

The Intrinsically Disordered Region of HBx and Virus–Host Interactions: Uncovering New Therapeutic Approaches for HBV and Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Human Viral Infections and Cancer
2. HBV Infection: A Global Health Concern
3. HBV Genome and Replication Cycle
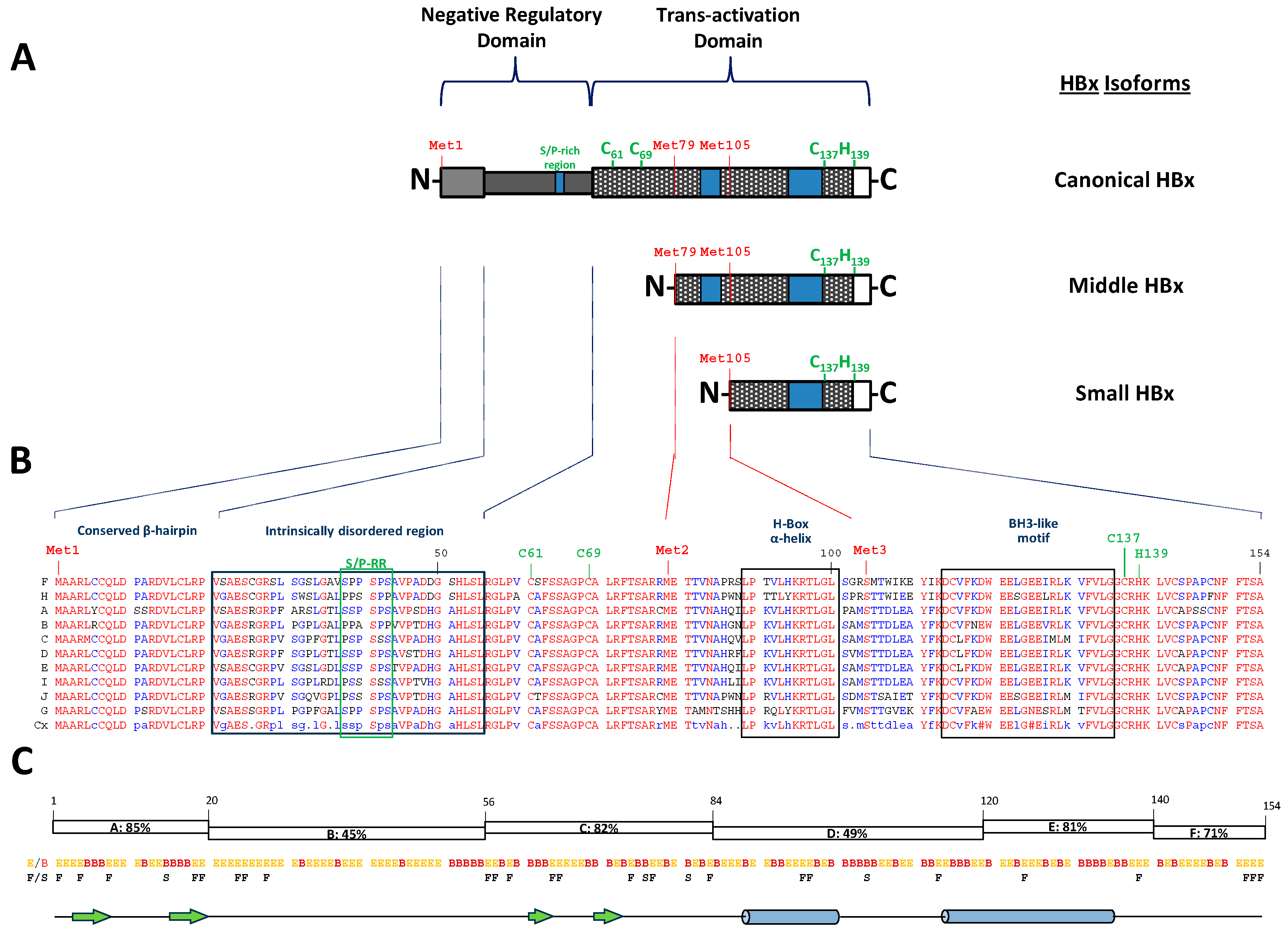
4. The Puzzle of Sequences, Partial Structures, and Different Functions of the HBV Canonical HBx
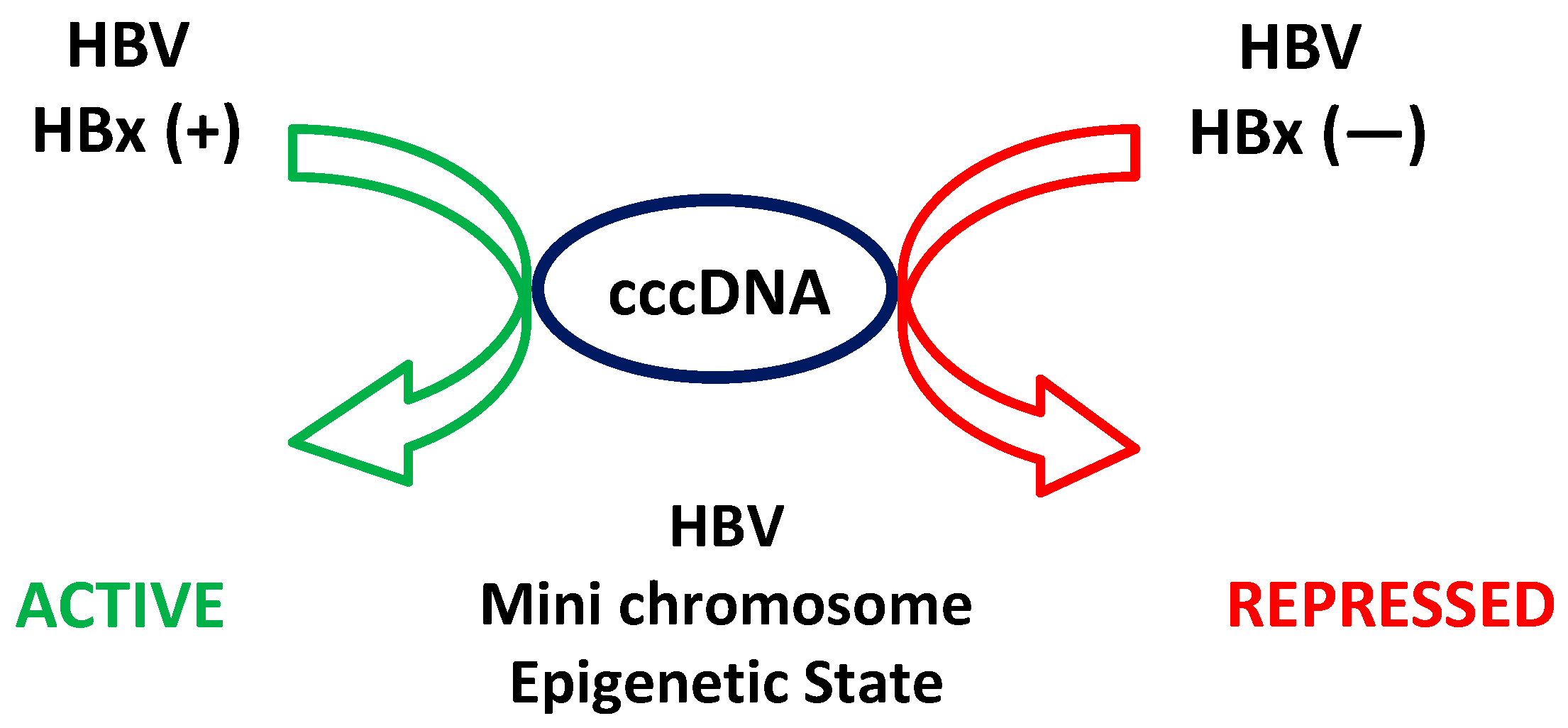
5. Epigenetic Regulation of HBV Transcription: Role of HBx Regarding cccDNA
6. The Role of Intrinsically Disordered Proteins (IDPs) and Regions (IDRs) in Cellular and Viral Interaction Networks
7. Disordered Regions Span Life Forms, Including Viruses
8. Disordered Proteins and Disease: Opportunities for Therapeutic Targeting
9. Protein–Protein Interactions Involving the Intrinsically Disordered Region of HBx
10. Identification of Key Host Proteins Directly Interacting with the HBx IDR
10.1. Par14/Par17 (PIN4 Gene)
10.2. Cortactin (CTTN, 61.6 kDa)
10.3. 14-3-3ζ Protein (YWHAZ Gene, 27.7 kDa)
10.4. NCOA3 (Nuclear Receptor Coactivator 3, 155 kDa)
10.5. Pin1 (Peptidyl-prolyl cis/trans Isomerase, 18.2 kDa)
11. Management of Chronic HBV Infection
12. Persistence of HBV and Challenges of Current Therapies
13. Targeting the HBx IDR and Interaction Partners with Direct-Acting Antiviral Agents (DAAs)
