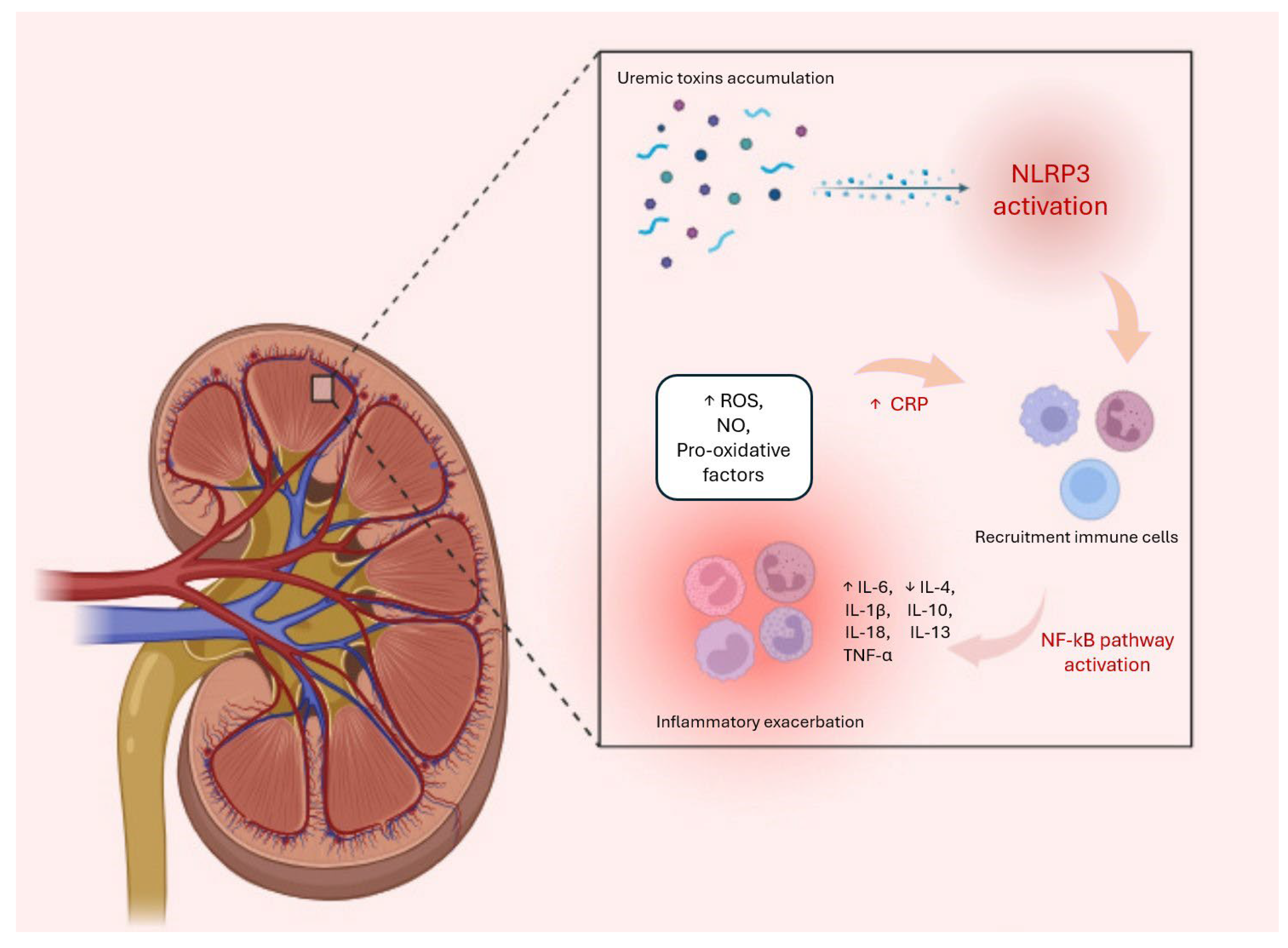