14. The Broader Potential of DAAs in HBV Treatment Results
15. The Future of HBV Therapy Through HBx-Directed DAAs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenner, F. A successful eradication campaign. Global eradication of smallpox. Rev. Infect. Dis. 1982, 4, 916–930. [Google Scholar] [CrossRef] [PubMed]
- Metzger, W.; Köhler, C.; Mordmüller, B. Lessons from a modern review of the smallpox eradication files. J. R. Soc. Med. 2015, 108, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G. Global burden of infection-related cancer revisited. Lancet Oncol. 2012, 13, 564–565. [Google Scholar] [CrossRef]
- Mui, U.; Haley, C.; Tyring, S. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Hausen, H.Z. The search for infectious causes of human cancers: Where and why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. WHO International Agency for Research on Cancer Monograph Working Group. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Capasso, M.; Cossiga, V.; Guarino, M.; Ranieri, L.; Morisco, F. The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis. Cancers 2024, 16, 1505. [Google Scholar] [CrossRef]
- Asandem, D.; Segbefia, S.; Kusi, K.; Bonney, J. Hepatitis B Virus Infection: A Mini Review. Viruses 2024, 16, 724. [Google Scholar] [CrossRef]
- WHO. Global Hepatitis Report, 2017. 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 15 June 2021).
- Revill, P.; Tu, T.; Netter, H.; Yuen, L.; Locarnini, S.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Revill, P. Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 2016, 64, S4–S16. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.; Jaspe, R.; Loureiro, C.; Chemin, I. Hepatitis B virus American genotypes: Pathogenic variants? Clin. Res. Hepatol. Gastroenterol. 2020, 44, 825–835. [Google Scholar] [CrossRef]
- Roman, S.; Panduro, A. HBV endemicity in Mexico is associated with HBV genotypes H and G. World. J. Gastroenterol. 2013, 19, 5446–5453. [Google Scholar] [CrossRef]
- Venegas, M.; Munoz, G.; Hurtado, C.; Alvarez, L.; Velasco, M.; Villanueva, R.A.; Brahm, J. Prevalence of hepatitis B virus genotypes in chronic carriers in Santiago, Chile. Arch. Virol. 2008, 153, 2129–2132. [Google Scholar] [CrossRef]
- Villanueva, R.; Loyola, A. Pre- and Post-Transcriptional Control of HBV Gene Expression: The Road Traveled towards the New Paradigm of HBx, Its Isoforms, and Their Diverse Functions. Biomedicines 2023, 11, 1674. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransportingpolypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Gómez-Moreno, A.; Ploss, A. Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription. Viruses 2024, 16, 609. [Google Scholar] [CrossRef]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef]
- Dias, J.; Sarica, N.; Neuveut, C. Early Steps of Hepatitis B Life Cycle: From Capsid Nuclear Import to cccDNA Formation. Viruses 2021, 13, 757. [Google Scholar] [CrossRef]
- Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.; Yuen, M.; Chan, H.; Gish, R.; Locarnini, S.; Chavez, D.; Ferrari, C.; Given, B.; Hamilton, J.; Kanner, S.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [PubMed]
- Giosa, D.; Lombardo, D.; Musolino, C.; Chines, V.; Raffa, G.; Tocco, F.C.d.; D’Aliberti, D.; Caminiti, G.; Saitta, C.; Alibrandi, A.; et al. Mitochondrial DNA is a target of HBV integration. Commun. Biol. 2023, 6, 684. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Kaneko, S.; Chung, C.; Girones, R.; Purcell, R. Compact organization of the hepatitis B virus genome. Hepatology 1989, 9, 322–327. [Google Scholar] [CrossRef]
- Murakami, S. Hepatitis B virus X protein: Structure, function and biology. Intervirology 1999, 42, 81–99. [Google Scholar] [CrossRef]
- Moolla, N.; Kew, M.; Arbuthnot, P. Regulatory elements of hepatitis B virus transcription. J. Viral Hepat. 2002, 9, 323–331. [Google Scholar] [CrossRef]
- Zhang, P.; Raney, A.; McLachlan, A. Characterization of the hepatitis B virus X- and nucleocapsid gene transcriptional regulatory elements. Virology 1992, 191, 31–41. [Google Scholar] [CrossRef]
- Treinin, M.; Laub, O. Identification of a promoter element located upstream from the hepatitis B virus X gene. Mol. Cell Biol. 1987, 7, 545–548. [Google Scholar]
- Kwee, L.; Lucito, R.; Aufiero, B.; Schneider, R. Alternate translation initiation on hepatitis B virus X mRNA produces multiple polypeptides that differentially transactivate class II and III promoters. J. Virol. 1992, 66, 4382–4389. [Google Scholar] [CrossRef]
- Leach, J.; Qiao, L.; Fang, Y.; Han, S.; Gilfor, D.; Fisher, P.; Grant, S.; Hylemon, P.; Peterson, D.; Dent, P. Regulation of p21 and p27 expression by the hepatitis B virus X protein and the alternate initiation site X proteins, AUG2 and AUG3. J. Gastroenterol. Hepatol. 2003, 18, 376–385. [Google Scholar] [CrossRef]
- Balsano, C.; Avantaggiati, M.; Natoli, G.; Marzio, E.D.; Will, H.; Perricaudet, M.; Levrero, M. Full-length and truncated versions of the hepatitis B virus (HBV) X protein (pX) transactivate the cmyc protooncogene at the transcriptional level. Biochem. Biophys. Res. Commun. 1991, 176, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Riegler, J.; Wu, J.; Yen, T. Novel short transcripts of hepatitis B virus X gene derived from intragenic promoter. J. Biol. Chem. 1994, 269, 22593–22598. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Hepatitis B virus X protein: A multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hamadalnil, Y.; Tu, T. Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis. Viruses 2024, 16, 1361. [Google Scholar] [CrossRef]
- Slagle, B.; Bouchard, M. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef]
- Melegari, M.; Scaglioni, P.; Wands, J. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 1998, 72, 1737–1743. [Google Scholar] [CrossRef]