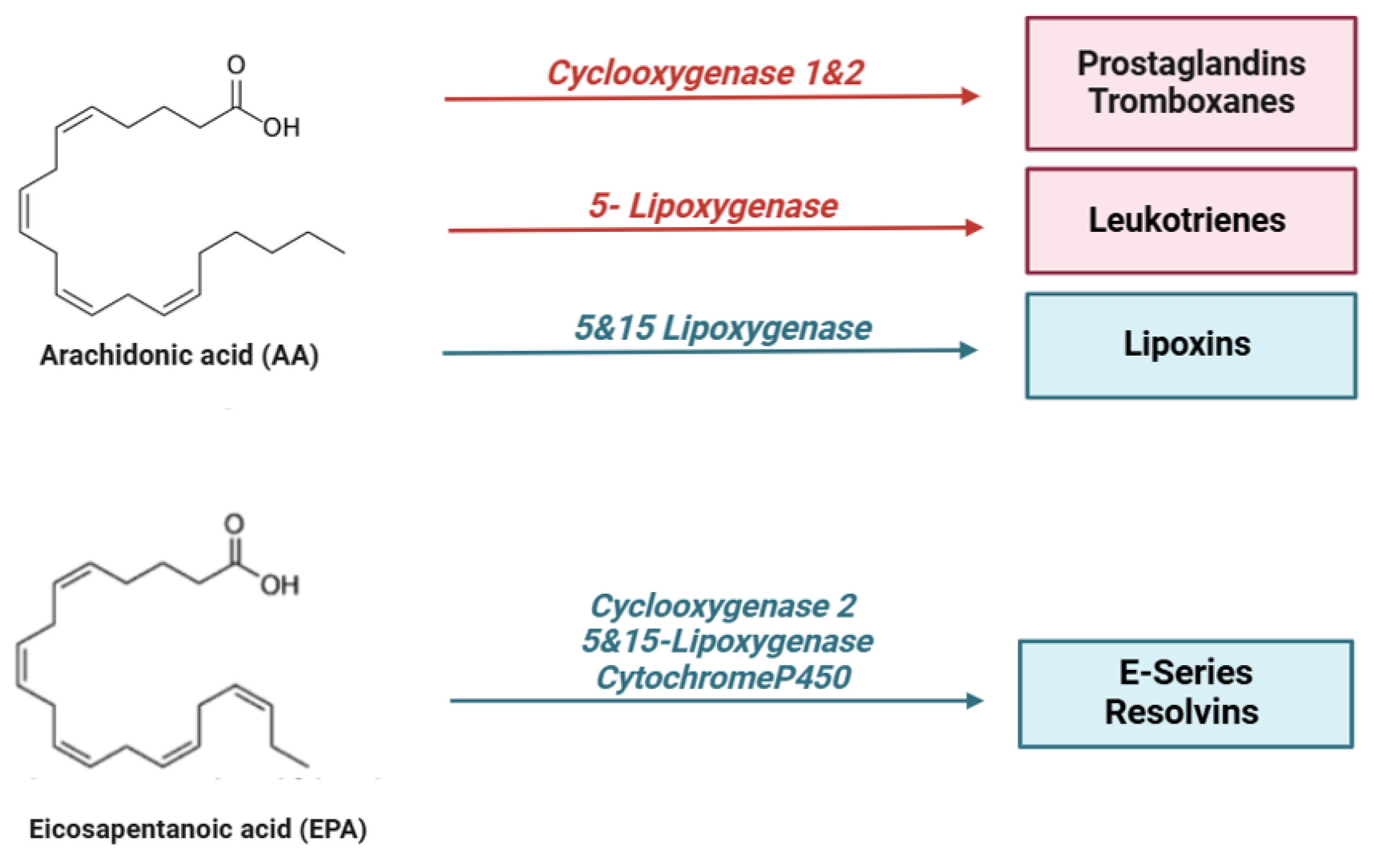
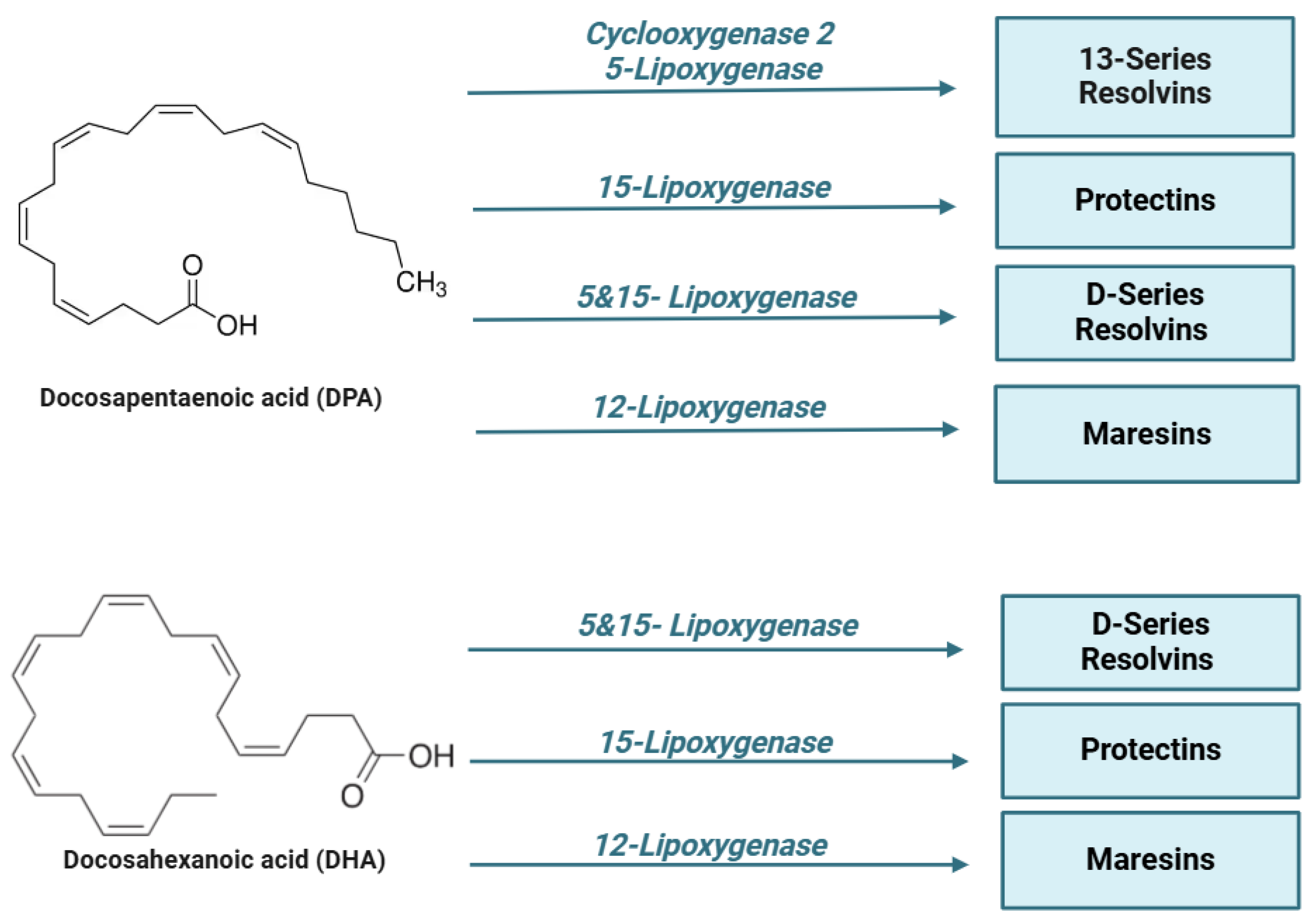