- Bouchard, M.; Wang, L.; Schneider, R. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef]
- Keasler, V.; Hodgson, A.; Madden, C.; Slagle, B. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Keasler, V.; Hodgson, A.; Madden, C.; Slagle, B. Hepatitis B virus HBx protein localized to the nucleus restores HBx-deficient virus replication in HepG2 cells and in vivo in hydrodynamically-injected mice. Virology 2009, 390, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; Nicola, F.D.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Torii, N.; Hu, Z.; Jacob, J.; Liang, T. X-deficient woodchuck hepatitis virus mutants behave like attenuated viruses and induce protective immunity in vivo. J. Clin. Investig. 2001, 108, 1523–1531. [Google Scholar] [CrossRef]
- Kumar, V.; Sarkar, D. Hepatitis B Virus X Protein: Structure–Function Relationships and Role in Viral Pathogenesis. In Transcription Factors. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2004; Volume 166, pp. 377–407. [Google Scholar]
- Murakami, S.; Cheong, J.; Kaneko, S. Human hepatitis B virus X gene encodes a regulatory domain which represses transactivation of X protein. J. Biol. Chem. 1994, 269, 15118–15123. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Gottlob, K.; Pagano, S.; Levrero, M.; Graessmann, A. Hepatitis B virus X protein transcription activation domains are neither required nor sufficient for cell transformation. Cancer Res. 1998, 58, 3566–3570. [Google Scholar]
- Lee, S.; Cha, E.; Lim, J.; Kwon, S.; Kim, D.; Cho, H.; Han, K. Structural characterization of an intrinsically unfolded mini-HBX protein from hepatitis B virus. Mol. Cells 2012, 34, 165–169. [Google Scholar] [CrossRef]
- Misra, K.; Mukherji, A.; Kumar, V. The conserved amino-terminal region (amino acids 1–20) of the hepatitis B virus X protein shows a transrepression function. Virus Res. 2004, 105, 157–165. [Google Scholar] [CrossRef]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A truncated mutant (residues 58–140) of the hepatitis B virus X protein retains transactivation function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef] [PubMed]
- Arii, M.; Takada, S.; Koike, K. Identification of three essential regions of hepatitis B virus X protein for trans-activation function. Oncogene 1992, 7, 397–403. [Google Scholar] [PubMed]
- Nijhara, R.; Jana, S.; Goswami, S.; Kumar, V.; Sarkar, D. An internal segment (residues 58–119) of the hepatitis B virus X protein is sufficient to activate MAP kinase pathways in mouse liver. FEBS Lett. 2001, 504, 59–64. [Google Scholar] [CrossRef]
- Reddi, H.; Kumar, R.; Jain, S.; Kumar, V. A carboxy-terminal region of the hepatitis B virus X protein promotes DNA interaction of CREB and mimics the native protein for transactivation function. Virus Genes 2003, 26, 227–238. [Google Scholar] [CrossRef]
- Lizzano, R.; Yang, B.; Clippinger, A.; Bouchard, M. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011, 155, 231–239. [Google Scholar] [CrossRef]
- Huh, K.; Siddiqui, A. Characterization of the mitochondrial association of hepatitis B virus X protein, HBx. Mitochondrion 2002, 1, 349–359. [Google Scholar] [CrossRef]
- McClain, S.; Clippinger, A.; Lizzano, R.; Bouchard, M. Hepatitis B virus replication is associated with an HBx-dependent mitrochondrion-regulated increase in cytosolic calcium levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef]
- Kornyeyev, D.; Ramakrishnan, D.; Voitenleitner, C.; Livingston, C.; Xing, W.; Hung, M.; Kwon, H.; Fletcher, S.; Beran, R. Spatiotemporal Analysis of Hepatitis B Virus X Protein in Primary Human Hepatocytes. J. Virol. 2019, 93, e00248-19. [Google Scholar] [CrossRef]
- Ramakrishnan, D.; Xing, W.; Beran, R.; Chemuru, S.; Rohrs, H.; Niedziela-Majka, A.; Marchand, B.; Mehra, U.; Zábranský, A.; Doležal, M.; et al. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93, e00250-19. [Google Scholar] [CrossRef]
- Liu, W.; Yao, Q.; Su, X.; Deng, Y.; Yang, M.; Peng, B.; Zhao, F.; Du, C.; Zhang, X.; Zhu, J.; et al. Molecular insights into Spindlin1-HBx interplay and its impact on HBV transcription from cccDNA minichromosome. Nat. Commun. 2023, 14, 4663. [Google Scholar] [CrossRef]
- Lee, W.; Lan, K.; Li, C.; Chao, Y.; Lin, H.; Lee, S. Pro-apoptotic or anti-apoptotic property of X protein of hepatitis B virus is determined by phosphorylation at Ser31 by Akt. Arch. Biochem. Biophys. 2012, 528, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.; Venegas, M.; Brahm, J.; Villanueva, R. The viral transactivator HBx protein exhibits a high potential for regulation via phosphorylation through an evolutionarily conserved mechanism. Infect. Agent. Cancer 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Prieto, C.; Montecinos, J.; Jiménez, G.; Riquelme, C.; Garrido, D.; Hernández, S.; Loyola, A.; Villanueva, R. Phosphorylation of Phylogenetically Conserved Amino Acid Residues Confines HBx within Different Cell Compartments of Human Hepatocarcinoma Cells. Molecules 2021, 26, 1254. [Google Scholar] [CrossRef]
- Li, T.; Robert, E.; Breugel, P.v.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Son, Y.; Ko, C.; Ryu, W. DDB1 Stimulates Viral Transcription of Hepatitis B Virus via HBx-Independent Mechanisms. J. Virol. 2016, 90, 9644–9653. [Google Scholar] [CrossRef]
- Kusunoki, H.; Tanaka, T.; Kohno, T.; Wakamatsu, K.; Hamaguchi, I. Structural characterization of the BH3-like motif of hepatitis B virus X protein. Biochem. Biophys. Res. Commun. 2014, 450, 741–745. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, M.; Wu, J.; Shi, Y. Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc. Natl. Acad. Sci. USA 2016, 113, 2074–2079. [Google Scholar] [CrossRef]
- Dandri, M.; Schirmache, P.; Rogler, C. Woodchuck hepatitis virus X protein is present in chronically infected woodchuck liver and woodchuck hepatocellular carcinomas which are permissive for viral replication. J. Virol. 1996, 70, 5246–5254. [Google Scholar] [CrossRef]
- Sirma, H.; Giannini, C.; Poussin, K.; Paterlini, P.; Kremsdorf, D.; Bréchot, C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene 1999, 18, 4848–4859. [Google Scholar] [CrossRef]
- Hoare, J.; Henkler, F.; Dowling, J.; Errington, W.; Goldin, R.; Fish, D.; McGarvey, M. Subcellular localisation of the X protein in HBV infected hepatocytes. J. Med. Virol. 2001, 64, 419–426. [Google Scholar] [CrossRef]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.; McGarvey, M.; Koshy, R.; King, I. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 2001, 82, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.; Ryu, D.; Jung, H.; Chang, H.; Ryu, W. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2009, 90, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Yun, Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J. Biol. Chem. 1998, 273, 25510–25515. [Google Scholar] [CrossRef]
- Feitelson, M.; Arzumanyan, A.; Spector, I.; Medhat, A. Hepatitis B x (HBx) as a Component of a Functional Cure for Chronic Hepatitis B. Biomedicines 2022, 10, 2210. [Google Scholar] [CrossRef]
- Rossner, M. Review: Hepatitis B virus X-gene product: A promiscuous transcriptional activator. J. Med. Virol. 1992, 36, 101–117. [Google Scholar] [CrossRef]
- Lin, Y.; Nomura, T.; Cheong, J.; Dorjsuren, D.; Iida, K.; Murakami, S. Hepatitis B virus X protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J. Biol. Chem. 1997, 272, 7132–7139. [Google Scholar] [CrossRef]
- Slagle, B.; Bouchard, M. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef]
- Decorsière, A.; Mueller, H.; Breugel, P.v.; Abdul, F.; Gerossier, L.; Beran, R.; Livingston, C.; Niu, C.; Fletcher, S.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Wyżewski, Z.; Świtlik, W.; Mielcarska, M.; Gregorczyk-Zboroch, K. The Role of Bcl-xL Protein in Viral Infections. Int. J. Mol. Sci. 2021, 22, 1956. [Google Scholar] [CrossRef]
- Bock, C.; Schranz, I.; Schroder, C.; Zentgraf, H. Hepatitis B Virus Genome Is Organized into Nucleosomes in the Nucleus of the Infected Cell. Virus Genes. 1994, 8, 215–229. [Google Scholar] [CrossRef]
- Newbold, J.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J. The Covalently Closed Duplex Form of the Hepadnavirus Genome Exists In Situ as a Heterogeneous Population of Viral Minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wu, C.; Xu, Z.; Teng, Y.; Zhao, L.; Zhao, K.; Wang, J.; Wang, W.; Zhan, Q.; Zhu, C.; et al. Hepatitis B Virus Core Protein Is Not Required for Covalently Closed Circular DNA Transcriptional Regulation. J. Virol. 2022, 96, e0136222. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M. Biological Roles of Hepatitis B Viral X Protein in the Viral Replication and Hepatocarcinogenesis. Acta Med. Okayama 2023, 77, 341–345. [Google Scholar]