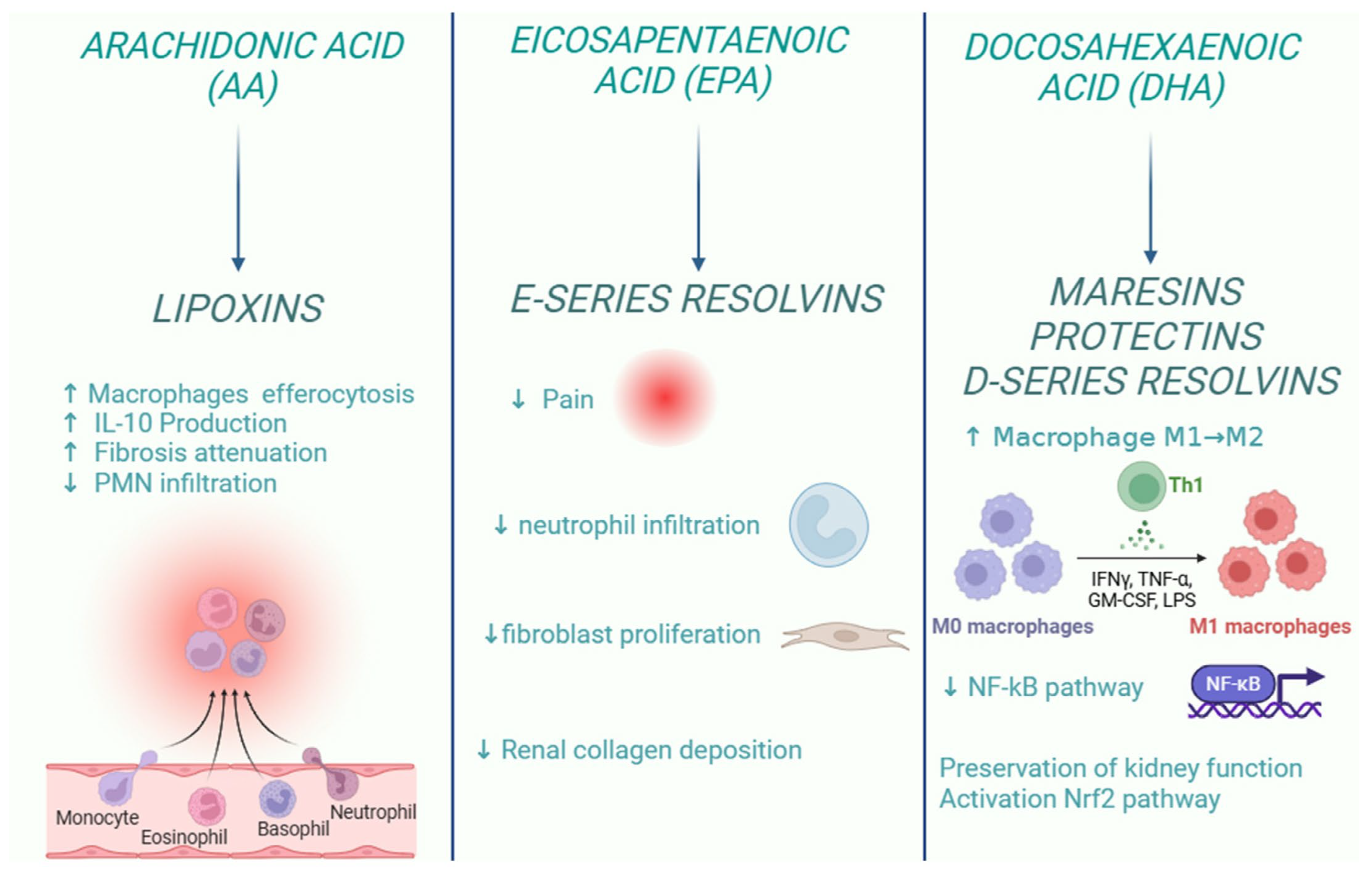
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).