- Andrisani, O. Epigenetic mechanisms in hepatitis B virus-associated hepatocellular carcinoma. Hepatoma Res. 2021, 7, 12. [Google Scholar] [CrossRef]
- Dandri, M.; Petersen, J. cccDNA Maintenance in Chronic Hepatitis B—Targeting the Matrix of Viral Replication. Infect. Drug Resist. 2020, 13, 3873–3886. [Google Scholar] [CrossRef]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef]
- Alarcon, V.; Hernández, S.; Rubio, L.; Alvarez, F.; Flores, Y.; Varas-Godoy, M.; Ferrari, G.D.; Kann, M.; Villanueva, R.; Loyola, A. The enzymes LSD1 and Set1A cooperate with the viral protein HBx to establish an active hepatitis B viral chromatin state. Sci. Rep. 2016, 6, 25901. [Google Scholar] [CrossRef]
- Rivière, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.; Buendia, M.-A.; Hantz, O.; Neuveut, C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef]
- Alvarez-Astudillo, F.; Garrido, D.; Varas-Godoy, M.; Gutiérrez, J.; Villanueva, R.; Loyola, A. The histone variant H3.3 regulates the transcription of the hepatitis B virus. Ann. Hepatol. 2021, 21, 100261. [Google Scholar] [CrossRef]
- Locatelli, M.; Quivy, J.; Chapus, F.; Michelet, M.; Fresquet, J.; Maadadi, S.; Aberkane, A.; Diederichs, A.; Lucifora, J.; Rivoire, M.; et al. HIRA Supports Hepatitis B Virus Minichromosome Establishment and Transcriptional Activity in Infected Hepatocytes. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 527–551. [Google Scholar] [CrossRef]
- Mirzaei, H.; Ghorbani, S.; Khanizadeh, S.; Namdari, H.; Faghihloo, E.; Akbari, A. Histone deacetylases in virus-associated cancers. Rev. Med. Virol. 2020, 30, e2085. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Y.; Ye, S.; Wu, Q.; Lin, Y.; Sheng, K.; Chen, W.; Lin, X.; Lin, X. Chromatin remodelling factor BAF155 protects hepatitis B virus X protein (HBx) from ubiquitin-independent proteasomal degradation. Emerg. Microbes Infect. 2019, 8, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhao, L.; Yuan, Y.; Yun, H.; Zheng, W.; Geng, Y.; Yang, G.; Wang, Y.; Zhao, M.; Zhang, X. HBx represses WDR77 to enhance HBV replication by DDB1-mediated WDR77 degradation in the liver. Theranostics 2021, 11, 8362–8378. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.; Obradovic, Z.; Romero, P.; Garner, E.; Brown, C. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Ferreon, J.; Martinez-Yamout, M.; Dyson, H.; Wright, P. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 13260–13265. [Google Scholar] [CrossRef]
- Ishiyama, N.; Lee, S.; Liu, S.; Li, G.; Smith, M.; Reichardt, L.; Ikura, M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell Mol. Gastroenterol. Hepatol. 2010, 141, 117–128. [Google Scholar] [CrossRef]
- Mitrea, D.; Yoon, M.; Ou, L.; Kriwacki, R. Disorder-function relationships for the cell cycle regulatory proteins p21 and p27. Biol. Chem. 2012, 393, 259–274. [Google Scholar] [CrossRef]
- Uversky, V. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef]
- Cortese, M.; Uversky, V.; Dunker, A. Intrinsic disorder in scaffold proteins: Getting more from less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef]
- Dunker, A.; Cortese, M.; Romero, P.; Iakoucheva, L.; Uversky, V. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Chinchilla, P.; Baum, J. Multifaceted interactions mediated by intrinsically disordered regions play key roles in alpha synuclein aggregation. Curr. Opin. Struct. Biol. 2023, 80, 102579. [Google Scholar] [CrossRef] [PubMed]
- Damme, E.V.; Vanhove, J.; Severyn, B.; Verschueren, L.; Pauwels, F. The Hepatitis B Virus Interactome: A Comprehensive Overview. Front. Microbiol. 2021, 12, 724877. [Google Scholar]
- Iakoucheva, L.; Brown, C.; Lawson, J.; Obradović, Z.; Dunker, A. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Selenko, P.; Frueh, D.; Elsaesser, S.; Haas, W.; Gygi, S.; Wagner, G. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat. Struct. Mol. Biol. 2008, 15, 321–329. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.; Kurgan, L.; Uversky, V.; Longhi, S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef]
- Tokuriki, N.; Oldfield, C.; Uversky, V.; Berezovsky, I.; Tawfik, D. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef]
- Dyson, H. Vital for Viruses: Intrinsically Disordered Proteins. J. Mol. Biol. 2023, 435, 167860. [Google Scholar] [CrossRef]
- Li, H.; Yang, C.; Lo, S. Hepatitis C Viral Replication Complex. Viruses 2021, 13, 520. [Google Scholar] [CrossRef]
- Han, Z.; Alves, C.; Gudima, S.; Taylor, J. Intracellular localization of hepatitis delta virus proteins in the presence and absence of viral RNA accumulation. J. Virol. 2009, 83, 6457–6463. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Cheng, H.; Roder, H.; Taylor, J. Intrinsic disorder and oligomerization of the hepatitis delta virus antigen. Virology 2010, 407, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Gianni, S. Function, Regulation, and Dysfunction of Intrinsically Disordered Proteins. Life 2021, 11, 140. [Google Scholar] [CrossRef]
- Mishra, P.; Verma, N.; Rao, C.; Uversky, V.; Nandi, C. Intrinsically disordered proteins of viruses: Involvement in the mechanism of cell regulation and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2020, 174, 1–78. [Google Scholar]
- Metallo, S. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert. Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef]
- Kim, K. PPIases Par14/Par17 Affect HBV Replication in Multiple Ways. Viruses 2023, 15, 457. [Google Scholar] [CrossRef]
- Saeed, U.; Kim, J.; Piracha, Z.; Kwon, H.; Jung, J.; Chwae, Y.; Park, S.; Shin, H.; Kim, K. Parvulin 14 and Parvulin 17 Bind to HBx and cccDNA and Upregulate Hepatitis B Virus Replication from cccDNA to Virion in an HBx-Dependent Manner. J. Virol. 2019, 93, e01840-18. [Google Scholar] [CrossRef]
- Kirkbride, K.; Sung, B.; Sinha, S.; Weaver, A. Cortactin: A multifunctional regulator of cellular invasiveness. Cell Adhes. Migr. 2011, 5, 187–198. [Google Scholar] [CrossRef]
- Feng, H.; Tan, T.; Niu, D.; Chen, W. HBV X protein interacts with cytoskeletal signaling proteins through SH3 binding. Front Biosci. (Elite Ed.) 2010, 2, 143–150. [Google Scholar]
- Li, Y.; Fu, Y.; Hu, X.; Sun, L.; Tang, D.; Li, N.; Peng, F.; Fan, X. The HBx-CTTN interaction promotes cell proliferation and migration of hepatocellular carcinoma via CREB1. Cell Death Dis. 2019, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Obsilova, V.; Obsil, T. Structural insights into the functional roles of 14-3-3 proteins. Front. Mol. Biosci. 2022, 9, 1016071. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, S.; Ding, G.; Wang, B.; Li, Y.; Zhao, Y.; Shao, Q.; Feng, J.; Liu, S.; Qin, L.; et al. The role of 14-3-3 proteins in cell signalling pathways and virus infection. J. Cell Mol. Med. 2021, 25, 4173–4182. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, Y.; Wang, C.; Sun, Z.; Li, L.; Dong, J.; Zhou, W. 14-3-3ζ binds to hepatitis B virus protein X and maintains its protein stability in hepatocellular carcinoma cells. Cancer Med. 2018, 7, 5543–5553. [Google Scholar] [CrossRef]
- Kiliti, A.; Sharif, G.; Martin, M.; Wellstein, A.; Riegel, A. AIB1/SRC-3/NCOA3 function in estrogen receptor alpha positive breast cancer. Front. Endocrinol. 2023, 14, 1250218. [Google Scholar] [CrossRef]
- Chang, A.; Wu, H. The role of AIB1 in breast cancer. Oncol. Lett. 2012, 4, 588–594. [Google Scholar] [CrossRef]
- Hong, A.; Han, D.; Wright, C.; Burch, T.; Piper, J.; Osiowy, C.; Gao, C.; Chiang, S.; Magill, T.; Dick, K.; et al. The interaction between hepatitis B virus X protein and AIB1 oncogene is required for the activation of NFκB signal transduction. Biochem. Biophys. Res. Commun. 2012, 423, 6–12. [Google Scholar] [CrossRef]
- Liu, Y.; Tong, Z.; Li, T.; Chen, Q.; Zhuo, L.; Li, W.; Wu, R.; Yu, C. Hepatitis B virus X protein stabilizes amplified in breast cancer 1 protein and cooperates with it to promote human hepatocellular carcinoma cell invasiveness. Hepatology 2012, 56, 1015–1024. [Google Scholar] [CrossRef]
- Kanna, M.; Nakatsu, Y.; Yamamotoya, T.; Encinas, J.; Ito, H.; Okabe, T.; Asano, T.; Sakaguchi, T. Roles of peptidyl prolyl isomerase Pin1 in viral propagation. Front. Cell Dev. Biol. 2022, 10, 1005325. [Google Scholar] [CrossRef]
- Lee, Y.; Teoh, D.; Yeung, K.; Liou, Y. The kingdom of the prolyl-isomerase Pin1: The structural and functional convergence and divergence of Pin1. Front. Cell Dev. Biol. 2022, 10, 956071. [Google Scholar] [CrossRef]
- Pang, R.; Lee, T.; Poon, R.; Fan, S.; Wong, K.; Kwong, Y.; Tse, E. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 2007, 132, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Banerjee, A.; Chandra, P.; Chakravarty, R. Pin1-HBx interaction: A step toward understanding the significance of hepatitis B virus genotypes in hepatocarcinogenesis. Gastroenterology 2007, 133, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Chien, R.; Liaw, Y. Current Trend in Antiviral Therapy for Chronic Hepatitis B. Viruses 2022, 14, 434. [Google Scholar] [CrossRef]
- Songtanin, B.; Chaisrimaneepan, N.; Mendóza, R.; Nugent, K. Burden, Outcome, and Comorbidities of Extrahepatic Manifestations in Hepatitis B Virus Infections. Viruses 2024, 16, 618. [Google Scholar] [CrossRef]
- Choi, J.; Choi, W.; Lim, Y. Are the New Nucleos(t)ide Analogs Better than the Old Nucleos(t)ide Analogs? Clin. Liver Dis. 2023, 27, 809–818. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, H.; Tang, L.; Wang, F.; Tolufashe, G.; Chang, J.; Guo, J. Mechanism of interferon alpha therapy for chronic hepatitis B and potential approaches to improve its therapeutic efficacy. Antivir. Res. 2024, 221, 105782. [Google Scholar] [CrossRef]
- Lok, A. Toward a Functional Cure for Hepatitis B. Gut Liver 2024, 18, 593–601. [Google Scholar] [CrossRef]
- Wong, G.; Gane, E.; Lok, A. How to achieve functional cure of HBV: Stopping NUCs, adding interferon or new drug development? J. Hepatol. 2022, 76, 1249–1262. [Google Scholar] [CrossRef]
- Bhat, S.; Kazim, S. HBV cccDNA-A Culprit and Stumbling Block for the Hepatitis B Virus Infection: Its Presence in Hepatocytes Perplexed the Possible Mission for a Functional Cure. ACS Omega 2022, 7, 24066–24081. [Google Scholar] [CrossRef]
- Ligat, G.; Goto, K.; Verrier, E.; Baumert, T. Targeting Viral cccDNA for Cure of Chronic Hepatitis B. Curr. Hepatol. Rep. 2020, 19, 235–244. [Google Scholar] [CrossRef]
- Kumar, V. HBx protein as a therapeutic target for functional cure of hepatitis B virus infection. Virology 2025, 604, 110438. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zheng, Z.; Zhao, Q.; Zhang, R.; Zheng, H. Targeting HBV cccDNA Levels: Key to Achieving Complete Cure of Chronic Hepatitis B. Pathogens 2024, 13, 1100. [Google Scholar] [CrossRef] [PubMed]
- Martinello, M.; Naggie, S.; Rockstroh, J.; Matthews, G. Direct-Acting Antiviral Therapy for Treatment of Acute and Recent Hepatitis C Virus Infection: A Narrative Review. Clin. Infect. Dis. 2023, 77, S238–S244. [Google Scholar] [CrossRef]
- Tabata, K.; Neufeldt, C.; Bartenschlager, R. Hepatitis C Virus Replication. Cold Spring Harb. Perspect. Med. 2020, 10, a037093. [Google Scholar] [CrossRef]
- Yau, A.; Marquez-Azalgara, V.; Yoshida, E. Hepatitis C (chronic). BMJ Clin. Evid. 2015, 2015, 0921. [Google Scholar]
- Alazard-Dany, N.; Denolly, S.; Boson, B.; Cosset, F. Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses 2019, 11, 30. [Google Scholar] [CrossRef]
- Brzdęk, M.; Zarębska-Michaluk, D.; Rzymski, P.; Lorenc, B.; Kazek, A.; Tudrujek-Zdunek, M.; Janocha-Litwin, J.; Mazur, W.; Dybowska, D.; Berak, H.; et al. Changes in characteristics of patients with hepatitis C virus-related cirrhosis from the beginning of the interferon-free era. World J. Gastroenterol. 2023, 29, 2015–2033. [Google Scholar] [CrossRef]
- Liu, C.; Chang, Y.; Kao, J. Cutting-edge pharmacotherapy for hepatitis C virus infection: A comprehensive review. Expert. Opin. Pharmacother. 2024, 25, 1691–1706. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villanueva, R.A.; Loyola, A. The Intrinsically Disordered Region of HBx and Virus–Host Interactions: Uncovering New Therapeutic Approaches for HBV and Cancer. Int. J. Mol. Sci. 2025, 26, 3552. https://doi.org/10.3390/ijms26083552
Villanueva RA, Loyola A. The Intrinsically Disordered Region of HBx and Virus–Host Interactions: Uncovering New Therapeutic Approaches for HBV and Cancer. International Journal of Molecular Sciences. 2025; 26(8):3552. https://doi.org/10.3390/ijms26083552
Chicago/Turabian StyleVillanueva, Rodrigo A., and Alejandra Loyola. 2025. "The Intrinsically Disordered Region of HBx and Virus–Host Interactions: Uncovering New Therapeutic Approaches for HBV and Cancer" International Journal of Molecular Sciences 26, no. 8: 3552. https://doi.org/10.3390/ijms26083552
APA StyleVillanueva, R. A., & Loyola, A. (2025). The Intrinsically Disordered Region of HBx and Virus–Host Interactions: Uncovering New Therapeutic Approaches for HBV and Cancer. International Journal of Molecular Sciences, 26(8), 3552. https://doi.org/10.3390/ijms26083552